

Abstract
The Aurora/Ipl1 family of protein kinases plays multiple roles in mitosis and cytokinesis. Here, we describe ZM447439, a novel selective Aurora kinase inhibitor. Cells treated with ZM447439 progress through interphase, enter mitosis normally, and assemble bipolar spindles. However, chromosome alignment, segregation, and cytokinesis all fail. Despite the presence of maloriented chromosomes, ZM447439-treated cells exit mitosis with normal kinetics, indicating that the spindle checkpoint is compromised. Indeed, ZM447439 prevents mitotic arrest after exposure to paclitaxel. RNA interference experiments suggest that these phenotypes are due to inhibition of Aurora B, not Aurora A or some other kinase. In the absence of Aurora B function, kinetochore localization of the spindle checkpoint components BubR1, Mad2, and Cenp-E is diminished. Furthermore, inhibition of Aurora B kinase activity prevents the rebinding of BubR1 to metaphase kinetochores after a reduction in centromeric tension. Aurora B kinase activity is also required for phosphorylation of BubR1 on entry into mitosis. Finally, we show that BubR1 is not only required for spindle checkpoint function, but is also required for chromosome alignment. Together, these results suggest that by targeting checkpoint proteins to kinetochores, Aurora B couples chromosome alignment with anaphase onset.
Keywords: mitosis; spindle checkpoint; chemical biology; aneuploidy; ZM447439
Introduction
Accurate chromosome segregation in mitosis requires that sister kinetochores attach to microtubules emanating from opposite spindle poles. Because kinetochore attachment is a stochastic process, it is error prone and can result in chromosome malorientation (Nicklas, 1997; Rieder and Salmon, 1998). Although it is well established that a surveillance mechanism, the spindle checkpoint, delays anaphase until all chromosomes correctly bi-orient (for review see Musacchio and Hardwick, 2002), the mechanisms that detect and resolve orientation errors are poorly understood.
The Ipl1/Aurora family of protein kinases plays multiple roles in mitosis (Bischoff and Plowman, 1999; Giet and Prigent, 1999; Adams et al., 2001a; Nigg, 2001). In budding yeast, Ipl1 ensures accurate chromosome segregation by resolving syntelic orientations, possibly by monitoring tension at centromeres and destabilizing inappropriately bound microtubules (Tanaka et al., 2002). Ipl1 phosphorylates the kinetochore component Ndc10 in vitro, suggesting that it may regulate kinetochore–microtubule interactions directly (Biggins et al., 1999). However, budding yeast are atypical in that centromeres connect to the unduplicated spindle pole body (SPB) in G1. Because centromeres replicate before SPB duplication, budding yeast cells then enter mitosis with both kinetochores attached to the old SPB (Tanaka et al., 2002). In higher eukaryotes, syntelic orientations are rare during mitosis (Nicklas, 1997), and therefore, it is unclear whether the kinetochore–SPB resolving activity exhibited by Ipl1 is a universal feature of the Aurora kinase family. Further evidence suggesting that Ipl1 monitors tension at centromeres comes from analyzing budding yeast mutants that lack sister chromatid cohesion or enter mitosis without replicating their DNA. In such mutants, anaphase is prevented in an Ipl1-dependent manner (Biggins and Murray, 2001). Because kinetochores in these cells lack sisters, they fail to come under tension despite microtubule attachment, arguing that Ipl1 is required for spindle checkpoint activation in response to loss of tension at centromeres. However, budding yeast kinetochores only attach a single microtubule. Therefore, if the primary role of Ipl1 is to destabilize bound microtubules, the apparent role of Ipl1 in checkpoint activation may be simply a secondary consequence of exposing microtubule binding sites (Tanaka et al., 2002). Furthermore, although the analysis of replication and cohesion mutants suggests that Ipl1 monitors tension at centromeres, these interpretations are complicated by the fact that centromeric localization of the fission yeast Aurora kinase, Ark1, requires sister chromatid cohesion (Morishita et al., 2001). Therefore, there is a clear need to analyze Aurora kinase function under conditions where replication and cohesion are normal.
Higher eukaryotes express two or more Aurora kinases. Aurora A and C localize to spindle poles, and Aurora A is required for bipolar spindle formation in a variety of systems (Bischoff and Plowman, 1999; Giet and Prigent, 1999; Adams et al., 2001a; Nigg, 2001). Inhibition of Aurora B, an inner centromere protein, affects multiple mitotic events including histone H3 phosphorylation, chromosome segregation, and cytokinesis (Bischoff and Plowman, 1999; Giet and Prigent, 1999; Adams et al., 2001a; Nigg, 2001). The role of Aurora B kinase activity has been addressed by ectopically expressing mutants in mammalian cells. However, these studies have yielded conflicting results. In two cases, cells expressing Aurora B K109R completed mitosis, but failed to undergo cytokinesis, suggesting that Aurora B activity is not required for chromosome segregation (Tatsuka et al., 1998; Terada et al., 1998). However, another report indicates that Aurora B K109R prevents chromosome alignment due to the failure of kinetochore–microtubule interactions (Murata-Hori and Wang, 2002). These experiments are complicated by the fact that expression of wild-type Aurora B can itself affect cell division (Tatsuka et al., 1998), and therefore, it is not clear whether these phenotypes are due to reduced Aurora B kinase activity or the disruption of Aurora B protein complexes. Xenopus cells injected with anti-Aurora B antibodies exit mitosis prematurely, consistent with a role for Aurora B in the spindle checkpoint (Kallio et al., 2002). However, in contrast to Ipl1 deficient strains, mitotic exit also occurred when microtubule polymerization was inhibited, suggesting that Aurora B monitors microtubule attachment, not just tension. Thus, although many roles have been attributed to Aurora B, the emerging picture is confusing, and molecular explanations for these phenotypes are currently lacking.
Aurora A and B are overexpressed in human tumors, and ectopic overexpression in cultured cells leads to transformation, centrosome abnormalities, and aneuploidy (Bischoff et al., 1998; Tatsuka et al., 1998; Zhou et al., 1998; Adams et al., 2001b; Meraldi et al., 2002). In addition, cells overexpressing Aurora A, but not a kinase mutant, readily form tumors in nude mice (Bischoff et al., 1998). Therefore, elevated Aurora kinase activity may promote tumor evolution either by providing a growth advantage or by promoting genetic instability. To develop novel anti-cancer drugs, we have generated small molecule inhibitors of Aurora kinase activity. Here, we describe ZM447439, which selectively inhibits the kinase activity of Aurora A and B. Using ZM447439 as a research tool, we directly address the role of Aurora kinase activity in human cells. We show that inhibition of Aurora kinase activity does not prevent progression through interphase, mitotic entry, bipolar spindle formation, or kinetochore–microtubule interactions. Rather, Aurora kinase activity is required for correct chromosome alignment and spindle checkpoint function. Using RNA interference (RNAi;* Elbashir et al., 2001), we demonstrate that these phenotypes are due to inhibition of Aurora B, not Aurora A.
Results
ZM447439, a novel inhibitor of Aurora A and Aurora B
To identify novel Aurora inhibitors, ∼250,000 compounds were screened for the ability to inhibit the kinase activity of recombinant human Aurora A against an artificial peptide substrate. One inhibitor identified was further modified to produce ZM447439 (4-(4-(N-benzoylamino)anilino)-6-methoxy-7-(3-(1-morpholino)propoxy)quinazoline; Fig. 1 A). In in vitro kinase assays using purified recombinant proteins, ZM447439 inhibited Aurora A and B with IC50 values of 110 and 130 nM, respectively (Fig. 1 B). In contrast, the majority of other protein kinases assayed were not inhibited by ZM447439. Based on data from model systems, we predicted that an Aurora inhibitor should prevent cell division and inhibit phosphorylation of histone H3 on serine 10. To determine whether ZM447439 inhibits cell division, a panel of human cell lines was treated with 2 μM ZM447439 for up to 96 h (Fig. 1 C). After 18 h, the vast majority of cells in all the lines had 4N DNA contents. All the lines analyzed then endoreduplicated, accumulating cells with DNA contents greater than 4N, demonstrating that ZM447439 completely inhibits cell division. To determine whether ZM447439 inhibits phosphorylation of histone H3 on serine 10, untreated and ZM447439-treated cells were stained with an anti-phosphohistone H3 antibody (Hsu et al., 2000). In untreated cells, chromosomes stained positive for phosphohistone H3 (Fig. 1 D). However, after brief exposures to ZM447439, phosphohistone H3 was not detectable, demonstrating that ZM447439 does inhibit mitotic phosphorylation of histone H3. Consistent with previous observations (Adams et al., 2001c), the lack of histone H3 phosphorylation did not appear to affect chromosome condensation.
