

Abstract
Previous in vitro studies using cGMP or cAMP revealed a cross-talk between signaling mechanisms activated by axonal guidance receptors. However, the molecular elements modulated by cyclic nucleotides in growth cones are not well understood. cGMP is a second messenger with several distinct targets including cGMP-dependent protein kinase I (cGKI). Our studies indicated that the α isoform of cGKI is predominantly expressed by sensory axons during developmental stages, whereas most spinal cord neurons are negative for cGKI. Analysis of the trajectories of axons within the spinal cord showed a longitudinal guidance defect of sensory axons within the developing dorsal root entry zone in the absence of cGKI. Consequently, in cGKI-deficient mice, fewer axons grow within the dorsal funiculus of the spinal cord, and lamina-specific innervation, especially by nociceptive sensory neurons, is strongly reduced as deduced from anti-trkA staining. These axon guidance defects in cGKI-deficient mice lead to a substantial impairment in nociceptive flexion reflexes, shown using electrophysiology. In vitro studies revealed that activation of cGKI in embryonic dorsal root ganglia counteracts semaphorin 3A–induced growth cone collapse. Our studies therefore reveal that cGMP signaling is important for axonal growth in vivo and in vitro.
Keywords: cGMP signaling; axonal pathfinding; cGMP-dependent protein kinase I; sensory axons; nociceptive flexion reflex
Introduction
The formation of precise and selective connections between neurons is essential for the function of the mature nervous system. During development, the integration of several distinct signals mediated by guidance receptors contributes to the pathfinding of axons toward their targets to establish synapses (Tessier-Lavigne and Goodman, 1996; Mueller, 1999; Raper, 2000; Klein, 2001). Previous in vitro studies using a growth cone turning assay indicated that signaling cascades activated by axonal guidance receptors could be modulated by cyclic nucleotides (Song et al., 1997; Hopker et al., 1999; Song and Poo, 1999). For example, increasing levels of cGMP converted a repulsive axonal growth of Xenopus spinal neurons induced by semaphorin 3A into an attractive extension and protected cultivated rat sensory growth cones from collapse (Song et al., 1998). However, the intracellular components activated by cGMP and whether cGMP signaling is also important for growth cone decisions in vivo remained unknown.
In many cell types, cGMP is a widely used second messenger that has several distinct targets (Hofmann et al., 2000; Lucas et al., 2000), including activation of cGMP-dependent protein kinase I (cGKI)* and cGKII, regulation of cAMP levels through the activation or inhibition of cAMP-dependent phosphodiesterases, and opening of cyclic nucleotide-gated cation channels. cGMP is generated by soluble and particulate guanylyl cyclases (Gibson and Garbers, 2000) and degraded by phosphodiesterase V. The serine/threonine kinases cGKI and II, of which cGKI is known to be expressed in the developing spinal cord (Qian et al., 1996), are composed of an NH2-terminal domain, a regulatory segment, and a catalytic domain. In mammals, cGKI is expressed in two alternatively spliced isoforms, termed α and β, that differ in their NH2-terminal domains (Pfeifer et al., 1999). These NH2-terminal stretches regulate homodimerization, cGMP affinity and activation of the catalytic domain, and the substrate binding/anchoring of the isoenzymes. cGKI, which has been investigated extensively in the context of regulation of smooth muscle tone, has a variety of pleiotropic cellular regulatory functions, including calcium release (Schlossmann et al., 2000) and myosin phosphatase activation (Surks et al., 1999). Here, we have analyzed whether cGKI is implicated in growth cone steering in vitro and in vivo. Our results indicate that cGKIα is important for growth cone extension in the dorsal root entry zone (DREZ) of the spinal cord and counteracts semaphorin 3A–induced growth cone collapse in vitro. We therefore demonstrated that cGMP signaling is important for axonal pathfinding in vivo and that cGMP signaling is mediated via cGKIα. Furthermore, physiological investigations indicated that pathfinding errors of sensory axons caused by the absence of cGKI resulted in a significant impairment of the nociceptive flexion reflex.
Results
cGKIα, but not cGKIβ, is selectively expressed in sensory axons of the developing spinal cord
To study a possible function of cGKI in axonal guidance in vivo, we first examined the expression of both cGKI isoforms in axons of the embryonic dorsal root ganglion (DRG) and the spinal cord. Western blots of extracts of embryonic day 13 (E13) DRGs using polyclonal antibodies directed to both isoforms or antibodies that distinguish between both isoforms demonstrated that the α isoform of cGKI is expressed whereas the β isoform remains undetectable (Fig. 1 a). Immunostaining of cryostat sections with these three different antibody preparations revealed a very restricted pattern of expression of cGKI. cGKIα is expressed in cell bodies and growing axons of DRG neurons (Fig. 1, b–g), and sensory axons at the DREZ were especially heavily labeled for cGKIα. At more advanced stages, the entire dorsal funiculus and the descending collaterals of the proprioceptive and, more weakly, of the nociceptive neurons were also stained at E14 (Fig. 1, e and f). The labeling intensity declined in the dorsal funiculus at more advanced developmental stages (Fig. 1 g). Spinal neurons and axonal tracts were not cGKI positive, however weak transient labeling was observed in motoneurons at earlier developmental stages (Fig. 1, b and c, arrowheads) and in preganglionic neurons. In addition, cGKIα appeared ideally situated to transduce signals in growing sensory axons, as punctate cGKI immunoreactivity could be observed in the filopodia and lamellipodia of DRG neuron growth cones in culture (Fig. 1 h). This very restricted spatio-temporal expression profile of cGKIα in sensory axons suggested that cGKIα might regulate growth and pathfinding of DRG axons in specific regions in vivo, in particular at the DREZ.
Figure 1.

cGKIα is selectively expressed in developing DRG axons. (a) Western blot analysis demonstrates the presence of the α, but not the β, isoform of cGKI in embryonic DRGs. Lane 1, antibodies to both cGKI isoforms (c, common); lane 2, antibodies to the α isoform; lane 3, antibodies to the β isoform. For comparison, a blot of antibodies to L1 is shown. (b–h) Immunohistochemical detection of cGKI. (b–d) cGKIα distribution in the E11–E13 spinal cord. cGKIα is expressed in the DREZ and the developing dorsal funiculus as well as weakly and transiently on motoneuron columns (arrowheads) and in preganglionic neurons. Bar, 100 μm. (e) cGKIα expression in proprioceptive collaterals in the dorso-medial spinal cord at E14. Bar, 50 μm. (f) cGKIα expression in nociceptive collaterals in the dorso-lateral spinal cord at E14. (g) cGKIα distribution in the E15 spinal cord. Bar, 100 μm. (h) cGKI is distributed throughout a cultivated DRG growth cone. Bar, 5 μm. Sections were either stained by antibodies recognizing both isoforms of cGKI (E11, E12, and E13) or only the α isoform (E14 and E15). Both antibody preparations gave identical results. Tissue sections of cGKI-deficient mice did not stain with antibodies against cGKI (unpublished data). D, drg; R, dorsal root; Z, DREZ.
