

Abstract
Myotonic dystrophy (DM) is caused by two similar noncoding repeat expansion mutations (DM1 and DM2). It is thought that both mutations produce pathogenic RNA molecules that accumulate in nuclear foci. The DM1 mutation is a CTG expansion in the 3′ untranslated region (3′-UTR) of dystrophia myotonica protein kinase (DMPK). In a cell culture model, mutant transcripts containing a (CUG)200 DMPK 3′-UTR disrupt C2C12 myoblast differentiation; a phenotype similar to what is observed in myoblast cultures derived from DM1 patient muscle. Here, we have used our cell culture model to investigate how the mutant 3′-UTR RNA disrupts differentiation. We show that MyoD protein levels are compromised in cells that express mutant DMPK 3′-UTR transcripts. MyoD, a transcription factor required for the differentiation of myoblasts during muscle regeneration, activates differentiation-specific genes by binding E-boxes. MyoD levels are significantly reduced in myoblasts expressing the mutant 3′-UTR RNA within the first 6 h under differentiation conditions. This reduction correlates with blunted E-box–mediated gene expression at time points that are critical for initiating differentiation. Importantly, restoring MyoD levels rescues the differentiation defect. We conclude that mutant DMPK 3′-UTR transcripts disrupt myoblast differentiation by reducing MyoD levels below a threshold required to activate the differentiation program.
Keywords: myotonic dystrophy; trinucleotide repeats; differentiation; MyoD; muscle development
Introduction
Myotonic dystrophy (DM)* is a common autosomal dominant form of muscular dystrophy that shows phenotypic and genetic heterogeneity. The distinguishing features of this disease are myotonia (delayed muscle relaxation after a voluntary contraction) and progressive skeletal muscle loss (Harper, 1989). Other frequent defects include cardiac arrhythmias, cataracts, insulin resistance, and frontal balding (Harper, 1989). The severity and age of onset of these symptoms can vary greatly from patient to patient, even within a single family. Patients affected by congenital DM are born with hypotonia, mental retardation, and muscle development defects, and later develop adult DM symptoms (Harper, 1989). To date, two mutations (DM1 and DM2), at unlinked loci, are associated with myotonic dystrophy.
The DM1 mutation, which accounts for 98% of DM cases, is a triplet repeat (CTG) expansion on human chromosome 19 located in the 3′ untranslated region (3′-UTR) of a protein kinase gene (dystrophia myotonica protein kinase [DMPK]) (Brook et al., 1992; Fu et al., 1992; Mahadevan et al., 1992). It is not known exactly how this mutation causes disease, but several mechanisms have been proposed (for review see Tapscott and Thornton, 2001). These include altered DMPK levels and/or isoforms, misexpression of neighboring genes due to chromatin reorganization at the DM1 locus, and a trans-dominant mutant DMPK mRNA. The DM2 mutation was recently identified as a CCTG expansion in the first intron of the zinc finger protein 9 (ZNF9) gene on human chromosome 3 (Liquori et al., 2001). The similarity of the nonprotein coding repeat expansion in both mutant ZNF9 and DMPK transcripts, plus the absence of genes analogous to DMPK or its neighbors at the DM2 locus, suggest that dominant effects mediated by CUG/CCUG expansion–containing RNA molecules play the predominant role in causing DM.
How the mutant RNA causes disease is under intense investigation. RNA FISH experiments show that both mutant DMPK and ZNF9 transcripts localize to distinct foci in the nuclei of DM patient cells (Taneja et al., 1995; Davis et al., 1997; Mankodi et al., 2001). It is thought that interactions between these transcripts and RNA binding proteins result in the aberrant expression of other genes. Several proteins bind the expanded DMPK 3′-UTR RNA in vitro (Timchenko et al., 1996; Lu et al., 1999; Miller et al., 2000; Tian et al., 2000; Tiscornia and Mahadevan, 2000), and one family of factors, MBNL, MBLL, and MBXL, colocalizes with RNA foci in both DM1 and DM2 patient cells (Fardaei et al., 2001, 2002; Mankodi et al., 2001). RNAs containing expanded CUG tracts have been shown to skew the alternative splicing of unrelated transcripts (Philips et al., 1998), including the insulin receptor mRNA, which likely explains insulin resistance seen in patients (Savkur et al., 2001). Perhaps the most compelling evidence implicating the mutant RNA in DM pathogenesis comes from transgenic mice that express heterologous transcripts containing ∼250 CUG repeats and develop myotonia and myopathy similar to what is seen in DM patients (Mankodi et al., 2000).
We have studied the effects of the mutant DMPK 3′-UTR RNA in a cell culture model. To establish this model, the human DMPK 3′-UTR sequence containing either a pathogenic (CUG)200 repeat tract or a wild-type (CUG)5 tract was fused to a reporter gene (lacZ or GFP) and expressed in C2C12 mouse myoblasts (Amack et al., 1999; Amack and Mahadevan, 2001). C2C12 are muscle precursor cells originally isolated from injured adult mouse muscle that have proven an excellent system to study myogenic differentiation (Yaffe and Saxel, 1977; Blau et al., 1983). When cultured in differentiation media lacking growth factors, C2C12 cells up-regulate muscle-specific genes, exit the cell cycle, and fuse into multinucleated myotubes. We have shown that transcripts containing the mutant DMPK 3′-UTR aggregate into nuclear foci and block C2C12 differentiation (Amack et al., 1999). This differentiation defect may represent muscle development abnormalities found in congenital DM patients (Harvey et al., 1976; Farkas-Bargeton et al., 1988) and/or point to defects in muscle regeneration, which could contribute to muscle wasting in adult patients. Recently, the differentiation defect was confirmed in cultured myogenic satellite cells taken from DM1 patients (Furling et al., 2001; Timchenko et al., 2001).
Here, we have used our cell culture model to investigate the molecular mechanisms that underlie how the mutant DMPK 3′-UTR RNA disrupts myoblast differentiation. The sequence of events in the differentiation pathway has been well characterized using C2C12 cells (Andres and Walsh, 1996; Fig. 1 A). MyoD and Myf5 are myogenic transcription factors expressed in committed myoblasts capable of inducing expression of myogenin and other genes required for differentiation. After myogenin expression, cells withdraw from the cell cycle and fuse into multinucleated myotubes. Previous RNA analyses using our cell culture system showed that the programmed up-regulation of myogenin and p21 is severely blunted in cells expressing the mutant DMPK 3′-UTR (Amack and Mahadevan, 2001). This suggested that the mutant 3′-UTR RNA interferes with the earliest stages of differentiation, perhaps by impeding the initiation of differentiation-specific gene expression. In this report, we show that MyoD protein levels are compromised in cells expressing mutant DMPK 3′-UTR transcripts at time points thought to be critical for the initiation of C2C12 differentiation.
Figure 1.



The myogenic machinery downstream of myogenin is functional in cells expressing the mutant DMPK 3′-UTR. (A) The temporal progression of major events in the C2C12 differentiation pathway. Cells proliferating in growth media express MyoD and Myf5. When cultured in differentiation media lacking growth factors, cells initiate myogenin expression, exit the cell cycle, turn on muscle-specific structural genes, such as MHC, and fuse into myotubes. (B) Uninfected GFP+mut 3′-UTR pool (mut 3′-UTR) cells show a differentiation defect, and do not fuse into myotubes (stained red by immunofluorescent staining of MHC) as effectively as GFP+wt 3′-UTR pool (wt 3′-UTR) and control C2C12 cells. However, GFP+mut 3′-UTR pool cells infected with a retrovirus that produces myogenin are capable of forming thick myotubes similar to those formed in GFP+wt 3′-UTR pool and C2C12 cells. Infection with a control retrovirus that expresses LacZ has no effect on the differentiation phenotype. Nuclei are stained blue with DAPI. (C) Western blot analysis of cultures in growth media shows exogenous myogenin expression only in cells infected with the myogenin retrovirus (+myg). These blots were also probed for dynein, which serves as a loading control.
Results
Mutant DMPK 3′-UTR RNA does not disable myogenic mechanisms downstream of myogenin
Myogenin is a key player in myoblast differentiation. In mice, the lack of myogenin inhibits myoblasts from differentiating into myotubes (Hasty et al., 1993; Nabeshima et al., 1993). However, myogenic cell lines that express myogenin and yet fail to differentiate show that defects in machinery downstream of myogenin can also block differentiation (Tapscott et al., 1993; Tedesco et al., 1995; Katagiri et al., 1997). Because myogenin up-regulation is blunted in cells expressing the mutant DMPK 3′-UTR RNA (Amack and Mahadevan, 2001), we wanted to determine whether adding exogenous myogenin is sufficient to push these cells through differentiation, or if downstream mechanisms/molecules in the pathway are also affected. To test this, myogenin was overexpressed via infection with a retroviral vector carrying a myogenin expression cassette. We tested the effects of exogenous myogenin in previously described pools of stable C2C12 clones (Amack and Mahadevan, 2001). Pools were generated by combining several individual clones that express GFP mRNA containing either a mutant DMPK 3′-UTR with ∼200 CUG repeats (termed GFP+mut 3′-UTR pool) or a wild-type 3′-UTR with five CUG repeats (GFP+wt 3′-UTR pool).
