

Abstract
Actin filament pointed-end dynamics are thought to play a critical role in cell motility, yet regulation of this process remains poorly understood. We describe here a previously uncharacterized tropomodulin (Tmod) isoform, Tmod3, which is widely expressed in human tissues and is present in human microvascular endothelial cells (HMEC-1). Tmod3 is present in sufficient quantity to cap pointed ends of actin filaments, localizes to actin filament structures in HMEC-1 cells, and appears enriched in leading edge ruffles and lamellipodia. Transient overexpression of GFP–Tmod3 leads to a depolarized cell morphology and decreased cell motility. A fivefold increase in Tmod3 results in an equivalent decrease in free pointed ends in the cells. Unexpectedly, a decrease in the relative amounts of F-actin, free barbed ends, and actin-related protein 2/3 (Arp2/3) complex in lamellipodia are also observed. Conversely, decreased expression of Tmod3 by RNA interference leads to faster average cell migration, along with increases in free pointed and barbed ends in lamellipodial actin filaments. These data collectively demonstrate that capping of actin filament pointed ends by Tmod3 inhibits cell migration and reveal a novel control mechanism for regulation of actin filaments in lamellipodia.
Keywords: cytoskeleton; angiogenesis; actin; tropomyosin; lamellipodia
Introduction
Cell migration is a complex, multistep process that is essential for many aspects of development in multicellular organisms. One step, which appears to be common to many types of cell migration, is the protrusion of lamella from cell bodies using an F-actin–rich lamellipodia (Small et al., 2002). Lamellipodial protrusion is driven by actin filament barbed-end assembly at the leading edge of migrating cells and is governed by several actin regulatory proteins. A key component is the actin-related protein 2/3 (Arp2/3)* complex, which nucleates the formation of new filaments in response to activated WASP or SCAR (for reviews see Carlier and Pantaloni, 1997; Pollard et al., 2000; Condeelis, 2001). In addition, cofilin/actin-depolymerizing factor (ADF) also provides new barbed ends for polymerization by filament severing (Chan et al., 2000; Ichetovkin et al., 2002). Together, these regulatory proteins cause rapid creation of actin filament barbed ends and assembly from these new barbed ends, which then provide force to push the membrane forward (Theriot, 2000).
Although the force of protrusion is provided by barbed-end polymerization, filaments must also be disassembled from their pointed ends to replenish the monomer pool and move the network forward. Indeed, analysis of polymerization and depolymerization rate constants indicates that filament disassembly from the pointed ends may be the rate-liming component in an actin filament treadmilling model (Pantaloni et al., 2001). To overcome this limitation, motile cells utilize several mechanisms to enhance the disassembly of the actin filaments in lamellipodia. First, filament severing by cofilin/ADF directly provides additional free pointed ends (Bamburg, 1999; Chan et al., 2000; Condeelis, 2001). Second, cofilin/ADF may also enhance subunit dissociation from existing pointed ends, which are occupied by ADP-actin subunits (Carlier et al., 1997; Maciver, 1998). Third, stochastic debranching of filaments linked by Arp2/3 can also provide pointed ends from which filaments can depolymerize. Debranching may be enhanced by nucleotide hydrolysis and phosphate release by actin subunits in daughter filaments at branch points (Blanchoin et al., 2000b). Conversely, cells may use several mechanisms to negatively regulate pointed-end disassembly and thus inhibit motility. For example, a well-characterized mechanism is the phosphorylation of cofilin/ADF, which inhibits its activity and prevents lamellipodial extension (Bamburg, 1999; Zebda et al., 2000).
An unexplored mechanism for negative regulation of pointed-end disassembly is pointed-end capping. Currently, tropomodulin (Tmod) is the only protein known whose sole function is to cap pointed ends of actin filaments (Weber et al., 1994; Littlefield et al., 2001). It was previously thought that Tmod was only involved in capping the pointed ends of filaments in stable, nondynamic cytoskeleton structures because it was originally found associated with actin filaments that are strictly regulated in length and persist over many days (e.g., erythrocyte membrane skeletons and striated muscle sarcomere thin filaments; Fowler, 1996). However, photobleaching experiments with GFP–Tmod and in vivo incorporation of fluorescently labeled actin in cardiac muscle cells have shown that Tmod capping is dynamic, and that the pointed ends that it occupies are sites of active assembly and disassembly (Littlefield et al., 2001). An interesting consequence of dynamic capping by Tmod is that the nucleotide at the pointed ends is converted from ADP-Pi to ADP (due to the slower rates of monomer association), thus leading to an increase in the pointed-end critical concentration and a decrease of F-actin at steady state in vitro (Weber et al., 1999).
We investigated the possibility that a Tmod isoform may be expressed in motile, nonmuscle cell types, as it would offer unique insights into the role of pointed-end dynamics in cell motility. We find that Tmod3 is expressed in motile endothelial cells, where it localizes to ruffles and lamellipodia. We demonstrate by modulation of Tmod3 levels that endothelial cell migration rates correlate inversely with the extent of pointed-end capping. Unexpectedly, F-actin and free barbed ends in the lamellipodia are also inversely correlated with Tmod3 levels, i.e., increased pointed-end capping decreases both lamellipodial free barbed ends and F-actin. These data indicate that regulation of pointed-end disassembly in lamellipodia is coordinated with the generation of free barbed ends and F-actin and is a key step in actin-mediated cell polarization and migration.
Results
Tmod3 is a pointed-end capping protein expressed in endothelial cells
Previous studies have identified a family of four canonical Tmod genes in vertebrate species, based on homology to known cDNA sequences (Cox and Zoghbi, 2000; Conley et al., 2001). A new isoform identified in vertebrates was designated as Tmod3. Like other Tmod isoforms, Tmod3 inhibits elongation from the pointed ends of gelsolin-capped actin filaments in a concentration-dependent manner (Fig. 1 A). When initial rates of elongation as a function of Tmod3 concentration are analyzed by double reciprocal plots, the Kd of Tmod3 for capping pointed ends of actin filaments is ∼110 nM. This is similar to the Kds for capping pointed ends of Tmod1 (Kd ≈ 180 nM) and Tmod4 (Kd ≈ 80 nM) (Fig. 1 A; Weber et al., 1999). However, whereas other Tmod isoforms are restricted to only a few differentiated cell types, Tmod3 was found in nearly all tissues tested (Cox and Zoghbi, 2000; Conley et al., 2001; unpublished data).
Figure 1.

Tmod3 is a novel Tmod isoform expressed in endothelial cells. (A) Tmod3 caps actin filament pointed ends with a similar affinity as Tmods 1 and 4. Pointed-end capping activities of human Tmod3 (□, large dash), human Tmod1 (▴, small dash), and chicken Tmod4 (•, solid). The extent of pointed-end capping is plotted as the initial rate of elongation in the presence of Tmod, divided by the initial rate of elongation for the control actin (R/CR), versus increasing Tmod concentration, such that when R/CR = 0, 100% of pointed ends are capped. (B) Cryosection of rat spleen stained with polyclonal anti-Tmod3 antibodies. Bar, 150 μm. (C) Immunoblot of total rat spleen protein with anti-Tmod3 antibody.
Upon further inspection of the tissues most enriched for Tmod3 expression, we observed that these tissues were also highly vascularized. Furthermore, sections of rat spleen, lung, or intestine stained with antibodies raised against recombinant human Tmod3 display prominent staining in the cells lining blood vessels (Fig. 1 B; unpublished data) and recognize a polypeptide of ∼40 kD in immunoblots of whole spleen (Fig. 1 C). Consistent with this, a human microvascular endothelial cell line (HMEC-1) contains a 40-kD polypeptide recognized by this antibody (Fig. 2 A). As these polyclonal antibodies to Tmod3 also cross-react weakly with purified recombinant human Tmod1 protein (Fig. 2 A, left), HMEC-1 cells were probed with monoclonal antibodies specific to Tmod1 (Fig. 2 A, right). However, no Tmod1 is detected in HMEC-1 cells (Fig. 2 A, right). Given the very narrow tissue distribution of Tmod2 and Tmod4 expression in neurons and skeletal muscle, respectively (Cox and Zoghbi, 2000; Conley et al., 2001), our data suggest that Tmod3 is the only known isoform expressed in these endothelial cells.
