

Abstract
Lysosome-related organelles are cell type–specific intracellular compartments with distinct morphologies and functions. The molecular mechanisms governing the formation of their unique structural features are not known. Melanosomes and their precursors are lysosome-related organelles that are characterized morphologically by intralumenal fibrous striations upon which melanins are polymerized. The integral membrane protein Pmel17 is a component of the fibrils and can nucleate their formation in the absence of other pigment cell–specific proteins. Here, we show that formation of intralumenal fibrils requires cleavage of Pmel17 by a furin-like proprotein convertase (PC). As in the generation of amyloid, proper cleavage of Pmel17 liberates a lumenal domain fragment that becomes incorporated into the fibrils; longer Pmel17 fragments generated in the absence of PC activity are unable to form organized fibrils. Our results demonstrate that PC-dependent cleavage regulates melanosome biogenesis by controlling the fibrillogenic activity of a resident protein. Like the pathologic process of amyloidogenesis, the formation of other tissue-specific organelle structures may be similarly dependent on proteolytic activation of physiological fibrillogenic substrates.
Keywords: furin; endosome; proteolysis; fibril; lysosome-related organelle
Introduction
Organelle function and morphology are intimately related, yet the molecular mechanisms that underlie organelle morphogenesis are largely uncharacterized. Organelle morphology within the central vacuolar system is controlled by extrinsic factors, such as cytoskeletal or “matrix” proteins (De Matteis and Morrow, 2000; Warren and Shorter, 2002), and by intrinsic factors, such as resident integral membrane and lumenal contents (Valladeau et al., 2000). Organelle morphogenesis must be regulated to ensure proper temporal and spatial control; disregulation may result in disease, as exemplified by the defects in lysosome-related organelles in patients with Hermansky-Pudlak Syndrome and corresponding mouse models (Swank et al., 1998; Spritz, 1999; Dell'Angelica et al., 2000). Understanding how morphogenesis of lysosome-related organelles is controlled may help to elucidate the etiology of these disorders.
Melanosomes serve as an excellent model to define biogenetic mechanisms among lysosome-related organelles. Melanosomes are specialized organelles within melanocytes and retinal pigment epithelial cells in which melanin pigments are generated and stored (Marks and Seabra, 2001; Raposo and Marks, 2002). As they are synthesized within maturing (stage III and IV) melanosomes, melanins are deposited along characteristic intralumenal fibers that appear before pigment deposition in stage II premelanosomes. Like the dense cores of secretory granules, the fibers serve to sequester and concentrate cargo—in this case melanins; they also likely function to detoxify oxidative melanin intermediates (Seiji et al., 1963; King et al., 1995). The fibers are thought to be proteinaceous because they lack the ultrastructural hallmarks of a lipid bilayer (Moyer, 1966; Maul, 1969; Hearing et al., 1973). Thus, they are similar to intralumenal fibrils that form in certain pathologies associated with amyloid deposition (Badman et al., 1996; Kim et al., 1999; Chen et al., 2001). The formation of fibrils represents a key step in melanosome biogenesis, but their composition and the molecular mechanisms underlying their formation are unknown.
Pmel17 (also known as gp100, ME20, and Silver) is a type I integral membrane protein, uniquely expressed by melanocytes and retinal pigment epithelial cells, that localizes to the premelanosome matrix (Kwon et al., 1987; Vennegoor et al., 1988; Adema et al., 1994; Kobayashi et al., 1994; Zhou et al., 1994; Lee et al., 1996; Raposo et al., 2001). A role for Pmel17 in melanosome function is supported by the pigmentation defect in silver mice (Dunn and Thigpen, 1930), in which the Pmel17 protein is truncated (Martínez-Esparza et al., 1999). Pmel17 immunolocalizes to the fibrils within stage II premelanosomes (Berson et al., 2001; Raposo et al., 2001), and Pmel17 expression is sufficient to induce the formation of premelanosome-like fibrils within multivesicular bodies (MVBs)* of nonpigment cells (Berson et al., 2001). Nevertheless, Pmel17 in nondenaturing detergent extracts of melanocytes or transfected HeLa cells remains covalently tethered to its transmembrane domain, even after the precursor polypeptide is cleaved into two fragments (Berson et al., 2001). This implies a continued association with membranes, belying the inferred nonmembranous nature of the striations. To probe the mechanism by which Pmel17 initiates fibril formation, we asked (1) what is the function of the intralumenal cleavage; and (2) what is the biochemical nature of Pmel17 on the intralumenal striations? Our results highlight a role for proprotein convertases (PCs) in organelle biogenesis, and reveal a striking parallel between the formation of the morphological hallmark of premelanosomes and the pathological fibril formation in amyloid disease.
Results
Identification of Pmel17 cleavage site and proteolysis by a PC
In human melanocytes, melanoma cells, and transfected nonpigment cells, Pmel17 is proteolytically cleaved within a post-Golgi compartment (Berson et al., 2001). Intralumenal cleavage generates a COOH-terminal 28-kD Mβ fragment containing part of the lumenal domain and the entire transmembrane and cytoplasmic domains, and an ∼80-kD Mα fragment consisting of the remaining NH2-terminal region of the lumenal domain (Fig. 1 A). Both fragments are stoichiometrically coprecipitated from cell lysates prepared with Triton X-100 (TX); a small amount of Mα, free of Mβ, is precipitated from cell supernatants (Berson et al., 2001). Mα migrates identically to ME-20s, consisting of Pmel17 residues 25–467, identified in melanoma cell supernatants (Maresh et al., 1994).
Figure 1.
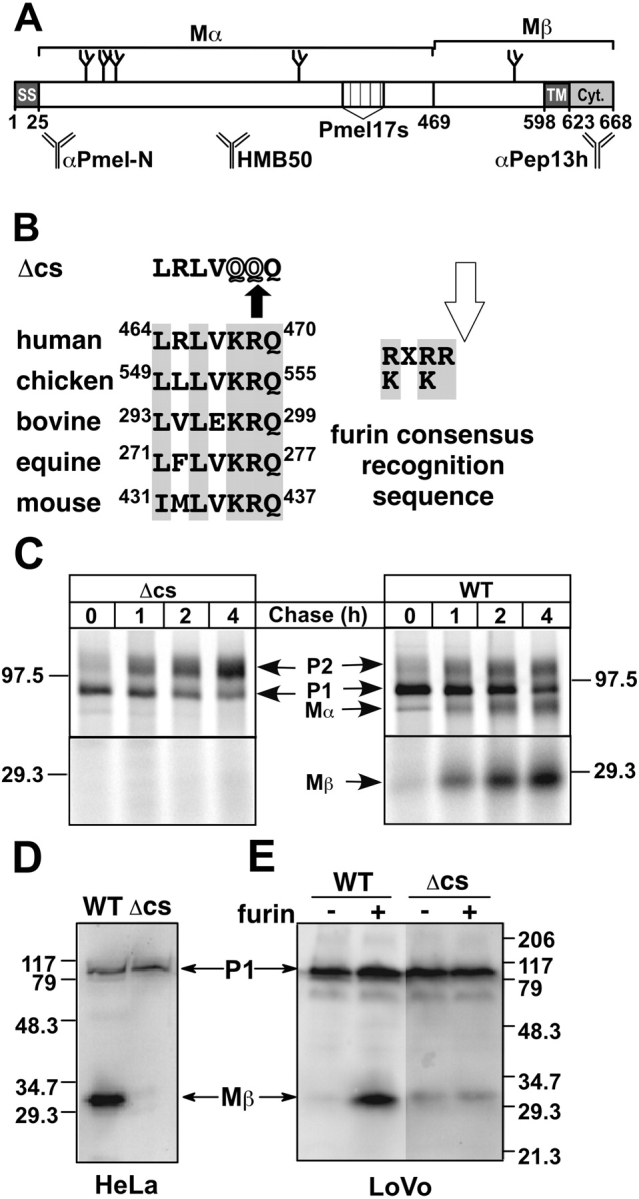
Identification of a conserved PC cleavage site in Pmel17. (A) Schematic diagram of the primary structure of Pmel17 drawn to scale. Numbers correspond to amino acid positions; positions of the signal sequence (SS), transmembrane (TM), and cytoplasmic (Cyt.) domains, aa 469, and a lumenal region deleted in the alternative spliced form, Pmel17-s, are indicated; N-linked glycans are represented by branched structures; regions corresponding to Mα and Mβ are shown above; and regions recognized by antibodies αPmel-N, HMB50, and αPep13h are shown below. (B) Sequence of the conserved dibasic consensus site from orthologues human Pmel17 (GenBank/EMBL/DDBJ accession no. NP_008859), chicken MMP115 (GenBank/EMBL/DDBJ accession no. Q98917), bovine RPE1 (GenBank/EMBL/DDBJ accession no. Q06154), equine Pmel17 (GenBank/EMBL/DDBJ accession no. AAC97108), and mouse Silver (GenBank/EMBL/DDBJ accession no. NP_068682). Amino acid positions are noted (equine and bovine sequences are from incomplete clones); conserved residues are in gray. Top, Pmel17Δcs (Δcs) sequence. Right, consensus recognition sequence for furin; cleavage site is indicated by arrow. (C) Cleavage is inhibited by mutagenesis of the dibasic site. Transiently transfected HeLa cells expressing wild-type Pmel17 (WT) or Pmel17Δcs (Δcs) were metabolically pulse labeled and chased as indicated. TX cell lysates at each time point were immunoprecipitated with αPEP13h, fractionated by SDS-PAGE, and analyzed by phosphorimaging. Shown are regions of the gels encompassing the relevant bands; the signal from the lower region was enhanced fourfold to emphasize the failure to detect Mβ from Pmel17Δcs. Migration of molecular weight markers and P1, P2, Mα, and Mβ forms of Pmel17 are indicated. (D and E) Western blot analysis of wild-type Pmel17 (WT) and Pmel17Δcs (Δcs) expressed transiently by transfection in (D) HeLa or (E) LoVo cells. In E, LoVo cells were cotransfected (+) or not (−) with a plasmid encoding rat furin. Whole-cell lysates from transfected cells were fractionated by SDS-PAGE, transferred to PVDF membranes, and probed using αPEP13h with enhanced chemifluorescence. Positions of molecular weight markers and P1 and Mβ forms of Pmel17 are indicated.