GFP+mut 3′-UTR pool cells infected with the myogenin retrovirus formed myotubes after 3 d in differentiation media, whereas cells infected with a control retrovirus expressing LacZ and uninfected cells failed to form myotubes (Fig. 1 B). Infection with the LacZ retrovirus demonstrated that myotube formation in these cells requires myogenin and is not simply a result of retroviral infection. The myotubes seen in GFP+mut 3′-UTR pool cultures infected with the myogenin retrovirus were similar to those seen in infected control C2C12 and GFP+wt 3′-UTR pool cells, which were larger than myotubes seen in uninfected C2C12 and GFP+wt 3′-UTR pool populations (Fig. 1 B). RNA FISH analysis showed that myotubes in the GFP+mut 3′-UTR pool were foci positive (unpublished data), indicating that expression of the mutant DMPK 3′-UTR RNA had not been lost. Exogenous myogenin expression in infected cells was confirmed by Western blot analysis (Fig. 1 C), and LacZ expression from the control retrovirus was detected by X-gal staining (unpublished data). These results show that myogenin is sufficient to correct the differentiation defect in cells expressing the mutant 3′-UTR, and that mechanisms downstream of myogenin required for myoblast differentiation in these cells are intact and functional.
MyoD protein levels are reduced in myoblasts expressing the mutant DMPK 3′-UTR
Our next step was to investigate the effects of the mutant DMPK 3′-UTR RNA on myogenic factors that act before myogenin induction. However, the initiation of myogenin expression and myoblast differentiation is governed by a complex regulatory network (for review see Puri and Sartorelli, 2000). Determining a priori which molecule(s) may be affected by the mutant RNA is daunting. Fortunately, we were presented a clue by an exceptional GFP+mut 3′-UTR clone (termed mut 3′-UTR-res) that expresses the mutant DMPK 3′-UTR RNA, but remains capable of differentiating into myotubes (Fig. 2 A). Any number of alterations in genetic background could account for this resistance, but further characterization revealed that steady-state MyoD mRNA levels were 2.6-fold higher than those in control C2C12 populations (Fig. 2 B). MyoD protein levels are also elevated in mut 3′-UTR-res cells, but only by 1.2-fold, as compared with control C2C12 cells (Fig. 2 C). This hinted to us that if increased MyoD is indeed responsible for “protecting” these myoblasts, then perhaps it is MyoD that is compromised by the mutant DMPK RNA.
Figure 2.

mut 3′-UTR-res cells express elevated levels of MyoD. (A) An individual GFP+mut 3′-UTR clone, named mut 3′-UTR-res, expresses mutant DMPK 3′-UTR transcripts, which are detected in aggregate foci (red) by RNA FISH, but retain the ability to fuse into multinucleated myotubes (stained green by GFP expression). Nuclei are stained blue with DAPI. (B) RNA slot blot analysis shows that steady-state MyoD levels are an average 2.6-fold higher in mut 3′-UTR-res cells than in C2C12 cells (n = 3 experiments). MyoD levels were normalized to gapdh levels. (C) Western blotting revealed that MyoD protein levels are elevated in mut 3′-UTR-res cells as compared with C2C12 cells, but only by an average of 1.2-fold (n = 3 experiments). MyoD protein levels were normalized to dynein.
MyoD is the founding member of the myogenic regulatory factor (MRF) family of transcription factors that also includes Myf5, myogenin, and MRF4 (Weintraub, 1993). At the onset of myoblast differentiation, MyoD directly transactivates myogenic genes, including myogenin (Hollenberg et al., 1993; Bergstrom et al., 2002). MyoD shares its role in myoblast determination with Myf5, and one can compensate for the lack of the other (Rudnicki et al., 1993). In our cell culture model, analyses of MyoD and Myf5 expression at the RNA level revealed no consistent differences in cells expressing the mutant DMPK 3′-UTR RNA when compared with control C2C12 cells (Amack and Mahadevan, 2001). However, it remained possible that MyoD and/or Myf5 are affected at the protein level. Therefore, we first tested whether the presence of the mutant RNA affects MyoD or Myf5 levels in proliferating myoblasts. Western blotting showed that both Myf5 and MyoD levels were variable in each of the cell populations tested (Fig. 3). This may be due to the fact that we assayed nonsynchronized cells in log phase growth, during which levels of both factors fluctuate with the cell cycle (Kitzmann et al., 1998; Lindon et al., 1998). Still, densitometric quantification of MyoD, normalized to a dynein loading control, showed that MyoD levels in the GFP+mut 3′-UTR pool were significantly lower than those in control cells. The graph in Fig. 3 A shows that the average MyoD level in the GFP+mut 3′-UTR pool was 32% of the levels found in C2C12 and GFP+wt 3′-UTR pool cells (P < 0.01; n = 6 experiments). Analysis of several individual GFP+mut 3′-UTR clones (n = 6) showed comparable results, and the average MyoD level among these clones was 40% of the average level in C2C12 (Fig. 3 A). There were no significant differences in Myf5 protein levels (Fig. 3 B), indicating that the effect on MyoD has some degree of specificity.
Figure 3.
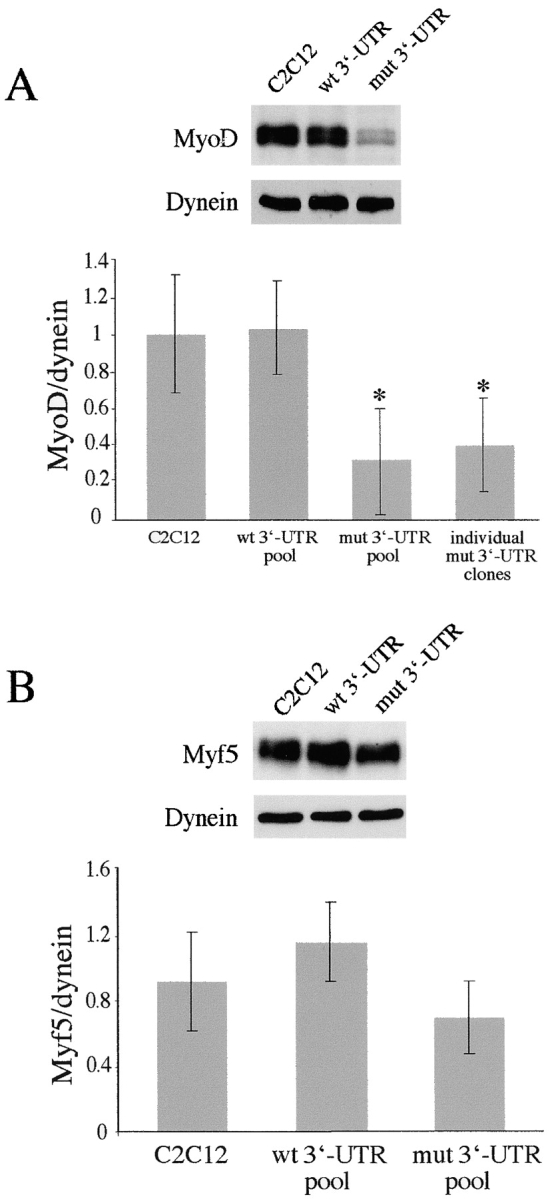
Protein levels of MyoD, but not Myf5, are low in proliferating GFP+mut 3′-UTR cells. (A) Despite variability, Western blots indicate that MyoD levels are significantly lower in proliferating GFP+mut 3′-UTR pool (mut 3′-UTR) cells as compared with GFP+wt 3′-UTR (wt 3′-UTR) and control C2C12 cells. Western blot results from six independent experiments were quantified by densitometry. The graph shows the average MyoD levels (normalized to dynein). Also shown is the average MyoD level among six individual GFP+mut 3′-UTR clones, which is similar to the average in the GFP+mut 3′-UTR pool. An asterisk (*) signifies a statistical difference (P < 0.05) when compared with both C2C12 and GFP+wt 3′-UTR pool cells. (B) Myf5 levels are also variable in proliferating cells, but quantification of Myf5 Western blots (n = 3 experiments) showed no significant differences among C2C12, GFP+wt 3′-UTR pool, and GFP+mut 3′-UTR pool cells. Error bars represent one standard deviation.