The longitudinal growth of sensory axons is impaired in the DREZ in the absence of cGKI
To investigate whether cGMP signaling via cGKI is important for axonal pathfinding in vivo, and because cGKIα is restricted to sensory axons at early developmental stages, we analyzed the trajectory of sensory axons within spinal cords of cGKI-deficient embryos (Wegener et al., 2002) (see Fig. 6 b) in whole mounts and in transverse as well as horizontal sections. Once sensory axons arrive at the DREZ of the developing spinal cord, each axon bifurcates into a rostral and caudal branch extending over several segments without growing into the gray matter (Davis et al., 1989; Eide and Glover, 1995; Mirnics and Koerber, 1995, 1997; Ozaki and Snider, 1997). After a waiting period, collaterals grow out from these longitudinal axons and form lamina-specific projections in the gray matter (Fig. 2 a). As expected from the localization, the absence of cGKI in cGKI-deficient mice did not affect the overall structure of the spinal cord and the DRGs or their dorsal roots (unpublished data). To visualize the growth in rostral/caudal direction within the spinal cord, we used the lipophilic axonal tracer DiI (Honig and Hume, 1989) to label primary afferents in preparations of E12 or E13 embryos fixed in paraformaldehyde. Whole mounts were analyzed after 2, 3, and 4 d of incubation with a fluorescence microscope. Whereas wild-type DRG axons extended a rostral and caudal branch with an equal frequency (24 DRGs analyzed), DRG axons in mutant mice had a strong preference for growing primarily in a rostral direction (67% of 24 DRGs from 13 mutant embryos) (Fig. 2 b; Table I). A confocal microscopic analysis of these DiI-labeled preparations revealed that wild-type fascicles of dorsal roots were split into a very fine meshwork within the DREZ, as would be expected from the formation of T-like branches that are laying on top of each other (Fig. 2 c, left). In contrast, in mutant embryos, thick fascicles of axons turning either caudally or, preferentially, in a rostral direction were observed (Fig. 2 c, middle and right). In summary, these observations indicate a defect of the mutant DRG axons within the DREZ in rostral/caudal growth.
Figure 6.
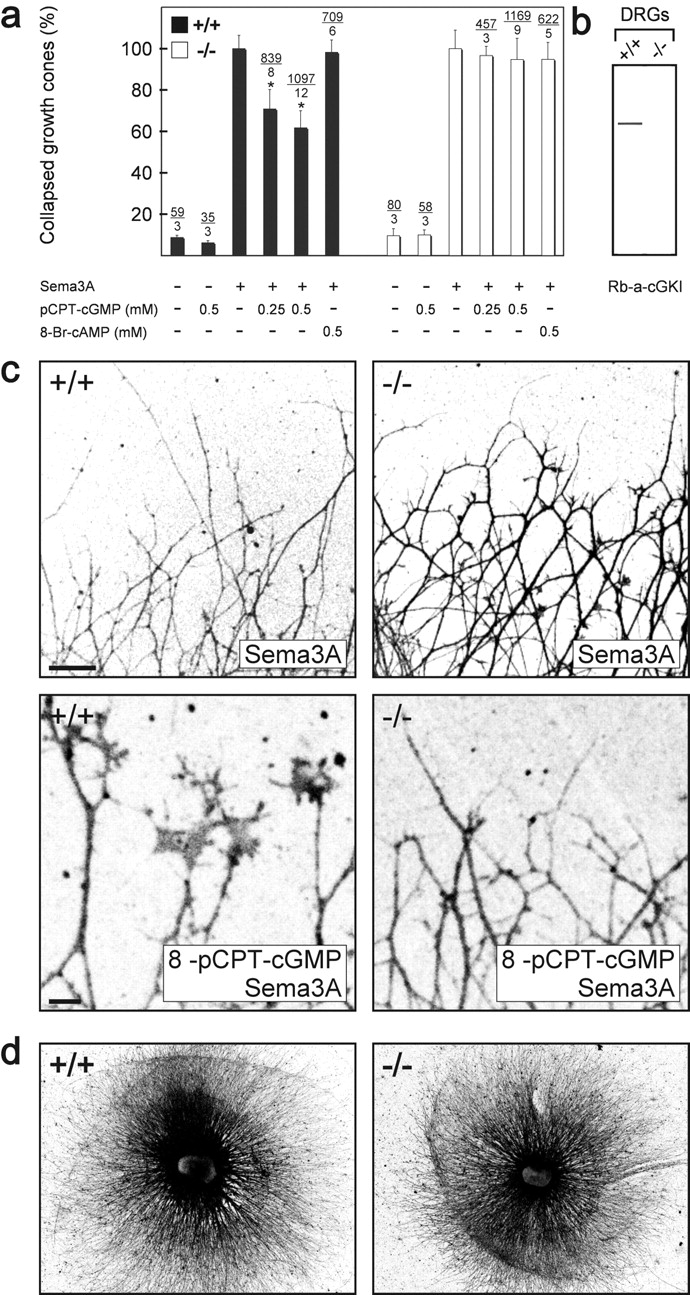
cGKI counteracts semaphorin 3A–induced growth cone collapse in vitro. (a) Growth cone collapse assay in the presence of 8-pCPT-cGMP or 8-Br-cAMP using DRG explants from litter-matched wild-type (+/+) or cGKI-deficient mice (−/−). The concentration and the presence of semaphorin 3A is indicated below the columns. The percentage of collapsed versus uncollapsed growth cones is presented and the number of counted collapsed growth cones and DRGs is given above the columns. The effect of semaphorin 3A alone was calculated as 100%. Error bars refer to SEM. Asterisk indicates significant difference from the samples without pretreatment with 8-pCPT-cGMP, P < 0.001 (t test). (b) cGKI protein could be detected in Western blots of DRG of wild-type (+/+) but not in DRGs of cGKI-deficient (−/−) embryos using polyclonal antibodies directed to both isoforms of cGKI. (c) Growth cones from wild-type (+/+) or cGKI-deficient (−/−) mice in the presence of semaphorin 3A alone or semaphorin 3A and 0.5 mM 8-pCPT-cGMP. Neurons were stained by mAb A2B5 for visualization and fluorescence images were inverted. DRGs from heterozygous (+/−) mice behaved as wild-type DRGs (unpublished data). Bar, 100 μm. Note that growth cones in the bottom images are shown at a higher magnification than in the top images. Bar, 10 μm. (d) Outgrowth of DRG explants of cGKI-deficient mice (−/−) is indistinguishable from those of wild-type mice (+/+). Explants from lumbar regions of litter-matched E13 embryos grown for 24 h on laminin-1 were silver stained (Rager et al., 1979).
Figure 2.

Longitudinal growth of sensory axons is impaired in the spinal cord of cGKI-deficient mice. (a) Schematic drawing of the three-dimensional morphology of a single sensory neuron within the spinal cord. (b) To visualize the longitudinal branching, primary afferents in preparations of E12 or E13 embryos were labeled with the lipophilic axonal tracer DiI (Honig and Hume, 1989). Whole mounts were analyzed laterally with a fluorescence microscope. Whereas wild-type (+/+) DRG axons branch equally in rostral and caudal direction, cGKI-deficient (−/−) DRG axons have a strong preference for growing primarily in rostral direction (to the right). Fluorescence images were inverted. Bar, 100 μm. (c) Three adjacent confocal images of DiI-labeled axons of wild-type (+/+) and mutant mice (−/−) within the DREZ are shown. Note that mutant axons turn preferentially rostrally as bundles. Bar, 100 μm. D, DRG; R, dorsal root.