Immediately after V467 in Pmel17 is a conserved dibasic pair within a consensus sequence that is similar to the recognition element for PCs of the furin family of proteases (Fig. 1 B; Hosaka et al., 1991). PCs and associated carboxypeptidases mediate regulated cleavage of proproteins within late secretory compartments and endosomes (Steiner, 1998; Varlamov and Fricker, 1998; Thomas, 2002). ME20s/Mα could potentially be generated by PC-dependent cleavage at R469 followed by removal of the dibasic pair by a carboxypeptidase D- or E-like activity. To determine whether Pmel17 cleavage requires the dibasic pair, KR468–469 was altered to QQ by site-directed mutagenesis, generating Pmel17Δcs (Fig. 1 B). Unlike wild-type Pmel17, Pmel17Δcs expressed in transfected HeLa cells was not processed to Mα and Mβ (Fig. 1, C and D). Instead, cells accumulated the P2 form, representing full-length Pmel17Δcs with Golgi-modified N-linked glycans (Berson et al., 2001). Furthermore, although cells expressing wild-type Pmel17 secrete low levels of Mα into the medium, cells expressing Pmel17Δcs did not (unpublished data).
To determine whether PCs are required for the cleavage, Pmel17 or the Δcs variant were expressed in LoVo, a human adenocarcinoma cell line lacking furin (Takahashi et al., 1995) and several other PCs (Miranda et al., 1996). Cleavage in transfected LoVo or control HeLa cells was assessed by Western blotting of whole-cell lysates with αPEP13h, an antibody to the cytoplasmic domain. Lysates from HeLa cells expressing wild-type Pmel17, like melanocytic cells, contain two immunoreactive bands with Mr 28,000 and ∼100,000, representing Mβ and the full-length, endoglycosidase H (endoH)-sensitive P1 precursor, respectively (Fig. 1 D; see Fig. 5 A). By contrast, in both HeLa cells expressing Pmel17Δcs (Fig. 1 D) and LoVo cells expressing wild-type Pmel17 (Fig. 1 E), only the ∼100-kD P1 band and not Mβ was observed, indicating that cleavage did not occur (a faint residual band that co-migrates with Mβ in LoVo cells expressing Pmel17Δcs may result from cleavage by NARC-1, a related protease that is active within the ER and for which a sequence similar to the consensus recognition site is generated by the KR468–469→QQ mutation; Seidah et al., 2003). Mβ was restored in LoVo cells by coexpression of both Pmel17 and furin, but not Pmel17Δcs and furin (Fig. 1 E). Thus, furin or a similar PC can cleave Pmel17 at the dibasic recognition site. Importantly, cleavage at the only other dibasic sites in Pmel17, KR147, or RR192, would not generate fragments of the appropriate size for Mα and Mβ.
Figure 5.

Mα accumulation in TX- insoluble fractions of MNT-1 cells and inhibition by α1-PDX. (A and B) Western blot analyses of MNT-1 cell fractions. Lysates fractionated by SDS-PAGE were probed with antibodies αPep13h (to COOH terminus) or αPmel-N (to NH2 terminus). (A) Whole-cell lysates were untreated or treated with endoH (H) or N-glycanase F (F). (B) Whole-cell lysates (T) and TX-soluble (S) or -insoluble (I) material were analyzed directly. Relevant bands are indicated; bands annotated with ′ are glycosidase cleavage products. (C–F) Pulse-chase/immunoprecipitation analyses of Mα formation in MNT-1 cells. Metabolically pulse-labeled and chased MNT-1 cells were extracted with TX, and TX-insoluble pellets were re-extracted in lysis buffer containing 8M urea at 60°C. TX-soluble and -insoluble/urea extracted fractions (after dilution of the urea) were sequentially immunoprecipitated first with normal rabbit serum, then with αPEP13h, and finally with αPmel-N. Immunoprecipitates were fractionated by SDS-PAGE and analyzed by phosphorimaging. Chase times (h) are indicated at top (C and F) or bottom (D and E); migration of molecular weight markers and relevant bands are indicated to the side. (C) αPep13h and αPmel-N immunoprecipitates from both fractions are shown. Panels on the right were exposed 22× longer than those on the left because of vastly reduced efficiency of immunoprecipitation after treatment of lysates with urea (unpublished data). Note the appearance of Mα in αPmel-N immunoprecipitates at later time points. (D) The intensity of indicated bands in each lane from the soluble fraction of C was quantitated by phosphorimaging, and ratios of the intensities of P1 to P2, Pmel17-s/Mα (Mα/X), and Mβ at each time point, precipitated by either αPep13h or αPmel-N, were calculated and plotted; the Pmel17-s and Mα bands were summed because they could not be easily resolved. Note that for each time point, ratios were similar for both antibodies. (E) Intensities of P1, P2, Pmel17-s, and Mα bands were summed at each time point for both antibodies in both soluble (sol) and insoluble (insol) fractions, and plotted as a percentage of the value at time 0. Note the loss of material from all fraction/antibody combinations except the insoluble fraction precipitated with αPmel-N, in which intensity increased over time. (F) Cells were uninfected or infected with recombinant adenoviruses expressing α1-PDX at moi of 5, PDX(5); or 20, PDX(20); or α1-AT at moi of 20, AT(20), before the experiment; shown are only the relevant regions of the αPmel-N immunoprecipitates from the TX insoluble fraction. Arrows, bands that co-migrate with Mα or a novel product (?) precipitated only from cells expressing α1-PDX.
To determine whether a furin-like PC cleaves Pmel17 in melanocytic cells, we inhibited PCs in the pigmented human melanoma, MNT-1, by expressing α1-antitrypsin Portland (α1-PDX), a PC-specific variant of α1-antitrypsin inhibitor (α1-AT; Anderson et al., 1993; Jean et al., 1998, 2000). FLAG epitope–tagged α1-PDX or α1-AT were expressed in MNT-1 cells by infection with recombinant adenoviruses. Transgene expression and Pmel17 processing were assessed by metabolic pulse/chase, immunoprecipitation, SDS-PAGE, and phosphorimaging analysis. Both transgenes were efficiently expressed, although a lower multiplicity of infection (moi) with the α1-PDX virus was required to obtain the same level of expression as α1-AT (Fig. 2 A; likely due to contaminating nonrecombinants in the α1-AT preparation). As described previously, Pmel17 in uninfected cells first appears as endoH-sensitive P1 and Pmel17-s forms (Berson et al., 2001). P1 is the full-length core glycosylated precursor, and Pmel17-s is the product of an alternatively spliced mRNA that is generated in melanocytic cells (Nichols et al., 2003), but not transfected HeLa cells (Berson et al., 2001); Pmel17-s is distinguished from Mα based on its direct reactivity with αPep13h, its appearance in the pulse-labeled samples, and its sensitivity to EndoH (see Fig. 5). P1 and Pmel17-s are subsequently modified in the Golgi to endoH-resistant P2, and P2 is then cleaved to Mα and Mβ (Fig. 2 B, lanes 1–5). All fragments disappeared from detergent cell lysates with a half-life of 2.5 h. In cells expressing α1-AT, the pattern and kinetics of Pmel17 processing were virtually unchanged (Fig. 2 B, lanes 6–10). However, in cells expressing α1-PDX, as in HeLa cells expressing Pmel17Δcs, P2 accumulated, and generation of Mα/Mβ was inhibited by at least 90% (Fig. 2 B, lanes 11–20; Fig. 2 C). Cleavage was equally inhibited in cells expressing two different levels of α1-PDX (Fig. 2, B and C), likely reflecting the secretion of α1-PDX by infected cells and uptake by uninfected cells. By contrast, α1-PDX did not affect the maturation and half-life of another melanosome protein, Tyrp1 (unpublished data). These data confirm that furin or a furin-like PC mediates Pmel17 cleavage in melanocytic cells.
Figure 2.