Figure 1.
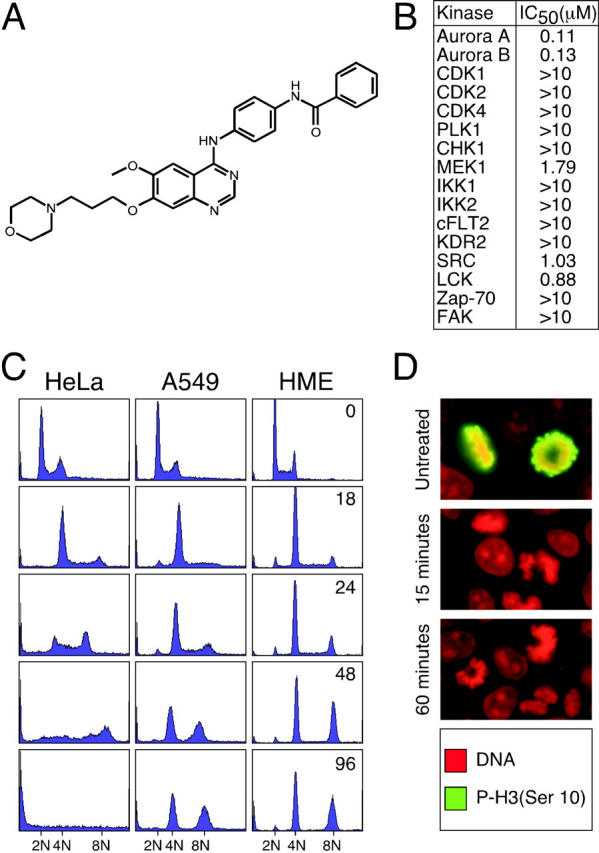
ZM447439 inhibits Aurora A and Aurora B. (A) Chemical structure of ZM447439. (B) Table showing the IC50 values (μM) of ZM447439 against a panel of protein kinases. (C) DNA content histograms of HeLa, A549, and HME cells treated with ZM447439 for the times indicated in hours. (D) Mitotic DLD-1 cells stained for phosphohistone H3 (green) and DNA (red).
ZM447439-induced endoreduplication is enhanced in the absence of p53 function
Although a significant fraction of A549 and HME cells endoreduplicated in the presence of ZM447439, after 48 h virtually all the cells arrested with either 4N or 8N DNA contents (Fig. 1 C). In contrast, HeLa cells, which lack a functional p53 response, continued DNA synthesis and rapidly lost viability. This raises the possibility that the 4N/8N arrest exhibited by the A549 and HME cells was not directly due to ZM447439, but was rather due to activation of the p53-dependent post-mitotic checkpoint that occurs after an aberrant mitosis and/or cytokinesis (Andreassen et al., 2001). Consistently, in the presence of ZM447439, U2OS cells expressing a dominant-negative p53 mutant endoreduplicated efficiently such that by 48 h, 78% had 8N DNA contents (Fig. 2 A). In contrast, only 38% of the p53-proficient parental U2OS cells had 8N DNA contents, demonstrating that p53 does restrain cell cycle progression after ZM447439-induced division failure. To test if cell cycle progression in the presence of ZM447439 may account for why the HeLa cells lost viability, we analyzed the colony-forming potential of cells after a 72-h exposure to ZM447439. MCF7 cells were selected for this experiment, as they can be arrested in G0 by treatment with anti-estrogens. Cells that were growth-arrested during exposure to ZM447439 gave rise to more colonies than those that were proliferating (Fig. 2 B). Consistently, at 1.25 and 2.5 μM ZM447439, the cloning efficiency of the proliferating cells was reduced to below 40%, whereas the cloning efficiency of the growth-arrested cells was largely unaffected (Fig. 2 C). Although continued cell cycle progression in the presence of ZM447439 leads to loss of viability, the p53-deficient U2OS cells did not appear to lose viability as rapidly as the HeLa cells. The reason for this is unclear, but it suggests that p53-independent mechanisms may also affect cell fate after ZM447439-induced tetraploidization.
Figure 2.
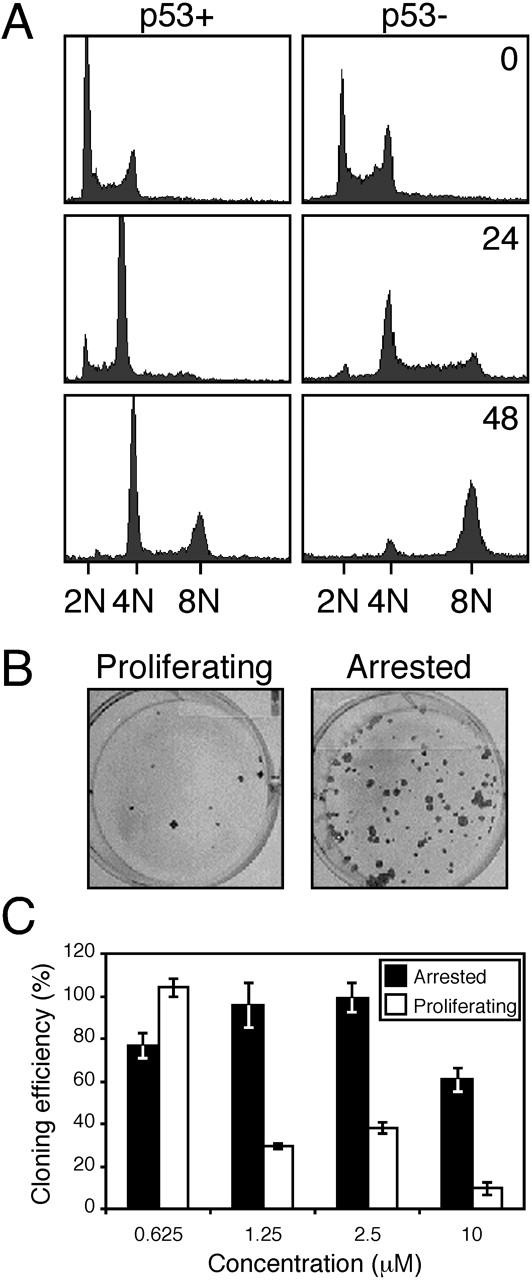
p53 restrains endoreduplication in the presence of ZM447439. (A) DNA content histograms of U20S cells with (p53+) or without (p53−) a functional p53 response treated with ZM447439 for the times indicated in hours. (B) Growth-arrested or proliferating MCF-7 cells were exposed to ZM447439 for 72 h, and were then assayed for the ability to form colonies in the absence of ZM447439. (C) Bar graph quantitating the cloning efficiency of MCF-7 cells treated with a range of ZM447439 concentrations. Each value represents the mean and SD from three independent experiments.
ZM447439-treated cells enter mitosis, but fail to divide
The observation that ZM447439-treated cells can enter additional S phases without dividing raises two possibilities: either the cells re-replicate their genomes without entering mitosis, or alternatively, the cells enter and exit mitosis without dividing and then enter a second S phase. To distinguish between these two possibilities, we analyzed HeLa cells after release from a G1/S block into fresh medium or media supplemented with either ZM447439 or nocodazole, a spindle toxin that prevents microtubule polymerization (Fig. 3) . At various times after G1/S, the cells were harvested to determine DNA content, mitotic index, and cyclin B1 levels. DNA content histograms show that the vast majority of control cells divided by 12 h then entered a second S phase such that by 18 h, the majority had DNA contents greater than 2N (Fig. 3 A). Consistently, the mitotic index peaked at 10 h and cyclin B1 levels decreased as the cells completed mitosis (Fig. 3, B and C). Nocodazole-treated cells entered mitosis, and then remained arrested with high cyclin B1 levels for the remainder of the experiment. Cells released into ZM447439 progressed through S phase, failed to divide, degraded cyclin B1 normally, and then entered a second S phase (Fig. 3, A and C). Significantly, the kinetics with which the mitotic index increased and decreased was very similar to the control culture (Fig. 3 B). Thus, in the presence of ZM447439, HeLa cells enter and exit mitosis normally, but fail to divide. A549 and HME cells exhibited a similar response (unpublished data).