Figure 2.

Tmod3 is expressed in HMEC-1 cells. (A) Immunoblot of cell lysates or purified recombinant proteins with either anti-Tmod3 polyclonal (left) or anti-Tmod1 monoclonal (right) antibodies. Mg2+ ghosts, human erythrocyte membranes (Babcock and Fowler, 1994); Tmod1, purified recombinant human Tmod1 protein; Tmod3, purified recombinant human Tmod3 protein. (B) Quantitation of Tmod3 in HMEC-1 cells. ♦, average counts from duplicate standards of purified recombinant human Tmod3; dashed lines indicate average counts from immunoblot bands of 2 × 105 and 5 × 105 HMEC-1 cells. Line fitted to standards by least squares method was used to determine the amount of Tmod in HMEC-1 cells in the linear range of the assay.
We determined the concentration of endogenous Tmod3 in HMEC-1 cells by quantitative immunoblotting. The amounts of total Tmod3 in known numbers of cells were compared with standards of recombinant purified human Tmod3 loaded on the same gel (Fig. 2 B, ♦). Using a mean cell volume determined from the mean cell diameter (measured by light microscopy of trypsinized cells), we estimate that the total intracellular Tmod3 concentration is ∼0.5 μM. Because the affinity of Tmod3 for actin filament pointed ends is ∼110 nM, this indicates that there is sufficient Tmod3 to cap pointed ends in HMEC-1 cells.
Tmod3 localizes to dynamic F-actin structures in HMEC-1 cells
Migrating HMEC-1 cells have small, transient lamellipodia, which often collapse into ruffles. Although these protrusions and ruffles are rich in F-actin, HMEC-1 cells also contain bundles of F-actin that are generally oriented perpendicular to the direction of movement (Fig. 3 , top, F-actin). To determine which populations of actin filaments in these cells might be targets for Tmod3 regulation, we localized endogenous Tmod3 in HMEC-1 cells by indirect immunofluorescence. In general, Tmod3 localizes preferentially to the F-actin–rich structures at the forward periphery of the cell, including lamellipodia (Fig. 3, top two rows, arrowheads) and subsequent ruffles (arrows) that form from their collapse. In contrast, Tmod3 does not localize prominently to the perpendicular F-actin bundles in the cell body. In most cells, a large diffuse staining component in the cytoplasm is also observed, likely due to fixation of a portion of the soluble pool (see below).
Figure 3.
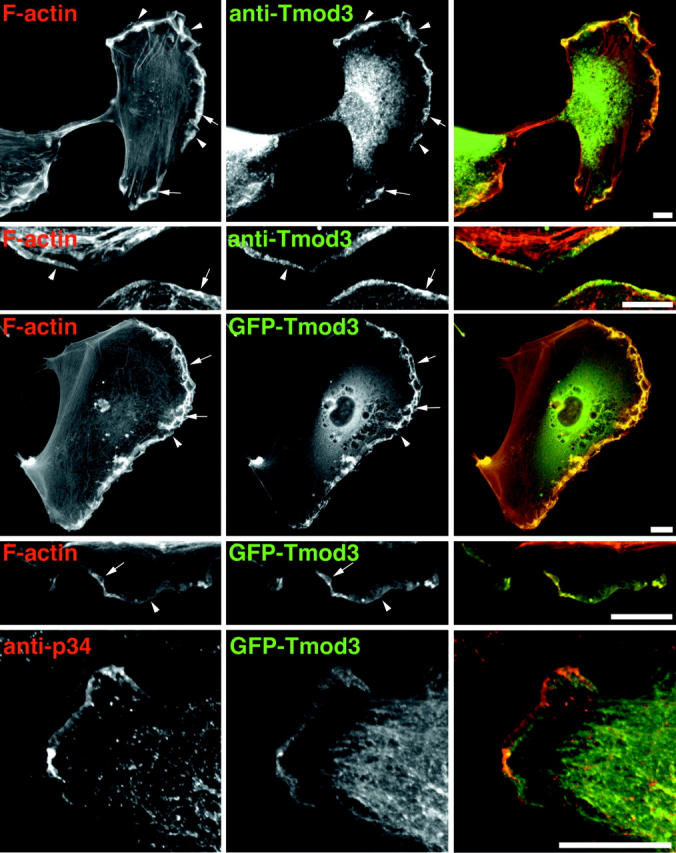
Localization of Tmod3 in HMEC-1 cells. HMEC-1 cells were fixed and stained with fluorescent phallacidin and anti-Tmod3 polyclonal antibodies followed by fluorescent anti–rabbit IgG antibodies. GFP–Tmod3 colocalizes similarly with fluorescent phalloidin. Arrows indicate ruffles, arrowheads indicate small lamellipodia. In the bottom row, Tmod3 localizes to the lamellipodia, including a region behind Arp2/3. Cells expressing low levels of GFP–Tmod3 were fixed and stained with anti-p34 polyclonal antibodies. Bars, 8 μm.
To further characterize Tmod localization, a fusion protein of GFP and Tmod3 was expressed transiently in HMEC-1 cells. As shown in Fig. 3, the localization of the fusion protein in lamellipodial protrusions and ruffles is similar to endogenous Tmod3 (Fig. 3, third and fourth rows), suggesting that GFP–Tmod3 can be used as a marker for endogenous Tmod3. Therefore, using transient expression of GFP–Tmod3, we compared its localization to the p34 subunit of the Arp2/3 complex in HMEC-1 cells. In extending lamellipodia, p34 localizes to the very edge of the cell (Fig. 3, bottom), as expected (Welch et al., 1997; Bailly et al., 1999). GFP–Tmod3 localizes throughout the lamellipodia but, interestingly, appears slightly enriched in a region immediately behind the p34 (Fig. 3).
Tmod3 localization to dynamic cellular structures is further supported by observations of live cells expressing GFP–Tmod3, where its localization in leading edge ruffles and protrusions is clearly evident (Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200209057/DC1). In cells expressing robust levels of GFP–Tmod3, there exists a large pool of soluble GFP–Tmod3, consistent with biochemical fractionation experiments (see below). Interestingly, GFP–Tmod3 also appears in highly motile dorsal protrusions and ruffles, similar to those observed with GFP-tagged capping protein or Arp2/3 subunits (Schafer et al., 1998). The significance of these structures with respect to Tmod3 function is not clear at present.
Increasing intracellular concentration of Tmod3 inhibits cell motility
HMEC-1 cells have been shown to have an average random migration velocity of ∼10–20 μm/h, depending on the substrate (Kiosses et al., 1999). Under our conditions, HMEC-1 cells were observed to translocate at a similar rate (Video 2, available at http://www.jcb.org/cgi/content/full/jcb.200209057/DC1; Fig. 4, A and B) . HMEC-1 cells displayed various motile behaviors, including polarization, protrusion, contraction, and detachment (Video 2; Fig. 4 A). When adenoviral vectors were used to transiently overexpress GFP–Tmod3, HMEC-1 cells were generally less polarized in morphology and adopted a more stationary, spreading morphology (Fig. 4, C and D), although protrusion and ruffling activity at the cell periphery continued in many of these cells (Video 3, available at http://www.jcb.org/cgi/content/full/jcb.200209057/DC1). However, whereas only a small fraction (∼4%) of control cells failed to exhibit protrusive activity over a 2-h window, ∼22% of GFP–Tmod3-overexpressing cells failed to exhibit significant protrusive activity (see Materials and methods) during the same time period. Those cells that do transiently polarize by protruding lamellipodia often quickly lose polarity and then attempt to polarize in another direction (Video 3). Furthermore, when rates of cell translocation were measured in the overexpressing cells, the average rate was approximately half of that observed for control cells (Fig. 4 B). Although there was some variability in individual cell velocities (as demonstrated by the large data range), the average velocity difference between control cells and GFP–Tmod3-overexpressing cells was statistically significant (P ≈ 3 × 10−7). Similar effects on cell motility and morphology were also observed with viruses expressing untagged Tmod3 (unpublished data).