PCs mediate Pmel17 cleavage in melanocytic cells. MNT-1 cells were uninfected (U) or infected with recombinant adenoviruses encoding tTA (moi of 20) and either FLAG-tagged α1-AT (moi of 20; A20) or α1-PDX at moi of 20 (P20) or 5 (P5). Two days after infection, cells were metabolically pulse labeled and chased for the indicated periods of time. Immunoprecipitates from TX cell lysates at indicated time points were fractionated by SDS-PAGE and analyzed by phosphorimaging. (A) Epitope-tagged α1-AT and α1-PDX immunoprecipitated from pulse-labeled cell lysates using an anti-FLAG antibody. Similar levels of transgene expression were obtained using α1-PDX at moi of 5 and α1-AT at moi of 20. (B) Pmel17 immunoprecipitated using αPEP13h. Only the relevant portion of the gels are shown; no other specific bands were consistently observed. The intensity of the lower part of the gel was amplified 2× to emphasize the Mβ band. Positions of molecular weight markers (right) and of P1, Pmel17-s, P2, Mα, and Mβ (left) are noted. Pmel17-s is the product of an alternatively spliced Pmel17 mRNA. (C) Quantitation of the signal intensity of P1, P2, and Mβ from B. Data represent the percentage of signal from each band relative to the values for P1 at 0 chase for each group (average from three experiments). Note that the predicted cysteine and methionine content of Mβ represents 42% of that present in full-length Pmel17.
PC cleavage is not required for localization of Pmel17 to multivesicular endosomes
Pmel17 transits through a multivesicular coated endosome en route to striated stage II premelanosomes in pigmented cells (Raposo et al., 2001). Similarly, Pmel17 in transfected HeLa cells localizes mainly to intralumenal vesicles (ILVs) of MVBs (Berson et al., 2001). To determine whether this localization step requires PC-dependent cleavage, HeLa cells expressing wild-type Pmel17 or Pmel17Δcs were analyzed by immunofluorescence microscopy (IFM) and immunoelectron microscopy (IEM). Pmel17Δcs, like wild-type Pmel17 (Berson et al., 2001), localized by IFM to vesicles that overlapped with a large fraction of vesicles that contained the late endosome/lysosome marker, LAMP1 (unpublished data). IEM of ultrathin cryosections identified these structures as MVBs, in which Pmel17Δcs accumulated largely in the ILVs (Fig. 3 A). Similarly, Pmel17 localization to multivesicular-coated endosomes in MNT-1 melanoma cells was not affected by expression of α1-PDX (Fig. 4 E). Together with the normal half-life in TX cell lysates and kinetics of conversion from P1 to P2 (Fig. 1 C and Fig. 2 B), these data indicate that ER export and post-Golgi trafficking of Pmel17 are not dramatically influenced by loss of PC cleavage.
Figure 3.
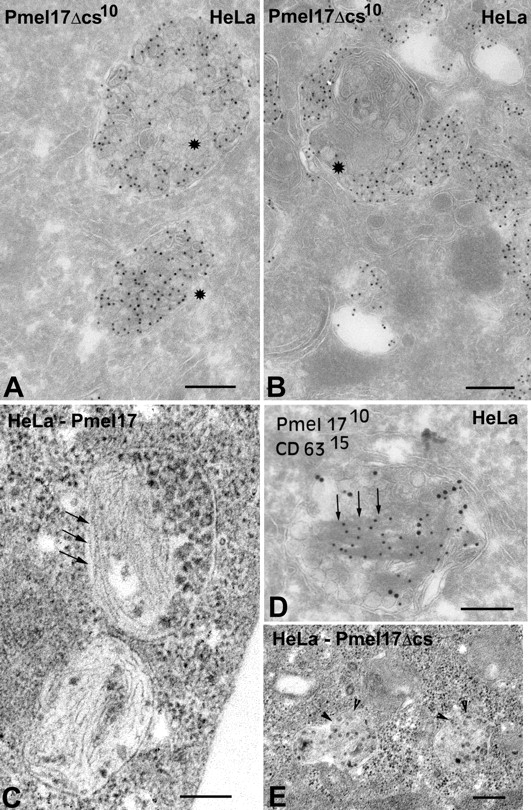
Cleavage site–deficient Pmel17 localizes to MVBs, but fails to form fibers. Transiently transfected HeLa cells expressing Pmel17Δcs (A, B, and E) or wild-type Pmel17 (C and D) were analyzed by either IEM (A, B, and D) or standard EM (C and E). (A, B, and D) Ultrathin cryosections were immunogold labeled with HMB50 and PAG-10; in D, sections were double labeled with anti-CD63 and PAG-15. (A) A typical Pmel17Δcs-expressing cell showing predominant labeling on ILVs of multivesicular late endosomes. (B) A cell with very high Pmel17Δcs expression; note the gold labeling over electron-dense aggregates in compartments with multiple layers of membranes (stars). (D) A cell with high wild-type Pmel17 expression; note the gold labeling over ILVs and fibrous structures within MVBs (arrows), and exclusion of CD63 labeling from the striated regions. (C) Standard EM of a cell expressing wild-type Pmel17. Note the fibers within MVBs (arrows). (E) Standard EM of a cell expressing Pmel17Δcs. Note the accumulation of electron-dense material in MVBs, but no obvious fibers (arrowheads). Bars, 200 nm.
Figure 4.
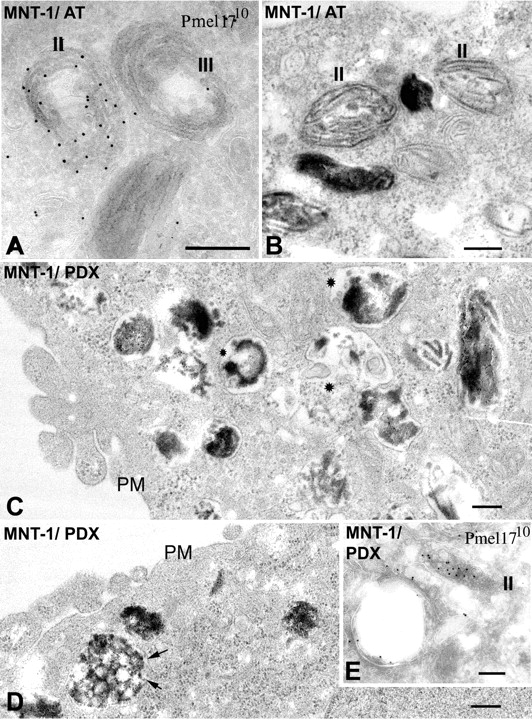
Disruption of melanosome morphology in MNT-1 cells expressing α1-PDX. MNT-1 cells infected with recombinant adenoviruses expressing tTA and either α1-AT (A and B) or α1-PDX (C–E) were analyzed by either IEM (A and E) or standard EM (B–D). (A and E) Ultrathin cryosections were immunogold labeled with HMB50 and PAG-10. (A and B) Note the characteristic striations of stage II and III premelanosomes (II and III) in cells expressing α1-AT. By IEM, these structures are densely labeled for Pmel17. (C and D) Note the absence of normal stage II and III premelanosomes in cells expressing α1-PDX, and the appearance of unusual melanin aggregates in disordered arrays (C, stars) and multivesicular structures (D, arrows). (E) By IEM, Pmel17 is detected normally in endosomes with a planar clathrin coat (bottom left), but dense labeling is limited to abnormal, nonstriated, multivesicular structures with the shape of stage II premelanosomes (II). PM, plasma membrane. Bars, 200 nm.
Formation of fibrous striations requires PC cleavage
Structures similar to the fibrous striations of premelanosomes are frequently observed in MVBs of transfected HeLa cells that express high levels of wild-type Pmel17 (Berson et al., 2001). By contrast, such structures were not observed in HeLa cells expressing comparable or higher levels of Pmel17Δcs (Table I and Fig. 3 E). Instead, Pmel17Δcs accumulated in irregular electron-dense aggregates within compartments that contained intralumenal membrane sheets rather than vesicles, and that thus resembled lysosomes (Fig. 3, B and E). Accordingly, these compartments labeled densely for LAMP1 by IEM (unpublished data). These data suggest that if Pmel17 is not cleaved by PCs, its ability to nucleate striation formation is inhibited concomitant with enhanced movement from late endosomes to lysosomes. The dense aggregates observed by IEM may represent intermediates in the degradation of Pmel17 by lysosomal proteases (see Fig. 4 and Fig. 5) .
Table I. Quantitation of Pmel17 localization to MVBs and striations in transfected HeLa cells.
| HeLa Pmel17 WT | HeLa Pmel17Δcs | |
|---|---|---|
| MVBs | 57% | 51% |
| MVBs + striations | 43% | 2%a |
| Lysosomes + aggregates | – | 47% |
HeLa cells expressing Pmel17 WT or Pmel17Δcs were fixed, and ultrathin cryosections were labeled with HMB50 anti-Pmel17 antibody and PAG-10. 50 cell profiles of each were analyzed for the presence of gold labeling in MVBs or lysosomes, and for the association of the gold particles with striation-like structures or aggregates. Percentages are relative to the total number of structures analyzed in all cell profiles (150 compartments for WT, 160 for Δcs).
These correspond to MVBs with non-organized proteinaceous material.