Figure 3.

ZM447439-treated cells enter mitosis, but fail to divide. HeLa cells were released from G1/S into various drug combinations, harvested at the times indicated in hours, and then analyzed by flow cytometry and immunoblotting. The data shown are representative of three independent experiments. The data in A and B are from the same experiment where the mitotic index peaked 10 h after release from G1/S. The data in C are from an independent experiment where the mitotic index peaked 12 h after release from G1/S. (A) DNA content histograms showing that ZM447439-treated cells fail to divide, but re-replicate their genomes with similar kinetics to the control cells. (B) Graph plotting mitotic index, as determined by MPM-2 reactivity, against time showing that ZM447439-treated cells enter and exit mitosis with similar kinetics to the controls. (C) Immunoblots showing that cyclin B1 levels rise and fall in ZM447439-treated cells with similar kinetics to the controls. Tubulin is used as a loading control.
ZM447439 inhibits chromosome alignment
The observation that ZM447439-treated cells enter and exit mitosis but fail to divide raises two possibilities: either chromosome segregation takes place normally but cytokinesis fails, or alternatively, chromosome segregation fails, preventing cytokinesis. To determine whether ZM447439 prevents chromosome segregation, we analyzed spindle morphology in ZM447439-treated cultures. In controls, prometaphase rosettes, and metaphase and anaphase spindles were readily apparent (Fig. 4, A and B) . In ZM447439-treated cultures, bipolar spindles were observed and the proportion of cells in prophase appeared normal. However, the proportion of metaphase and anaphase spindles was markedly reduced, indicating that ZM447439 inhibits chromosome segregation (Fig. 4 B). Despite the lack of anaphases, cells treated with ZM447439 alone did not accumulate in mitosis (Fig. 4 C). Cells treated with ZM447439 and nocodazole did however accumulate in mitosis, confirming that ZM447439-treated cells enter and exit mitosis without undergoing chromosome segregation. Chromosome alignment also appeared abnormal in the presence of ZM447439. In particular, the chromosomes either splayed out throughout the cell or, rather than aligning at the spindle equator, lined up along the length of the spindle (Fig. 4 A). In these latter cases, the kinetochores were oriented toward the spindle, suggesting that kinetochore–microtubule interactions were taking place. Indeed, kinetochores and kinetochore fibers were observed in thin sections analyzed by electron microscopy (Fig. 4 D). In 19 control cells, fibers containing multiple microtubules were observed in 12 cases. Eight of these could be traced to kinetochores. In 20 ZM447439-treated cells, fibers containing multiple microtubules were observed in 15 cases, and six were traced to kinetochores. Although we cannot rule out the possibility that ZM447439 effects kinetochore structure and/or microtubule binding capacity, ZM447439 clearly does not prevent kinetochore–microtubule interactions or bipolar spindle formation. However, ZM447439 does inhibit chromosome alignment and segregation. To determine whether ZM447439 prevents sister chromatid separation, we allowed cells to pass through mitosis in the presence of ZM447439, and then analyzed chromosome spreads in the following mitosis. Diplochromosomes were never observed (unpublished data), suggesting that ZM447439 does not prevent loss of sister chromatid cohesion. It is unclear whether ZM447439 directly inhibits cytokinesis or whether cytokinesis is prevented as a secondary consequence of the block to chromosome segregation.
Figure 4.
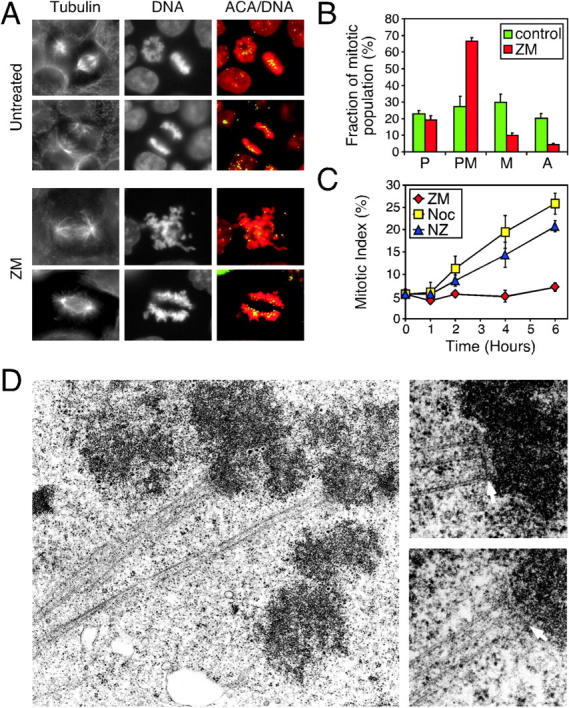
ZM447439 prevents chromosome alignment and segregation. (A) Mitotic DLD-1 cells stained for tubulin, kinetochores/centromeres (ACA, green), and DNA (red). Bottom panels show examples of abnormal prometaphases frequently observed after treatment with ZM447439 for 1 h. (B) Graph quantitating the number of prophase (P), prometaphase (PM), metaphase (M), and anaphase (A) cells after exposure of DLD-1 cells to ZM447439 for 1 h, showing that chromosome alignment and segregation are inhibited. (C) Graph showing the mitotic index of DLD-1 cultures at the times indicated after treatment with either ZM447439 alone (red diamonds), nocodazole (yellow squares), or ZM447439 plus nocodazole (blue triangles). Values in B and C represent the mean and SEM derived from three independent experiments in which at least 1,000 cells were counted. (D) Electron micrographs of ZM447439-treated HeLa cells showing kinetochores (arrows) and kinetochore fibers.
ZM447439 compromises spindle checkpoint function
The observations described above present a paradox: chromosome alignment defects should activate the spindle checkpoint, and thus cause mitotic arrest. However, ZM447439-treated cells exit mitosis with normal kinetics (Fig. 3 B), suggesting that ZM447439 may compromise checkpoint function. To test this possibility, we analyzed the effect of ZM447439 on cells after release from a nocodazole block. Nocodazole-arrested mitotic cells were isolated by selective detachment, washed to remove the nocodazole, and were then replated in various drug combinations (Fig. 5 , A–C). At various times the cells were harvested to determine DNA content, mitotic index, and cyclin B1 levels. 2 h after release, the majority of the control cells had completed chromosome segregation and divided (Fig. 5 A). In contrast, ZM447439, nocodazole, and ZM447439 plus nocodazole-treated cells failed to divide (unpublished data). The mitotic index of the control cells was initially high, but then fell to 15% 2 h after release. In contrast, in the continued presence of nocodazole the cells remained arrested in mitosis (Fig. 5 B). Consistently, in control cells, cyclin B1 levels fell after 2 h but remained high in the presence of nocodazole (Fig. 5 C). Strikingly, the mitotic index of ZM447439-treated cells fell extremely rapidly: after 1 h, only 4% of the ZM447439-treated cells were still in mitosis, compared with 67% of the controls (Fig. 5 B). Consistently, cyclin B1 was undetectable after 1 h. Thus, ZM447439-treated cells rapidly exit mitosis, consistent with the notion that ZM447439 overrides the spindle checkpoint.
Figure 5.

ZM447439 compromises spindle checkpoint function. (A–C) Mitotic HeLa cells were isolated by selective detachment after a 12-h nocodazole block, replated in various drugs combinations, and then harvested at the times indicated in hours and analyzed. (A) DNA content histograms showing that the control cells divide after 1 h. (B) Graph plotting the mitotic index, as determined by MPM-2 reactivity, showing that ZM447439 accelerates mitotic exit. (C) Immunoblots showing that cyclin B1 and phosphohistone H3 levels fall rapidly in ZM447439-treated cells. (D and E) DLD-1 cells were treated with drug combinations and were then fixed and analyzed by fluorescence microscopy. (D) Bar graph plotting the mitotic index after 6 h, showing that ZM447439 compromises paclitaxel- but not nocodazole-induced mitotic arrest. (E) Line graph plotting the mitotic index after 8 h, showing that ZM447439 resolves the effects of nocodazole and paclitaxel with respect to spindle checkpoint function. Values in D and E represent the mean and SEM derived from three independent experiments in which at least 1,000 cells were counted. Drug combinations used were nocodazole alone (Noc, yellow squares); paclitaxel alone (Tax); ZM447439 alone (ZM, red diamonds); nocodazole plus ZM447439 (NZ, blue triangles); paclitaxel plus ZM447439 (TZ, green squares); and no drug (Control, green circles). (F) Mitotic HeLa cells harvested after a 2- or 12-h nocodazole block were replated in nocodazole (black bar) or nocodazole plus ZM447439 (white bars), and the mitotic index was determined by measuring MPM-2 reactivity after 4 h. Bar graph shows that ZM447439 compromises nocodazole-induced checkpoint activation after prolonged mitotic arrest.