Figure 4.

HMEC-1 cell morphology and migration rate are altered by transient overexpression of GFP–Tmod3. Stills of supplemental videos (available at http://www.jcb.org/cgi/content/full/jcb.200209057/DC1) of control cells (A, Video 2) or GFP–Tmod3 cells (C and D, Video 3). Bar, 10 μm. (B) Graph of average cell migration rate of control-infected or GFP–Tmod3-overexpressing HMEC-1 cells. Box and whisker plots were generated as described in the Materials and methods. n ≥ 22 cells for each measurement.
Increasing Tmod3 concentration decreases total free pointed ends in vivo
To determine whether an increase in Tmod3 leads to increased capping of existing free pointed ends in HMEC-1 cells, we used DNase I as a marker for free pointed ends in cells in situ (Chan et al., 2000). In control HMEC-1 cells, DNase I–stained free pointed ends are located throughout the cell, with a concentration at the cell periphery, both at the anterior edge and in apparent tail extensions (Fig. 5 D). As expected, DNase I also prominently stained cell nuclei, which were identified with DAPI staining (Fig. 5, B and E). In cells in which GFP–Tmod3 was overexpressed, there was a decrease in DNase I staining (Fig. 5, compare A with B) that was particularly significant in regions where GFP–Tmod3 was found (e.g., at the cell periphery; Fig. 5 C). Note that the extraction of monomers before staining with this procedure causes some cytoplasmic GFP–Tmod3 to collapse onto the nucleus (Fig. 5 C), but this is not normally observed in live or fixed cells (e.g., compare with Fig. 3). When regions of nuclear staining are excluded, the DNase I staining intensity can thus be used to quantitate the relative levels of free pointed ends in the cells. When compared with control-infected cells, cells overexpressing GFP–Tmod3 exhibit a fivefold reduction in free pointed ends (Fig. 5 F), roughly proportional to the increase in cytoskeleton-associated Tmod3 in the cells (see below). These data suggest that GFP–Tmod3 caps a large proportion of the available pointed ends in actin structures throughout the cell, including at the leading edge.
Figure 5.

Tmod3 caps actin filament pointed ends in vivo. (A–C) Permeabilized cell expressing GFP–Tmod3 stained with DNase I or coumarin–phalloidin and DAPI. (A) Rhodamine–DNase I; (B) DAPI + phallacidin; (C) GFP–Tmod3. (D and E) Permeabilized control-infected cell stained with DNase I (D) and coumarin–phalloidin (E). DAPI staining labels nuclei in the same channel as coumarin–phalloidin. Bar, 15 μm. (F) Quantitation of nonnuclear DNase I staining in control and GFP–Tmod3-expressing cells. Values are averages from >20 cells for each cell type, standard deviation is shown by error bars.
Transient overexpression of Tmod3 does not alter cytoskeleton-associated F-actin levels in whole cells
Because the adenovirus (Ad)-mediated expression of GFP–Tmod3 results in consistent overexpression in virtually all cells in an experiment, we were able to use a biochemical approach to compare the relative level of Tmod3 expression in both control and overexpressing cells by quantitative immunoblot analyses. This showed that overexpression of GFP–Tmod3 results in approximately a fivefold increase in total Tmod3 expression (Fig. 6, A and B) . Fractionation studies using Triton X-100 revealed that ∼30–40% of endogenous Tmod3 was associated with the cytoskeleton in control cells (Fig. 6 B). When GFP–Tmod3 was overexpressed, a five- to sixfold increase in total cytoskeleton-associated Tmod3 was observed (Fig. 6 B), with a proportional increase in the soluble pool as well. These data are consistent with the relative decrease observed in free pointed ends in the cell (Fig. 5 F). Interestingly, the overall distribution (soluble versus insoluble) of Tmod3 in overexpressing cells was not substantially different from control cells.
Figure 6.

Overexpression of GFP–Tmod3 and fractionation of HMEC-1 cells. (A) Immunoblot of total cell lysate of cells transiently expressing either GFP or GFP–Tmod. For both endogenous Tmod3 and GFP–Tmod3, some proteolysis was observed. When GFP–Tmod is overexpressed, an apparent decrease in the endogenous Tmod3 is observed, but overall levels of Tmod3 are increased approximately sixfold. (B) Representative experiment showing [125I]protein A–probed immunoblot quantitation of Tmod3 and actin in cells overexpressing GFP or GFP–Tmod3. cpm, 125I counts per minute (detection of anti-Tmod3 anti-actin antibodies). Dark bars, Triton-insoluble fraction; light bars, Triton-soluble fraction. For GFP–Tmod3 cells, data shown are the total of both GFP–Tmod (arrowhead) and endogenous Tmod (*) bands in a given fraction.
Despite a significant increase in the total amount of Tmod3 associated with the cytoskeleton and a corresponding decrease in free pointed ends upon overexpression of GFP–Tmod3, the proportion of actin in the Triton-insoluble fraction in HMEC-1 cells was not significantly altered (Fig. 6 B). Furthermore, overall F-actin staining patterns and levels were not observably different (Fig. 3; unpublished data). These data suggest that although increased Tmod3 caps a large proportion of previously free pointed ends in cells, most of the filamentous actin in the cell is not dramatically affected by increased pointed-end capping.
Excess Tmod3 in HMEC-1 cells decreases F-actin but not G-actin in the lamellipodia
Because Tmod3 localizes to lamellipodia, we investigated the F-actin and G-actin content of this cell compartment in closer detail by quantitative fluorescence microscopy. Fluorescently labeled vitamin D–binding protein (Alexa®–DBP), which binds to G-actin and not pointed ends of actin filaments (Goldschmidt-Clermont et al., 1985), was used as a reporter for local G-actin (Cao et al., 1993). When cells are fixed to retain G-actin and stained with Alexa®–DBP and fluorescent phalloidin, relative G- or F-actin contents of lamellipodia from control or GFP–Tmod3-overexpressing cells can be compared (see Materials and methods). In cells overexpressing GFP–Tmod3, no significant change was observed in the G-actin levels in their lamellipodia, relative to those in control cells (Table I), consistent with our biochemical fractionation data for whole cells. However, as determined by phalloidin staining, the relative level of F-actin in the lamellipodia of cells overexpressing GFP–Tmod3 was about threefold less than in the lamellipodia of control cells (Table I). Thus, although no alteration in global F-actin is observed (Fig. 6), the dynamic actin structures in the lamellipodia are significantly affected. Previous studies have found some differences when comparing phalloidin staining using different permeabilization/fixation protocols (Small et al., 1995). Under conditions used here to stain for free barbed ends, we also observed a similar decrease in the relative level of F-actin in the lamellipodia of cells overexpressing Tmod3 compared with those of control cells (unpublished data). This is reassuring because the two protocols are performed under different conditions, i.e., on unfixed, permeabilized cells (barbed ends) or on paraformaldehyde-fixed cells (G-actin). Interestingly, these data would suggest that the G/F ratio in the lamellipodia of cells overexpressing GFP–Tmod3 could be significantly higher than that found in control cell lamellipodia.
Table I. Lamellipodial actin analyses.
| Actin parameter | Control cells | GFP–Tmod3 cells |
|---|---|---|
| F-actina (n) | 309 ± 84 (23) | 104 ± 46 (33) |
| G-actin (n) | 104 ± 46 (23) | 98 ± 39 (33) |
| Barbed ends/Fb (n) | 2.98 ± 1.20 (48) | 1.43 ± 0.81 (40) |
| p34c (n) | 186 ± 117 (28) | 57.6 ± 31.4 (41) |
Average values reported are in arbitrary units, with standard deviations given. n denotes number of cells. Barbed ends/F were measured from ratiometric images that were the result of rhodamine–actin/phalloidin.
P = 8.2 × 10−9.