MyoD exists as a phosphoprotein that can be resolved into hypophosphorylated and hyperphosphorylated forms on Western blots (Fig. 3 A). The slower migrating hyperphosphorylated form has been shown to be a target for degradation (Song et al., 1998; Kitzmann et al., 1999). It was possible that decreased MyoD protein levels in GFP+mut 3′-UTR cells were due to increased phosphorylation and increased turnover. We examined the relative ratios of hypophosphorylated versus hyperphosphorylated bands, but found no evidence of differences between the GFP+mut 3′-UTR pool and control C2C12 cells (unpublished data). Additionally, immunohistochemical analysis showed no differences in MyoD intracellular localization between the various pools of cells (unpublished data).
MyoD levels are further reduced under differentiation conditions
Although MyoD protein levels are lower in proliferating myoblasts that express mutant DMPK 3′-UTR transcripts, this difference may not have deleterious consequences in proliferating cells. The transcriptional activity of MyoD and Myf5 is held in check until cells are stimulated to enter the differentiation program by the removal of growth factors (Wei and Paterson, 2001). Therefore, we characterized the pattern of MyoD and Myf5 expression in GFP+mut 3′-UTR cells under differentiation conditions. In control C2C12 cells, MyoD levels decreased by an average of 28% relative to the starting levels in proliferating cells after 24 h in differentiation media, and then remained between 50–60% of the starting amount over the next 48 h (Fig. 4 A). A similar pattern was seen in GFP+wt 3′-UTR pool cells, in which MyoD levels decreased by 36% after 24 h. In the GFP+mut 3′-UTR pool, however, the average initial drop in MyoD after 24 h was significantly greater at 72% (Fig. 4 A). MyoD remained low at 48 h in these cells, but then rebounded by 72 h to levels similar to those in C2C12 cells (Fig. 4 A). The sharp drop in MyoD in GFP+mut 3′-UTR cells, coupled with already low starting levels, results in nearly undetectable levels of MyoD after 24 h in differentiation media.
Figure 4.

MyoD protein levels are reduced and E-box–luciferase activation is blunted in GFP+mut 3′-UTR cells after 24 h in differentiation media. (A) The pattern of MyoD expression in control C2C12, GFP+wt 3′-UTR pool, and GFP+mut 3′-UTR pool cells cultured in differentiation media was analyzed by Western blots. A representative Western blot is shown, and the graph reports the average behavior of MyoD expression in each cell population, as determined by quantifying results from five independent experiments. MyoD levels are normalized to dynein levels, which do not change during differentiation, and are analyzed relative to the levels measured in proliferating cells (0 h in differentiation media). The difference in the MyoD expression pattern between GFP+mut 3′-UTR pool cells and both C2C12 and GFP+wt 3′-UTR pool cells is statistically significant (P < 0.05) only after 24 h (*). (B) A parallel analysis of the Myf5 expression pattern revealed no statistically significant differences at any time point between the GFP+mut 3′-UTR pool and the control C2C12 and GFP+wt 3′-UTR pool populations. (C) A luciferase (LUC) reporter construct containing three E-boxes was used to measure E-box–mediated gene expression in control C2C12, GFP+wt 3′-UTR pool, and GFP+mut 3′-UTR pool cells. The graph shows the average fold induction of E-box–luciferase expression relative to luciferase levels in proliferating cells (from three independent experiments). All error bars represent one standard deviation.
Analysis of Myf5 expression revealed that concomitant with the decrease in MyoD levels, Myf5 levels increased ∼140% after 24 h in differentiation media (Fig. 4 B). This is consistent with previous studies of MyoD and Myf5 expression patterns during the cell cycle (Kitzmann et al., 1998; Lindon et al., 1998). Unlike our analysis of MyoD, we found no significant differences in the Myf5 expression pattern between GFP+mut 3′-UTR pool and control cells (Fig. 4 B), indicating that the negative effect after 24 h has some specificity to MyoD.
Activation of E-box–mediated gene expression is blunted in GFP+mut 3′-UTR cells
The blunted up-regulation of genes such as myogenin and p21 in GFP+mut 3′-UTR cells (Amack and Mahadevan, 2001) suggests defects in early differentiation-specific transcription. MyoD and Myf5 regulate transcription at DNA sequences (CANNTG) called E-boxes in promoter regions of muscle-specific genes (Lassar et al., 1989). Do diminished MyoD levels adversely affect E-box–directed gene expression, or can Myf5, which is unaffected, compensate for the loss of MyoD? We measured E-box activation using a reporter construct from which expression of a luciferase gene is driven by a thymidine kinase (TK) promoter containing three tandem E-boxes (pE-boxLuc). To study the fold activation of E-box expression over several days, cells were cotransfected with a luciferase reporter gene and a puromycin resistance marker that was used to select for stable transfectants. We determined the amount of luciferase activation that was E-box mediated by subtracting the contribution made by the TK promoter sequences, as measured using a control plasmid lacking the E-boxes (pTKLuc). In transfected control C2C12 and GFP+wt 3′-UTR pool cells, an average of three experiments showed that E-box–luciferase expression was activated ∼1.7-fold after 24 h in differentiation media and approximately twofold after 72 h (Fig. 4 C). In contrast, no activation was detected at any time point in transfected GFP+mut 3′-UTR pool cells (Fig. 4 C). This suggested that the consequence of reduced MyoD levels after 24 h in differentiation media may be blunted E-box expression, and indicates that Myf5 cannot compensate for the lack of MyoD.
MyoD protein levels and E-box activation are compromised within the first 6 h under differentiation conditions
Because each of the MRFs can transactivate E-box–containing promoters, we cannot separate out the individual contributions of MyoD, Myf5, and myogenin (MRF4 is expressed only in mature C2C12 myotubes). It is possible that the difference observed in E-box activation is due to unequal myogenin expression, which first starts to appear after 24 h in differentiation media in control C2C12 and GFP+wt 3′-UTR cells, but not in GFP+mut 3′-UTR cells (see Fig. 7 C, uninfected cells). To determine whether blunted E-box expression correlates directly with reduced MyoD levels, we analyzed luciferase expression in transfected cultures before myogenin expression. After 6 h in differentiation media, E-box–luciferase expression was up-regulated ∼1.5-fold in C2C12 and GFP+wt 3′-UTR pool cells, but no up-regualtion was seen in the GFP+mut 3′-UTR pool (Fig. 5 A). This is a time point at which no myogenin was detected by Western blotting (unpublished data). Analysis of MyoD at these time points showed that the reduction of MyoD protein levels in GFP+mut 3′-UTR pool cells occurs within 6 h of withdrawing growth factors (Fig. 5 B). Thus, the lack of E-box–luciferase expression in these cells correlates with reduced MyoD levels, and, at least initially, is not due to differential expression of myogenin. This suggests that the reduction of MyoD results in blunted E-box–mediated gene activation.
Figure 7.



Exogenous MyoD rescues the differentiation defect in cells expressing the mutant DMPK 3′-UTR RNA. (A) GFP+mut 3′-UTR pool cells infected with a retrovirus that expresses MyoD regain the ability to form myotubes (stained red by immunofluorescent staining of MHC) as effectively as infected control C2C12 and GFP+wt 3′-UTR pool cultures. Nuclei are stained blue with DAPI. (B) Western blotting shows increased expression of MyoD protein in proliferating cells infected with the MyoD retrovirus. The graph shows relative MyoD levels (normalized to dynein) in infected and uninfected cells. Similar results were seen in three separate infections. (C) Analysis of myogenin protein expression in infected and uninfected cells cultured in differentiation media shows that exogenous MyoD expression corrects the defect in myogenin up-regulation in GFP+mut 3′-UTR pool cells.
Figure 5.