However, asynchronous cultures treated with ZM447439 and nocodazole accumulated cells in mitosis (Fig. 4 C), indicating that ZM447439 does not override the checkpoint under all circumstances. To explain this, we reasoned that perhaps ZM447439 only compromises the checkpoint when microtubules are allowed to polymerize. To test this, we analyzed the effect of ZM447439 on the accumulation of mitotic cells in response to paclitaxel, a spindle toxin that activates the checkpoint by stabilizing microtubules. In the presence of paclitaxel, nocodazole, or nocodazole plus ZM447439, the mitotic index reached ∼25% after 6 h (Fig. 5 D). In contrast, the mitotic index of the ZM447439-treated culture remained low at ∼7%. Significantly, the mitotic index of the culture treated with paclitaxel plus ZM447439 reached only 12%. To confirm this differential effect, we determined the mitotic index of nocodazole- and paclitaxel-treated cultures over a range of ZM447439 concentrations. Fig. 5 E clearly shows that ZM447439 resolves the pharmacological effects nocodazole and paclitaxel have on checkpoint function. Thus, these observations confirm that ZM447439 can efficiently override the spindle checkpoint when microtubules are allowed to polymerize.
Although asynchronous cultures treated with ZM447439 and nocodazole accumulate mitotic cells in a manner similar to cultures treated with nocodazole alone (Fig. 4 C), after a 12-h nocodazole block, mitotic cells released into nocodazole plus ZM447439 exit mitosis faster than cells released into nocodazole alone (Fig. 5 B). This suggests that perhaps after prolonged mitotic arrest, ZM447439 can also compromise the checkpoint induced by microtubule depolymerization. To test this, mitotic HeLa cells were harvested after a 2- or 12-h nocodazole block, then replated for 4 h in either nocodazole alone or nocodazole plus ZM447439. Although ∼80% of the cells harvested after a 2-h block remained arrested in the presence of ZM447439 and nocodazole, only ∼20% of the cells isolated after a 12-h block remained arrested. Thus, after prolonged mitotic arrest, ZM447439 can compromise checkpoint arrest induced by microtubule depolymerization. However, what is clearly evident from this analysis is that in the short term, ZM447439-treated cells do undergo mitotic arrest when microtubules are depolymerized, but fail to arrest when microtubules are stabilized (Fig. 5 E).
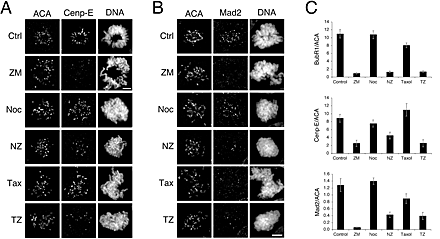
ZM447439 inhibits kinetochore localization of BubR1, Mad2, and Cenp-E
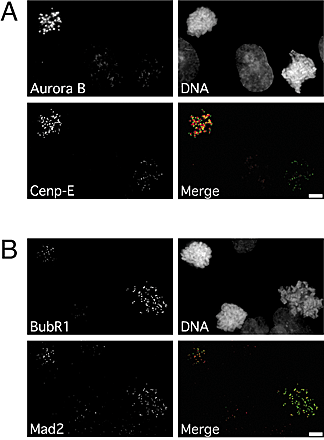
To gain insight into how ZM447439 compromises chromosome alignment and checkpoint function, we analyzed its effect on the localization of Aurora A, Aurora B, Survivin, the spindle checkpoint components BubR1 and Mad2, and the kinesin-related motor protein Cenp-E. ZM447439 did not prevent localization of Aurora A to spindle poles (unpublished data) or the localization of Aurora B and Survivin to centromeres (Fig. 6 A). However, ZM447439 did reduce kinetochore bound BubR1, Cenp-E, and Mad2 (Fig. 7 A and Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200208091/DC1). Quantitation of pixel intensities shows that ZM447439 reduced kinetochore-associated BubR1 to ∼10%, both in the presence and absence of microtubule toxins (Fig. 7 B, Fig. S1, and Table SI). ZM447439 reduced kinetochore-bound Mad2 to <10% in prometaphase cells and to 30 and 43% in the presence of nocodazole and paclitaxel, respectively (Fig. S1 C). Kinetochore-bound Cenp-E was reduced to 28 and 23% in cells treated with ZM447439 and ZM447439 plus paclitaxel, respectively. However, in the presence of nocodazole and ZM447439, Cenp-E was only reduced to 59%. Interestingly, inspection of the fluorescence intensity histograms (Fig. S2) shows that in the presence of nocodazole, although ZM447439 reduced kinetochore-bound Cenp-E at the majority of kinetochores, a significant number retained normal Cenp-E levels (30% have a Cenp-E/ACA ratio greater than the mean value for kinetochores in cells treated with nocodazole only). In contrast, no kinetochores had normal BubR1 levels in the presence of ZM447439 and nocodazole (0% have a BubR1/ACA ratio greater than the mean nocodazole only value).
Figure 6.
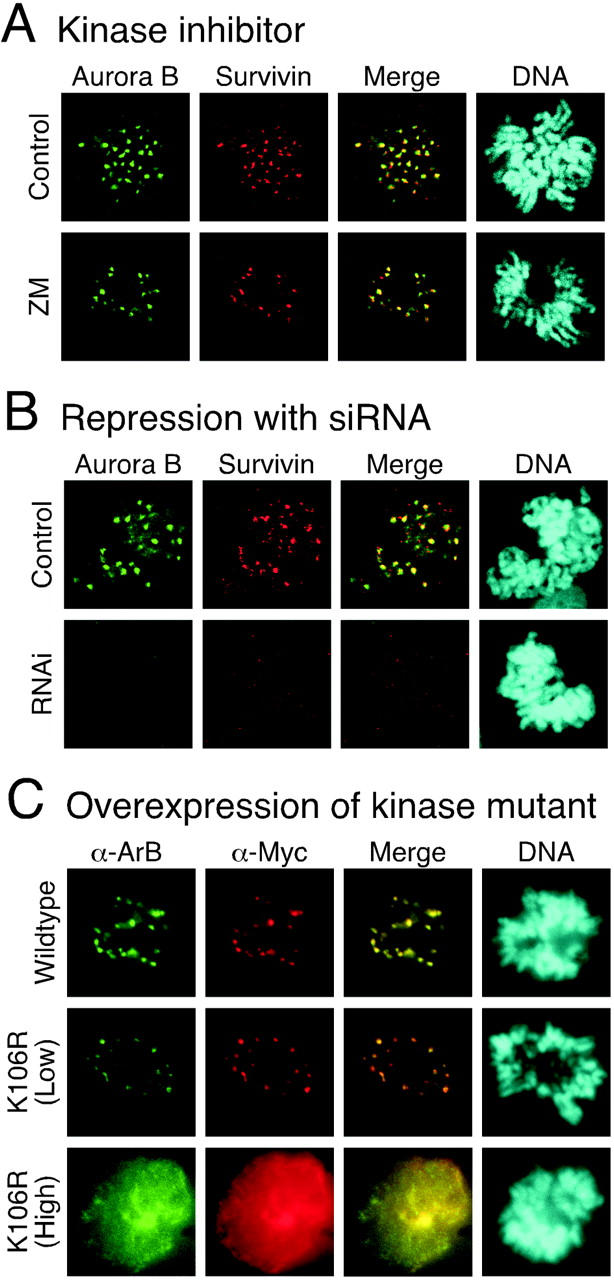
ZM447439 does not prevent localization of Aurora B to centromeres. Immunofluorescence images of prometaphase DLD-1 cells stained to detect Aurora B (green), Survivin (red), and DNA (blue) after (A) exposure to ZM447439 for 1 h or (B) transfection of Aurora B siRNA duplexes. (C) Transiently transfected HeLa cells expressing either Myc-tagged Aurora B or Myc-tagged Aurora B K106R stained to detect Aurora B (green), Myc-tagged proteins (red), and DNA (blue).
