

Abstract
We utilize structurally targeted peptides to identify a “tC fusion switch” inherent to the coil domains of the neuronal t-SNARE that pairs with the cognate v-SNARE. The tC fusion switch is located in the membrane-proximal portion of the t-SNARE and controls the rate at which the helical bundle that forms the SNAREpin can zip up to drive bilayer fusion. When the fusion switch is “off” (the intrinsic state of the t-SNARE), zippering of the helices from their membrane-distal ends is impeded and fusion is slow. When the tC fusion switch is “on,” fusion is much faster. The tC fusion switch can be thrown by a peptide that corresponds to the membrane-proximal half of the cognate v-SNARE, and binds reversibly to the cognate region of the t-SNARE. This structures the coil in the membrane-proximal domain of the t-SNARE and accelerates fusion, implying that the intrinsically unstable coil in that region is a natural impediment to the completion of zippering, and thus, fusion. Proteins that stabilize or destabilize one or the other state of the tC fusion switch would exert fine temporal control over the rate of fusion after SNAREs have already partly zippered up.
Keywords: SNAP-25; liposome; syntaxin; vesicle; VAMP
Introduction
SNAREpins assembling between cellular membranes from cognate v- and t-SNAREs drive lipid bilayer fusion and dictate its inherent specificity (Söllner et al., 1993; Weber et al., 1998; Nickel et al., 1999; McNew et al., 2000a). Fusion results from a protein-folding reaction that is mechanistically coupled to two closely adherent lipid bilayers held together by the folding SNAREpin itself. Energy made available during folding is likely transmitted to the bilayers via the membrane anchor and linker sequences that tether the folding cytoplasmic domains of v- and t-SNAREs. This is indicated by the observation that although the precise chemical nature of these anchors is not critical, their lengths and those of the linkers, and their topological patterns of insertion into the bilayers, are all highly constrained (McNew et al., 1999, 2000b; Parlati et al., 2000). After fusion, the ATPase NSF, together with its cofactor, α-SNAP, then invest energy from ATP hydrolysis to unfold the now fully assembled cis-SNARE complex, returning the SNAREs to their initial high energy states and recycling them for repeated use (Block et al., 1988; Söllner et al., 1993; Mayer et al., 1996).
The core of the SNARE complex is a bundle of four helices: three from the t-SNARE and one from the v-SNARE (Fasshauer et al., 1998; Poirier et al., 1998b; Sutton et al., 1998). Each helix consists of a conserved SNARE motif, a specialized heptad repeat domain (Weimbs et al., 1998). In neurons, the principal v-SNARE responsible for regulated exocytosis is vesicle-associated membrane protein 2 (VAMP2);* the principal t-SNARE is a heterodimer of syntaxin 1A and the peripheral membrane protein, synaptosomal-associated protein of 25 kD (SNAP-25; Söllner et al., 1993). SNAP-25 contains two SNARE motifs; syntaxin 1A and VAMP2 each contain one such domain.
Deeper understanding of the fusion reaction and the basis for its regulation will require further insights into the choreographed pathway by which v-SNAREs and t-SNAREs assemble. At least two steps of the SNARE assembly pathway are under the control of syntaxin's NH2-terminal regulatory domain (NRD) which is itself composed of two parts: a separately folding, three-helix bundle termed Habc (Fernandez et al., 1998), and an ∼40-residue linker domain that connects Habc to the coil domain of syntaxin 1A. The NRD of isolated syntaxin folds back onto the SNARE motif (Dulubova et al., 1999), and this closed conformation is stabilized by the binding of members of the Sec1 family (Misura et al., 2000). Mutations in the linker region of the NRD destabilize the closed conformation (Dulubova et al., 1999; Misura et al., 2000). Studies on the homologous plasma membrane yeast t-SNARE subunits have demonstrated that the closed conformation of the syntaxin (Sso1) is a kinetic deterrent to t-SNARE complex formation with Sec9 (Munson et al., 2000). Likewise, in the known crystal structure of the n-Sec1–Syntaxin complex, critical SNAP-25 binding sites are occluded (Misura et al., 2000), suggesting that the NRD may also play a role in this system in regulating t-SNARE assembly that we call the “NRD assembly switch.”
In addition to regulating t-SNARE assembly, the NRD also controls SNAREpin (v-/t-SNARE trans-complex) formation and thereby, the rate of the fusion reaction. Because this step links up two membranes, we refer to it as the “NRD docking switch.” Like the NRD assembly switch, the docking switch is constitutively “off,” and as a result, fusion mediated by isolated v- and t-SNAREs is intrinsically very slow. However, when the NRD is removed, the docking switch is now constitutively “on,” so fusion is now greatly accelerated and its rate is now limited by steps after docking (Parlati et al., 1999).