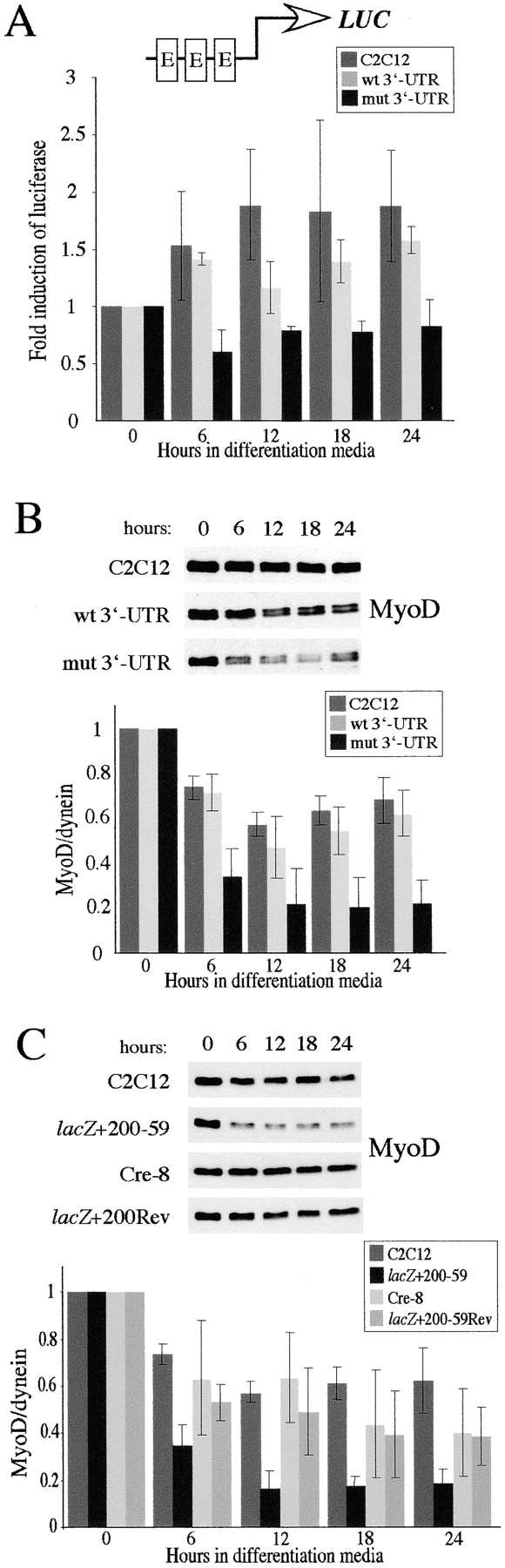
E-box–luciferase activation and MyoD protein levels are compromised in cells expressing the mutant DMPK 3′-UTR within the first 6 h in differentiation media. (A) The average fold induction of E-box–luciferase expression in C2C12, GFP+wt 3′-UTR pool, and GFP+mut 3′-UTR pool cells during the first 24 h in differentiation media (from three experiments). (B) Western analysis at these time points revealed a significant reduction in MyoD protein levels in the GFP+mut 3′-UTR pool as compared with control cells. The graph shows averaged results from three experiments. (C) Analysis of MyoD protein levels in lacZ+200–59 cells, which express the mutant DMPK 3′-UTR RNA, and lacZ+200–59Rev and Cre-8 cells, which have had expression of the RNA ablated, demonstrates that the reduction of MyoD is due to the presence of the mutant DMPK 3′-UTR transcript. Again, the graph shows results from three experiments. All error bars represent one standard deviation.
To ensure that the 70% reduction of MyoD was a true consequence of the mutant DMPK 3′-UTR RNA and not an artifact specific to expression of our GFP constructs, we next analyzed MyoD levels in another previously described set of C2C12 clones (Amack et al., 1999). In this case, cells either express the mutant DMPK 3′-UTR fused to a lacZ reporter transcript and fail to differentiate into myotubes (lacZ+200–59), or have had expression of this RNA ablated by a spontaneous deletion and have regained the ability to differentiate (lacZ+200–59Rev and Cre-8). After 6 h in differentiation media, MyoD levels decreased by 65% in lacZ+200–59 cells, which is similar to the reduction seen in GFP+mut 3′-UTR cells (Fig. 5 C). This reduction was not seen in lacZ+200–59Rev and Cre-8 cells, in which MyoD expression patterns more closely resembled control C2C12 cells (Fig. 5 C). These results provide additional evidence that the presence of the mutant DMPK 3′-UTR RNA compromises MyoD protein levels.
MyoD reduction is posttranscriptional
To determine whether the reduction of MyoD protein is due to transcriptional or posttranscriptional defects, we analyzed steady-state MyoD RNA levels during the first 12 h in differentiation media. In control C2C12 cells, MyoD RNA levels dropped to 52% of the starting levels in proliferating cells during the first 6 h under differentiation conditions (Fig. 6). Similar decreases were observed in GFP+wt 3′-UTR pool (46%) and GFP+mut 3′-UTR pool cells (44%) (Fig. 6). Even after 12 h there were no significant differences in MyoD RNA levels among the three cell populations (Fig. 6). Similar results were obtained when comparing MyoD RNA levels in the lacZ+200–59, lacZ+200–59Rev, and Cre-8 clones (unpublished data). This demonstrates that reduced MyoD protein levels in GFP+mut 3′-UTR cells within the first 6 h in differentiation media are not due to decreased MyoD transcription or unstable MyoD mRNA.
Figure 6.
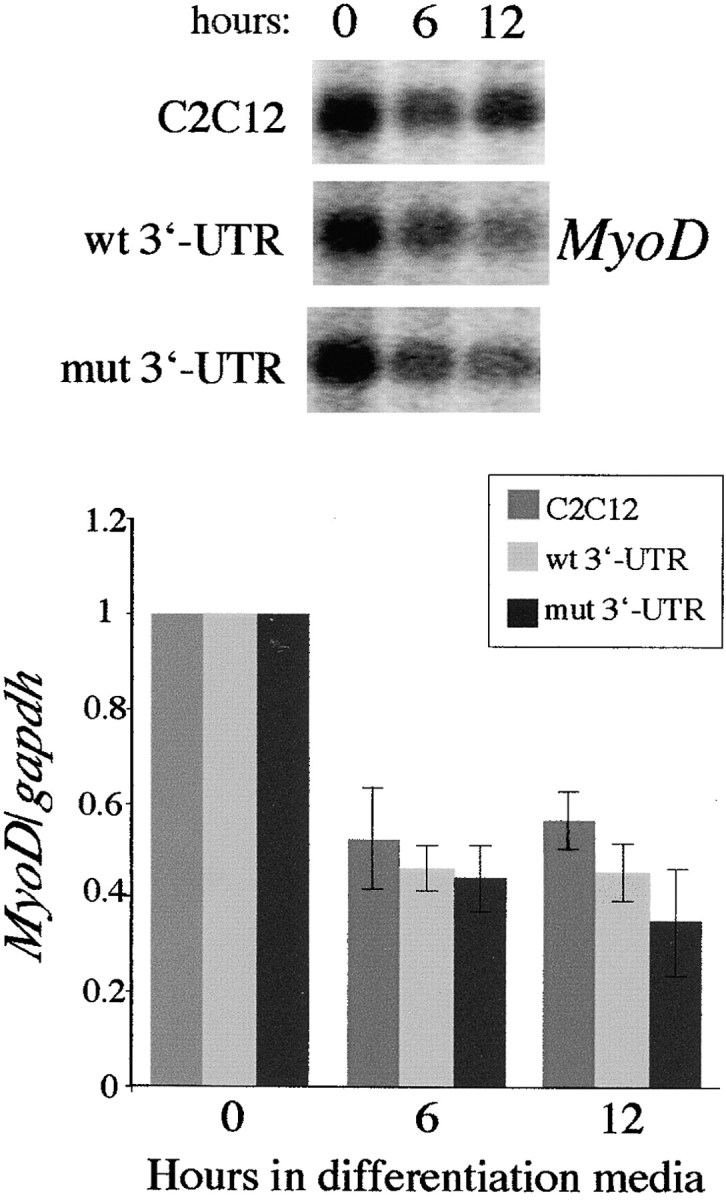
Reduced MyoD protein levels in GFP+mut 3′-UTR cells do not result from differential MyoD transcription or RNA stability. MyoD expression in control C2C12 cells, the GFP+wt 3′-UTR pool, and the GFP+mut 3′-UTR pool after 0, 6, and 12 h in differentiation media were analyzed on Northern blots and RNA slot blots. A Northern blot is shown, and the graph shows the behavior of MyoD steady-state RNA levels (normalized to gapdh levels) after 6 and 12 h in differentiation media relative to levels present in proliferating cells. These results were quantified from three slot blot experiments.