Table I. Sensory axons of cGKI-deficient embryos reveal a longitudinal branching error within the DREZ.
| Embryo No. | Genotype | No. of injected DRGs | No. of DRGs with primarily rostral growth | No. of DRGs with equal bidirectional branching |
|---|---|---|---|---|
| E12 | ||||
| 498-10 | −/− | 3 | 3 | 0 |
| 498-11 | −/− | 4 | 4 | 0 |
| 517-1 | −/− | 1 | 1 | 0 |
| 519-2 | −/− | 2 | 2 | 0 |
| 519-9 | −/− | 3 | 0 | 1 |
| 520-1 | −/− | 1 | 0 | 1 |
| 520-7 | −/− | 2 | 2 | 0 |
| 522-10 | −/− | 2 | 2 | 0 |
| 524-5 | −/− | 1 | 1 | 0 |
| 524-8 | −/− | 1 | 0 | 1 |
| 527-6 | −/− | 2 | 0 | 2 |
| 528-1 | −/− | 1 | 0 | 1 |
| 498-1 | +/+ | 2 | 0 | 2 |
| 498-2 | +/+ | 4 | 0 | 4 |
| 498-6 | +/+ | 2 | 0 | 2 |
| 498-9 | +/+ | 3 | 0 | 3 |
| 517-8 | +/+ | 1 | 0 | 1 |
| 519-1 | +/+ | 1 | 0 | 1 |
| 519-4 | +/+ | 2 | 0 | 2 |
| 522-7 | +/+ | 2 | 0 | 2 |
| 522-8 | +/+ | 1 | 0 | 1 |
| 524-4 | +/+ | 3 | 0 | 3 |
| 524-6 | +/+ | 2 | 0 | 2 |
| 524-7 | +/+ | 1 | 0 | 1 |
| E13 | ||||
| 483-1 | −/− | 1 | 1 | 0 |
| 483-4 | +/− | 2 | 1 | 1 |
| 483-5 | +/− | 2 | 0 | 2 |
Number of DRGs labeled by DiI is given. The intensity of rostral and caudal segments within the spinal cord was measured in identical fields using NIH image software. The ratio of rostral/caudal staining varied from 1:0.51 to 1:0.85 in labeled DRGs with a primarily rostral growth in the absence of cGKI (mean value, 1:0.66 ± 0.11). All others were considered as equal bidirectional growth. In wild-type DRGs, we observed a 1:1.02 ratio (mean).
To describe selectively the trajectory of nociceptive sensory axons within the spinal cord, we analyzed serial transverse as well as horizontal sections of mutant and wild-type embryos stained with antibodies to the NGF receptor trkA. Axon bundles positive for trkA, observed at E10.5 at trunk levels where sensory axons had just arrived at the DREZs, were disorganized and divided into several spots in cGKI-deficient mice, which represent axon fascicles in a cross section. In contrast, in wild type, trkA-positive fascicles had a compact and oval-shaped appearance (Fig. 3, a and b, arrowheads). In horizontal sections through the most ventral part at the same trunk level of the mutant E10.5 DREZ, axon fascicles had grown into the gray matter toward the midline and then turned rostrally (Fig. 3 c). These fascicles lacked a caudal arm of similar size if traced back in serial sections (unpublished data). This indicated that trkA-positive axons in cGKI mutant mice have difficulties identifying their correct path as soon as they reach the DREZ. At more rostral levels of the body where more axons had already arrived and growth in the rostro/caudal direction is more advanced at E10.5, many trkA-positive axon fascicles were observed in horizontal sections to grow directly to the central canal from the DREZ in mutants (Fig. 3 e, arrowheads). Later in development (E12–E13), the DREZ appeared more similar to wild-type in transverse sections but had a reduced dorsal medial extension in the cGKI-deficient spinal cord (Fig. 3, f and g). At E14, the dorsal funiculus flattened out further in wild-type embryos, whereas in cGKI-deficient mice, a dorsally bifurcated appearance of the dorsal funiculus increased (Fig. 3, h and i) and trkA-positive axon fascicles left the lower arm of the developing dorsal funiculus, growing directly dorsal medially to the central canal (Fig. 3 i, arrowheads), most likely without branches in the rostral/caudal direction. Taken together, the anti-trkA staining indicates pathfinding errors of nociceptive axons at the DREZ. Other axon systems within the spinal cord appear not to be affected by the absence of cGKI. For example, commissural axons labeled by a mAb to TAG-1 grow correctly to the floor plate at E11 (Fig. 3, l and m), and staining with anti-L1 antibodies, a more general axon marker, indicated that the pattern of the lateral, as well as of the ventral funiculus, is not affected in mutant embryos (Fig. 3, j and k). The anti-L1 staining, however, revealed the errors of nociceptive axons that extended directly to the central canal (Fig. 3 k, arrowheads).
Figure 3.

Pathfinding of nociceptive neurons is impaired in the DREZ of cGKI-deficient mice. trkA-positive axons were analyzed in transverse or horizontal sections of wild-type (+/+) and cGKI-deficient mice (−/−) at E12–E14. (a, b, f, g, h, i, j, k, l, and m) Transverse sections; (c, d and e) horizontal sections. The plane of transverse section used in a and b is shown in a cartoon in n. The plane of horizontal sections in c is indicated in o (the most ventral part of the DREZ), and the plane in d and e is indicated in p (center of the DREZ). (j–m) Staining of transverse sections using polyclonal antibodies to L1 as a more general axon marker (j and k) or a mAb to TAG-1 to label commissural axons (l and m). These stainings indicate that the pattern of the lateral as well as the ventral funiculus and the growth of the commissural axons to the floor plate are not affected by the absence of cGKI. Arrowheads in e, g, i, and k point to axons or axon fascicles leaving the primordium of the dorsal funiculus of cGKI-deficient embryos to extend directly to the central canal. In transverse sections, dorsal is up, and in horizontal sections, rostral is up. Bars: (a–e) 10 μm; (f and g) 50 μm; (h–m) 100 μm. C, central canal.
An incorrect bifurcation at the DREZ, as observed by the DiI tracing and anti-trkA staining, either due to an inability to branch correctly or to maintain one axon from the T-like branch point would result in a significant reduction of axon numbers in the developing dorsal funiculus. We therefore quantified the anti-trkA staining as a measure of axon numbers in transverse as well as in horizontal sections at different trunk levels. These measurements indicated a 50–60% reduction of the trkA labeling in cGKI-deficient embryos in comparison to wild type. Although axons have not been directly counted, this result suggested that substantially fewer axons grew within the developing dorsal funiculus at E13 (Fig. 4 e), as would be expected from an incorrect T-like branch formation or retention. At more advanced stages (E15 and E16), the mutant dorsal funiculus was also consistently thinner than in similar regions of wild-type mice. Moreover, in mutant mice, the dorso-medial part of the dorsal funiculus (Fig. 4 d, arrows) never became populated by trkA-positive axons throughout all spinal cord levels, which again might reflect a reduction in axon numbers. To exclude the possibility that this reduction of trkA-positive axons within the spinal cord was caused by a loss of cells in the DRG or by a decreased expression of trkA protein, cell numbers at E13 and E14 in L4 and L5 were determined and Western blots of DRG extracts were analyzed with antibodies to trkA. Neither a reduction in cell number nor a decrease in trkA protein was observed in DRGs of cGKI-deficient embryos in comparison to wild-type embryos (Fig. 4, f and g).
Figure 4.