Figure 7.

ZM447439 inhibits kinetochore localization and phosphorylation of BubR1. (A) Projections of deconvolved image stacks showing mitotic DLD-1 cells treated for 1 h with the drugs indicated and then stained to detect kinetochores/centromeres (ACA), BubR1, and DNA. (B) Bar graph plotting the fluorescence ratio of BubR1 to ACA signal under various conditions, showing that ZM447439 reduces the BubR1 signal by ∼10-fold. Values represent the mean and SEM of at least 26 different kinetochore/centromere pairs analyzed in at least three different cells. (C) Plot of BubR1 signal intensity versus interkinetochore distance at metaphase kinetochores in untreated cells (green circles) or cells exposed to ZM447439 (red diamonds) or paclitaxel (blue squares) for 40 min. ACA foci were used to determine kinetochore position. The ovals encompass at least 75% of the data points. (D) Mitotic HeLa cells collected by selective detachment after release from G1/S into the drugs indicated were analyzed by immunoblotting. In the presence of ZM447439, the phosphorylated form of BubR1 is not detectable. (E) BubR1 was affinity purified form nocodazole-arrested mitotic HeLa cells and assayed for kinase activity in the presence ZM447439. Each value represents the mean and SEM derived from three independent experiments.
Although kinetochore-bound BubR1 diminishes after chromosome alignment (Chan et al., 1999; Taylor et al., 2001), it is recruited back to metaphase kinetochores after the loss of tension induced by low levels of vinblastine (Skoufias et al., 2001). To determine whether ZM447439 inhibits the reassociation of BubR1 with aligned kinetochores after loss of tension, we measured BubR1 signal intensity and interkinetochore distance at metaphase kinetochores after a 40-min treatment with either ZM447439 or paclitaxel (Fig. 7 C and Table SII). After paclitaxel treatment, the mean interkinetochore distance was reduced from 0.89 ± 0.20 μm to 0.58 ± 0.15 μm (P < 0.001), and the mean BubR1/ACA fluorescence ratio increased from 2.23 ± 1.21 to 3.86 ± 2.41 (P < 0.001). In contrast, although exposure to ZM447439 reduced the mean interkinetochore distance to 0.55 ± 0.14 μm, the BubR1/ACA fluorescence ratio decreased to 1.11 ± 0 0.88 μm, which is statistically different from the paclitaxel-treated and control cells (P < 0.001).
BubR1 is phosphorylated in response to spindle damage (Chan et al., 1999; Taylor et al., 2001). To determine whether ZM447439 prevents BubR1 phosphorylation, mitotic cells were harvested ∼9.5 h after release from a G1/S block into various drug combinations. In the presence of ZM447439, the phosphorylated form of BubR1 was not detectable, either in the presence or absence of nocodazole (Fig. 7 D). To rule out the possibility that ZM447439 inhibits BubR1 directly, BubR1 immunoprecipitates were assayed for kinase activity in the presence and absence of ZM447439. Although ZM447439 inhibited Aurora A, it did not inhibit BubR1 (Fig. 7 E).
Repression of Aurora B inhibits kinetochore localization of BubR1, Cenp-E, and Mad2
The ZM447439 data suggest that Aurora kinase activity is required for chromosome alignment and spindle checkpoint function. To determine which Aurora is required for these functions, and to rule out the possibility that the ZM447439 phenotypes might be due to inhibition of another kinase, we repressed Aurora A and B by RNAi (Fig. 8 A). Although control and Aurora A RNAi cultures had robust spindle checkpoints, repression of Aurora B reduced the accumulation of mitotic cells after spindle damage (Fig. 8 B). Repression of Aurora B (but not Aurora A) inhibited kinetochore localization of BubR1, Cenp-E, and Mad2 (Fig. 8 C and Fig. S3). Thus, these observations indicate that the ZM447439-induced phenotypes described above are due to inhibition of Aurora B, not Aurora A or some other kinase.
Figure 8.

Repression of Aurora B prevents kinetochore localization of BubR1. HeLa and DLD-1 cells were transfected with siRNA duplexes to repress either Aurora A or B. (A) Immunoblots of HeLa cell lysates showing repression of Aurora A and B. (B) Transfected DLD-1 cells were exposed to spindle toxins, and the mitotic index was measured over time. Values represent the mean and SEM from three independent experiments in which at least 1,000 cells were counted. (C) Projected deconvolved image stacks of nocodazole-treated mitotic DLD-1 cells after transfection of siRNAs targeting Aurora A (top) and Aurora B (bottom). (D) Examples of spindle morphology in control and Aurora B RNAi cultures. (E) Plot of interkinetochore distance in control and Aurora B–repressed cells.
Overexpression of Aurora B K106R mislocalizes the endogenous protein
The Aurora B RNAi phenotypes appear more severe than those induced by ZM447439. In particular, Aurora B RNAi compromised checkpoint arrest in response to nocodazole as well as paclitaxel (Fig. 8 B). Furthermore, the chromosomes were frequently adjacent to the spindle (Fig. 8 D), suggesting the absence of kinetochore–microtubule interactions. Accordingly, the mean interkinetochore distance was markedly reduced (Fig. 8 E). One possible explanation for these differences is that in addition to its catalytic role, Aurora B may also play a structural role at centromeres. Significantly, Aurora B, INCENP, and Survivin form a complex that is required for multiple aspects of mitosis and cytokinesis (Adams et al., 2001a). Disruption of this complex may have more extensive consequences rather than simply inhibiting Aurora B kinase activity. Consistently, although Survivin localizes to centromeres after ZM447439 treatment (Fig. 6 A), it does not after Aurora B RNAi (Fig. 6 B). If the above argument is valid, ectopic expression of an Aurora B kinase mutant should produce a phenotype similar to ZM447439. However, overexpression of Aurora B K109R abolished kinetochore–microtubule interactions (Murata-Hori and Wang, 2002), resulting in a phenotype that is more consistent with our RNAi data. Therefore, we tested whether Aurora B localized to centromeres after overexpression of the Aurora B kinase mutant. After transient transfections of HeLa cells, the exogenous wild-type Aurora B localized to centromeres in prometaphase (Fig. 6 C) and midbodies after cell division (unpublished data). In addition, at low levels of expression, the Aurora B K106R mutant also localized to centromeres (note that human Aurora B K106R mutant is equivalent to rat K109R mutant described previously in Murata-Hori and Wang [2002]). However, in prometaphase cells expressing moderate to high levels of the K106R mutant, Aurora B was not apparent at centromeres (Fig. 6 C). Thus, in contrast to treatment with the Aurora kinase inhibitor, overexpression of an Aurora B kinase mutant prevents localization of the endogenous protein to centromeres.
BubR1 is required for chromosome alignment
Our observations are consistent with the notion that Aurora B kinase activity regulates the spindle checkpoint, at least in part, by targeting BubR1 to kinetochores. Because BubR1 binds Cenp-E (Chan et al., 1999; Yao et al., 2000), we reasoned that the requirement for Aurora B in promoting correct chromosome alignment might also be mediated, at least in part, via its affect on BubR1. To test this, we used RNAi to determine whether repression of BubR1 inhibited chromosome alignment. Consistent with antibody injection experiments (Chan et al., 1999), repression of BubR1 compromised spindle checkpoint function. In particular, in asynchronous cultures, the number of metaphases was reduced and the anaphases frequently displayed lagging chromosomes (Fig. S4). Furthermore, BubR1 RNAi cultures did not accumulate mitotic cells on exposure to spindle toxins (unpublished data). Strikingly, prometaphase cells in BubR1 RNAi cultures often appeared abnormal with the chromosomes aligned along the length of the spindle rather than at the metaphase plate (Fig. 9 A). Although these chromosomes appear to be attached to the spindle, the mean interkinetochore distance was reduced compared with control cells (Fig. 9 B), consistent with a reduction in pulling forces.
Figure 9.