Exogenous MyoD rescues the differentiation defect
Is increasing MyoD levels in cells that express the mutant DMPK 3′-UTR RNA sufficient to rescue the differentiation defect? To test this, we infected cells with a retrovirus that produces MyoD. GFP+mut 3′-UTR pool cells infected with the MyoD retrovirus formed myotubes as effectively as infected GFP+wt 3′-UTR pool and control C2C12 cells after 3 d in differentiation media (Fig. 7 A). This was also observed in several individual GFP+mut 3′-UTR clones and in lacZ+200–59 cells (unpublished data). As shown previously, infection with the LacZ control retrovirus had no effect on differentiation. Myotubes in GFP+mut 3′-UTR cultures infected with the MyoD retrovirus were RNA foci positive, indicating that MyoD acts by overriding the effects of the mutant RNA rather than by silencing its expression (unpublished data). Western blotting confirmed elevated MyoD expression in these cells to levels similar to those found in uninfected C2C12 and GFP+wt 3′-UTR cells (Fig. 7 B). Western blots also showed that the defect in myogenin up-regulation in GFP+mut 3′-UTR pool cells was corrected by increased levels of MyoD (Fig. 7 C). In fact, infected GFP+mut 3′-UTR cells show accelerated myogenin expression, similar to that seen in infected control C2C12 and GFP+wt 3′-UTR cells (Fig. 7 C). These results, which are consistent with mut 3′-UTR-res cells, demonstrate that increasing levels of MyoD is sufficient to override the deleterious effects of the mutant DMPK 3′-UTR and rescue the differentiation defect.
Discussion
We have used a malleable cell culture system to investigate the molecular basis for how RNA molecules containing DMPK 3′-UTR sequences with 200 CUG repeats interfere with myoblast differentiation. Recently, it was discovered that myoblasts isolated from DM1 patients show differentiation defects similar to those we have observed in our cell culture system (Furling et al., 2001; Timchenko et al., 2001). Using primary skeletal muscle cell lines from DM patients, Timchenko et al. (2001) found that p21 up-regulation and cell cycle withdrawal are compromised. However, this study did not analyze earlier myogenic events such as myogenin induction, which occurs before cell cycle withdrawal in C2C12 cells (Andres and Walsh, 1996). Because myogenin is not properly up-regulated in cells that express mutant DMPK 3′-UTR in our model system, we have focused on the affects of the mutant RNA on the earliest stages in the myogenic pathway. To be sure we were looking in the right place, we tested whether the machinery downstream of myogenin was functional in these cells. Ectopic myogenin expression enabled cells to overcome the deleterious effects of mutant 3′-UTR transcripts and effectively fuse into myotubes (Fig. 1 B), indicating that downstream myogenic molecules and cascades are capable of responding to myogenin and properly regulating differentiation. This led us to hypothesize that the mutant 3′-UTR RNA may compromise upstream factor(s) involved in the induction of myogenin and other early differentiation genes.
MyoD is compromised by mutant DMPK 3′-UTR RNA
Genetic studies using knock-out mice have placed MyoD and Myf5 upstream of myogenin (for review see Rudnicki and Jaenisch, 1995). This made MyoD and Myf5 candidate targets of the mutant DMPK RNA, but there are several other factors and signaling cascades that help regulate the initiation of the differentiation pathway (Puri and Sartorelli, 2000). Fortunately, analysis of C2C12 myoblasts resistant to the effects of the mutant DMPK 3′-UTR RNA demonstrated that MyoD is overexpressed in these cells, providing us with a hint that MyoD levels may be linked to the RNA-mediated block of differentiation. Although MyoD mRNA levels are not affected by mutant DMPK 3′-UTR transcripts in our cell culture system (Amack and Mahadevan, 2001), we investigated whether MyoD protein levels were altered in proliferating or differentiating myoblasts. We have identified two separate effects on MyoD protein levels in cells expressing the mutant 3′-UTR. First, MyoD levels in proliferating GFP+mut 3′-UTR cells were approximately three times lower than in control cells (Fig. 3 A). Second, MyoD levels were reduced an additional ∼70% in these cells during the first 24 h in differentiation media, as compared with only an ∼30% decrease in control C2C12 and GFP+wt 3′-UTR cells (Fig. 4 A; Fig. 5 B). No significant differences were found in Myf5 protein levels at either time point, indicating that these negative effects have some specificity for MyoD, and are not due to a general effect on protein levels. MyoD levels are also reduced in cells expressing a (CUG)200 DMPK 3′-UTR fused to a lacZ transcript (lacZ+200–59 cells). Significantly, this affect is abrogated when expression of the RNA is silenced (Fig. 5 C). Taken together, these results demonstrate that the presence of DMPK 3′-UTR transcripts containing 200 CUG repeats, but not five CUG repeats, compromises MyoD protein levels.
The loss of MyoD function has been studied using MyoD−/− mice. Cultured MyoD−/− myoblasts show differentiation defects and blunted up-regulation of differentiation-specific genes (Sabourin et al., 1999; Cornelison et al., 2000), a phenotype strikingly similar to what we observe in cells expressing the mutant 3′-UTR. This is also reminiscent of the phenotype described for DM1 patient myoblasts in culture (Furling et al., 2001; Timchenko et al., 2001). Importantly, DM1 patient cells show reduced MyoD protein levels as compared with control cells (Timchenko et al., 2001), suggesting that the molecular defects that reduce MyoD levels in GFP+mut 3′-UTR cells may also compromise MyoD in patient cells. Exogenous MyoD is capable of rescuing the defects seen in MyoD−/− cells (Sabourin et al., 1999) and overriding the negative effects of the mutant DMPK 3′-UTR RNA on differentiation (Fig. 7). This is consistent with the idea that spontaneous overexpression of MyoD provides the “resistance” in mut 3′-UTR-res cells (Fig. 2). Because the differentiation defect can be rescued by elevating MyoD levels, or by adding exogenous myogenin, which acts downstream of MyoD, we conclude that compromised MyoD levels are responsible for inhibiting myoblast differentiation.
Loss of MyoD function in GFP+mut 3′-UTR cells is consistent with the blunted induction of differentiation-specific genes such as myogenin and p21. The expression of these genes is regulated in part through E-boxes (Yee and Rigby, 1993; Halevy et al., 1995), providing evidence of a defect in MyoD and/or Myf5 activity. Consistent with the lack of activation of endogenous genes containing E-boxes, we found that E-box–luciferase expression was not induced in GFP+mut 3′-UTR pool cells under differentiation conditions (Fig. 4 C; Fig. 5 A). During the first 24 h in differentiation media, the lack of E-box activation correlated with the sharp reduction of MyoD protein levels. Elevating MyoD levels in these cells restores the induction of myogenin (Fig. 7 C) and E-box–luciferase (unpublished data). This suggests that the amount of MyoD in GFP+mut 3′-UTR pool cells is not sufficient to initiate and/or sustain E-box–mediated gene expression, which explains why myogenin, p21, and other differentiation-specific genes are not properly expressed.
Why can't the normal levels of Myf5 up-regulate E-box genes in these cells? Consistent with our results that Myf5 cannot compensate for the lack of MyoD, myoblasts derived from adult MyoD−/− mice show dampened up-regulation of differentiation-specific genes despite a >10-fold up-regulation of Myf5 (Sabourin et al., 1999). This indicates that MyoD has essential functions that Myf5 is unable to perform. Additional studies have shown that during differentiation of both C2C12 and primary mouse myoblasts, MyoD is found exclusively in differentiating cells and Myf5 is expressed only in the cells that do not differentiate (Kitzmann et al., 1998; Lindon et al., 1998). These observations argue that proper MyoD expression and activity are absolutely required for myoblast differentiation.
How do mutant DMPK 3′-UTR transcripts compromise MyoD?
It is thought that mutant DMPK RNA disrupts cellular processes by interacting aberrantly with RNA binding proteins. The aggregation of DMPK transcripts in nuclear foci could either activate or sequester proteins, leading to a variety of downstream consequences. Characterization of one such protein, CUGBP1, has shown that RNAs containing expanded CUG repeats affect CUGBP1 function in both mRNA splicing and translation (Philips et al., 1998; Timchenko et al., 1999). Defects in any one of several processes could account for the reduction of MyoD protein levels in cells expressing the mutant DMPK 3′-UTR (Fig. 8). We first tested MyoD expression at the RNA level and found that transcription and mRNA stability are not affected in the GFP+mut 3′-UTR pool during the first 12 h in differentiation media (Fig. 6). An intriguing possibility downstream of transcription was that MyoD transcripts get “caught” in the nuclear RNA foci and are not transported to the cytoplasm for translation. However, nuclear and cytoplasmic fractionation of RNA from GFP+mut 3′-UTR cells showed no evidence that MyoD mRNA was retained in the nucleus (unpublished data). RNA splicing and/or processing defects could reduce MyoD protein levels, but gross changes in transcript length were not apparent on Northern blots. Any splicing alterations would have to be subtle and could not change the size or stability of the transcript. Other possibilities include mislocalization of MyoD RNA in the cytoplasm, poor translation efficiency, and decreased MyoD protein stability (Fig. 8). Our cell culture model will provide an excellent system to test each of these possibilities and further characterize the trans effect on MyoD protein levels.
Figure 8.