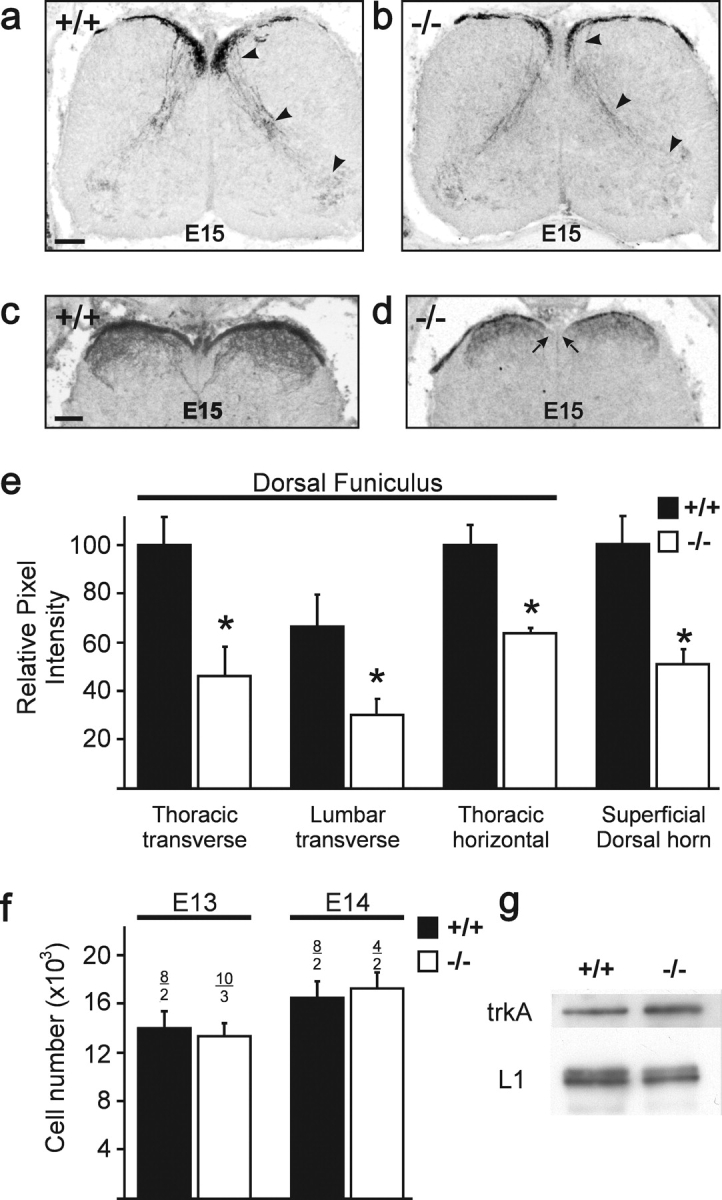
Quantitative analysis of proprioceptive and nociceptive axons within the developing spinal cord. (a–d) Less proprioceptive and nociceptive axons and their collaterals are observed in transverse sections of E15 cGKI-deficient than wild-type mice using parvalbumin (a and b) or trkA staining (c and d). Fluorescence images were inverted. In c and d, only the dorsal region of the spinal cord is shown. Arrowheads in a and b indicate parvalbumin-positive collaterals. In d, the two arrows indicate dorso-medial regions not populated by trkA-positive axons. Bars, 100 μm. (e) Quantification of trkA staining in transverse and horizontal sections at different levels of the spinal cord, which was taken as measure for axons growing in the developing dorsal funiculus at E13. The mean of the total pixel intensity of sections from wild-type embryos at the thoracic level was taken as 100%. In horizontal sections, the staining intensity of serial sections of the total developing dorsal funiculus in three neighboring regions was summed. The intensity of staining in wild-type embryos was taken as 100%. The last two columns indicate quantitative measurements of four fields in the E15 dorsal superficial horn, the termination zone for nociceptive C-fibers. Error bars indicate standard deviations. (f) Sum of cell numbers in L4 and L5 DRG at E13 and E14 (black, wild type; white, cGKI-deficient embryos). The number of analyzed DRGs and embryos is indicated above each column. Error bars indicate standard deviations. (g) Western blot analysis of DRG extracts from wild-type and cGKI-deficient E13 embryos using polyclonal antibodies to trkA or, for comparison, to the Ig cell adhesion protein L1. The 145-kD component of trkA and the 200-kD doublet of L1 are shown.
Collaterals of trkA-positive axons extended within the superficial dorsal horn and displayed a similar pattern to that of wild type. However, the intensity of trkA-positive fibers within the superficial dorsal horn was substantially (50%) reduced in mutant mice (Fig. 4 e). The reduced innervation of superficial laminae is probably a consequence of the reduced axon numbers available for the production of collaterals from the stem axon within the dorsal funiculus. Because cGKIα was also expressed in the collaterals of proprioceptive axons (Fig. 1 e), we followed the development of the proprioceptive axon collaterals in transverse serial sections using parvalbumin as a marker. Although pathfinding appeared unaffected in mutant embryos, fewer parvalbumin-positive proprioceptive collaterals arrived in ventral spinal cord to innervate motoneurons (Fig. 4, a and b).
Functional connectivity of spinal reflexes are impaired in cGKI-deficient mice
The longitudinal branching error, the decreased anti-trkA staining, which might reflect a reduced axon number in the dorsal funiculus, and the reduced innervation of the nociceptive superficial dorsal horn by trkA-positive sensory fibers in cGKI mutant mice prompted us to examine the functional connectivity of sensory afferents in mutant mice. We did this by using an in vitro spinal cord preparation (Fig. 5 a; Heppenstall and Lewin, 2001) to measure the integrated ventral root potential in isolated postnatal day 5 (P5)–P9 spinal cord. We found that the fast motoneuron potential evoked by electrical stimulation of large diameter afferents was reduced in magnitude in cGKI mutant mice, but this reduction of 40% did not reach statistical significance (Fig. 5 c; t test, P = 0.06). At higher stimulation intensities, a nociceptor-specific long-lasting ventral root potential is evoked that represents activation of the nociceptive flexion reflex. This potential was substantially (50%) and significantly reduced in cGKI mutant mice compared with wild-type controls (Fig. 5, b–d) (P values and percentages refer to integrated area of A-fiber and C-fiber response). We also measured the activity-dependent facilitation of the ventral root potential evoked by repetitive (1 Hz) stimulation of nociceptive axons, a phenomenon termed wind-up. The magnitude of the facilitation (percentage increase from baseline) was unaltered in mutants, indicating that this post-synaptic plasticity was largely intact (Fig. 5, e and f). Thus, the defect in cGKIα-mediated regulation of axon guidance at the DREZ early in development leads to a loss of functional connectivity that is selective for a pain-related spinal reflex. The striking correlation we observed between the anatomical and functional lack of sensory connectivity suggested that the magnitude of the flexion reflex response is limited by nociceptor connectivity within the superficial spinal cord.
Figure 5.