P = 1.2 × 10−9.
P = 5.3 × 10−9.
Excess Tmod3 alters availability of barbed ends at cell periphery and in lamellipodia
Given the migration and polarization defects exhibited by the GFP–Tmod3-overexpressing cells, we analyzed the localization and relative levels of free barbed ends in these cells compared with control cells. When cells are permeabilized and incubated with labeled actin monomers at concentrations just above the barbed-end critical concentration, polymerization in situ from available barbed ends can be visualized (see Materials and methods; Symons and Mitchison, 1991). Incorporation of rhodamine–ATP-actin in permeabilized HMEC-1 cells demonstrates that free barbed ends are located throughout the cell, but that the most prominent population occurs at cell edges (Fig. 7 A), as expected from previous studies (Symons and Mitchison, 1991). In cells overexpressing GFP–Tmod3, this distribution is altered such that the width and intensity at cell edges are dramatically decreased (Fig. 7 B). The observed decrease in free barbed ends is not simply due to the decrease in lamellipodial F-actin (Table I), because the ratio of free barbed ends to F-actin also displays a decrease in intensity and width at the cell edge. This is demonstrated clearly by quantitative line scans (along radial lines perpendicular to the membrane) performed on ratiometric images (Fig. 7 C). Furthermore, quantitation of the ratio of free barbed ends to F-actin in whole lamellipodia indicates that cells overexpressing Tmod3 exhibit a twofold decrease in free barbed ends per relative unit of filamentous actin when compared with cells expressing endogenous levels of Tmod3 (Table I). In contrast, when the total amount of rhodamine–actin incorporation per cell is measured as a function of total cell area (to eliminate the effects of cell size and spreading), only an ∼16% decrease is observed between control cells and GFP–Tmod3-overexpressing cells (unpublished data). These data indicate that changes in free barbed ends upon overexpression of GFP–Tmod3 are confined to the cell periphery.
Figure 7.

Analyses of free barbed ends in HMEC-1 cells by rhodamine–actin incorporation in situ. (A and B) Control or GFP–Tmod3-expressing HMEC-1 cells permeabilized and labeled with rhodamine–actin. (C) Representative line scans of rhodamine–actin/fluorescent phallacidin ratio at the edge of the cell. Traces were taken from outside cell (near y axis) to inside cell (representative regions of line scans shown as white bars in A and B). See Table I for quantitation of ratiometric data from whole lamellipodia.
Previous studies using in vitro polymerization assays have shown that Tmod1 does not interact with actin filament barbed ends, nor does it bind to monomers (Weber et al., 1994, 1999). Thus, the surprising result that increased levels of Tmod3 reduced free barbed ends in the lamellipodia suggested that Tmod3 might affect other components involved in the generation of free barbed ends in lamellipodia. Using a polyclonal antibody to the p34 subunit of the Arp2/3 complex, we measured the relative levels of endogenous Arp2/3 in cell lamellipodia. In both control and GFP–Tmod3-overexpressing cells, p34 is present in the lamellipodia (Fig. 8) . However, in cells overexpressing GFP–Tmod3, the band of p34 staining was considerably less intense and narrower (Fig. 8), similar to the decrease in rhodamine–actin incorporation (Fig. 7). When the average intensity per unit area in the lamellipodia is quantitated, there is approximately threefold less Arp2/3 in the lamellipodia of GFP–Tmod3-overexpressing cells than in those of control cells (Table I). The total amount of Arp2/3 per cell is not significantly different (unpublished data). Thus, by increasing Tmod3 levels transiently in HMEC-1 cells, Arp2/3 complex appears to be displaced from the leading lamellipodia, which may result in the generation of fewer barbed ends in these structures.
Figure 8.
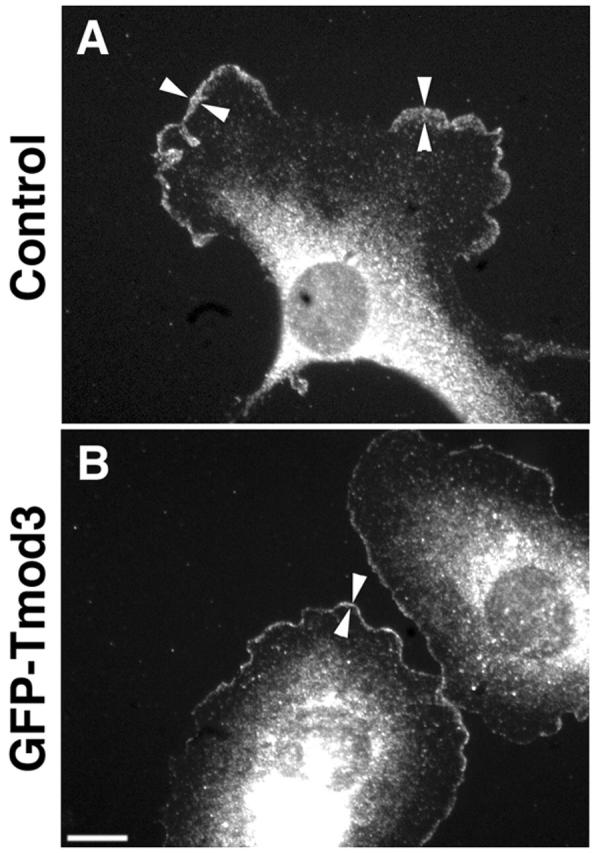
GFP–Tmod3 overexpression decreases Arp2/3 complex staining in lamellipodia. Control cells (A) or GFP–Tmod3- overexpressing cells (B) were stained for Arp2/3 using anti-p34 polyclonal antibodies. Arrowheads mark width of p34 staining in the lamellipodia. Bar, 10 μm.
Decreased Tmod3 in HMEC-1 cells results in faster migration rates
To determine whether endogenous Tmod3 similarly antagonizes cell migration in HMEC-1 cells, we decreased Tmod3 expression levels using small interfering RNA (siRNA) duplexes, as previously described (Elbashir et al., 2002). Transfection of siRNA duplexes onto HMEC-1 cells decreased Tmod3 levels to ∼15% (or less) of control transfected cells (Fig. 9 A). Examination of cells by immunofluorescence staining for Tmod3 revealed that at least 70% of cells were transfected (see Materials and methods; unpublished data). Cells transfected with siRNA(Tmod3) were observed by time-lapse photomicroscopy to determine if the decrease in Tmod3 levels had an effect on cell migration rates. Indeed, these cells exhibited a significantly increased average cell velocity compared with control transfected cells (Fig. 9 B). Thus, reduced Tmod3 levels lead to the opposite effects induced by Tmod3–GFP overexpression, indicating that both endogenous and overexpressed Tmod3 exert an inhibitory effect on cell migration.
Figure 9.