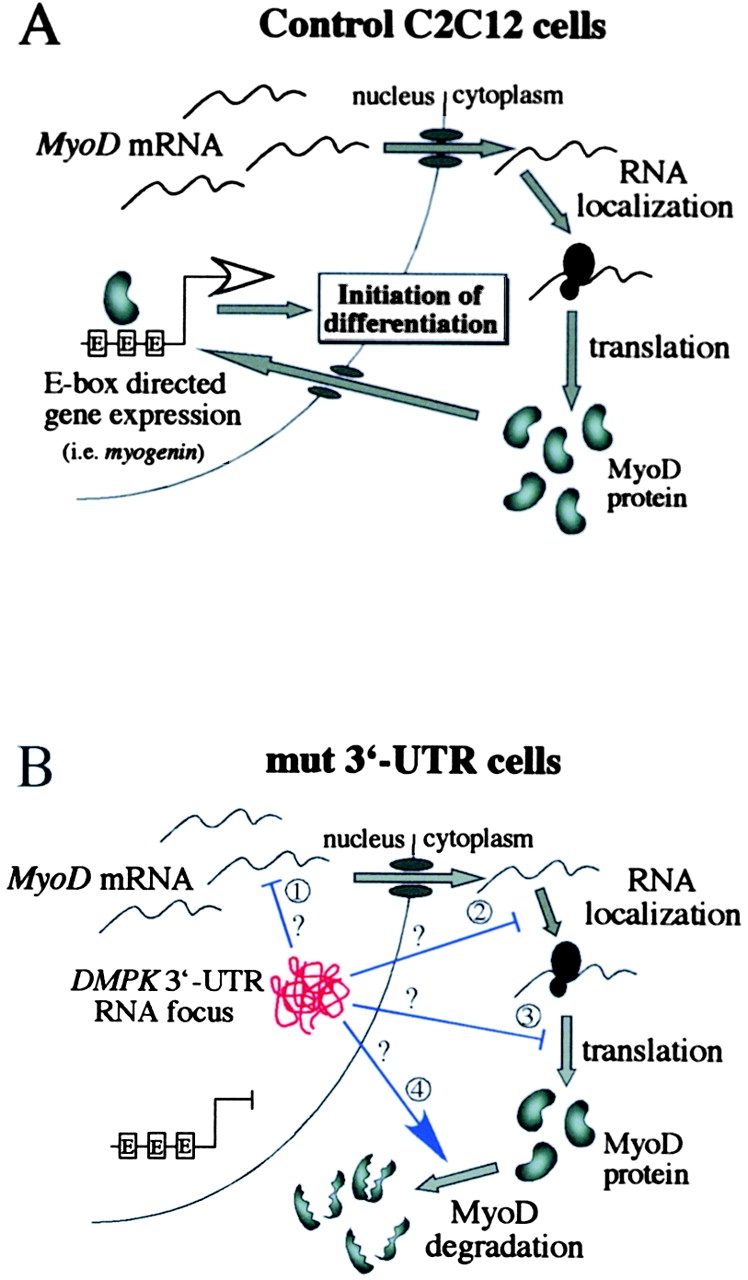
A model for how mutant DMPK 3′-UTR transcripts reduce MyoD protein levels and disrupt C2C12 differentiation. (A) A schematic of steps involved in producing MyoD in C2C12 myoblasts. MyoD, which is required for differentiation of adult myoblasts, ultimately initiates differentiation genes, such as myogenin, by binding E-boxes. (B) Potential steps where mutant DMPK 3′-UTR RNA (red knot in the nucleus) could interfere with MyoD protein production. MyoD mRNA transcription, stability, and transport are unaffected, but the mutant 3′-UTR RNA could alter splicing or processing of the MyoD transcript (1), RNA localization in the cytoplasm (2), translation efficiency (3), or protein stability (4). The end result is a reduction of MyoD protein levels, which fall below a critical threshold required to effectively activate gene expression and initiate differentiation.
The results presented here support the model shown in Fig. 8 for how mutant transcripts disrupt C2C12 differentiation. First, MyoD protein levels are compromised as a result of aberrant interactions between RNA binding proteins and nuclear aggregates of the mutant DMPK 3′-UTR RNA. Links between MyoD level and differentiation efficiency suggest that there is a threshold level of MyoD required for differentiation (Montarras et al., 1996; Kitzmann et al., 1998). Our data suggest that during the first 24 h in differentiation media, MyoD levels drop below the threshold that is required to transactivate target genes. This time window is thought to be critical for the induction of differentiation genes that commit cells to differentiation. MyoD binds E-boxes and induces several genes within 24 h of being activated (Bergstrom et al., 2002). For example, myogenin up-regulation begins within 12 h of MyoD activation and is maximal after 24 h (Bergstrom et al., 2002). In our model, MyoD protein expression is reduced to levels that are insufficient to activate myogenin and other differentiation genes, and in the absence of these gene products, cells fail to initiate the differentiation program.
In conclusion, we have identified MyoD as a target of a trans effect mediated by mutant DMPK 3′-UTR RNA. The reduction of MyoD protein levels provides a molecular explanation for why cells expressing mutant 3′-UTR transcripts fail to differentiate. Future projects, some of which are already underway, are designed to determine how the mutant DMPK RNA compromises MyoD. In addition, as demonstrated by the successful rescue of the differentiation defect by retroviral expression of myogenin and MyoD, our cell culture model may be useful in developing strategies to override the effects of the mutant DMPK RNA. Understanding how mutant DMPK RNA causes deleterious effects at the molecular level may provide insight into how muscle defects arise in DM patients and how to correct them.
Materials and methods
Cell culture
C2C12 myoblasts were maintained at subconfluence in growth media consisting of DME (Cellgro®) plus 10% cosmic calf serum (HyClone). Stable transfected GFP+wt 3′-UTR and GFP+mut 3′-UTR cells (described in Amack and Mahadevan, 2001) were maintained in selective media containing 0.5 μg/ml G418 (GIBCO BRL). The generation of clone pools was previously described (Amack and Mahadevan, 2001). To induce myoblast differentiation, cells were grown to ∼90% confluence and then cultured in differentiation media containing DME supplemented with 2% horse serum (HyClone). Differentiation media was replaced every 24 h.
Retroviral infection
For transduction with retroviral vectors, cells were plated at low density (2 × 105 cells) in 10-cm dishes. The next day, cells were exposed to 4 ml of media harvested from confluent cells producing the retrovirus, supplemented with 8 μg/ml polybrene (Sigma-Aldrich), for 3 h. 24 h later, the cells were exposed to a second infection. Mock infections (referred to as uninfected cells) were conducted in parallel using regular growth media plus polybrene. For differentiation studies, infected and uninfected cells were switched into differentiation media 1 d after the second infection. The following retroviruses were used: LMDSN expressed MyoD (Weintraub et al., 1989); and JR-gal expressed LacZ (Wang et al., 1991). Media containing the myogenin retrovirus was a gift from R. Ilaria (University of Texas Southwestern, Dallas, TX).
Immunostaining and RNA FISH
To detect myosin heavy chain (MHC) expression, cells cultured in differentiation media for 3 d were fixed in 4% paraformaldehyde for 15 min at room temperature and stored in 70% ethanol at 4°C. Cells rehydrated in PBS were incubated with MY32 primary antibody (Sigma-Aldrich) diluted in PBS + 1% BSA for 1 h at 37°C. After washing, cells were incubated with a Texas red–conjugated secondary antibody (Jackson ImmunoResearch Laboratories) for 1 h at 37°C. Nuclei were stained with DAPI (Sigma-Aldrich).
RNA FISH experiments were performed as previously described (Amack et al., 1999). In brief, fixed cells were incubated with a CY3-conjugated (CAG)10 oligonucleotide probe (Operon) diluted to 0.1 μg/ml in RNA hybridization buffer for 2 h at 37°C. After three washes in PBS, nuclei were stained with DAPI.
Fluorescent signals were visualized using an Olympus IX 50 microscope with epifluorescence. Pictures were taken with a SPOT II digital camera (Diagnostic Instruments), and images were assembled using Adobe Photoshop® and Canvas (Deneba Systems) software.
Western blot analysis
Cells were lysed in 50 mM Hepes, pH 7.6, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM MgCl2, 0.5 mM DTT, and 0.1 mM EDTA supplemented a with protease inhibitor cocktail (Roche). 50 μg of total protein was fractionated by SDS-PAGE and transferred onto a nitrocellulose membrane (Schleicher & Schuell). Membranes were blocked overnight in PBS containing 10% skimmed milk, and then incubated for 1–3 h with primary antibodies diluted in PBS containing 1% BSA. The following primary antibodies were used: MyoD (BD Biosciences), Myf5 (Santa Cruz Biotechnology, Inc.), myogenin (Santa Cruz Biotechnology, Inc.), and dynein (Santa Cruz Biotechnology, Inc.). After incubation with the primary antibody, membranes were washed in PBS containing 0.3% Tween-20, and then incubated with a secondary antibody conjugated to HRP (Santa Cruz Biotechnology, Inc.) for 1 h. After several washes, proteins were detected using ECL reagents. Films were scanned and protein bands were quantified using ImageQuant software (Molecular Dynamics). The Mann-Whitney test was used for all statistical analyses.