To determine whether Pmel17 cleavage was required for striation formation in melanocytic cells, MNT-1 cells expressing α1-PDX or α1-AT were analyzed by EM (Fig. 4). Because α1-PDX or α1-AT are secreted, even uninfected cells in the cultures were partially subject to their effects. Although expression of α1-AT had little effect on the abundance or morphology of melanosomes or on the localization of Pmel17 (Fig. 4, A and B), many cells expressing α1-PDX were depleted of striated stage II premelanosomes, and most accumulated irregular pigment-containing structures (Fig. 4, C–E). Some of these structures were multivesicular (Fig. 4 D), and others contained irregular melanin aggregates (Fig. 4 C). Ultrastructurally normal stage II, III, and IV melanosomes in these cells were reduced in number; quantitative analyses of 50 cell profiles in Epon sections indicated that 16% lacked any stage II premelanosomes, and an additional 72% had disorganized melanosome structures. By IEM, Pmel17 in these cells accumulated in membrane-enclosed structures with disordered aggregates of fuzzy electron-dense material, similar to those observed in Pmel17Δcs-expressing HeLa cells (compare Fig. 4 A with Fig. 4 E); these likely represent malformed stage II premelanosomes, late endosomes, or lysosomes with mistargeted Pmel17. By contrast, Pmel17 labeling in both the limiting and intralumenal membranes of coated endosomes in MNT-1 cells was not affected by α1-PDX expression (Fig. 4 E). These results confirm the importance of Pmel17 cleavage in the formation of characteristic intralumenal striations of melanosomes.
Association of the lumenal domain fragment with TX-insoluble complexes
The dependence of striation formation on PC cleavage of Pmel17 would support a model in which Mα fragments form intralumenal proteinaceous fibrils upon their release from membranes. By analogy to amyloid, such fibrils might be insoluble in nondenaturing detergents such as TX. Indeed, Mα was observed in TX-insoluble fractions of MNT-1 cells. By Western blotting, an endoH-resistant, N-glycanase F–sensitive Mα band was detected in whole MNT-1 cell lysates with αPmel-N (antibody to the lumenally exposed NH2 terminus of Pmel17), but not with αPep13h (antibody to the cytoplasmically exposed COOH terminus; Fig. 5 A), distinguishing it from the endoH-sensitive, αPep13h-reactive alternative splice product, Pmel17-s. Although most Mβ, Pmel17-s, and P1 isoforms were soluble in TX, a significant fraction of Mα was detected in the TX-insoluble pool (Fig. 5 B).
Formation of TX-insoluble Mα was kinetically linked to both cleavage and dissociation from Mβ by a metabolic pulse/chase assay in which TX-insoluble material from cell lysates was extracted by heating in 8M urea. Pmel17 isoforms from this TX-insoluble pool (after dilution of the urea) and from the soluble fraction were sequentially immunoprecipitated with αPEP13h and then αPmel-N, then analyzed by SDS-PAGE and phosphorimaging. From the soluble pool, αPmel-N only immunoprecipitated residual P1, P2, Pmel17-s, Mα, and Mβ in similar proportions as in the initial αPep13h immunoprecipitates (Fig. 5, C and D; compare lanes 1–4 with lanes 5–8). From the TX-insoluble pool, although αPep13h immunoprecipitated only full-length P1 and Pmel17-s (Fig. 5 C, lanes 9–12), subsequent αPmel-N immunoprecipitates additionally contained an ∼80-kD band (lanes 13–16); its failure to be depleted by αPep13h suggests that this band is Mα. The intensity of this band increased over time from undetectable at the pulse to most intense by 4 h of chase (Fig. 5, C and E). Thus, at least a portion of Mα was released from Mβ during the chase and accumulated in TX-insoluble complexes.
To confirm the identity of this TX-insoluble band as Mα and to determine whether its accumulation required PC cleavage, MNT-1 cells expressing α1-PDX were similarly analyzed by pulse/chase and immunoprecipitation. As shown in Fig. 5 F, expression of α1-PDX (but not α1-AT) inhibited the formation of TX insoluble Mα. Appearance of the fragment was both delayed and reduced in efficiency by expression of α1-PDX. Instead, these cells accumulated a larger fragment of Pmel17, lacking the COOH terminus and possibly comprising the entire lumenal domain (Fig. 5 F; band marked “?”). Appearance of this band correlates with the disordered aggregates observed by IEM (Fig. 4 E), and likely results from cleavage within the juxtamembrane region by lysosomal proteases. These data show that formation of intact fibrils correlates with appearance of TX-insoluble Mα.
The premelanosome fibrils contain TX-insoluble Mα
If the fibrils consist of Mα fragments, they should label with antibodies to the Pmel17 lumenal domain, but not to the cytoplasmic domain. IEM analyses confirm this prediction. In MNT-1 cells, αPEP13h labels the coated endosome precursor to stage II premelanosomes, including the ILVs within them, but not the striated stage II premelanosomes themselves (Fig. 6 A; also see supplemental materials in Raposo et al., 2001, available at http://www.jcb.org/cgi/content/full/152/4/809/DC1/3). Concordantly, in transfected HeLa cells expressing high levels of Pmel17, αPEP13h labeling is observed on the ILVs of MVBs (Fig. 6 B), but not on the striated regions that form within the same multivesicular structures (Fig. 6 C). By contrast, antibodies to the Pmel17 lumenal domain label both the ILVs and the striations (Fig. 6, B and C). These data confirm that the fibrils lack the cytoplasmic domain present on full-length Pmel17 and the Mβ fragment.
Figure 6.
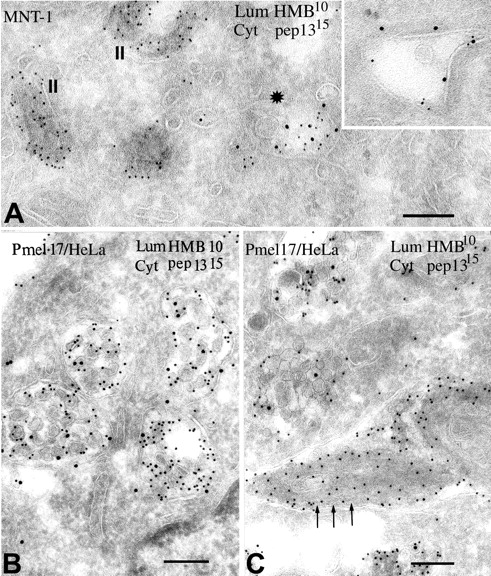
Failure to detect Pmel17 COOH terminus in striated regions of premelanosomes and MVBs. Ultrathin cryosections of (A) MNT-1 or (B and C) transiently transfected HeLa cells expressing wild-type Pmel17 were double immunogold–labeled with HMB50 and PAG-10 and with αPep13h and PAG-15. (A) A section of MNT-1 cells showing labeling by both antibodies on multivesicular coated endosomes (asterisk and inset), but only by HMB50 on stage II premelanosomes (II). Note the intralumenal labeling by αPep13h in the marked coated endosome (asterisk). (B) A typical section of transfected HeLa cells containing abundant multivesicular compartments. Note the labeling with both antibodies, with HMB50 labeling on the lumenal face and αPEP13h labeling primarily on the interior of the vesicles. (C) A section from a transfected HeLa cell demonstrating nascent striations. Note the absence of αPEP13h labeling in the striated regions (arrows) and on closely apposed membranes (arrows). Bars, 200 nm.
To determine directly whether the premelanosome fibrils contain TX-insoluble Mα fragments, MNT-1 cells were analyzed by subcellular fractionation. Light and dense fractions were harvested from post-nuclear cell homogenates by differential centrifugation, and membranes isolated from each fraction were subfractionated into TX-soluble and -insoluble pools (Fig. 7 A). The TX-soluble pools of both fractions contained P1 and Pmel17-s (representing pre-Golgi–localized Pmel17) and Mβ (Fig. 7 B, lanes 6 and 9); these products were nearly completely excluded from the TX-insoluble pool (lanes 7 and 10). By contrast, although Mα was also distributed among both membrane fractions, within the dense membrane fraction it was enriched in the TX-insoluble pool (Fig. 7 B, lane 7). IEM analyses of the dense membrane fraction revealed melanosomes of all stages and other dense organelles such as lysosomes and late endosomes (Fig. 7, C and D). The limiting membranes of mature melanosomes labeled densely for Tyrp1 (Fig. 7 C); very little labeling for Pmel17 was observed on these nonpermeabilized, fixed samples (Fig. 7 D), likely due to sequestration within the lumen at a distance from the limiting membrane. As expected, no intact structures were recovered from the TX-soluble pool by IEM (unpublished data). By contrast, the insoluble pool consisted primarily of masses of pigment and nonpigmented fibrous structures, largely free of limiting membranes, that appeared to derive from stage II–IV melanosomes (Fig. 7, E–G). The fibers labeled densely with antibody to the Pmel17 lumenal domain (Fig. 7, E and G), but not with αPep13h (not depicted) or antibody to Tyrp1 (Fig. 7 F); by contrast, remaining limiting membrane surrounding the pigment labeled with antibody to Tyrp1 but not to Pmel17 (Fig. 7 F). Thus, TX-insoluble fibers contain TX-insoluble Mα fragments. Together, the data show that formation of striations results at least in part from the incorporation of Mα fragments into nonmembrane associated, TX-insoluble fibrous structures.
Figure 7.