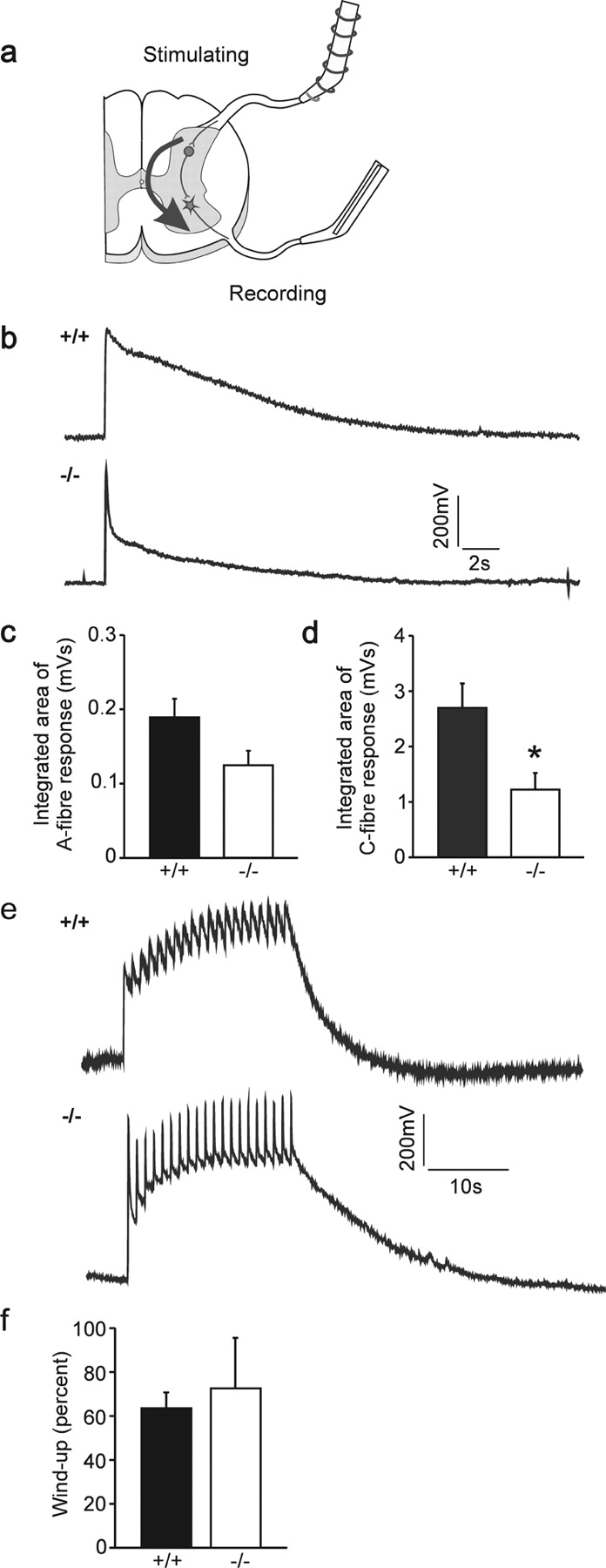
Impairment of the VRP of the spinal cord in the absence of cGKI. (a) In vitro–hemisected spinal cord preparation to measure monosynaptic and polysynaptic reflexes evoked by primary afferents. A scheme of a cross section of the spinal cord is shown and stimulating and recording electrodes are indicated. Dorsal is up. (b) VRPs of wild-type and cGKI-deficient neonatal mice. Electrical stimulation of dorsal root fibers generates a potential that can be recorded at the ventral root. The VRP is composed of a fast component evoked by A-fibers and a slower one activated by C-fibers (Heppenstall and Lewin, 2001). (c and d) Quantification of the VRP in wild-type and cGKI-deficient mice. The integrated area of the C-fiber area is significantly reduced in cGKI-deficient mice (P < 0.05; n = 6 per group). The integrated area of the A-fiber response was reduced but did not reach significance (P = 0.06). (e) Wind-up in wild-type and cGKI-deficient neonatal mice. Repetitive electrical stimulation of dorsal root fibers (20 times, 1 Hz) evokes an increase in the potential recorded from the ventral root. (f) Quantification of wind-up in wild-type and cGKI-deficient mice. Wind-up, measured as the percentage increase in the potential, is not different between wild-type and cGKI-deficient mice (P = 0.7; n = 6 per group).
cGMP counteracts semaphorin 3A–induced growth cone collapse in vitro
Previous in vitro studies revealed that enhanced cGMP levels modulate the collapse and turning behavior of growth cones in response to semaphorin 3A (Song et al., 1998). To determine whether cGKI participates in the transduction of these cGMP-mediated signals, DRGs explanted from wild-type and cGKI-deficient mice were analyzed for semaphorin 3A–mediated growth cone collapse. Under control conditions, growth cone morphology of DRG neurons of cGKI-deficient mice was indistinguishable from that of wild-type mice. They spread well and have lamellipodia and filopodia. We also did not observe any significant differences in neurite length and fasciculation between explants from cGKI−/− mice and wild-type littermates (Fig. 6 d; unpublished data). Furthermore, the efficiency of the semaphorin 3A–induced growth cone collapse was indistinguishable in wild-type and cGKI-deficient DRG explants (Fig. 6, a and c). Using membrane-permeable cyclic nucleotide analogues, we found that raising cytosolic cGMP levels (with 8-pCPT-cGMP) inhibited semaphorin 3A–mediated growth cone collapse in wild-type, as described by Song et al. (1998), but not in cGKI-deficient DRGs, whereas raising cAMP levels with the analogue 8-Br-cAMP was without effect (Fig. 6, a and c). Similar results were obtained with 8-Br-cGMP (1–5 mM; unpublished data). Neither 8-Br-cAMP nor 8-pCPT-cGMP alone had any detectable effect on growth cones of both genotypes (Fig. 6 a). Taken together, these in vitro studies indicate that cGKI is not part of the semaphorin 3A–induced signaling cascade that results in a collapse of growth cones, but cGKI activation can counteract semaphorin 3A–induced growth cone collapse of sensory axons.
Discussion
In this study, we have identified cGKIα as a component of the sensory axon growth cone machinery that transduces endogenous cGMP changes required for correct sensory axon guidance within the developing DREZ of the spinal cord. Tracing with DiI and anti-trkA staining demonstrated that sensory axons are unable to identify their correct path at the entrance zone of the spinal cord and, as a consequence, are unable to generate a rostral and caudal branch or, alternatively, are unable to maintain one of the two branches. Evidence for mistargeting of sensory axons in cGKI mutant mice was observed by anti-trkA staining, as many axon fascicles grow directly into the dorsal horn inappropriately in the direction of the central canal most likely without the formation of a rostral or caudal axon. It is not possible with present methods to distinguish whether cGKI-deficient sensory axons are unable to branch at the DREZ or sustain two branches indefinitely. As a consequence of this branching or retention failure, the developing dorsal funiculus of mutant mice was found to be smaller, and less collaterals were observed that can project to lamina-specific targets in the dorsal and ventral horn. The longitudinal growth error is most likely not due to a lack of axonal defasciculation in the DREZ, because sensory nerves of mutant mice do not appear more fasciculated outside the cord and in vitro–cultivated DRG axons do not generate thicker fascicles. The preference for rostral growth within the DREZ in the absence of cGKI remains currently speculative. However, a striking correlation was found between our anatomical and functional investigations, and therefore, perhaps as a result of early errors in sensory axon targeting in the DREZ, the amplitude of the nociceptive flexion reflex is selectively and substantially reduced in mutant mice. Thus axon targeting errors have clear functional consequences for spinal reflex circuit connectivity. The reduction in the sensory neuron to dorsal horn connectivity we observed functionally probably indicates that the reduction in the innervation of lamina II by nociceptive sensory neurons is not compensated by an increased number of synapses per sensory neuron. Interestingly, plasticity of the nociceptive flexion reflex, termed wind-up, is preserved, indicating that this largely post-synaptic phenomenon (Herrero et al., 2000) is unaffected by the reduced anatomical connectivity of C-fiber afferents found in cGKI mutant mice. Early in development, cGKI shows also a very restricted localization within the spinal cord, and, consequently, pathfinding of other axon systems was not affected by the absence of cGKI.
Previous in vitro assays indicated that cGMP can antagonize the semaphorin 3A–induced growth cone collapse (Song et al., 1998). Our analysis of growth cone collapse using cGKI-deficient DRGs indicated that the activation of cGKIα mediates, in part, this counteracting effect of cGMP. Because growth cones of cGKI-deficient DRGs collapse as efficiently as wild-type DRGs, it can also be concluded that cGKIα is not part of the semaphorin 3A signaling pathway that results in the collapse of sensory growth cones. This contrasts with conclusions drawn from in vitro studies using exclusively pharmacological reagents (Dontchev and Letourneau, 2002). Although semaphorin 3A has been described as an in vitro branching factor for Xenopus retinal growth cones but not for rat E17 DRG neurons (Wang et al., 1999; Campbell et al., 2001), a possible counteracting effect by activation of cGKIα might not play a role for sensory pathfinding at the DREZ, as studies on semaphorin 3A– or neuropilin-1–deficient mice have not reported sensory neuron guidance defects similar to those we describe here (Kitsukawa et al., 1997; Taniguchi et al., 1997).