Repression of BubR1 prevents chromosome alignment. DLD-1 cells were transfected with siRNA duplexes to repress BubR1 and were then stained to detect Bub1 (green), tubulin (red), and DNA (blue). (A) Examples of abnormal prometaphase cells with chromosomes aligned along the length of the spindle rather than along the metaphase plate. Bar graph shows that ∼40% of prometaphases appear abnormal with chromosomes aligned along the length of the spindle. (B) Plot of interkinetochore distances measured using Bub1 foci to determine kinetochore position. (C) Proportion of mitotic cells exhibiting either prometaphase or metaphase chromosome alignment after a 1-h exposure to MG132. Values represent the mean and SEM derived from three independent experiments in which at least 100 mitotic cells were counted.
To rule out the possibility that this alignment defect was simply due to the cells prematurely entering anaphase, we treated control and BubR1 RNAi cultures with the proteosome inhibitor MG132 to prevent anaphase onset. Over a 3-h time course, the number of prophases and anaphases fell to almost zero in both control and BubR1 RNAi cultures (Fig. S4). In control cultures, the proportion of prometaphases remained roughly constant at ∼25% and the proportion of metaphases rose to ∼66% (Fig. 9 C), consistent with MG132 preventing the metaphase–anaphase transition. However, in BubR1-repressed cultures, the proportion of prometaphases rose from ∼32 to ∼49%, and the number of metaphases reached only ∼44% (Fig. 9 B). Thus, when the metaphase–anaphase transition is blocked downstream of spindle checkpoint activation, repression of BubR1 reduces the accumulation of cells in metaphase, suggesting that BubR1 is indeed required for chromosome alignment.
Discussion
ZM447439 is a novel specific inhibitor of Aurora kinase activity
The usefulness of protein kinase inhibitors as therapeutic agents and experimental research tools depends on the selectivity of the inhibitor. ZM447439 inhibits Aurora A and B in vitro with IC50 values in the 100 nM range. In contrast, when tested against a panel of 14 other protein kinases of diverse structural types, 11, including the mitotic kinases CDK1 and PLK1, were inhibited with IC50 values >10 μM. Although MEK1 and SRC, which have been implicated in mitotic progression, were inhibited in the 1 μM range, the phenotypes described here are unlikely to be due to inhibition of these enzymes because, in contrast to ZM447439, selective SRC and MEK1 inhibitors delay G2/M progression (Moasser et al., 1999; Wright et al., 1999). Insight into the mechanism underlying the selectivity of ZM447439 comes from the crystal structure of Aurora A complexed with a related inhibitor (Andrew Pannifer and Richard Pauptit, AstraZeneca Pharmaceuticals, personal communication). This shows that the inhibitor occupies the ATP binding pocket and an adjacent cleft that is not present in other kinases. The sequence homology between the Aurora kinases (Bischoff and Plowman, 1999) suggests that ZM447439 probably inhibits Aurora B, and possibly Aurora C, in a similar manner. Although we cannot rule out the possibility that ZM447439 inhibits other protein kinases not tested, the RNAi data (Fig. 8) provide compelling evidence that the phenotypes described here are due to inhibition of Aurora B, not Aurora A or any other protein kinase. Aurora A is required for bipolar spindle formation in Drosophila embryos and Xenopus egg extracts and centrosome maturation in C. elegans embryos (Hannak et al., 2001). Although we have not tested the effect of ZM447439 on centrosome maturation, bipolar spindles do form in the presence of ZM447439. However, bipolar spindles also form when Aurora A is repressed by RNAi (unpublished data), suggesting that perhaps Aurora A is not required for spindle bipolarity in human somatic cells.
Aurora B kinase activity regulates chromosome alignment and the spindle checkpoint
Consistent with previous reports (Adams et al., 2001c; Kallio et al., 2002; Murata-Hori and Wang, 2002), our observations show that Aurora B is required for both spindle checkpoint function and metaphase chromosome alignment in human cells. By using a novel selective protein kinase inhibitor, we have been able to directly address, for the first time, the requirement for Aurora B kinase activity in these processes. Because the phenotypes derived from protein repression and overexpression appear more extensive than those induced by ZM447439, our data demonstrate the usefulness of small molecule inhibitors in dissecting complex cellular processes. Indeed, kinetochore fibers form in the presence of ZM447439, suggesting that Aurora B kinase activity is not required for kinetochore–microtubule interactions, but rather regulates these interactions to promote correct chromosome alignment. Such a role for Aurora B is entirely consistent with the role Ipl1 plays in budding yeast: Ipl1 is not required for the attachment of chromosomes to the spindle, but rather resolves inappropriate kinetochore–microtubule interactions to ensure correct bi-orientation (Tanaka et al., 2002).
Ipl1 has also been implicated in spindle checkpoint function (Biggins and Murray, 2001) as well as chromosome alignment. Because Aurora B also promotes chromosome alignment, is it possible that its role in checkpoint activation is a secondary consequence of generating unattached kinetochores, as has been argued for Ipl1 (Tanaka et al., 2002)? If this were the case, we would predict that in the absence of Aurora B kinase activity, BubR1 and Mad2 should localize to kinetochores that lack bound microtubules. However, in the presence of nocodazole and ZM447439, localization of both BubR1 and Mad2 to kinetochores is severely reduced. Thus, in human cells at least, Aurora B kinase activity does appear to be directly required for checkpoint function.
The most striking observation in this work is the differential effect that ZM447439 has on nocodazole- and paclitaxel-induced mitotic arrest. Below, we consider two possible explanations for this observation, one of which is qualitative in nature, the other quantitative. The “qualitative” interpretation is that the checkpoint must be composed of multiple pathways, one of which does not require Aurora B kinase activity. If this is the case, then Aurora B kinase activity is not essential for checkpoint activation in response to loss of kinetochore–microtubule interactions and/or spindle destruction, but is essential under conditions where kinetochores can capture microtubules, but not correctly align on the spindle. This would indicate that, like Ipl1 in budding yeast (Biggins and Murray, 2001), Aurora B does not directly monitor kinetochore–microtubule interactions, but rather activates the checkpoint in response to loss of tension at centromeres.
An alternative “quantitative” explanation is that Aurora B simply targets BubR1 and Mad2 to kinetochores irrespective of tension. Although ZM447439 clearly reduces kinetochore-associated BubR1 and Mad2 (by ∼90 and ∼70%, respectively), perhaps the residual bound protein is sufficient to sustain mitotic arrest in the absence of kinetochore–microtubule interactions. If microtubule occupancy is sufficient to then inactivate the remaining bound protein, this may explain why cells cannot arrest in the presence of ZM447439 and paclitaxel. Interestingly, kinetochore-bound Cenp-E is almost twofold higher in cells treated with ZM447439 and nocodazole compared with cells treated with ZM447439 plus paclitaxel. Because Cenp-E is thought to activate the checkpoint via BubR1 (Chan et al., 1999; Yao et al., 2000), perhaps this elevated level of Cenp-E is sufficient to activate the residual BubR1 and thus maintain mitotic arrest in the presence of nocodazole.
At present, it is difficult to imagine how one could distinguish between these two possibilities. Indeed, whether the spindle checkpoint monitors tension, microtubule attachment, or both remains unsolved (Musacchio and Hardwick, 2002). Although evidence from budding yeast suggests that Ipl1 does activate the checkpoint in response to the lack of tension (Biggins and Murray, 2001), there is compelling evidence to argue that the checkpoint in mammalian somatic cells only monitors microtubule attachment (Rieder et al., 1995). Yet mammalian kinetochores are clearly sensitive to changes in tension (Waters et al., 1998), and in particular BubR1, but not Mad2, is recruited to aligned kinetochores after loss of tension (Skoufias et al., 2001; Shannon et al., 2002). However, it is possible that tension and attachment are not separable events in terms of checkpoint function. Indeed, it was shown many years ago that tension stabilizes microtubule attachment (Nicklas and Koch, 1969). Furthermore, our observation showing that BubR1 plays a dual role in congression and checkpoint function indicates that the mechanisms which regulate and monitor chromosome alignment are interweaved at the molecular level.