Because the fusion reaction is driven by the assembly of unfolded or partially unfolded helical domains in the individual SNARE proteins, it will be critical to relate structurally defined folding intermediates to functionally defined stages in the fusion process. Structural arguments have suggested that the helical bundle that comprises the SNAREpin “zips” progressively toward the bilayers as its heptad-repeat v- and t-SNARE cytoplasmic domains fold up, beginning at their membrane-distal NH2-termini and progressing to their membrane-proximal COOH termini (Fasshauer et al., 1998; Fiebig et al., 1999; Lin and Scheller, 2000). Currently, the most direct evidence for this mechanism comes from the selective effects of conformationally specific proteolytic neurotoxins and antibodies on the kinetics and physiology of exocytosis in situ (Hua and Charlton, 1999; Xu et al., 1999). These results strongly imply that the SNAREpins are partly (but not completely) zipped-up only one or a few milliseconds before the opening of the fusion pore, and in doing so, provide a powerful confirmation of the direct role SNARE assembly plays in bilayer merger in vivo, ruling out alternative mechanisms.
Here, we provide the first functional test of the zippering model using structurally targeted peptides as inhibitors of SNARE assembly. If fusion requires zippering in the N→C direction, then a reversibly dissociating peptide that binds to the NH2-terminal portion of the t-SNARE will block the first contact between v- and t-SNAREs, and thus inhibit fusion initially. However, once zippering has already begun, fusion will be resistant to the NH2-terminally directed peptide. By contrast, a peptide that binds reversibly to the COOH-terminal portion of the t-SNARE will still allow zippering to begin and will allow fusion to proceed at a rate determined by how fast it is displaced as the SNAREs zip-up in what is now an intramolecular process. Other models have distinct outcomes. For example, if zippering were in the opposite C→N direction, then the NH2- and COOH-peptide would have the opposite properties.
Our results further support that SNAREpin assembly between bilayers is polarized, and that zippering must occur in the N→C direction for fusion to result. But unexpectedly, the COOH-peptide dramatically increases the rate of fusion, revealing an intrinsic regulatory switch on the t-SNARE that controls the rate of zippering that we term the “tC fusion switch.” When the tC fusion switch is off, zippering of the SNAREpin is blocked approximately midway, potentially corresponding to the state of SNAREpins in vesicles that are available for ready release after a signal for exocytosis (Hua and Charlton, 1999; Xu et al., 1999). When the tC fusion switch is turned on, zippering can be rapidly completed and fusion results. Furthermore, while we were in the process of characterizing this tC fusion switch for the mammalian exocytic SNAREs, we established that a similar COOH-peptide–dependent increase in fusion could be observed with yeast endosomal or Golgi-related SNARE complexes (Paumet et al., 2001; Parlati et al., 2002), demonstrating the conservation of this regulatory switch. We suggest how regulatory proteins could control the tC fusion switch and therefore the rate of membrane fusion.
Results
Peptides derived from the sequences of viral fusion proteins have proven useful as probes of the structure and activation mechanism of these proteins (Wild et al., 1994; Chan and Kim, 1998). SNAREs are especially attractive targets for the design of specific peptide ligands. v-SNAREs are unfolded before binding to t-SNAREs (Fasshauer et al., 1998; Fiebig et al., 1999; Hazzard et al., 1999) and, unlike viral fusion, each round of cell membrane fusion requires cognate, sequence-specific pairing of v- and t-SNAREs. Consequently, a peptide derived from the coil region of a v-SNARE can potentially bind with great specificity to the same site on the cognate t-SNARE that is normally occupied by the corresponding portion of the v-SNARE in the SNARE complex. If the peptide is short enough, its binding will be readily reversible, so the peptide will be a specific, well-behaved ligand.
To explore the hypothesis of polarized zippering, which predicts that the membrane-distal and membrane-proximal regions of the SNAREpins are functionally asymmetric, we initially chose peptides corresponding to the NH2-terminal and COOH-terminal halves of the SNARE motif of the exocytic v-SNARE VAMP2, termed vN-pep and vC-pep, respectively (Fig. 1). Together, the sequences of vN-pep and vC-pep comprise the complete sequence of the coil region of VAMP2. The dividing line between them is after arginine 56, chosen to correspond to the hydrophilic “zero layer” in the otherwise hydrophobic core of the coiled coil (Sutton et al., 1998).
Figure 1.

VAMP2 domain organization and recombinant constructs. The borders of the various constructs are defined by amino acid positions. Together, the synthetic peptides vN-pep and vC-pep (shaded gray) encompass the entire coiled-coil domain of VAMP2 as determined by crystal structure analysis (Sutton et al., 1998). TM, transmembrane region.
The VAMP2-derived peptides bind to cognate sites in the t-SNARE syntaxin 1/SNAP-25
First, we tested the specificity and stability of binding of vN-pep and vC-pep to the cognate t-SNARE to see if they were well-behaved ligands. The cytoplasmic domain of the v-SNARE VAMP2 inhibits fusion between liposomes by occupying the VAMP2 binding site on the reconstituted t-SNARE (Weber et al., 1998). Together, vN-pep and vC-pep block membrane fusion as efficiently as if they were contiguous (Fig. 2 A), which suggests that vN-pep and vC-pep assemble into the fourth of a four-helix bundle as if they were covalently joined in the v-SNARE.