The guidance signals within the DREZ and the responding guidance receptor(s) on sensory axons whose activation elevates intracellular cGMP and subsequently cGKIα remain to be determined. Slit2, a large extracellular matrix glycoprotein that acts as an axonal guidance signal for several axon systems (Erskine et al., 2000; Niclou et al., 2000; Ringstedt et al., 2000; Bagri et al., 2002; Plump et al., 2002), has also been described as an in vitro branching factor for DRG neurons growing on collagen (Wang et al., 1999). Although the more general limitation of monolayer cell cultures does not allow a definitive conclusion, most likely collateral branching and not the formation of T-like branches were analyzed under the experimental conditions described by Wang et al. (1999), because relatively advanced DRG neurons from E17 rats were assayed. A recent study also suggested that cGMP might modulate Slit2 signaling via cGKs (Nguyen-Ba-Charvet et al., 2001). However, in these investigations, the pharmacological reagent KT5823 was used, which does not function as an inhibitor of cGKI in living cells, in contrast to in vitro conditions (Burkhardt et al., 2000).
The analysis of downstream targets of cGKIα might also help to define the exact function of cGKIα in sensory growth cone steering at the DREZ further. In other cell types outside the nervous system, cGKI lowers [Ca2+]i by interfering with intracellular calcium release (Schlossmann et al., 2000) and/or activation of maxi KCa channels (Sausbier et al., 2000), and may decrease myosin light chain phosphorylation by interacting with the myosin targeting subunit of myosin phosphatase (Surks et al., 1999) or by phosphorylation of RhoA, leading to its inactivation (Sauzeau et al., 2000). Phosphorylation/dephosphorylation of the myosin light chain regulates the cytoskeletal organization in many cell types (Hall, 1998; Burridge, 1999) and could affect filopodial extension in growth cones by modulating retrograde flow of actin filaments (Lin et al., 1996). In addition, organization of cytoskeletal actin by cGKIα in growth cones of sensory axons might also occur by affecting the activity of the actin organizing proteins VASP (Reinhard et al., 2001) or CRP2 (Huber et al., 2000). Further studies will reveal which, or whether a combination of these multiple downstream targets is regulated by cGKIα in the DREZ.
Materials and methods
cGKI-deficient mice and quantification of the ventral root potential
The generation of a cGKI-null allele and mouse genotyping has been previously described (Wegener et al., 2002). Wild-type (+/+), heterozygous (+/−), and cGKI-deficient (−/−) embryos were obtained from staged matings of heterozygous mice on a pure 129/Sv or mixed 129Sv/C57BL6 genetic background. The measurement of the functional connectivity of sensory afferents in spinal cord preparations was done using an in vitro–hemisected spinal cord preparation (Heppenstall and Lewin, 2001). Spinal cords were removed from neonatal mice (P5–P9), hemisected down the midline, placed in a Perspex recording chamber, and superfused with oxygenated modified Krebs solution (138 mM NaCl, 1.35 mM KCl, 21 mM NaHCO3, 0.58 mM NaH2PO4, 1.16 mM MgCl2, 1.26 mM CaCl2, 10 mM glucose). DC recordings were made after a 2-h recovery period with a close-fitting glass suction electrode attached to the L5 ventral root. Low intensity electrical stimuli of the L5 dorsal root via a glass suction electrode (50 μA, 50 μs) evoke fast short duration ventral root potential, as this stimulus is selective for larger diameter A-fiber afferents. With higher intensity stimuli that activate both A- and C-fiber afferents (500 μA, 500 μs), a long-lasting slow ventral root potential (VRP) is evoked. Repetitive stimulation at 1 Hz for 20 s produces a marked facilitation of the ventral root potential amplitude (Fig. 5 e), and this is only observed when C-fiber afferents are stimulated (Heppenstall and Lewin, 2001). The A-fiber– and C-fiber–mediated components of the VRP were measured as the integrated area of the response at 0–2 and 2–20 s, respectively. Wind-up was determined by normalizing the increase in size of the VRP to the initial magnitude and expressing it as a percentage increase. Traces were acquired with the Powerlab 4.0 system using Superscope software.
Immunodetection, DiI tracing, and DRG cell counting
For Western blotting, DRGs from E13 mice were collected, solubilized in SDS-PAGE sample buffer, and separated by 10% SDS-PAGE. Equal loading of lanes was confirmed by staining with antibodies to L1 (Rathjen and Schachner, 1984). Rabbit antibodies specific to both isoforms, to the α or β isoform of cGKI, were generated as detailed elsewhere (Schlossmann et al., 2000). For immunohistochemical detection, cryostat sections of formaldehyde-fixed embryos were stained by indirect immunofluorescence using rabbit antibodies to cGKI (1:500 to 1:1,000), trkA (1:2,000; provided by L. F. Reichardt, University of California San Francisco, San Francisco, CA), parvalbumin (1:1,000; Calbiochem), TAG-1 (Dodd et al., 1988), or L1 (Rathjen and Schachner, 1984) followed by goat anti–rabbit or rabbit anti–mouse antibodies coupled to Cy3 (Dianova). To detect intracellular localized cGKI, cultivated DRG neurons were fixed and permeabilized with 0.1% Triton X-100 before incubation with antibodies. For DiI (Molecular Probes) tracing, mouse embryos were immersion fixed in 4% paraformaldehyde in PBS and stored in fixative until the application of axon tracer. DiI dissolved in 100% ethanol was injected into single DRGs. The preparations were then incubated in 4% paraformaldehyde buffer at 37°C for 2–4 d. Whole mounts were viewed laterally using a confocal fluorescence microscope (Bio-Rad Laboratories).
The intensity of trkA staining was quantified in serial horizontal sections at E13 in three different defined fields in thoracic regions of the spinal cord using NIH image software. The pixel intensity of each field of the complete primordium of the dorsal funiculus (eight to nine serial sections of 20 μm thickness) was integrated, and the mean calculated and presented as relative units. In transverse sections, the trkA-stained area was delineated with a mouse cursor to measure the area and the intensity in throracic and lumbar regions. Data from four to five adjacent sections of each level were collected and the mean was calculated.
Cells from L4 and L5 DRG of E13 and E14 embryos were counted in serial cryostat sections following methods previously described (ElShamy and Ernfors, 1996; Farinas et al., 1996; Riethmacher et al., 1997). Sections (14 μm) were stained by the fluorescent Hoechst 33258 nuclear stain (Spaltmann and Brummendorf, 1996). The maximum diameter observed by this stain was 10 μm, suggesting that the actual cell numbers might be underestimated.
Growth cone collapse assay
For the growth cone collapse assay, we followed methods developed by Raper and Kapfhammer (1990) using E13 DRG explants from the lumbar region grown on poly-l-lysine/laminin-1 (GIBCO BRL) for 24 h in F-12 (GIBCO BRL) supplemented with hNGF-β (20 ng/ml; Boehringer), N2 supplement (GIBCO BRL), and bovine pituitary extract (200 μg/ml; GIBCO BRL) (Luo et al., 1993). Explants were pretreated with the indicated concentrations of 8-pCPT-cGMP, 8-Br-cGMP, or 8-Br-cAMP (BioLog) for 30 min followed by incubation with semaphorin 3A–containing culture supernatants of transiently transfected COS7 cells for 1 h. Culture supernatants were titrated to induce a collapse of 90–100% of growth cones after 1 h. Glutaraldehyde-fixed explants were stained by mAb A2B5 (Developmental Studies Hybridoma Bank) for visualization. cDNA encoding chick semaphorin 3A (collapsin-1) (Luo et al., 1993) was obtained by RT-PCR and subcloned without the signal sequence into a modified pSG5 vector (Stratagene) containing the signal sequence of neurofascin (Volkmer et al., 1992) followed by a myc and his tag. The cDNA sequence of the construct was verified by DNA sequencing. A cDNA construct obtained from J.A. Raper (University of Philadelphia, Philadelphia, PA) served as a control in some experiments.