Aurora inhibitors as potential therapeutic agents
The Auroras represent a new family of protein kinases with oncogenic potential (Bischoff et al., 1998; Tatsuka et al., 1998; Zhou et al., 1998; Adams et al., 2001a). Our observations show that it is possible to selectively inhibit Aurora kinase activity in cells with a small molecule inhibitor. Furthermore, we have shown that relative to cells with a functional p53 response, p53-deficient cells are more likely to continue cell cycle progression in the presence of ZM447439. In addition, cycling cells rapidly lose viability in the presence of ZM447439, whereas nondividing cells retain viability. Together, these observations suggest that Aurora kinase inhibitors may be selectively toxic to proliferating tumor cells, and therefore open up new opportunities to develop novel anti-cancer agents.
Materials and methods
In vitro kinase assays
Recombinant Aurora A and B were expressed as NH2-terminal His6-tagged fusion proteins using a baculovirus expression system (FastBac™; GIBCO BRL) according to the manufacturer's instructions. Aurora A was purified by affinity chromatography using Ni-NTA agarose, and Aurora B was purified by ion exchange chromatography using CM Sepharose Fast Flow (Amersham Biosciences). 1 ng purified recombinant enzyme was added to a reaction cocktail containing 25 mM Tris-HCl, pH 7.5, 12.5 mM KCl, 2.5 mM NaF, 0.6 mM DTT, 6.25 mM MnCl2, 10 μM peptide substrate (Biotinyl-Ahx-tetra (LRRWSLG); Bachem), 10 μM for Aurora A or 5 μM ATP for Aurora B, and 0.2 μCi γ[33P]ATP (specific activity ≥2,500 Ci/mmol; Amersham Biosciences), and was then incubated at RT for 60 min. Reactions were stopped by addition of 20% phosphoric acid, and the products were captured on P30 nitrocellulose filters (Whatman) and assayed for incorporation of 33P with a Betaplate™ counter (Wallac). No enzyme and no compound control values were used to determine the concentration of ZM447439, which gave 50% inhibition of enzyme activity. Further details are available on request from Nicholas Keen (AstraZeneca Pharmaceuticals, Cheshire, UK).
Cell culture
A549, MCF-7, and DLD-1 cells (American Type Culture Collection), TA-HeLa cells (Taylor and McKeon, 1997), U2OS cells stably transfected with pCMVp53 143A, and parental U2OS cells (both from Karen Vousden, Beatson Institute for Cancer Research, Glasgow, UK) were all cultured as described previously (Taylor et al., 2001). Nontransformed human mammary epithelial cells expressing hTERT (CLONTECH Laboratories, Inc.) were cultured in MEGM (Clonetics). Nocodazole, paclitaxel, and thymidine were used as described previously (Taylor et al., 2001). MG132 (Calbiochem) was dissolved in DMSO and used at a final concentration of 20 μM. ZM447439 was dissolved in DMSO at 10 mM and stored at −20°C for up to 9 mo in individual aliquots to avoid freeze-thaw cycles, and was then freshly diluted in media. The IC50 values for Aurora A and B (∼100 nM) were determined at the Km for ATP (see above). However, because the cellular ATP concentration is ∼200-fold higher, and because ZM447439 is an ATP competitor, we used ZM447439 at a concentration of 2 μM in all the cell assays unless stated otherwise. DMSO was added to drug-free cultures to account for the solvent.
Cell cycle analysis and cloning assays
DNA content and mitotic index measurements and synchronization of TA-HeLa cells at G1/S using a double thymidine block were done as described previously (Taylor and McKeon, 1997). To determine cloning efficiency, MCF7 cells were plated in phenol red free DME plus 5% stripped serum (HyClone), and were then treated with or without the anti-estrogen ICI 182780 at 1 μM for 48 h. ZM447439 was then added at the indicated concentrations for 72 h. The cells were harvested, washed, and ∼400 cells plated in each well of a 6-well plate in complete media without ZM447439. After 10 d, the colonies were fixed, stained with crystal violet, and counted. The cloning efficiency represents the number of colonies on ZM447439-treated plates compared with DMSO-treated controls.
Antibody techniques
Immunofluorescence, immunoblotting, and immunoprecipitations were all done as described previously (Taylor et al., 2001) using antibodies against the following: phosphohistone H3 (Upstate Biotechnology); cyclin B1 (Upstate Biotechnology); tubulin (TAT1); centromere/kinetochores (human ACA); BubR1 (SBR1.1); Bub1 (4B12); Aurora A (RAA.1); and Myc-tag (9E10). Aurora B was detected using either the anti-AIM-1 mouse mAb (Transduction Laboratories) or a sheep anti–human Aurora B pAb (unpublished data). For localization of Mad2 and Survivin, we used DLD-1 cell lines stably expressing Myc-tagged hMad2 or hSurvivin ORFs (unpublished data). For IP kinase assays, beads were equilibrated in kinase buffer (10 mM Tris, pH7.5, 5 mM KCl, 1 mM NaF, 0.24 mM DTT, and 2.5 mM MnCl) and were then incubated at RT for 1 h in kinase buffer supplemented with 2.5 μM ATP, 5 μCi γ[32P]ATP, and 3 μM biotinyl-Ahx-tetra (LRRWSLG) peptide substrate. Reactions were stopped with 20% phosphoric acid, and were then spotted onto P30 filtermat (Whatman). After five washes in 0.5% phosphoric acid, bound radiolabel was quantitated by scintillation counting. Deconvolution microscopy and pixel intensity quantitation were performed as described previously (Taylor et al., 2001). In brief, kinetochore fluorescence values were determined using softWoRx imaging software (Applied Precision). Background readings were subtracted, and the values were then normalized against the ACA signal to account for any variations in staining or image acquisition. SoftWoRx was used to measure interkinetochore distances using either ACA or Bub1 foci as indicated to determine kinetochore position.
Electron microscopy
Cells were fixed with 2.5% glutaraldehyde in phosphate buffer for 2 h at RT, and were then pelleted and post-fixed in 1% osmium tetroxide for 1 h at RT. After washes, samples were stained en bloc in 1% uranyl acetate for 16 h at 4°C, dehydrated with acetone, and then embedded in Spurr's resin. 70-nm sections were cut, stained with uranyl acetate and lead citrate, then examined on an electron microscope (Tecnai 12 BioTWIN; FEI Company).
RNAi
siRNA duplexes (Dharmacon Research) designed to repress Aurora A (5′-AAGCACAAAAGCUUGUCUCCA-3′), Aurora B (5′-AACGCGGCACUUCACAAUUGA-3′), and BubR1 (5′-AACGGGCAUUUGAAUAUGAAA-3′) were transfected using OligofectAMINE™ (Invitrogen) according to the manufacturer's instructions. In brief, 105 cells were seeded in wells of a 24-well plate 24 h before transfection. siRNA duplexes and OligofectAMINE™ were diluted in media, mixed, and incubated for 20 min. siRNA/lipid complexes were then added to cells for 4 h followed by addition of complete media. 24 h later, the cells were replated and then analyzed 48–72 h after transfection.
Transient transfections
The human Aurora B ORF (Bischoff et al., 1998) was amplified by PCR, cloned into pcDNA-3 Myc (Taylor and McKeon, 1997), mutated to create K106R, and sequenced, all after standard procedures. Plasmid DNA was then transfected into TA-HeLa cells on coverslips using the ProFection® calcium phosphate kit (Promega). 24 h after transfection, the cells were fixed with 1% formaldehyde in PBS and processed for immunofluorescence as described above.
Online supplemental material
The supplemental figures and tables show (1) the effects of ZM447439 and Aurora B RNAi on Cenp-E and Mad2 localization; (2) the effect of BubR1 RNAi on the spindle checkpoint; and (3) quantitation of the data from the experiment examining the effect of ZM447439 on interkinetochore distance and BubR1 binding. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200208091/DC1
Supplemental Material
Acknowledgments
We are grateful to Karen Vousden for p53 +/− U2OS cells; Sam Newby for assistance with electron microscopy; members of the Taylor lab, including Gordon Turner, Sarah Holt and Claire Bullock, for reagents; Iain Hagan and Rene Medema for comments on the manuscript; and all our colleagues at AstraZeneca, especially Stephen Green, without whom this work would not have been possible.
C. Ditchfield is funded by AstraZeneca, and V.L. Johnson by Cancer Research UK and The Biotechnology and Biological Sciences Research Council (BBSRC). S.S. Taylor is a BBSRC David Phillips Research Fellow.
The online version of this article includes supplemental material.
Footnotes
Abbreviation used in this paper: RNAi, RNA interference.