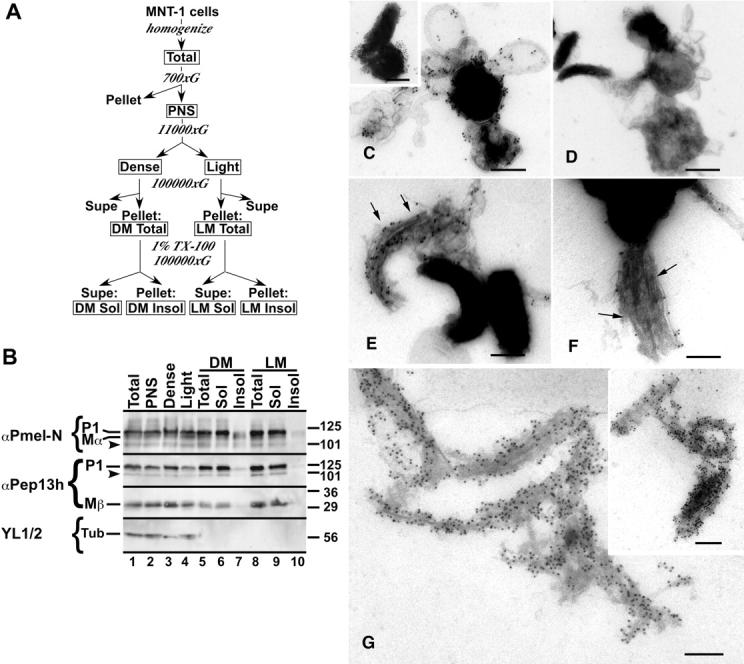
Cofractionation of TX-insoluble Mα and premelanosome fibers. (A) Scheme for fractionation of post-nuclear supernatants (PNS) from homogenates of MNT-1 cells. Boxed fractions were analyzed by Western blotting in B. (B) Equal cell equivalents from each fraction were analyzed by SDS-PAGE and Western blotting with αPmel-N, αPep13h, or the anti-tubulin antibody YL 1/2, as indicated. Shown are the relevant portions of each gel; positions of molecular weight markers (right) and relevant proteins (left) are indicated. Note the enrichment of only Mα in the TX-insoluble pool of the DM fraction (lane 7). (C–G) IEM analyses of relevant fractions using HMB45 (to Pmel17 lumenal domain) or TA99 (to Tyrp1) and PAG-10. (C and D) Nonsolubilized membranes from the dense fraction (lane 5 in B) labeled with TA99 (C) or HMB45 (D). Note the labeling for Tyrp1 on the limiting membrane of highly pigmented structures (particularly in inset), and the absence of HMB45 labeling on these nonpermeabilized, whole-mounted fractions. (E–G) TX-insoluble membranes from the dense fraction (lane 7 in B) labeled with HMB45 (E and G) or TA99 (F). Note the dense labeling of fibrous structures (arrows), including relatively intact contents of a stage II premelanosome (G, inset), by HMB45. TA99 only sparsely labels residual limiting membranes (F, arrows). Bars, 200 nm.
Discussion
Morphogenetic properties of resident intraorganellar proteins must be tightly regulated to ensure that organelle biogenesis proceeds at the proper place and time. One way to regulate these properties is by specific modifications of resident proteins during passage through transport intermediates. Our results demonstrate that premelanosome morphogenesis is regulated at the level of initiating intraorganellar fibrillogenesis through controlled PC-dependent proteolysis of a resident glycoprotein. These observations may reflect a more general requirement for regulated proteolysis of resident proteins by PCs and other proteases in the biogenesis of lysosome-related organelles; a similar model has been invoked for the formation of Weibel-Palade bodies in endothelial cells (Journet et al., 1993) and lamellar bodies in lung epithelial cells (Guttentag et al., 2003). Furthermore, our results extend the parallels between lysosome-related organelles and conventional dense core secretory granules, for which a biogenetic role for PCs is well established (Arvan and Castle, 1998). Finally, our data elucidate the molecular basis for a well-known but enigmatic feature of melanosome morphology, the intralumenal fibrous striations, and show a striking parallel between the physiologic formation of these fibrils and the pathogenic formation of amyloid fibers.
Role of PCs in organelle morphogenesis
PC cleavage is required to activate numerous secreted and cell surface proproteins and viral fusion proteins (Thomas, 2002), but few intracellular substrates are known. Here, we used two approaches to show that cleavage of Pmel17 by furin or a related PC is required to generate premelanosome fibrils. The formation of both fibrils and the mature Pmel17-derived fragments, Mα and Mβ, was blocked by (1) mutagenesis of the PC recognition site of Pmel17 in an exogenous expression system, and (2) inhibition of PC activity in a melanocytic cell line. Previous reports demonstrated defects in Weibel-Palade bodylike structures in an ectopic expression system upon mutagenesis of a PC site in von Willebrand factor (Journet et al., 1993), and in lamellar body formation in lung epithelial type II cells upon treatment with a cysteine protease inhibitor (Guttentag et al., 2003). However, this is the first time that both approaches have been used to conclusively demonstrate a role for PCs or other proteases in the activation of a single substrate protein to initiate morphogenesis of a lysosome-related organelle.
The relevant PC in melanocytic cells may be furin; furin is expressed in melanocytic cells (unpublished results), is capable of cleaving Pmel17 in reconstituted LoVo cells (Fig. 1 E), and is somewhat selectively inhibited by α1-PDX (Jean et al., 1998). On the other hand, the complement of PCs expressed by pigment cells is not known, and a different PC may function physiologically because α1-PDX can inhibit other PCs (Jean et al., 1998), and the Pmel17 cleavage site differs from the consensus furin recognition site (Fig. 1 B). We have shown that cleavage occurs within an acidic, post-Golgi, prelysosomal compartment that kinetically precedes stage II premelanosomes (Berson et al., 2001), consistent with the known localization of furin and other PCs to the TGN, endosomes, and secretory granules (Molloy et al., 1999; Thomas, 2002). The residual cleavage that occurs in the presence of brefeldin A and that results in a small pool of endoH-sensitive Mβ (Berson et al., 2001), like the residual cleavage of Pmel17Δcs in LoVo cells (Fig. 1 E), is likely mediated by NARC-1 or a related protease (Seidah et al., 2003). Of note, the accumulation of Golgi-processed Pmel17 observed upon inhibition of PC cleavage adds to the growing evidence that premelanosomes derive from post-Golgi compartments, and not from smooth ER (Kushimoto et al., 2001).
In MNT-1 cells expressing α1-PDX, striated structures were less abundant than in control cells, and many cells displayed numerous abnormal pigment-containing structures (Fig. 4). Because melanogenic enzymes such as tyrosinase and Tyrp1 are normally delivered to preformed stage II premelanosomes (Raposo et al., 2001), these unusual pigmented structures likely represent the products of malformed premelanosomes, depleted of fibrils, to which melanogenic enzymes were still delivered. The failure to completely deplete stage II premelanosomes in α1-PDX–treated cells was likely due to the short duration of expression after adenovirus infection; “normal” melanosomes of all stages present within α1-PDX–expressing cells likely represent later stage melanosomes that were in the process of forming, and/or partial fibril formation by a pool of properly cleaved Pmel17 that was present within abundant stage I premelanosomes, before a threshold of α1-PDX expression. Tonic exposure to α1-PDX for extended periods of time would likely further reduce the level of normal melanosomal structures in these cells, and may permit a timeline for melanosome maturation to be derived.
Composition and formation of striations and parallels with fibrillary diseases
The striated fibrils of stage II and III melanosomes have been thought to consist of protein with no underlying membrane based on their periodicity of electron lucence and density and the frequent appearance of a “zigzag” shape by EM (Birbeck et al., 1956; Moyer, 1966; Maul, 1969; Hearing et al., 1973). This interpretation seemed inconsistent with the ability of the integral membrane protein, Pmel17, to induce formation of and become incorporated into the striations (Berson et al., 2001; Raposo et al., 2001). We now reconcile these observations by showing that the PC cleavage product, Mα, is eventually released from membranes and accumulates in TX-insoluble complexes. The insolubility of these complexes explains our previous inability to observe Mα released from Mβ in the TX-soluble fraction (Berson et al., 2001) and the short half-life of TX-soluble Pmel17 (Kobayashi et al., 1994; Donatien and Orlow, 1995; Berson et al., 2001), and supports earlier evidence for Pmel17-derived fragments in detergent-insoluble fractions of partially purified melanosomes (Orlow et al., 1993; Zhou et al., 1994). The identity of Mα is confirmed by (1) its immunoreactivity with αPmel-N but not αPep13h and its insensitivity to endoH; (2) the failure to generate Mα and Mβ upon mutagenesis of KR469, inhibition of PCs by α1-PDX, or expression in PC-deficient LoVo cells; (3) the lack of any other potential dibasic cleavage sites in Pmel17 that could generate the appropriate sized fragments; and (4) the co-migration of intracellular Mα with secreted Pmel17 (Berson et al., 2001), for which the sequence is known to end at V467 (Maresh et al., 1994). The presence of TX-insoluble, Mβ-dissociated Mα on isolated fibrils and the dependence of striation formation on PC cleavage of Pmel17 indicates that (1) Pmel17 is not only sufficient, but also necessary for the generation of striations; and (2) the striations consist of insoluble complexes composed at least in part of Mα without Mβ and underlying membrane.