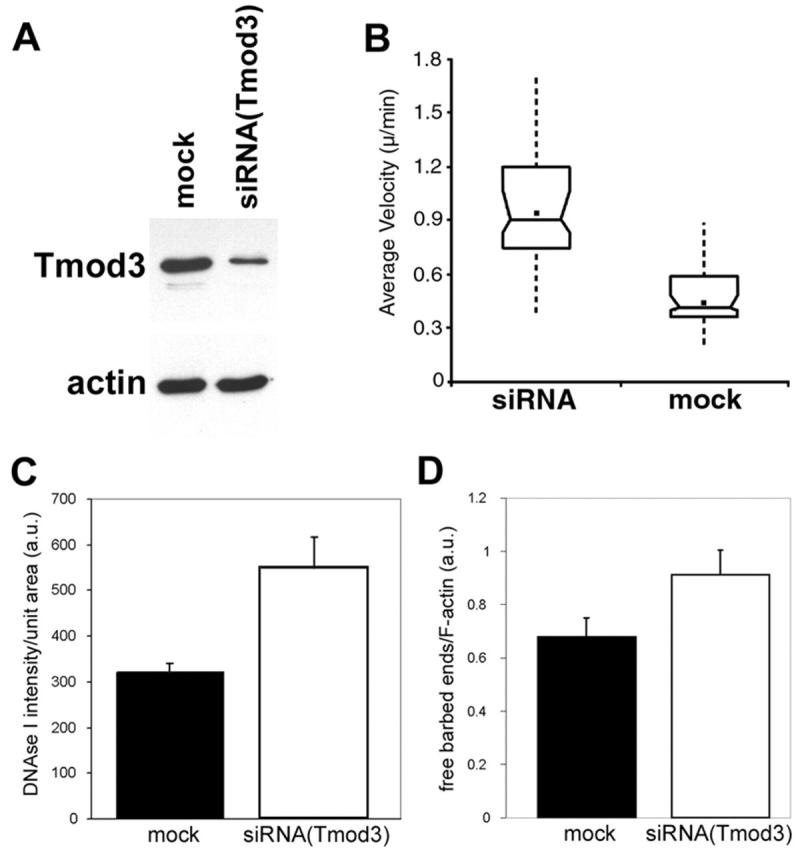
Effects of decreased Tmod3 levels by siRNA(Tmod3). HMEC-1 cells were transfected with siRNA duplex oligos against Tmod3 or mock transfected. Cells were later trypsinized and seeded onto either dishes for immunoblot analyses or onto coverslips for live cell motility analyses. (A) Immunoblots of equal numbers of mock- or siRNA-transfected cells, probed with either anti-Tmod3 antibodies (top) or C4 anti-actin monoclonal antibodies (bottom). (B) Average cell migration velocities measured over 4 h on either mock-transfected (n = 72) or siRNA-transfected (n = 64) cells. Box and whisker plots were generated as described in the Materials and methods. siRNA(Tmod3)-transfected cells migrate ∼2× faster than mock cells (P < 0.001). (C) Quantitation of total free pointed ends per cell by DNase I staining. (D) Quantitation of lamellipodial free barbed ends by rhodamine–actin incorporation as a ratio of fluorescent phalloidin. siRNA(Tmod3)-transfected cells exhibit a 45% increase in free barbed ends/F in lamellipodia (n = 30 per category; P < 0.005) and a 70% increase in total free pointed ends (n = 22 per category; P = 0.0069). In C and D, standard error of the mean is indicated.
To determine if endogenous Tmod3 inhibits HMEC-1 cell motility by a similar mechanism to overexpressed Tmod3, we first measured free pointed ends using DNase I staining, as above, in either mock-transfected or siRNA (Tmod3)-transfected cells. Significantly, HMEC-1 cells transfected with siRNA(Tmod3) exhibited an approximately two-fold increase in free pointed ends per cell compared with mock-transfected cells (Fig. 9 C). Furthermore, using the rhodamine–actin incorporation assay described above, we observed that the siRNA(Tmod3)-transfected cells displayed a modest (∼1.5-fold), but significant, increase in free barbed ends (measured as a ratio to F-actin) in their lamellipodia (Fig. 9 D). Thus, when Tmod3 levels are reduced, more free pointed ends and free lamellipodial barbed ends are created, concomitant with faster average cell migration rates.
Discussion
Disassembly of actin filaments is critical to the turnover of filament networks in lamellipodia and is required for cell migration (Cramer, 1999). Current treadmilling models for filament turnover predict that disassembly of actin filament pointed ends is a rate-limiting step (Borisy and Svitkina, 2000; Pantaloni et al., 2001), yet no pointed-end regulators have been identified previously in motile cells. We demonstrate that Tmod3 is a bona fide actin filament pointed-end capping protein present in motile cells in sufficient concentration to cap actin filament pointed ends. Furthermore, Tmod3 is associated with dynamic actin structures in lamellipodia. Functional analyses of cell migration rates and pointed-end capping in permeabilized cells show that increases in levels of Tmod3 lead to more pointed-end capping, resulting in slower cells, whereas decreases in levels of Tmod3 lead to less pointed-end capping, resulting in faster cells. The simplest interpretation of our observations is that pointed-end capping by Tmod3 reduces the rates of actin pointed-end disassembly in lamellipodia, consistent with the hypothesis that this is a rate-limiting step in the turnover of lamellipodial actin filaments in migrating cells.
The unexpected finding that Tmod3 levels are inversely correlated with the amount of F-actin and free barbed ends in the lamellipodia is intriguing. One possible explanation is that increased Tmod3 levels transiently cap existing pointed ends with greater frequency, slowing their depolymerization rate (Weber et al., 1999; Littlefield et al., 2001). This would lead to longer-lived filaments, thus increasing the proportion of ADP-actin subunits within these filaments, particularly near their pointed ends. Indeed, dynamic capping by high levels of Tmod1 in vitro has been shown to increase the proportion of ADP subunits at pointed ends at steady state (Weber et al., 1999). Because Arp2/3 branching and nucleation is favored from ATP or ADP-Pi subunits on filaments (Ichetovkin et al., 2002), longer-lived filaments with more ADP-actin subunits would likely reduce Arp2/3-mediated branching of the network. A reduced amount of branching would result in fewer free barbed ends at any given time, consistent with our observations here.
Another mechanism to account for the observed decreases in free barbed ends in the presence of increased Tmod3 levels is that Tmod3 could compete directly with Arp2/3 for the capture of free pointed ends of actin filaments and oligomers. Both Tmods and Arp2/3 cap ATP-actin with similar affinities as determined by in vitro pointed-end elongation assays (this study; Weber et al., 1994, 1999; Blanchoin et al., 2000b). However, unlike Tmod, which shows little preference for ATP-actin over ADP-actin (Weber et al., 1999), Arp2/3 has a 25-fold lower affinity for ADP-actin pointed ends (Blanchoin et al., 2000b). Thus, endogenous levels of Tmod3 (0.5 μM in HMEC-1 cells) could strongly compete with Arp2/3 for ADP-occupied pointed ends, and higher levels of Tmod3 overexpressed in cells could also compete with Arp2/3 for ATP or ADP-Pi pointed ends, inhibiting capture of oligomers by Arp2/3. Interestingly, recent studies to directly observe actin filament polymerization in vitro provide evidence that Arp2/3 can capture short actin filaments or oligomers to form new branches (Fujiwara et al., 2002a), in addition to de novo nucleation of filaments from the sides of existing ones (Blanchoin et al., 2000a; Amann and Pollard, 2001).
Cofilin/ADF has been proposed to accelerate the rate of actin pointed-end disassembly in treadmilling models for lamellipodia filament turnover by two alternative mechanisms (Pollard et al., 2000). The first is by directly enhancing the actin off rate from the pointed end (Carlier et al., 1997), whereas the second is by severing filaments, leading to more free pointed ends that can depolymerize (Maciver et al., 1991, Chan et al., 2000), in addition to synergizing with Arp2/3 to create new barbed ends (Ichetovkin et al., 2002). In the first case, the ability of Tmod3 to cap pointed ends and slow down actin disassembly raises the possibility that Tmod3 might directly compete for binding to pointed ends with cofilin/ADF and antagonize its actin pointed-end depolymerizing activity. In the second case, the presence of excess Tmod3 would be expected to cap the newly appearing pointed ends, thus slowing down actin disassembly. It is difficult to see how transient capping of pointed ends by Tmod3 could directly affect cofilin severing of filaments, which can take place many subunits distant from the pointed end (Chan et al., 2000). However, if Tmod3 were to antagonize cofilin/ADF severing, this could also explain the decrease in the proportion of barbed ends in lamellipodia, which is observed here with increasing Tmod3 levels. In support of a mechanism where Tmod3 may antagonize cofilin/ADF activity, recently Dawe et al. (2003) reported that inhibition of cofilin/ADF activity in fibroblasts causes cell depolarization in a phenotype strikingly similar to the phenotype observed by overexpression of Tmod3.