Luciferase assays
The reporter plasmid pE-boxLuc was generated by subcloning a HinDIII-XhoI fragment from p3R-CAT (Weintraub et al., 1990), which contains three tandem “right” E-boxes from the muscle-specific creatine kinase enhancer inserted 5′ of TK promoter sequences, upstream of the luciferase gene in pTKLuc (a gift from E. Olson, University of Texas Southwestern). pTKLuc serves as a control construct that contains the TK promoter and luciferase, but lacks E-boxes.
Control C2C12, GFP+wt 3′-UTR pool, and GFP+mut 3′-UTR pool cells were transfected as previously described (Amack and Mahadevan, 2001) with pE-boxLuc and a plasmid conferring puromycin resistance, and stable transfectants were selected in media containing 4 μg/ml puromycin. After selection, puromycin-resistant colonies (an average of ∼270 per transfection) were trypsinized, combined, and analyzed as a collection of independent transfection events. Visualization of GFP and RNA FISH experiments demonstrated that transfected GFP+wt 3′-UTR and GFP+mut 3′-UTR cells retained expression of the 3′-UTR transcripts (not depicted). These cells were grown in selective media containing both G418 and puromycin. In parallel, each population of cells was separately transfected with the control pTKLuc construct.
To measure luciferase, samples were prepared using the luciferase assay system (Promega), and relative light units were determined by a MLX luminometer (Dynex Technologies). Raw relative light units were normalized to protein concentration, which was measured using the Bio-Rad Laboratories protein assay. The amount of luciferase produced in proliferating cells, which varied among the different stably transfected cell lines, was set equal to one, and changes in luciferase expression during differentiation were analyzed relative to these starting levels. To determine the fold change in luciferase expression that was due strictly to the E-boxes, we subtracted changes that were due to the TK promoter sequences, as observed in cells transfected with the control pTKLuc construct. The fold induction of E-box–mediated luciferase expression was analyzed in three independent differentiation experiments.
RNA analysis
Total RNA was prepared by lysing cells in 4 M guanidinium thiocyanate, 20 mM sodium acetate, pH 5.4, and 0.5% sarkosyl and then isolating RNA by ultracentrifugation through a 5.7 CsCl density gradient. Northern and RNA slot blots were generated and analyzed as previously described (Amack and Mahadevan, 2001). Results were quantified using ImageQuant software (Molecular Dynamics).
Acknowledgments
We thank Dr. E.N. Olson's laboratory for the PTKLUC plasmid, Dr. Y. Lee (University of Wisconsin-Madison) for the p3R-CAT plasmid, Dr. G. Lyons (University of Wisconsin-Madison) for the MyoD cDNA probe, Dr. D. Miller (University of Washington, Seattle, WA) for the MyoD retrovirus (LMDSN), Dr. M. Gould (University of Wisconsin-Madison) for the LacZ retrovirus (JR-gal), and Dr. R. Ilaria for sharing his myogenin retrovirus construct.
This work was supported by grants from the Muscular Dystrophy Association and the National Institutes of Health grant R01 AR45992-01. This is paper no. 3602 from the University of Wisconsin-Madison Laboratory of Genetics.
J.D. Amack's present address is Huntsman Cancer Institute, University of Utah, 2000 Circle of Hope, Salt Lake City, UT 84112.
Footnotes
Abbreviations used in this paper: DM, myotonic dystrophy; DMPK, dystrophia myotonica protein kinase; MHC, myosin heavy chain; MRF, myogenic regulatory factor; TK, thymidine kinase; 3′-UTR, 3′ untranslated region; ZNF9, zinc finger protein 9.
References
- Amack, J.D., and M.S. Mahadevan. 2001. The myotonic dystrophy expanded CUG repeat tract is necessary but not sufficient to disrupt C2C12 myoblast differentiation. Hum. Mol. Genet. 10:1879–1887. [DOI] [PubMed] [Google Scholar]
- Amack, J.D., A.P. Paguio, and M.S. Mahadevan. 1999. Cis and trans effects of the myotonic dystrophy (DM) mutation in a cell culture model. Hum. Mol. Genet. 8:1975–1984. [DOI] [PubMed] [Google Scholar]
- Andres, V., and K. Walsh. 1996. Myogenin expression, cell cycle withdrawal, and phenotypic differentiation are temporally separable events that precede cell fusion upon myogenesis. J. Cell Biol. 132:657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom, D.A., B.H. Penn, A. Strand, R.L. Perry, M.A. Rudnicki, and S.J. Tapscott. 2002. Promoter-specific regulation of MyoD binding and signal transduction cooperate to pattern gene expression. Mol. Cell. 9:587–600. [DOI] [PubMed] [Google Scholar]
- Blau, H.M., C.P. Chiu, and C. Webster. 1983. Cytoplasmic activation of human nuclear genes in stable heterocaryons. Cell. 32:1171–1180. [DOI] [PubMed] [Google Scholar]
- Brook, J.D., M.E. McCurrach, H.G. Harley, A.J. Buckler, D. Church, H. Aburatani, K. Hunter, V.P. Stanton, J.P. Thirion, T. Hudson, et al. 1992. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 69:385. [DOI] [PubMed] [Google Scholar]
- Cornelison, D.D., B.B. Olwin, M.A. Rudnicki, and B.J. Wold. 2000. MyoD(−/−) satellite cells in single-fiber culture are differentiation defective and MRF4 deficient. Dev. Biol. 224:122–137. [DOI] [PubMed] [Google Scholar]
- Davis, B.M., M.E. McCurrach, K.L. Taneja, R.H. Singer, and D.E. Housman. 1997. Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc. Natl. Acad. Sci. USA. 94:7388–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fardaei, M., K. Larkin, J.D. Brook, and M.G. Hamshere. 2001. In vivo co-localisation of MBNL protein with DMPK expanded-repeat transcripts. Nucleic Acids Res. 29:2766–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fardaei, M., M.T. Rogers, H.M. Thorpe, K. Larkin, M.G. Hamshere, P.S. Harper, and J.D. Brook. 2002. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet. 11:805–814. [DOI] [PubMed] [Google Scholar]
- Farkas-Bargeton, E., J.P. Barbet, S. Dancea, R. Wehrle, A. Checouri, and O. Dulac. 1988. Immaturity of muscle fibers in the congenital form of myotonic dystrophy: its consequences and its origin. J. Neurol. Sci. 83:145–159. [DOI] [PubMed] [Google Scholar]
- Fu, Y.H., A. Pizzuti, R.G. Fenwick, Jr., J. King, S. Rajnarayan, P.W. Dunne, J. Dubel, G.A. Nasser, T. Ashizawa, P. de Jong, et al. 1992. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 255:1256–1258. [DOI] [PubMed] [Google Scholar]
- Furling, D., L. Coiffier, V. Mouly, J.P. Barbet, J.L. St Guily, K. Taneja, G. Gourdon, C. Junien, and G.S. Butler-Browne. 2001. Defective satellite cells in congenital myotonic dystrophy. Hum. Mol. Genet. 10:2079–2087. [DOI] [PubMed] [Google Scholar]
- Halevy, O., B.G. Novitch, D.B. Spicer, S.X. Skapek, J. Rhee, G.J. Hannon, D. Beach, and A.B. Lassar. 1995. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 267:1018–1021. [DOI] [PubMed] [Google Scholar]
- Harper, P.S. 1989. Myotonic Dystrophy. Second edition. W.B. Saunders Co., London. 384 pp.