How does Mα become incorporated into detergent-insoluble complexes? Mα must first be released from the integral membrane Mβ fragment, to which it is disulfide bonded on ILVs of multivesicular endosomes (Berson et al., 2001; Raposo and Marks, 2002). By analogy to the “sorting by retention” model of secretory granule formation (Arvan and Castle, 1998), the MVB may provide a structural framework to sequester Pmel17 and other molecules required for striation formation from other endosome components. Release of Mα from the ILV must be mediated by either reduction of the disulfide bonds or proteolytic digestion of Mβ; the latter is more likely, given the oxidative nature of melanin intermediates, the presence of lysosomal hydrolases in melanosomes (Orlow, 1995), and evidence for proteolytic maturation of Pmel17 in stage II premelanosome-enriched subcellular fractions (Chiamenti et al., 1996; Kushimoto et al., 2001). We speculate that release of Mα from Mβ is accompanied by a conformational change that favors fibrillogenic activity, perhaps facilitated by the low pH of forming premelanosomes (Raposo et al., 2001). Lumenal segments within Mβ may serve to inhibit fibrillogenesis because longer Pmel17 fragments generated upon inhibition of PCs fail to properly form fibrils.
This model for physiological fibril formation in premelanosome biogenesis is reminiscent of the pathological process of fibril formation in a number of amyloid diseases. A specific requirement for PC cleavage and/or acidification in the formation of fibrils is observed in several pathogenic fibrillogenic substrates, including gelsolin in familial amyloidoses of Finnish type (Chen et al., 2001), BRI-L in familial British dementia (Kim et al., 1999), and pro-islet amyloid polypeptide in type II diabetes (Badman et al., 1996). Proteolytic maturation, acidification, and/or association with detergent-insoluble lipid rafts—the lipid contents of which are similar to the ILVs of MVBs (Mobius et al., 2002; Wubbolts et al., 2003)—are common features in fibril formation by the amyloidogenic peptide in Alzheimer's disease (Esler and Wolfe, 2001; Ehehalt et al., 2003) and by prions (Taraboulos et al., 1995; Mayer et al., 1996; Zou and Cashman, 2002). Defining the fibrillogenic determinant within the Pmel17 lumenal domain and characterizing its properties will likely provide insights into the mechanism by which fibril formation is favored by proteolytic maturation, not only in premelanosomes, but also in these clinically important substrates.
Implications for biogenesis of lysosome-related organelles
PCs are well known to regulate the formation of dense cores in conventional secretory granules by permitting ordered aggregation and dense packing of granule contents (Arvan and Castle, 1998). Our work, together with the presumed role of PCs or other proteases in the formation of Weibel-Palade bodies or lamellar bodies, respectively, demonstrates that proteolytic activation plays a general role in lysosome-related organelle biogenesis as well. Other fibrillogenic substrates may include Langerin, a C-type lectin that is involved in the formation of Birbeck granules in Langerhans cells (Valladeau et al., 2000). Future studies will likely confirm the generality of this biogenetic function of proteases, and may reveal whether fibrillogenic activation steps are disrupted by genetic diseases of organelle biogenesis, such as Hermansky-Pudlak Syndrome (Spritz, 1999; Swank et al., 2000).
Materials and methods
Cell culture and transfections
MNT-1 human melanoma cells and furin-deficient LoVo adenocarcinoma cells (American Type Culture Collection) were maintained in DME containing 10% AIM-V medium (Life Technologies), 20% FBS, nonessential amino acids, sodium pyruvate, and antibiotics. HeLa cells were cultured as described previously (Berson et al., 2001). Cells were transiently transfected using either calcium phosphate precipitation (Marks et al., 1996) or FuGENE™ 6 (Roche) for HeLa or LipofectAMINE™ (Life Technologies) for LoVo according to manufacturer's instructions. Cells were analyzed 2–3 d after transfection.
Antibodies
mAb specific for Tyrp1 (TA99/Mel-5; Thomson et al., 1985) and affinity-purified rabbit antibody to the Pmel17 COOH terminus (αPEP13h; Berson et al., 2001) were described previously. Purchased mAbs were HMB50 and HMB45 (anti-Pmel17; Lab Vision), M2 (anti-FLAG; Sigma-Aldrich), H4A3 (anti-LAMP1; Developmental Studies Hybridoma Bank, Iowa City, IA), anti-CD63 (Immunotech), and YL 1/2 (anti-tubulin; Accurate Chemical). Rabbit antiserum αPmel-N was generated by Genemed Synthesis, Inc. to a synthetic peptide (CTKVPRNQDWLGVSRQLR-CO2H) corresponding to the human Pmel17 NH2 terminus (residues 24–40), and affinity purified on peptide coupled to SulfoLink beads (Pierce Chemical Co.). αPmel-N had no reactivity with untransfected HeLa cells. Protein A gold conjugates were purchased from Dr. J. Slot (Utrecht Medical School, Utrecht, Netherlands).
Plasmids
pCI-Pmel17 encoding the Pmel17 long form in pCI (Promega) has been described previously (Berson et al., 2001). Pmel17Δcs was generated by site-directed mutagenesis using two-step amplification (Higuchi et al., 1988), replacing the endogenous sequence using unique KasI and NsiI sites, and sequence verified by automated dideoxy sequencing. HA11 epitope-tagged furin in pSX (Bosshart et al., 1994) was a gift of Dr. J.S. Bonifacino (National Institutes of Health, Bethesda, MD).
Recombinant virus production and infection
Recombinant adenoviruses encoding the tetracycline-responsive transactivator (tTA) and tetracycline-responsive, FLAG-epitope tagged α1-PDX and α1-AT were gifts of Drs. G. Thomas and L. Thomas (Vollum Institute, Oregon Health Sciences University, Portland, OR; Jean et al., 2000). Viruses were amplified and titred using the Tissue Culture Infectious Dose 50 method according to the AdEasy™ vector system application manual (Qbiogene). Crude virus preparations in PBS were used directly for double infections of MNT-1 cells, according to the AdEasy™ manual, using tTA virus at moi of 20 and α1-PDX or α1-AT virus at moi of 5 or 20. Infections were not cytotoxic for MNT-1 cells as judged by trypan blue exclusion, and resulted in transgene expression in ≥50% of the cells by IFM. Cells were harvested 2 d after infection for analyses.
Metabolic labeling and immunoprecipitation
Cells were metabolically pulse labeled for 15–30 min with [35S]methionine/cysteine and chased for indicated times as described previously (Marks et al., 1996). Fresh or frozen cell pellets were lysed in 1% (wt/vol) TX for 30 min as described previously (Berson et al., 2000), and lysates were clarified by centrifugation for 15 min at 20,000 g. Where noted, the resulting TX-insoluble pellets were resuspended in 50 μl TX lysis buffer containing 8M urea and heated at 60°C for 10 min. After cooling, samples were diluted 20× with TX lysis buffer for immunoprecipitation. Immunoprecipitations, SDS-PAGE on 10% polyacrylamide gels, and phosphorimaging analysis were performed as described previously (Berson et al., 2000). For sequential immunoprecipitations, unbound material from the first precipitation was clarified by centrifugation at 20,000 g for 5 min before incubation with the subsequent immobilized antibody.
Immunoblotting
Western blotting using whole-cell lysates prepared with 1% SDS, and endoH/ N-glycanase F treatments were as described previously (Berson et al., 2000). For fractionation into TX-soluble and -insoluble fractions, 106 cells were resuspended in 200 μl lysis buffer containing 1% TX and protease inhibitors at 4°C for 2 h. After centrifugation at 20,000 g for 15 min, supernatants (TX soluble) were harvested and 6× SDS-PAGE sample buffer was added before analysis. Pellets (TX insoluble) were washed once with 1% TX lysis buffer and then resuspended in 1× SDS-PAGE sample buffer for analysis. 8% PAGE/5% methanol transfer buffer was used for αPmel-N and anti-tubulin blots, and 12% PAGE/15% methanol transfer buffer for αPep13h blots. Immobilon-P membranes (Millipore) with transferred proteins were probed with indicated antibodies, and bands were detected with alkaline phosphatase–conjugated goat anti–rabbit Ig, enhanced chemifluorescence, and phosphorimaging analysis using a fluorescence imaging system (Storm® 860; Molecular Dynamics, Inc.) and ImageQUANT software (Amersham Biosciences).
Subcellular fractionation
See Fig. 7 A for schematic. MNT-1 cells (∼108) were homogenized with a hand-held Dounce homogenizer in 4 volumes of buffer I (25 mM Hepes, 1 mM EDTA, 0.1 mM EGTA, and 0.02% sodium azide, pH 7.4) containing 0.25 M sucrose and a cocktail of protease inhibitors (Berson et al., 2000). Cell debris and nuclei were pelleted at 700 g for 10 min. Post-nuclear supernatants were layered over a cushion of 2.0 M sucrose in buffer I and centrifuged at 11,000 g for 10 min. The black interface at the cushion (dense membrane fraction) and the diffuse supernatant (light membrane fraction) were harvested, and each was subjected to 100,000 g for 60 min to recover a pellet (total membrane fraction). Pellets were resuspended in 600 μl 150 mM NaCl/0.1 M Tris-HCl, pH 8.0; after a sample was removed for EM analysis, TX was added to a final concentration of 1%, and samples were rotated at 4°C for 2 h. Samples were then spun at 20,000 g for 15 min, and supernatants (TX-soluble fraction) and pellets (TX insoluble fraction) were harvested. Approximately 3% of the material in each fraction was used for each lane for Western blotting analyses. An aliquot of each fraction was fixed for EM analysis as described below.