Acknowledgments
We thank L.F. Reichardt and J.A. Raper for providing antibodies to trkA and collapsin-1 expression plasmid, respectively, J. Schlossmann for generation of cGKI antibodies, U. Zacharias for help with NIH image software, and T. Brümmendorf and J. Cohen for the critical reading of an earlier version of the manuscript.
This work was supported by grants from Deutsche Forschungsgemeinschaft, Boehringer Ingelheim Stiftung, VolkswagenStiftung, and Fond der Chemischen Industrie.
H. Schmidt's present address is King's College London, Max-Delbrück-Centrum Centre for Developmental Neurobiology, Guy's Hospital Campus, London SE1 1UL, UK.
Footnotes
Abbreviations used in this paper: cGKI, cGMP-dependent protein kinase I; DREZ, dorsal root entry zone; DRG, dorsal root ganglion; E, embryonic day; P, postnatal day; VRP, ventral root potential.
References
- Bagri, A., O. Marin, A.S. Plump, J. Mak, S.J. Pleasure, J.L.R. Rubenstein, and M. Tessier-Lavigne. 2002. Slit proteins prevent midline crossing and determine the dorsoventral position of major axonal pathways in the mammalian forebrain. Neuron. 33:233–248. [DOI] [PubMed] [Google Scholar]
- Burkhardt, M., M. Glazova, S. Gambaryan, T. Vollkommer, E. Butt, B. Bader, K. Heermeier, T.M. Lincoln, U. Walter, and A. Palmetshofer. 2000. KT5823 inhibits cGMP-dependent protein kinase activity in vitro but not in intact human platelets and rat mesangial cells. J. Biol. Chem. 275:33536–33541. [DOI] [PubMed] [Google Scholar]
- Burridge, K. 1999. Crosstalk between Rac and Rho. Science. 283:2028–2029. [DOI] [PubMed] [Google Scholar]
- Campbell, D.S., A.G. Regan, J.S. Lopez, D. Tannahill, W.A. Harris, and C.E. Holt. 2001. Semaphorin 3A elicits stage-dependent collapse, turning, and branching in Xenopus retinal growth cones. J. Neurosci. 21:8538–8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, B.M., E. Frank, F.A. Johnson, and S.A. Scott. 1989. Development of central projections of lumbosacral sensory neurons in the chick. J. Comp. Neurol. 279:556–566. [DOI] [PubMed] [Google Scholar]
- Dodd, J., S.B. Morton, D. Karagogeos, M. Yamamoto, and T.M. Jessell. 1988. Spatial regulation of axonal glycoprotein expression on subsets of embryonic spinal neurons. Neuron. 1:105–116. [DOI] [PubMed] [Google Scholar]
- Dontchev, V.D., and P.C. Letourneau. 2002. Nerve growth factor and semaphorin 3A signaling pathways interact in regulating sensory neuronal growth cone motility. J. Neurosci. 22:6659–6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide, A.L., and J.C. Glover. 1995. Development of the longitudinal projection patterns of lumbar primary sensory afferents in the chicken embryo. J. Comp. Neurol. 353:247–259. [DOI] [PubMed] [Google Scholar]
- ElShamy, W.M., and P. Ernfors. 1996. A local action of neurotrophin-3 prevents the death of proliferating sensory neuron precursor cells. Neuron. 16:963–972. [DOI] [PubMed] [Google Scholar]
- Erskine, L., S.E. Williams, K. Brose, T. Kidd, R.A. Rachel, C.S. Goodman, M. Tessier-Lavigne, and C.A. Mason. 2000. Retinal ganglion cell axon guidance in the mouse optic chiasm: expression and function of robos and slits. J. Neurosci. 20:4975–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinas, I., C.K. Yoshida, C. Backus, and L.F. Reichardt. 1996. Lack of neurotrophin-3 results in death of spinal sensory neurons and premature differentiation of their precursors. Neuron. 17:1065–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, A.D., and D.L. Garbers. 2000. Guanylyl cyclases as a family of putative odorant receptors. Annu. Rev. Neurosci. 23:417–439. [DOI] [PubMed] [Google Scholar]
- Hall, A. 1998. Rho GTPases and the actin cytoskeleton. Science. 279:509–514. [DOI] [PubMed] [Google Scholar]
- Heppenstall, P.A., and G.R. Lewin. 2001. BDNF but not NT-4 is required for normal flexion reflex plasticity and function. Proc. Natl. Acad. Sci. USA. 98:8107–8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero, J.F., J.M. Laird, and J.A. Lopez-Garcia. 2000. Wind-up of spinal cord neurones and pain sensation: much ado about something? Prog. Neurobiol. 61:169–203. [DOI] [PubMed] [Google Scholar]
- Hofmann, F., A. Ammendola, and J. Schlossmann. 2000. Rising behind NO: cGMP-dependent protein kinases. J. Cell Sci. 113:1671–1676. [DOI] [PubMed] [Google Scholar]
- Honig, M.G., and R.I. Hume. 1989. Dil and diO: versatile fluorescent dyes for neuronal labelling and pathway tracing. Trends Neurosci. 12:333–335, 340–341. [PubMed] [Google Scholar]
- Hopker, V.H., D. Shewan, L.M. Tessier, M. Poo, and C. Holt. 1999. Growth-cone attraction to netrin-1 is converted to repulsion by laminin-1. Nature. 401:69–73. [DOI] [PubMed] [Google Scholar]
- Huber, A., W.L. Neuhuber, N. Klugbauer, P. Ruth, and H.D. Allescher. 2000. Cysteine-rich protein 2, a novel substrate for cGMP kinase I in enteric neurons and intestinal smooth muscle. J. Biol. Chem. 275:5504–5511. [DOI] [PubMed] [Google Scholar]
- Kitsukawa, T., M. Shimizu, M. Sanbo, T. Hirata, M. Taniguchi, Y. Bekku, T. Yagi, and H. Fujisawa. 1997. Neuropilin-semaphorin III/D-mediated chemorepulsive signals play a crucial role in peripheral nerve projection in mice. Neuron. 19:995–1005. [DOI] [PubMed] [Google Scholar]
- Klein, R. 2001. Excitatory Eph receptors and adhesive ephrin ligands. Curr. Opin. Cell Biol. 13:196–203. [DOI] [PubMed] [Google Scholar]
- Lin, C.H., E.M. Espreafico, M.S. Mooseker, and P. Forscher. 1996. Myosin drives retrograde F-actin flow in neuronal growth cones. Neuron. 16:769–782. [DOI] [PubMed] [Google Scholar]
- Lucas, K.A., G.M. Pitari, S. Kazerounian, I. Ruiz-Stewart, J. Park, S. Schulz, K.P. Chepenik, and S.A. Waldman. 2000. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol. Rev. 52:375–414. [PubMed] [Google Scholar]
- Luo, Y., D. Raible, and J.A. Raper. 1993. Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell. 75:217–227. [DOI] [PubMed] [Google Scholar]
- Mirnics, K., and H.R. Koerber. 1995. Prenatal development of rat primary afferent fibers: II. Central projections. J. Comp. Neurol. 355:601–614. [DOI] [PubMed] [Google Scholar]