References
- Adams, R.R., M. Carmena, and W.C. Earnshaw. 2001. a. Chromosomal passengers and the (aurora) ABCs of mitosis. Trends Cell Biol. 11:49–54. [DOI] [PubMed] [Google Scholar]
- Adams, R.R., D.M. Eckley, P. Vagnarelli, S.P. Wheatley, D.L. Gerloff, A.M. Mackay, P.A. Svingen, S.H. Kaufmann, and W.C. Earnshaw. 2001. b. Human INCENP colocalizes with the Aurora-B/AIRK2 kinase on chromosomes and is overexpressed in tumour cells. Chromosoma. 110:65–74. [DOI] [PubMed] [Google Scholar]
- Adams, R.R., H. Maiato, W.C. Earnshaw, and M. Carmena. 2001. c. Essential roles of Drosophila inner centromere protein (INCENP) and aurora B in histone H3 phosphorylation, metaphase chromosome alignment, kinetochore disjunction, and chromosome segregation. J. Cell Biol. 153:865–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen, P.R., O.D. Lohez, F.B. Lacroix, and R.L. Margolis. 2001. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol. Biol. Cell. 12:1315–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins, S., and A.W. Murray. 2001. The budding yeast protein kinase Ipl1/Aurora allows the absence of tension to activate the spindle checkpoint. Genes Dev. 15:3118–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins, S., F.F. Severin, N. Bhalla, I. Sassoon, A.A. Hyman, and A.W. Murray. 1999. The conserved protein kinase Ipl1 regulates microtubule binding to kinetochores in budding yeast. Genes Dev. 13:532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff, J.R., and G.D. Plowman. 1999. The Aurora/Ipl1p kinase family: regulators of chromosome segregation and cytokinesis. Trends Cell Biol. 9:454–459. [DOI] [PubMed] [Google Scholar]
- Bischoff, J.R., L. Anderson, Y. Zhu, K. Mossie, L. Ng, B. Souza, B. Schryver, P. Flanagan, F. Clairvoyant, C. Ginther, et al. 1998. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 17:3052–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, G.K., S.A. Jablonski, V. Sudakin, J.C. Hittle, and T.J. Yen. 1999. Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J. Cell Biol. 146:941–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir, S.M., J. Harborth, W. Lendeckel, A. Yalcin, K. Weber, and T. Tuschl. 2001. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 411:494–498. [DOI] [PubMed] [Google Scholar]
- Giet, R., and C. Prigent. 1999. Aurora/Ipl1p-related kinases, a new oncogenic family of mitotic serine-threonine kinases. J. Cell Sci. 112:3591–3601. [DOI] [PubMed] [Google Scholar]
- Hannak, E., M. Kirkham, A.A. Hyman, and K. Oegema. 2001. Aurora-A kinase is required for centrosome maturation in Caenorhabditis elegans. J. Cell Biol. 155:1109–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, J.Y., Z.W. Sun, X. Li, M. Reuben, K. Tatchell, D.K. Bishop, J.M. Grushcow, C.J. Brame, J.A. Caldwell, D.F. Hunt, et al. 2000. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 102:279–291. [DOI] [PubMed] [Google Scholar]
- Kallio, M.J., M.L. McCleland, P.T. Stukenberg, and G.J. Gorbsky. 2002. Inhibition of aurora B kinase blocks chromosome segregation, overrides the spindle checkpoint, and perturbs microtubule dynamics in mitosis. Curr. Biol. 12:900–905. [DOI] [PubMed] [Google Scholar]
- Meraldi, P., R. Honda, and E.A. Nigg. 2002. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. EMBO J. 21:483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moasser, M.M., M. Srethapakdi, K.S. Sachar, A.J. Kraker, and N. Rosen. 1999. Inhibition of Src kinases by a selective tyrosine kinase inhibitor causes mitotic arrest. Cancer Res. 59:6145–6152. [PubMed] [Google Scholar]
- Morishita, J., T. Matsusaka, G. Goshima, T. Nakamura, H. Tatebe, and M. Yanagida. 2001. Bir1/Cut17 moving from chromosome to spindle upon the loss of cohesion is required for condensation, spindle elongation and repair. Genes Cells. 6:743–763. [DOI] [PubMed] [Google Scholar]
- Murata-Hori, M., and Y. Wang. 2002. The kinase activity of Aurora B is required for kinetochore-microtubule interactions during mitosis. Curr. Biol. 12:894–899. [DOI] [PubMed] [Google Scholar]
- Musacchio, A., and K.G. Hardwick. 2002. The spindle checkpoint: structural insights into dynamic signalling. Nat. Rev. Mol. Cell Biol. 3:731–741. [DOI] [PubMed] [Google Scholar]
- Nicklas, R.B. 1997. How cells get the right chromosomes. Science. 275:632–637. [DOI] [PubMed] [Google Scholar]
- Nicklas, R.B., and C.A. Koch. 1969. Chromosome micromanipulation. 3. Spindle fiber tension and the reorientation of mal-oriented chromosomes. J. Cell Biol. 43:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg, E.A. 2001. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2:21–32. [DOI] [PubMed] [Google Scholar]
- Rieder, C.L., and E.D. Salmon. 1998. The vertebrate cell kinetochore and it's roles during mitosis. Trends Cell Biol. 8:310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder, C.L., R.W. Cole, A. Khodjakov, and G. Sluder. 1995. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J. Cell Biol. 130:941–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon, K.B., J.C. Canman, and E.D. Salmon. 2002. Mad2 and BubR1 function in a single checkpoint pathway that responds to a loss of tension. Mol. Biol. Cell. 13:3706–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoufias, D.A., P.R. Andreassen, F.B. Lacroix, L. Wilson, and R.L. Margolis. 2001. Mammalian mad2 and bub1/bubR1 recognize distinct spindle-attachment and kinetochore-tension checkpoints. Proc. Natl. Acad. Sci. USA. 98:4492–4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, T.U., N. Rachidi, C. Janke, G. Pereira, M. Galova, E. Schiebel, M.J. Stark, and K. Nasmyth. 2002. Evidence that the Ipl1-Sli15 (Aurora kinase-INCENP) complex promotes chromosome bi-orientation by altering kinetochore-spindle pole connections. Cell. 108:317–329. [DOI] [PubMed] [Google Scholar]
- Tatsuka, M., H. Katayama, T. Ota, T. Tanaka, S. Odashima, F. Suzuki, and Y. Terada. 1998. Multinuclearity and increased ploidy caused by overexpression of the aurora- and Ipl1-like midbody-associated protein mitotic kinase in human cancer cells. Cancer Res. 58:4811–4816. [PubMed] [Google Scholar]
- Taylor, S.S., and F. McKeon. 1997. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 89:727–735. [DOI] [PubMed] [Google Scholar]
- Taylor, S.S., D. Hussein, Y. Wang, S. Elderkin, and C.J. Morrow. 2001. Kinetochore localisation and phosphorylation of the mitotic checkpoint components Bub1 and BubR1 are differentially regulated by spindle events in human cells. J. Cell Sci. 114:4385–4395. [DOI] [PubMed] [Google Scholar]
- Terada, Y., M. Tatsuka, F. Suzuki, Y. Yasuda, S. Fujita, and M. Otsu. 1998. AIM-1: a mammalian midbody-associated protein required for cytokinesis. EMBO J. 17:667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters, J.C., R.H. Chen, A.W. Murray, and E.D. Salmon. 1998. Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J. Cell Biol. 141:1181–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, J.H., E. Munar, D.R. Jameson, P.R. Andreassen, R.L. Margolis, R. Seger, and E.G. Krebs. 1999. Mitogen-activated protein kinase kinase activity is required for the G(2)/M transition of the cell cycle in mammalian fibroblasts. Proc. Natl. Acad. Sci. USA. 96:11335–11340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, X., A. Abrieu, Y. Zheng, K.F. Sullivan, and D.W. Cleveland. 2000. CENP-E forms a link between attachment of spindle microtubules to kinetochores and the mitotic checkpoint. Nat. Cell Biol. 2:484–491. [DOI] [PubMed] [Google Scholar]
- Zhou, H., J. Kuang, L. Zhong, W.L. Kuo, J.W. Gray, A. Sahin, B.R. Brinkley, and S. Sen. 1998. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat. Genet. 20:189–193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}