A mechanism for regulation of Tmod3 in endothelial cells and its effects on lamellipodial actin could be stabilization of tropomyosin (TM) on Tmod3-capped actin filaments. This could account for the decrease in lamellipodial free barbed ends and F-actin upon increased Tmod3 expression, because TM is known to inhibit both Arp2/3-mediated filament branching (Blanchoin et al., 2001) and cofilin/ADF-mediated filament severing (DesMarais et al., 2002). Indeed, in vitro filament depolymerization data suggest that Tmod1 may be able to stabilize TM on the ends of actin filaments (Weber et al., 1994, 1999). However, neither endogenous Tmod3 nor GFP–Tmod3 show strong localization to stress fiber bundles, which are the major TM-coated F-actin structures in cells (Fig. 3; unpublished data). Conversely, lamellipodia have been shown to be lacking in TM (DesMarais et al., 2002) where Tmod3 was most prominent. In fact, we have observed that lamellipodia containing GFP–Tmod3 exhibit a paucity of at least some TM isoforms (unpublished data). These data would suggest that TM does not significantly regulate Tmod3 localization, and vice versa.
One of the most conspicuous features of the phenotype resulting from increased Tmod3 levels is the depolarization of cells accompanying the decrease in migration rate. Creation of a protruding leading lamellipodia is critical for the initiation of polarized cell migration (Borisy and Svitkina, 2000). Although the data presented here demonstrate that many of the overexpressing cells continue to extend protrusions (Video 3), quantitative analyses of protrusion kymographs indicate that these protrusions show a marked decrease in the persistence and efficiency of forward movement (unpublished data). Recent studies show that effective forward protrusion of the lamellipodium requires coordinated generation of optimum levels of barbed ends together with adhesion of cell protrusions (Krause et al., 2002; Mogilner and Edelstein-Keshet, 2002). Our data suggest that the generation of free barbed ends is coordinated with pointed-end disassembly in lamellipodial actin networks. Thus, the loss of free barbed ends, as an indirect consequence of excess Tmod3, may lead to loss of polarization by decreasing the efficiency of forward protrusion. However, despite clear evidence of lamellipodial actin defects in Tmod3-overexpressing cells, we cannot formally rule out additional effects of Tmod3 on cell adhesion. This is in part due to the difficulty of parsing the functions of cell adhesion from cell spreading and protrusion, as both functions are mutually dependent. Future studies using methods with increased time resolution to alter capping activity levels (e.g., uncaging experiments, etc.) coupled with real-time analyses of actin turnover (e.g., fluorescent speckle microscopy) may allow better correlation of actin turnover with lamellipodial protrusion, adhesion, and polarization.
Materials and methods
Cell culture
HMEC-1 cells were cultured as previously described (Kiosses et al., 1999), with a few modifications. Cells were maintained in EGM-2 medium (Clonetics) at 37°C in 7% CO2. For microscopy experiments, cells were plated on glass coverslips coated overnight with 2 μg/ml fibronectin (Sigma-Aldrich).
Expression of recombinant Tmod proteins and adenoviral vectors
Full-length human Tmod3 (U-Tmod) (accession no. AF177172) and human Tmod1 (E-Tmod) (accession no. AF131836) cDNAs were inserted in frame in the pGEX-KG vector. GST–Tmods were expressed in Escherichia coli and affinity purified on a glutathione column, and then Tmods were released from the GST moiety and purified to homogeneity as previously described (Babcock and Fowler, 1994). For overexpression studies in HMEC-1 cells, the full-length cDNA for human Tmod3 was subcloned into pEGFP (CLONTECH Laboratories, Inc.) to generate the GFP–Tmod3 fusion cDNA. Adenoviral (Ad) vectors were made as previously described (Altschuler et al., 1998). HMEC-1 cells were infected 12–14 h after plating with the GFP–Tmod3 Ads together with Ads expressing the tet transcriptional activator protein (tTA) under control of the murine CMV promoter. Expression of GFP–Tmod3 was observed by ∼20 h after infection, and all biochemical assays and in situ stains were performed at 24 h after infection; live cell data were collected from 22 to 30 h after infection. Cells overexpressing GFP–Tmod3 were viable for at least 3 d after onset of expression, indicating that cytotoxicity was not an issue in our experiments. Overexpression controls for the Ad vector were either a virus construct expressing GFP with the tTA-expressing virus or tTA virus alone. Neither control infection regime produced observable effects on cell morphology or behavior (not depicted). In our experiments, free GFP (unfused) tended to form bright aggregates in some HMEC-1 cell bodies (not depicted), whereas GFP fusion proteins did not (Fig. 3). Therefore, for fluorescence experiments, tTA virus alone was used in control infections.
Suppression of Tmod3 expression by RNA interference
Target specific siRNA duplexes were designed as described previously (Elbashir et al., 2002). Candidate siRNA sequences were compared against the human genome by BLAST searches to ensure that Tmod3 was the only sequence targeted. Selected siRNA duplexes were from Dharmacon Research Inc. For experiments described here, the sequence used was AATTGTGTGACCTCGCAGCAATT (437–459 relative to the start codon). To observe uptake of the duplexes by cells, duplexes were modified with 5′-fluorescein. Cells were transfected with duplexes using Oligofectamine (Invitrogen) at 12 and 60 h after plating. Typical transfection rates observed via the 5′-fluorescein were ∼70%. Cells were replated onto fibronectin-coated coverslips (for immunofluorescence or motility studies) or plastic dishes (for biochemistry) 10–12 h before experiments. As negative controls, cell were either mock transfected (with reagent only) or transfected with single-stranded DNA oligonucleotides (also 5′-fluorescein labeled; purchased from Proligo.com).
Antibodies and reagents
Rabbit polyclonal antibodies to gel-purified recombinant human Tmod3 were prepared and affinity purified according to standard procedures. Monoclonal antibodies (mAb9) to Tmod1 (E-Tmod) have been previously described (Gregorio and Fowler, 1995). Fluorescence-labeled secondary goat antibodies were from Jackson ImmunoResearch Laboratories. Monoclonal antibody to actin (C4) was a gift from J. Lessard (University of Cincinnati, Cincinnati, OH). Polyclonal sera to Arp2/3 was a gift from M. Welch (University of California, Berkley, CA). Labeled phalloidin, phallacidin, and fluorescent DNase I were from Molecular Probes. Unlabeled phallacidin was from Sigma-Aldrich. Rhodamine-labeled actin was prepared as previously described (Littlefield et al., 2001). Fluorescence-labeled DBP was prepared using purified DBP (Calbiochem) and Alexa Fluor®546–maleimide (Molecular Probes), according to the manufacturer's suggestions.
Actin polymerization measurements
Measurements of elongation rates at the pointed end were performed using 8–16% pyrenyl-labeled rabbit skeletal muscle actin and gelsolin-capped actin filaments as nuclei for polymerization (Weber et al., 1994, 1999). Capping activities for Tmods were obtained from the initial elongation rates, measured directly from the slopes of the polymerization traces over the first 30 s to 1 min. Rates in the presence of Tmod were divided by the rate for actin in the absence of Tmod, giving a rate/control rate. The Kds for Tmods were calculated from the x intercept of a double reciprocal plot of 1/(1 − [rate/control rate]) versus 1/Tmod concentration.
Cell fractionation and immunoblot analyses
For whole cell lysates, cells were harvested by scraping directly into warm Laemmli sample buffer and analyzed by SDS-PAGE. For fractionation of cells into cytoskeleton-associated and soluble pools, cells were lysed with cold CSK buffer as previously described (Gregorio and Fowler, 1995). Cell fractions or whole cell lysates were analyzed in immunoblot assays as previously described (Kuhlman et al., 1996).
Fluorescence staining and quantitation
For general immunofluorescence staining, cells were fixed with fresh 3.7% paraformaldehyde in cytoskeleton buffer (CB) (Small et al., 1978) for 20 min at room temperature, after which cells were permeabilized with 0.25% Triton in CB buffer for 20 min at room temperature. For p34 staining, cells were postfixed for 5 min in −20°C methanol before permeabilization. Cells were blocked and stained in blocking buffer (2% goat serum, 1% BSA in CB). F-actin was stained by including BODIPY–phallacidin, rhodamine–phalloidin, or coumarin–phalloidin (Molecular Probes) with the secondary antibodies.