Electron microscopy
Conventional EM of MNT-1 or transfected HeLa cells was performed as described previously (Raposo et al., 2001). For IEM, cells were fixed with 2% PFA/0.2% glutaraldehyde and single- or double-immunogold labeling of ultrathin cryosections was performed as described previously (Raposo et al., 1997, 2001) using protein A conjugated to 10-nm or 15-nm gold particles (PAG-10 or -15). For transfected HeLa cells, only cells expressing comparable levels of wild-type Pmel17 and Pmel17Δcs were analyzed in parallel. Expression per cell was assessed as total expression level (judged by Western blotting) divided by percentage of transfected cells (judged by IFM). Relative quantitation as described in the text was performed directly under the electron microscope by counting the number of affected and unaffected relevant compartments among 50 cell profiles. For EM analysis of subcellular fractions, 20-μl drops of each fraction, previously fixed with 2% PFA in PHEM buffer, as described in Raposo et al. (2001), were placed on formvar-carbon–coated EM grids for 30 min. The grids were then immunogold labeled, contrasted, and embedded in a mixture of uranyl acetate and methylcellulose as described for ultrathin cryosections (Raposo et al., 1997).
Acknowledgments
We thank G. Thomas and L. Thomas for gifts of α1-PDX, α1-AT, and tTA-encoding adenoviruses, P. Arvan for suggesting their use, and P. Bates for technical help with adenovirus protocols. We also thank N. Seidah for helpful discussions and G. Thomas and E. Dell'Angelica for critical review of the manuscript.
This work was supported by National Institutes of Health grants R01 EY 12207 and R01 AR 48155 (to M.S. Marks) and by Association de Recherche pour le Cancer and Vaincre les Maladies Lysosomales (to G. Raposo). J.F. Berson was supported in part by training grant T32 CA 09140 from the National Cancer Institute and by American Cancer Society Fellowship PF-99-336-01-CIM.
The Marks and Raposo laboratories contributed equally to this work.
Footnotes
Abbreviations used in this paper: α1-AT, α1-antitrypsin inhibitor; α1-PDX, α1-antitrypsin Portland; endoH, endoglycosidase H; IEM, immunoelectron microscopy; IFM, immunofluorescence microscopy; ILV, intralumenal vesicle; moi, multiplicity of infection; MVB, multivesicular body; PC, proprotein convertase; tTA, tetracycline-repressible transactivator; TX, Triton X-100.
References
- Adema, G.J., A.J. de Boer, A.M. Vogel, W.A.M. Loenen, and C.G. Figdor. 1994. Molecular characterization of the melanocyte lineage-specific antigen gp100. J. Biol. Chem. 269:20126–20133. [PubMed] [Google Scholar]
- Anderson, E.D., L. Thomas, J.S. Hayflick, and G. Thomas. 1993. Inhibition of HIV-1 gp160-dependent membrane fusion by a furin-directed α1-antitrypsin variant. J. Biol. Chem. 268:24887–24891. [PubMed] [Google Scholar]
- Arvan, P., and D. Castle. 1998. Sorting and storage during secretory granule biogenesis: looking backward and looking forward. Biochem. J. 332:593–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badman, M.K., K.I. Shennan, J.L. Jermany, K. Docherty, and A. Clark. 1996. Processing of pro-islet amyloid polypeptide (proIAPP) by the prohormone convertase PC2. FEBS Lett. 378:227–231. [DOI] [PubMed] [Google Scholar]
- Berson, J.F., D.W. Frank, P.A. Calvo, B.M. Bieler, and M.S. Marks. 2000. A common temperature-sensitive allelic form of human tyrosinase is retained in the endoplasmic reticulum at the nonpermissive temperature. J. Biol. Chem. 275:12281–12289. [DOI] [PubMed] [Google Scholar]
- Berson, J.F., D. Harper, D. Tenza, G. Raposo, and M.S. Marks. 2001. Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol. Biol. Cell. 12:3451–3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbeck, M.S.C., E.H. Mercer, and N.A. Barnicot. 1956. The structure and formation of pigment granules in human hair. Exp. Cell Res. 10:505–514. [DOI] [PubMed] [Google Scholar]
- Bosshart, H., J. Humphrey, E. Deignan, J. Davidson, J. Drazba, L. Yuan, V. Oorschot, P.J. Peters, and J.S. Bonifacino. 1994. The cytoplasmic domain mediates localization of furin to the trans-Golgi network en route to the endosomal/lysosomal system. J. Cell Biol. 126:1157–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C.-D., M.E. Huff, J. Matteson, L.J. Page, R. Phillips, J.W. Kelly, and W.E. Balch. 2001. Furin initiates gelsolin familial amyloidosis in the Golgi through a defect in Ca2+ stabilization. EMBO J. 20:6277–6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiamenti, A.M., F. Vella, F. Bonetti, M. Pea, S. Ferrari, G. Martignoni, A. Benedetti, and H. Suzuki. 1996. Anti-melanoma monoclonal antibody HMB-45 on enhanced chemiluminescence-western blotting recognizes a 30-35 kDa melanosome-associated sialated glycoprotein. Melanoma Res. 6:291–298. [DOI] [PubMed] [Google Scholar]
- De Matteis, M.A., and J.S. Morrow. 2000. Spectrin tethers and mesh in the biosynthetic pathway. J. Cell Sci. 113:2331–2343. [DOI] [PubMed] [Google Scholar]
- Dell'Angelica, E.C., C. Mullins, S. Caplan, and J.S. Bonifacino. 2000. Lysosome-related organelles. FASEB J. 14:1265–1278. [DOI] [PubMed] [Google Scholar]
- Donatien, P.D., and S.J. Orlow. 1995. Interaction of melanosomal proteins with melanin. Eur. J. Biochem. 232:159–164. [DOI] [PubMed] [Google Scholar]
- Dunn, L.C., and L.W. Thigpen. 1930. The silver mouse: a recessive color variation. J. Hered. 21:495–498. [Google Scholar]
- Ehehalt, R., P. Keller, C. Haass, C. Thiele, and K. Simons. 2003. Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 160:113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esler, W.P., and M.S. Wolfe. 2001. A portrait of Alzheimer secretases–new features and familiar faces. Science. 293:1449–1454. [DOI] [PubMed] [Google Scholar]
- Guttentag, S., L. Robinson, P. Zhang, F. Brasch, F. Buhling, and M. Beers. 2003. Cysteine protease activity is required for surfactant protein B processing and lamellar body genesis. Am. J. Respir. Cell Mol. Biol. 28:69–79. [DOI] [PubMed] [Google Scholar]
- Hearing, V.J., P. Phillips, and M.A. Lutzner. 1973. The fine structure of melanogenesis in coat color mutants of the mouse. J. Ultrastruct. Res. 43:88–106. [DOI] [PubMed] [Google Scholar]
- Higuchi, R., B. Krummel, and R.K. Saiki. 1988. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 16:7351–7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka, M., M. Nagahama, W.S. Kim, T. Watanabe, K. Hatsuzawa, J. Ikemizu, K. Murakami, and K. Nakayama. 1991. Arg-X-Lys/Arg-Arg motif as a signal for precursor cleavage catalyzed by furin within the constitutive secretory pathway. J. Biol. Chem. 266:12127–12130. [PubMed] [Google Scholar]
- Jean, F., K. Stella, L. Thomas, G. Liu, Y. Xiang, A.J. Reason, and G. Thomas. 1998. α1-antitrypsin Portland, a bioengineered serpin highly selective for furin: application as an antipathogenic agent. Proc. Natl. Acad. Sci. USA. 95:7293–7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean, F., L. Thomas, S.S. Molloy, G. Liu, M.A. Jarvis, J.A. Nelson, and G. Thomas. 2000. A protein-based therapeutic for human cytomegalovirus infection. Proc. Natl. Acad. Sci. USA. 97:2864–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Journet, A.M., S. Saffaripour, E.M. Cramer, D. Tenza, and D.D. Wagner. 1993. von Willebrand factor storage requires intact prosequence cleavage site. Eur. J. Cell Biol. 60:31–41. [PubMed] [Google Scholar]
- Kim, S.H., R. Wang, D.J. Gordon, J. Bass, D.F. Steiner, D.G. Lynn, G. Thinakaran, S.C. Meredith, and S.S. Sisodia. 1999. Furin mediates enhanced production of fibrillogenic ABri peptides in familial British dementia. Nat. Neurosci. 2:984–988. [DOI] [PubMed] [Google Scholar]
- King, R.A., V.J. Hearing, D.J. Creel, and W.S. Oetting. 1995. Albinism. The Metabolic and Molecular Bases of Inherited Disease. Vol. 3. C.R. Scriver, A.L. Beaudet, W.S. Sly, and D. Valle, editors. McGraw-Hill, Inc., New York. 4353–4392.