- Harvey, B., M.D. Sarnat, W. Shirley, M.D. Silbert. 1976. Maturational arrest of fetal muscle in neonatal myotonic dystrophy. Arch. Neurol. 33:466-474. [DOI] [PubMed] [Google Scholar]
- Hasty, P., A. Bradley, J.H. Morris, D.G. Edmondson, J.M. Venuti, E.N. Olson, and W.H. Klein. 1993. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature. 364:501–506. [DOI] [PubMed] [Google Scholar]
- Hollenberg, S.M., P.F. Cheng, and H. Weintraub. 1993. Use of a conditional MyoD transcription factor in studies of MyoD trans-activation and muscle determination. Proc. Natl. Acad. Sci. USA. 90:8028–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri, T., S. Akiyama, M. Namiki, M. Komaki, A. Yamaguchi, V. Rosen, J.M. Wozney, A. Fujisawa-Sehara, and T. Suda. 1997. Bone morphogenetic protein-2 inhibits terminal differentiation of myogenic cells by suppressing the transcriptional activity of MyoD and myogenin. Exp. Cell Res. 230:342–351. [DOI] [PubMed] [Google Scholar]
- Kitzmann, M., G. Carnac, M. Vandromme, M. Primig, N.J. Lamb, and A. Fernandez. 1998. The muscle regulatory factors MyoD and myf-5 undergo distinct cell cycle–specific expression in muscle cells. J. Cell Biol. 142:1447–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzmann, M., M. Vandromme, V. Schaeffer, G. Carnac, J.C. Labbe, N. Lamb, and A. Fernandez. 1999. Cdk1- and cdk2-mediated phosphorylation of MyoD Ser200 in growing C2 myoblasts: role in modulating MyoD half-life and myogenic activity. Mol. Cell. Biol. 19:3167–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassar, A.B., J.N. Buskin, D. Lockshon, R.L. Davis, S. Apone, S.D. Hauschka, and H. Weintraub. 1989. MyoD is a sequence-specific DNA binding protein requiring a region of myc homology to bind to the muscle creatine kinase enhancer. Cell. 58:823–831. [DOI] [PubMed] [Google Scholar]
- Lindon, C., D. Montarras, and C. Pinset. 1998. Cell cycle–regulated expression of the muscle determination factor Myf5 in proliferating myoblasts. J. Cell Biol. 140:111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liquori, C.L., K. Ricker, M.L. Moseley, J.F. Jacobsen, W. Kress, S.L. Naylor, J.W. Day, and L.P. Ranum. 2001. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 293:864–867. [DOI] [PubMed] [Google Scholar]
- Lu, X., N.A. Timchenko, and L.T. Timchenko. 1999. Cardiac elav-type RNA-binding protein (ETR-3) binds to RNA CUG repeats expanded in myotonic dystrophy. Hum. Mol. Genet. 8:53–60. [DOI] [PubMed] [Google Scholar]
- Mahadevan, M., C. Tsilfidis, L. Sabourin, G. Shutler, C. Amemiya, G. Jansen, C. Neville, M. Narang, J. Barcelo, K. O'Hoy, et al. 1992. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 255:1253–1255. [DOI] [PubMed] [Google Scholar]
- Mankodi, A., E. Logigian, L. Callahan, C. McClain, R. White, D. Henderson, M. Krym, and C.A. Thornton. 2000. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 289:1769–1773. [DOI] [PubMed] [Google Scholar]
- Mankodi, A., C.R. Urbinati, Q.P. Yuan, R.T. Moxley, V. Sansone, M. Krym, D. Henderson, M. Schalling, M.S. Swanson, and C.A. Thornton. 2001. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum. Mol. Genet. 10:2165–2170. [DOI] [PubMed] [Google Scholar]
- Miller, J.W., C.R. Urbinati, P. Teng-Umnuay, M.G. Stenberg, B.J. Byrne, C.A. Thornton, and M.S. Swanson. 2000. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 19:4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montarras, D., F. Aurade, T. Johnson, J. IIan, F. Gros, and C. Pinset. 1996. Autonomous differentiation in the mouse myogenic cell line, C2, involves a mutual positive control between insulin-like growth factor II and MyoD, operating as early as at the myoblast stage. J. Cell Sci. 109:551–560. [DOI] [PubMed] [Google Scholar]
- Nabeshima, Y., K. Hanaoka, M. Hayasaka, E. Esumi, S. Li, and I. Nonaka. 1993. Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature. 364:532–535. [DOI] [PubMed] [Google Scholar]
- Philips, A.V., L.T. Timchenko, and T.A. Cooper. 1998. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 280:737–741. [DOI] [PubMed] [Google Scholar]
- Puri, P.L., and V. Sartorelli. 2000. Regulation of muscle regulatory factors by DNA-binding, interacting proteins, and post-transcriptional modifications. J. Cell. Physiol. 185:155–173. [DOI] [PubMed] [Google Scholar]
- Rudnicki, M.A., and R. Jaenisch. 1995. The MyoD family of transcription factors and skeletal myogenesis. Bioessays. 17:203–209. [DOI] [PubMed] [Google Scholar]
- Rudnicki, M.A., P.N. Schnegelsberg, R.H. Stead, T. Braun, H.H. Arnold, and R. Jaenisch. 1993. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell. 75:1351–1359. [DOI] [PubMed] [Google Scholar]
- Sabourin, L.A., A. Girgis-Gabardo, P. Seale, A. Asakura, and M.A. Rudnicki. 1999. Reduced differentiation potential of primary MyoD−/− myogenic cells derived from adult skeletal muscle. J. Cell Biol. 144:631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savkur, R.S., A.V. Philips, and T.A. Cooper. 2001. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 29:40–47. [DOI] [PubMed] [Google Scholar]
- Song, A., Q. Wang, M.G. Goebl, and M.A. Harrington. 1998. Phosphorylation of nuclear MyoD is required for its rapid degradation. Mol. Cell. Biol. 18:4994–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja, K.L., M. McCurrach, M. Schalling, D. Housman, and R.H. Singer. 1995. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol. 128:995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapscott, S.J., and C.A. Thornton. 2001. Biomedicine. Reconstructing myotonic dystrophy. Science. 293:816–817. [DOI] [PubMed] [Google Scholar]
- Tapscott, S.J., M.J. Thayer, and H. Weintraub. 1993. Deficiency in rhabdomyosarcomas of a factor required for MyoD activity and myogenesis. Science. 259:1450–1453. [DOI] [PubMed] [Google Scholar]
- Tedesco, D., M. Caruso, L. Fischer-Fantuzzi, and C. Vesco. 1995. The inhibition of cultured myoblast differentiation by the simian virus 40 large T antigen occurs after myogenin expression and Rb up-regulation and is not exerted by transformation-competent cytoplasmic mutants. J. Virol. 69:6947–6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, B., R.J. White, T. Xia, S. Welle, D.H. Turner, M.B. Mathews, and C.A. Thornton. 2000. Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA. 6:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timchenko, L.T., J.W. Miller, N.A. Timchenko, D.R. DeVore, K.V. Datar, L. Lin, R. Roberts, C.T. Caskey, and M.S. Swanson. 1996. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res. 24:4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timchenko, N.A., A.L. Welm, X. Lu, and L.T. Timchenko. 1999. CUG repeat binding protein (CUGBP1) interacts with the 5′ region of C/EBPβ mRNA and regulates translation of C/EBPβ isoforms. Nucleic Acids Res. 27:4517–4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timchenko, N.A., P. Iakova, Z.J. Cai, J.R. Smith, and L.T. Timchenko. 2001. Molecular basis for impaired muscle differentiation in myotonic dystrophy. Mol. Cell. Biol. 21:6927–6938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiscornia, G., and M.S. Mahadevan. 2000. Myotonic dystrophy: the role of the CUG triplet repeats in splicing of a novel DMPK exon and altered cytoplasmic DMPK mRNA isoform ratios. Mol. Cell. 5:959–967. [DOI] [PubMed] [Google Scholar]
- Wang, B.C., W.S. Kennan, J. Yasukawa-Barnes, M.J. Lindstrom, and M.N. Gould. 1991. Carcinoma induction following direct in situ transfer of v-Ha-ras into rat mammary epithelial cells using replication-defective retrovirus vectors. Cancer Res. 51:2642–2648. [PubMed] [Google Scholar]
- Wei, Q., and B.M. Paterson. 2001. Regulation of MyoD function in the dividing myoblast. FEBS Lett. 490:171–178. [DOI] [PubMed] [Google Scholar]
- Weintraub, H. 1993. The MyoD family and myogenesis: redundancy, networks, and thresholds. Cell. 75:1241–1244. [DOI] [PubMed] [Google Scholar]
- Weintraub, H., S.J. Tapscott, R.L. Davis, M.J. Thayer, M.A. Adam, A.B. Lassar, and A.D. Miller. 1989. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc. Natl. Acad. Sci. USA. 86:5434–5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub, H., R. Davis, D. Lockshon, and A. Lassar. 1990. MyoD binds cooperatively to two sites in a target enhancer sequence: occupancy of two sites is required for activation. Proc. Natl. Acad. Sci. USA. 87:5623–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe, D., and O. Saxel. 1977. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 270:725–727. [DOI] [PubMed] [Google Scholar]
- Yee, S.P., and P.W. Rigby. 1993. The regulation of myogenin gene expression during the embryonic development of the mouse. Genes Dev. 7:1277–1289. [DOI] [PubMed] [Google Scholar]