- Mirnics, K., and H.R. Koerber. 1997. Properties of individual embryonic primary afferents and their spinal projections in the rat. J. Neurophysiol. 78:1590–1600. [DOI] [PubMed] [Google Scholar]
- Mueller, B.K. 1999. Growth cone guidance: first steps towards a deeper understanding. Annu. Rev. Neurosci. 22:351–388. [DOI] [PubMed] [Google Scholar]
- Nguyen-Ba-Charvet, K.T., K. Brose, V. Marillat, C. Sotelo, M. Tessier-Lavigne, and A. Chedotal. 2001. Sensory axon response to substrate-bound Slit2 is modulated by laminin and cyclic GMP. Mol. Cell. Neurosci. 17:1048–1058. [DOI] [PubMed] [Google Scholar]
- Niclou, S.P., L. Jia, and J.A. Raper. 2000. Slit2 is a repellent for retinal ganglion cell axons. J. Neurosci. 20:4962–4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki, S., and W.D. Snider. 1997. Initial trajectories of sensory axons toward laminar targets in the developing mouse spinal cord. J. Comp. Neurol. 380:215–229. [PubMed] [Google Scholar]
- Pfeifer, A., P. Ruth, W. Dostmann, M. Sausbier, P. Klatt, and F. Hofmann. 1999. Structure and function of cGMP-dependent protein kinases. Rev. Physiol. Biochem. Pharmacol. 135:105–149. [DOI] [PubMed] [Google Scholar]
- Plump, A.S., L. Erskine, C. Sabatier, K. Brose, C.J. Epstein, C.S. Goodman, C.A. Mason, and M. Tessier-Lavigne. 2002. Slit1 and Slit2 cooperate to prevent premature midline crossing of retinal axons in the mouse visual system. Neuron. 33:219–232. [DOI] [PubMed] [Google Scholar]
- Qian, Y., D.S. Chao, D.R. Santillano, T.L. Cornwell, A.C. Nairn, P. Greengard, T.M. Lincoln, and D.S. Bredt. 1996. cGMP-dependent protein kinase in dorsal root ganglion: relationship with nitric oxide synthase and nociceptive neurons. J. Neurosci. 16:3130–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rager, G., S. Lausmann, and F. Gallyas. 1979. An improved silver stain for developing nervous tissue. Stain Technol. 54:193–200. [DOI] [PubMed] [Google Scholar]
- Raper, J.A. 2000. Semaphorins and their receptors in vertebrates and invertebrates. Curr. Opin. Neurobiol. 10:88–94. [DOI] [PubMed] [Google Scholar]
- Raper, J.A., and J.P. Kapfhammer. 1990. The enrichment of a neuronal growth cone collapsing activity from embryonic chick brain. Neuron. 4:21–29. [DOI] [PubMed] [Google Scholar]
- Rathjen, F.G., and M. Schachner. 1984. Immunocytological and biochemical characterization of a new neuronal cell surface component (L1 antigen) which is involved in cell adhesion. EMBO J. 3:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard, M., T. Jarchau, and U. Walter. 2001. Actin-based motility: stop and go with Ena/VASP proteins. Trends Biochem. Sci. 26:243–249. [DOI] [PubMed] [Google Scholar]
- Riethmacher, D., E. Sonnenberg-Riethmacher, V. Brinkmann, T. Yamaai, G.R. Lewin, and C. Birchmeier. 1997. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature. 389:725–730. [DOI] [PubMed] [Google Scholar]
- Ringstedt, T., J.E. Braisted, K. Brose, T. Kidd, C. Goodman, M. Tessier-Lavigne, and D.D. O'Leary. 2000. Slit inhibition of retinal axon growth and its role in retinal axon pathfinding and innervation patterns in the diencephalon. J. Neurosci. 20:4983–4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sausbier, M., R. Schubert, V. Voigt, C. Hirneiss, A. Pfeifer, M. Korth, T. Kleppisch, P. Ruth, and F. Hofmann. 2000. Mechanisms of NO/cGMP-dependent vasorelaxation. Circ. Res. 87:825–830. [DOI] [PubMed] [Google Scholar]
- Sauzeau, V., H. Le Jeune, C. Cario-Toumaniantz, A. Smolenski, S.M. Lohmann, J. Bertoglio, P. Chardin, P. Pacaud, and G. Loirand. 2000. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J. Biol. Chem. 275:21722–21729. [DOI] [PubMed] [Google Scholar]
- Schlossmann, J., A. Ammendola, K. Ashman, X. Zong, A. Huber, G. Neubauer, G.X. Wang, H.D. Allescher, M. Korth, M. Wilm, et al. 2000. Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Iβ. Nature. 404:197–201. [DOI] [PubMed] [Google Scholar]
- Song, H., G. Ming, Z. He, M. Lehmann, M. Tessier-Lavigne, and M. Poo. 1998. Conversion of neuronal growth cone responses from repulsion to attraction by cyclic nucleotides. Science. 281:1515–1518. [DOI] [PubMed] [Google Scholar]
- Song, H.J., and M.M. Poo. 1999. Signal transduction underlying growth cone guidance by diffusible factors. Curr. Opin. Neurobiol. 9:355–363. [DOI] [PubMed] [Google Scholar]
- Song, H.J., G.L. Ming, and M.M. Poo. 1997. cAMP-induced switching in turning direction of nerve growth cones. Nature. 388:275–279. [DOI] [PubMed] [Google Scholar]
- Spaltmann, F., and T. Brummendorf. 1996. CEPU-1, a novel immunoglobulin superfamily molecule, is expressed by developing cerebellar Purkinje cells. J. Neurosci. 16:1770–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surks, H.K., N. Mochizuki, Y. Kasai, S.P. Georgescu, K.M. Tang, M. Ito, T.M. Lincoln, and M.E. Mendelsohn. 1999. Regulation of myosin phosphatase by a specific interaction with cGMP-dependent protein kinase Iα. Science. 286:1583–1587. [DOI] [PubMed] [Google Scholar]
- Taniguchi, M., S. Yuasa, H. Fujisawa, I. Naruse, S. Saga, M. Mishina, and T. Yagi. 1997. Disruption of semaphorin III/D gene causes severe abnormality in peripheral nerve projection. Neuron. 19:519–530. [DOI] [PubMed] [Google Scholar]
- Tessier-Lavigne, M., and C.S. Goodman. 1996. The molecular biology of axon guidance. Science. 274:1123–1133. [DOI] [PubMed] [Google Scholar]
- Volkmer, H., B. Hassel, J.M. Wolff, R. Frank, and F.G. Rathjen. 1992. Structure of the axonal surface recognition molecule neurofascin and its relationship to a neural subgroup of the immunoglobulin superfamily. J. Cell Biol. 118:149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K.H., K. Brose, D. Arnott, T. Kidd, C.S. Goodman, W. Henzel, and M. Tessier-Lavigne. 1999. Biochemical purification of a mammalian slit protein as a positive regulator of sensory axon elongation and branching. Cell. 96:771–784. [DOI] [PubMed] [Google Scholar]
- Wegener, J.W., H. Nawrath, W. Wolfsgruber, S. Kühbander, C. Werner, F. Hofmann, and R. Feil. 2002. cGMP-dependent protein kinase I mediates the negative inotropic effect of cGMP in the murine myocardium. Circ. Res. 90:18–20. [DOI] [PubMed] [Google Scholar]