Fluorescent DNase I was used to quantitate free pointed ends in HMEC-1 cells as described previously (Chan et al., 2000). To eliminate the DNase I nuclear staining component, DAPI or TOPRO-3 (Molecular Probes) were included to identify nuclear regions. The total amount of free pointed ends per cell observed in the DNase I image was then calculated as the average intensity (per unit area) of the total cell area minus the nuclear region.
Free barbed ends were visualized using a protocol developed by Symons and Mitchison (1991). In our hands, >88% of the rhodamine–actin incorporation per cell was blocked by coincubation of permeabilized cells with 1 μM cytochalasin D (not depicted), indicating that the incorporation was predominately at free barbed ends, as expected (Symons and Mitchison, 1991). After background correction, intensity data from rhodamine–actin images was computed as a ratio to intensity data from corresponding fluorescent phalloidin images. The resulting ratiometric images were used for line scans or for quantitation of lamellipodial free barbed ends.
Fluorescence-labeled DBP was used as a probe for G-actin (Cao et al., 1993). Cells were fixed as for immunofluorescence staining described above, similar to other previously described protocols for G-actin staining (Cramer et al., 2002). Cells were incubated with 10 μg/ml Alexa®–DBP and coumarin–phalloidin in blocking buffer for 30 min, followed by rinses in TBS. In controls in which Alexa®–DBP was preincubated with stoichiometric amounts of monomeric actin, staining was completely blocked (not depicted), demonstrating that the stain is specific for actin monomers.
Images were obtained using a Carl Zeiss MicroImaging, Inc. Axioskop microscope with a Carl Zeiss MicroImaging, Inc. Plan Apo 63X (1.4 n.a.) objective and a Princeton Instruments cooled CCD camera as previously described (Littlefield et al., 2001). For quantitative analyses, images were imported into Metamorph 6.0 software (Universal Imaging Corp.). To quantitate the amount of G-actin, F-actin, or free barbed ends in the lamellipodia, regions of individual lamellipodia were selected by hand using tools in Metamorph, taking care to avoid selection of ruffled areas. A region within 0.7 μm from the cell edge was defined as the leading edge. Fluorescence intensity per unit area for each region was then recorded, and statistical analyses were performed in Excel. To eliminate small variations in absolute stain intensities, we compared data from within single experiments only and report representatives of each type of experiment.
Live cell microscopy
For all live cell experiments, cells were filmed on a Nikon Quantum TE300 inverted microscope using phase contrast optics with Nikon objectives. Cells were maintained in a heated chamber at 37°C on the microscope stage, in standard culture media supplemented with 10 mM Hepes, pH 7.5, lacking phenol red dye. Images were collected using a Hamamatsu ORCA II cooled CCD camera (Hamamatsu Corp.) and a Sutter Filter wheel and shutter mechanism (Sutter Instrument Co.).
Cell motility analyses were performed using low magnification time-lapse microscopy of random fields of cells over 4 h. Total distance traveled by each cell was determined by following nuclear positions over time. Data shown are from a representative experiment of multiple independent experiments. For all migration experiments, controls and experimentals from a single experiment were filmed over the same times after plating to eliminate differences in basal migration rates, as the cells tend to have slightly lower migration rates with increased time in culture. To score for protrusive activity, cell boundary regions from frames 30 min apart were compared (Kiosses et al., 1999). Cells exhibiting less than a 10% change in occupied area over the observation period were scored as nonprotrusive. Box and whisker plots of data were generated using software by Analyze-It Software Ltd., essentially as described previously (Bear et al., 2000). Boxes show median values (horizontal center line), lower and upper quartiles (boxes), and confidence intervals around the median (notch in box) (Fig. 4 B; Fig. 9 B). The mean value is represented by ▪.
Online supplemental material
The supplemental movies (Videos 1–3) are available online at http://www.jcb.org/cgi/content/full/jcb.200209057/DC1. Video 1 shows GFP–Tmod3 localizing to the advancing edge of motile HMEC-1 cells (fluorescence time-lapse). Video 2 demonstrates control-infected HMEC-1 cell migration in vitro (phase time-lapse). Video 3 shows GFP–Tmod3-overexpressing HMEC-1 migration in vitro (phase time-lapse).
Supplemental Material
Acknowledgments
The authors are grateful to Dr. Clare Waterman-Storer and Wendy Salmon for invaluable advice and assistance on live cell microscopy and analyses. The technical assistance of Jeanette Moyer and Lorena Puto is also appreciated, as are the insightful comments of Dr. Ryan Littlefield and Dr. Clare Waterman-Storer. in the preparation of this manuscript.
This work was supported by National Institutes of Health (NIH) grants GM34225 and EY10814 to V.M. Fowler and by The Scripps Research Institute NEI Eye Core (NIH EY 12598) Live Cell Microscopy Facility.
The online version of this article includes supplemental material.
Footnotes
Abbreviations used in this paper: Ad, adenovirus; ADF, actin-depolymerizing factor; Arp2/3, actin-related protein 2/3; CB, cytoskeleton buffer; DBP, vitamin D–binding protein; HMEC-1, human microvascular endothelial cells; siRNA, small interfering RNA; TM, tropomyosin; Tmod, tropomodulin; tTA, tet transcriptional activator.
References
- Altschuler, Y., S.M. Barbas, L.J. Terlecky, K. Tang, S. Hardy, K.E. Mostov, and S.L. Schmid. 1998. Redundant and distinct functions for dynamin-1 and dynamin-2 isoforms. J. Cell Biol. 143:1871–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann, K.J., and T.D. Pollard. 2001. Direct real-time observation of actin filament branching mediated by Arp2/3 complex using total internal reflection fluorescence microscopy. Proc. Natl. Acad. Sci. USA. 98:15009–15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock, G.G., and V.M. Fowler. 1994. Isoform-specific interaction of tropomodulin with skeletal muscle and erythrocyte tropomyosins. J. Biol. Chem. 269:27510–27518. [PubMed] [Google Scholar]
- Bailly, M., F. Macaluso, M. Cammer, A. Chan, J.E. Segall, and J.S. Condeelis. 1999. Relationship between Arp2/3 complex and the barbed ends of actin filaments at the leading edge of carcinoma cells after epidermal growth factor stimulation. J. Cell Biol. 145:331–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamburg, J.R. 1999. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu. Rev. Cell Dev. Biol. 15:185–230. [DOI] [PubMed] [Google Scholar]
- Bear, J.E., J.J. Loureiro, I. Libova, R. Fassler, J. Wehland, and F.B. Gertler. 2000. Negative regulation of fibroblast motility by Ena/VASP proteins. Cell. 101:717–728. [DOI] [PubMed] [Google Scholar]
- Blanchoin, L., K.J. Amann, H.N. Higgs, J.B. Marchand, D.A. Kaiser, and T.D. Pollard. 2000. a. Direct observation of dendritic actin filament networks nucleated by Arp2/3 complex and WASP/Scar proteins. Nature. 404:1007–1011. [DOI] [PubMed] [Google Scholar]
- Blanchoin, L., T.D. Pollard, and R.D. Mullins. 2000. b. Interactions of ADF/cofilin, Arp2/3 complex, capping protein and profilin in remodeling of branched actin filament networks. Curr. Biol. 10:1273–1282. [DOI] [PubMed] [Google Scholar]