- Kobayashi, T., K. Urabe, S.J. Orlow, K. Higashi, G. Imokawa, B.S. Kwon, B. Potterf, and V.J. Hearing. 1994. The Pmel 17/silver locus protein. Characterization and investigation of its melanogenic function. J. Biol. Chem. 269:29198–29205. [PubMed] [Google Scholar]
- Kushimoto, T., V. Basrur, J. Valencia, J. Matsunaga, W.D. Vieira, V.J. Ferrans, J. Muller, E. Appella, and V.J. Hearing. 2001. A model for melanosome biogenesis based on the purification and analysis of early melanosomes. Proc. Natl. Acad. Sci. USA. 98:10698–10703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon, B.S., R. Halaban, G.S. Kim, L. Usack, S. Pomerantz, and A.K. Haq. 1987. A melanocyte-specific complementary DNA clone whose expression is inducible by melanotropin and isobutylmethyl xanthine. Mol. Biol. Med. 4:339–355. [PubMed] [Google Scholar]
- Lee, Z.H., L. Hou, G. Moellmann, E. Kuklinska, K. Antol, M. Fraser, R. Halaban, and B.S. Kwon. 1996. Characterization and subcellular localization of human Pmel 17/silver, a 100-kDa (pre)melanosomal membrane protein associated with 5,6,-dihydroxyindole-2-carboxylic acid (DHICA) converting activity. J. Invest. Dermatol. 106:605–610. [DOI] [PubMed] [Google Scholar]
- Maresh, G.A., W.-C. Wang, K.S. Beam, A.R. Malacko, I. Hellström, K.E. Hellström, and H. Marquardt. 1994. Differential processing and secretion of the melanoma-associated ME20 antigen. Arch. Biochem. Biophys. 311:95–102. [DOI] [PubMed] [Google Scholar]
- Marks, M.S., and M.C. Seabra. 2001. The melanosome: membrane dynamics in black and white. Nat. Rev. Mol. Cell Biol. 2:738–748. [DOI] [PubMed] [Google Scholar]
- Marks, M.S., L. Woodruff, H. Ohno, and J.S. Bonifacino. 1996. Protein targeting by tyrosine- and di-leucine-based signals: evidence for distinct saturable components. J. Cell Biol. 135:341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Esparza, M., C. Jiménez-Cervantes, D.C. Bennett, J.A. Lozano, F. Solano, and J.C. García-Borrón. 1999. The mouse silver locus encodes a single transcript truncated by the silver mutation. Mamm. Genome. 10:1168–1171. [DOI] [PubMed] [Google Scholar]
- Maul, G.G. 1969. Golgi-melanosome relationship in human melanoma in vitro. J. Ultrastruct. Res. 26:163–176. [DOI] [PubMed] [Google Scholar]
- Mayer, R.J., C. Tipler, J. Arnold, L. Laszlo, A. Al-Khedhairy, J. Lowe, and M. Landon. 1996. Endosome-lysosomes, ubiquitin and neurodegeneration. Adv. Exp. Med. Biol. 389:261–269. [DOI] [PubMed] [Google Scholar]
- Miranda, L., J. Wolf, S. Pichuantes, R. Duke, and A. Franzusoff. 1996. Isolation of the human PC6 gene encoding the putative host protease for HIV-1 gp160 processing in CD4+ T lymphocytes. Proc. Natl. Acad. Sci. USA. 93:7695–7700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobius, W., Y. Ohno-Iwashita, E.G. van Donselaar, V.M. Oorschot, Y. Shimada, T. Fujimoto, H.F. Heijnen, H.J. Geuze, and J.W. Slot. 2002. Immunoelectron microscopic localization of cholesterol using biotinylated and non-cytolytic perfringolysin O. J. Histochem. Cytochem. 50:43–55. [DOI] [PubMed] [Google Scholar]
- Molloy, S.S., E.D. Anderson, F. Jean, and G. Thomas. 1999. Bi-cycling the furin pathway: from TGN localization to pathogen activation and embryogenesis. Trends Cell Biol. 9:28–35. [DOI] [PubMed] [Google Scholar]
- Moyer, F.H. 1966. Genetic variations in the fine structure and ontogeny of mouse melanin granules. Am. Zool. 6:43–66. [DOI] [PubMed] [Google Scholar]
- Nichols, S.E., D.C. Harper, J.F. Berson, and M.S. Marks. 2003. A novel splice variant of Pmel17 expressed by human melanocytes and melanoma cells lacking some of the internal repeats. J. Invest. Dermatol. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlow, S.J. 1995. Melanosomes are specialized members of the lysosomal lineage of organelles. J. Invest. Dermatol. 105:3–7. [DOI] [PubMed] [Google Scholar]
- Orlow, S.J., B.-K. Zhou, R.E. Boissy, and S. Pifko-Hirst. 1993. Identification of a mammalian melanosomal matrix glycoprotein. J. Invest. Dermatol. 101:141–144. [DOI] [PubMed] [Google Scholar]
- Raposo, G., and M.S. Marks. 2002. The dark side of lysosome-related organelles: specialization of the endocytic pathway for melanosome biogenesis. Traffic. 3:237–248. [DOI] [PubMed] [Google Scholar]
- Raposo, G., M.J. Kleijmeer, G. Posthuma, J.W. Slot, and H.J. Geuze. 1997. Immunogold labeling of ultrathin cryosections: application in immunology. Handbook of Experimental Immunology. Vol. 4. L.A. Herzenberg, D. Weir, L.A. Herzenberg, and C. Blackwell, editors. Blackwell Science, Inc., Cambridge, MA. 1–11.
- Raposo, G., D. Tenza, D.M. Murphy, J.F. Berson, and M.S. Marks. 2001. Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J. Cell Biol. 152:809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidah, N.G., S. Benjannet, L. Wickham, J. Marcinkiewicz, S.B. Jasmin, S. Stifani, A. Basak, A. Prat, and M. Chrétien. 2003. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. USA. 100:928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiji, M., T.B. Fitzpatrick, R.T. Simpsom, and M.S.C. Birkbeck. 1963. Chemical composition and terminology of specialized organelles (melanosomes and melanin granules) in mammalian melanocytes. Nature. 197:1082–1084. [DOI] [PubMed] [Google Scholar]
- Spritz, R.A. 1999. Multi-organellar disorders of pigmentation: intracellular traffic jams in mammals, flies and yeast. Trends Genet. 15:337–340. [DOI] [PubMed] [Google Scholar]
- Steiner, D.F. 1998. The proprotein convertases. Curr. Opin. Chem. Biol. 2:31–39. [DOI] [PubMed] [Google Scholar]
- Swank, R.T., E.K. Novak, M.P. McGarry, M.E. Rusiniak, and L. Feng. 1998. Mouse models of Hermansky Pudlak syndrome: a review. Pigment Cell Res. 11:60–80. [DOI] [PubMed] [Google Scholar]
- Swank, R.T., E.K. Novak, M.P. McGarry, Y. Zhang, W. Li, Q. Zhang, and L. Feng. 2000. Abnormal vesicular trafficking in mouse models of Hermansky-Pudlak Syndrome. Pigment Cell Res. 13:59–67. [DOI] [PubMed] [Google Scholar]
- Takahashi, S., T. Nakagawa, K. Kasai, T. Banno, S.J. Duguay, W.J. Van de Ven, K. Murakami, and K. Nakayama. 1995. A second mutant allele of furin in the processing-incompetent cell line, LoVo. Evidence for involvement of the homo B domain in autocatalytic activation. J. Biol. Chem. 270:26565–26569. [DOI] [PubMed] [Google Scholar]
- Taraboulos, A., M. Scott, A. Semenov, D. Avrahami, L. Laszlo, S.B. Prusiner, and D. Avrahami. 1995. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol. 129:121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, G. 2002. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 3:753–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson, T.M., M.J. Mattes, L. Roux, L.J. Old, and K.O. Lloyd. 1985. Pigmentation-associated glycoprotein of human melanomas and melanocytes: definition with a mouse monoclonal antibody. J. Invest. Dermatol. 85:169–174. [DOI] [PubMed] [Google Scholar]
- Valladeau, J., O. Ravel, C. Dezutter-Dambuyant, K. Moore, M. Kleijmeer, Y. Liu, V. Duvert-Frances, C. Vincent, D. Schmitt, J. Davoust, et al. 2000. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity. 12:71–81. [DOI] [PubMed] [Google Scholar]
- Varlamov, O., and L.D. Fricker. 1998. Intracellular trafficking of metallocarboxypeptidase D in AtT-20 cells: localization to the trans-Golgi network and recycling from the cell surface. J. Cell Sci. 111:877–885. [DOI] [PubMed] [Google Scholar]
- Vennegoor, C., P. Hageman, H. van Nouhuijs, D.J. Ruiter, J. Calafat, P.J. Ringens, and P. Rümke. 1988. A monoclonal antibody specific for cells of the melanocytic lineage. Am. J. Pathol. 130:179–192. [PMC free article] [PubMed] [Google Scholar]
- Warren, G., and J. Shorter. 2002. Golgi architecture and inheritance. Annu. Rev. Cell Dev. Biol. 18:379–420. [DOI] [PubMed] [Google Scholar]
- Wubbolts, R.W., R.S. Leckie, P.T. Veenhuizen, G. Schwartzmann, W. Möbius, J. Hoernschemeyer, J.W. Slot, H.J. Geuze, and W. Stoorvogel. 2003. Proteomic and biochemical analyses of human B cell-derived exosomes. Potential implications for their function and multivesicular body formation. J. Biol. Chem. 278:10963–10972. [DOI] [PubMed] [Google Scholar]
- Zhou, B.K., T. Kobayashi, P.D. Donatien, D.C. Bennett, V.J. Hearing, and S.J. Orlow. 1994. Identification of a melanosomal matrix protein encoded by the murine si (silver) locus using “organelle scanning”. Proc. Natl. Acad. Sci. USA. 91:7076–7080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, W.-Q., and N.R. Cashman. 2002. Acidic pH and detergents enhance in vitro conversion of human brain PrPC to a PrPSc-like form. J. Biol. Chem. 277:43942–43947. [DOI] [PubMed] [Google Scholar]