- Blanchoin, L., T.D. Pollard, and S.E. Hitchcock-DeGregori. 2001. Inhibition of the Arp2/3 complex-nucleated actin polymerization and branch formation by tropomyosin. Curr. Biol. 11:1300–1304. [DOI] [PubMed] [Google Scholar]
- Borisy, G.G., and T.M. Svitkina. 2000. Actin machinery: pushing the envelope. Curr. Opin. Cell Biol. 12:104–112. [DOI] [PubMed] [Google Scholar]
- Cao, L.G., D.J. Fishkind, and Y.L. Wang. 1993. Localization and dynamics of nonfilamentous actin in cultured cells. J. Cell Biol. 123:173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier, M.F., and D. Pantaloni. 1997. Control of actin dynamics in cell motility. J. Mol. Biol. 269:459–467. [DOI] [PubMed] [Google Scholar]
- Carlier, M.F., V. Laurent, J. Santolini, R. Melki, D. Didry, G.X. Xia, Y. Hong, N.H. Chua, and D. Pantaloni. 1997. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J. Cell Biol. 136:1307–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, A.Y., M. Bailly, N. Zebda, J.E. Segall, and J.S. Condeelis. 2000. Role of cofilin in epidermal growth factor–stimulated actin polymerization and lamellipod protrusion. J. Cell Biol. 148:531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condeelis, J. 2001. How is actin polymerization nucleated in vivo? Trends Cell Biol. 11:288–293. [DOI] [PubMed] [Google Scholar]
- Conley, C.A., K.L. Fritz-Six, A. Almenar-Queralt, and V.M. Fowler. 2001. Leiomodins: larger members of the tropomodulin (Tmod) gene family. Genomics. 73:127–139. [DOI] [PubMed] [Google Scholar]
- Cox, P.R., and H.Y. Zoghbi. 2000. Sequencing, expression analysis, and mapping of three unique human tropomodulin genes and their mouse orthologs. Genomics. 63:97–107. [DOI] [PubMed] [Google Scholar]
- Cramer, L.P. 1999. Role of actin-filament disassembly in lamellipodium protrusion in motile cells revealed using the drug jasplakinolide. Curr. Biol. 9:1095–1105. [DOI] [PubMed] [Google Scholar]
- Cramer, L.P., L.J. Briggs, and H.R. Dawe. 2002. Use of fluorescently labelled deoxyribonuclease I to spatially measure G-actin levels in migrating and non-migrating cells. Cell Motil. Cytoskeleton. 51:27–38. [DOI] [PubMed] [Google Scholar]
- Dawe, H.R., L.S. Minamide, J.R. Bamburg, and L.P. Cramer. 2003. ADF/cofilin controls cell polarity during fibroblast migration. Curr. Biol. 13:252–257. [DOI] [PubMed] [Google Scholar]
- DesMarais, V., I. Ichetovkin, J. Condeelis, and S.E. Hitchcock-DeGregori. 2002. Spatial regulation of actin dynamics: a tropomyosin-free, actin-rich compartment at the leading edge. J. Cell Sci. 115:4649–4660. [DOI] [PubMed] [Google Scholar]
- Elbashir, S.M., J. Harborth, K. Weber, and T. Tuschl. 2002. Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods. 26:199–213. [DOI] [PubMed] [Google Scholar]
- Fowler, V.M. 1996. Regulation of actin filament length in erythrocytes and striated muscle. Curr. Opin. Cell Biol. 8:86–96. [DOI] [PubMed] [Google Scholar]
- Fujiwara, I., S. Suetsugu, S. Uemura, T. Takenawa, and S. Ishiwata. 2002. a. Visualization and force measurement of branching by Arp2/3 complex and N-WASP in actin filament. Biochem. Biophys. Res. Commun. 293:1550–1555. [DOI] [PubMed] [Google Scholar]
- Goldschmidt-Clermont, P.J., R.M. Galbraith, D.L. Emerson, P.A. Werner, A.E. Nel, and W.M. Lee. 1985. Accurate quantitation of native Gc in serum and estimation of endogenous Gc: G-actin complexes by rocket immunoelectrophoresis. Clin. Chim. Acta. 148:173–183. [DOI] [PubMed] [Google Scholar]
- Gregorio, C.C., and V.M. Fowler. 1995. Mechanisms of thin filament assembly in embryonic chick cardiac myocytes: tropomodulin requires tropomyosin for assembly. J. Cell Biol. 129:683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichetovkin, I., W. Grant, and J. Condeelis. 2002. Cofilin produces newly polymerized actin filaments that are preferred for dendritic nucleation by the Arp2/3 complex. Curr. Biol. 12:79–84. [DOI] [PubMed] [Google Scholar]
- Kiosses, W.B., R.H. Daniels, C. Otey, G.M. Bokoch, and M.A. Schwartz. 1999. A role for p21-activated kinase in endothelial cell migration. J. Cell Biol. 147:831–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause, M., J.E. Bear, J.J. Loureiro, and F.B. Gertler. 2002. The Ena/VASP enigma. J. Cell Sci. 115:4721–4726. [DOI] [PubMed] [Google Scholar]
- Kuhlman, P.A., C.A. Hughes, V. Bennett, and V.M. Fowler. 1996. A new function for adducin. Calcium/calmodulin-regulated capping of the barbed ends of actin filaments. J. Biol. Chem. 271:7986–7991. [DOI] [PubMed] [Google Scholar]
- Littlefield, R., A. Almenar-Queralt, and V.M. Fowler. 2001. Actin dynamics at pointed ends regulates thin filament length in striated muscle. Nat. Cell Biol. 3:544–551. [DOI] [PubMed] [Google Scholar]
- Maciver, S.K. 1998. How ADF/cofilin depolymerizes actin filaments. Curr. Opin. Cell Biol. 10:140–144. [DOI] [PubMed] [Google Scholar]
- Maciver, S.K., H.G. Zot, and T.D. Pollard. 1991. Characterization of actin filament severing by actophorin from Acanthamoeba castellanii. J. Cell Biol. 115:1611–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogilner, A., and L. Edelstein-Keshet. 2002. Regulation of actin dynamics in rapidly moving cells: a quantitative analysis. Biophys. J. 83:1237–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaloni, D., C. Le Clainche, and M.F. Carlier. 2001. Mechanism of actin-based motility. Science. 292:1502–1506. [DOI] [PubMed] [Google Scholar]
- Pollard, T.D., L. Blanchoin, and R.D. Mullins. 2000. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 29:545–576. [DOI] [PubMed] [Google Scholar]
- Schafer, D.A., M.D. Welch, L.M. Machesky, P.C. Bridgman, S.M. Meyer, and J.A. Cooper. 1998. Visualization and molecular analysis of actin assembly in living cells. J. Cell Biol. 143:1919–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small, J.V., M. Herzog, and K. Anderson. 1995. Actin filament organization in the fish keratocyte lamellipodium. J. Cell Biol. 129:1275–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small, J.V., T. Stradal, E. Vignal, and K. Rottner. 2002. The lamellipodium: where motility begins. Trends Cell Biol. 12:112–120. [DOI] [PubMed] [Google Scholar]
- Small, J.V., G. Isenberg, and J.E. Celis. 1978. Polarity of actin at the leading edge of cultured cells. Nature. 272:638–639. [DOI] [PubMed] [Google Scholar]
- Symons, M.H., and T.J. Mitchison. 1991. Control of actin polymerization in live and permeabilized fibroblasts. J. Cell Biol. 114:503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot, J.A. 2000. The polymerization motor. Traffic. 1:19–28. [DOI] [PubMed] [Google Scholar]
- Weber, A., C.R. Pennise, G.G. Babcock, and V.M. Fowler. 1994. Tropomodulin caps the pointed ends of actin filaments. J. Cell Biol. 127:1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, A., C.R. Pennise, and V.M. Fowler. 1999. Tropomodulin increases the critical concentration of barbed end-capped actin filaments by converting ADP.P(i)-actin to ADP-actin at all pointed filament ends. J. Biol. Chem. 274:34637–34645. [DOI] [PubMed] [Google Scholar]
- Welch, M.D., A.H. DePace, S. Verma, A. Iwamatsu, and T.J. Mitchison. 1997. The human Arp2/3 complex is composed of evolutionarily conserved subunits and is localized to cellular regions of dynamic actin filament assembly. J. Cell Biol. 138:375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebda, N., O. Bernard, M. Bailly, S. Welti, D.S. Lawrence, and J.S. Condeelis. 2000. Phosphorylation of ADF/cofilin abolishes EGF-induced actin nucleation at the leading edge and subsequent lamellipod extension. J. Cell Biol. 151:1119–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.