Figure 2.
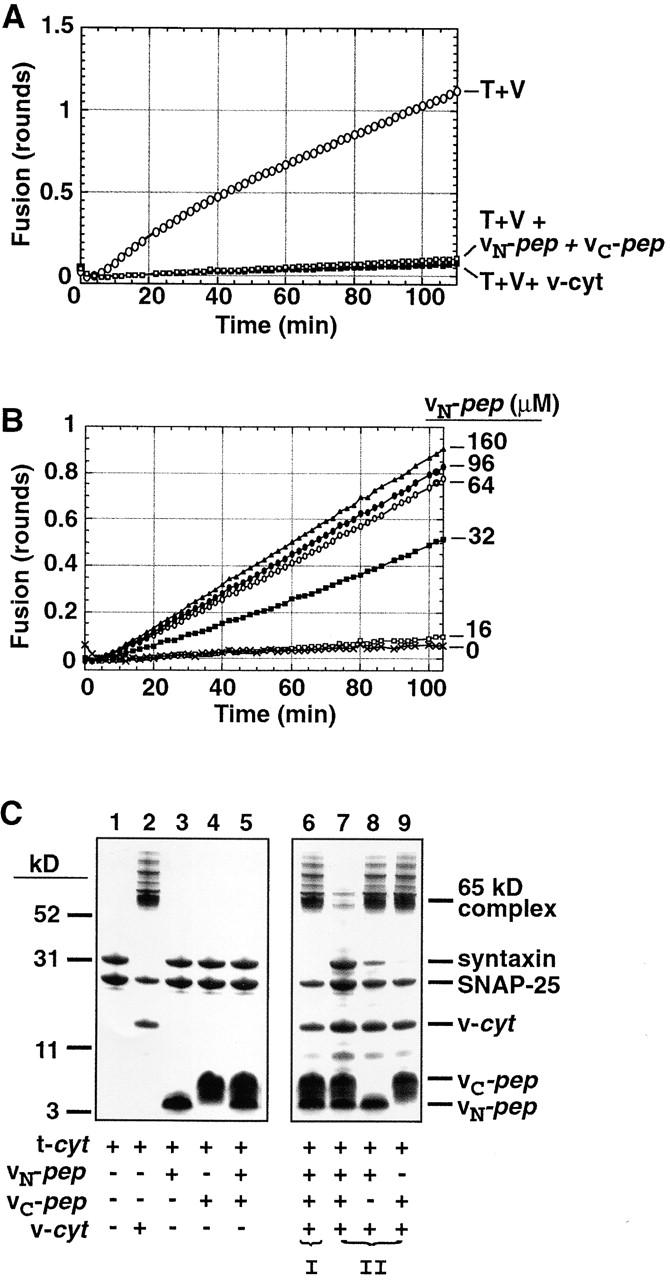
vN-pep and vC-pep bind the t-SNARE via the functionally relevant VAMP2 binding site. (A) vN-pep and vC-pep, when added together, compete with v-liposomes to inhibit fusion as effectively as v-cyt. Lipid-mixing fusion assays of t- and v-liposomes (T + V) were performed at 37°C as described in Materials and methods. Protein- and peptide-mediated inhibitions were tested by adding buffer (○), 50 μM vN-pep + 38 μM vC-pep (□), or 31 μM v-cyt (▪). (B) The VAMP2 coiled coil can be broken at the zero layer and still function in liposome fusion. vC-pep-cys was covalently modified with a C45 isoprenoid and incorporated into labeled liposomes. vC-pep-cys-C45 liposome fusion is dependent upon the concentration of vN-pep (no vN-pep, X; 16 μM, □; 32 μM, ▪; 64 μM, ○; 96 μM, •; 160 μM, ▵). (C) Together, the two peptides block SNARE complex formation. To test for the ability to form SDS-resistant complexes (indicated by the presence of the 65-kD band), 10 μg t-cyt was incubated with buffer (lane 1), 6 μg v-cyt (lane 2), 10 μg vN-pep (lane 3), 10 μg vC-pep (lane 4), or both peptides (lane 5) for 20 min at RT, and then immediately mixed with buffer without boiling and analyzed by SDS-PAGE and Coomassie blue staining. In order of addition experiments, to test for competition of binding at the VAMP2 binding site (lanes 6–9), t-cyt was preincubated with one component for 20 min before addition of the second component for 20 additional min. (I) t-cyt was incubated with v-cyt, and then both peptides. (II) t-cyt was incubated with one or both peptides, and then v-cyt. (Note: the SNAP-25 that does not become incorporated into the 65-kD SDS-resistant complex is derived from an excess of free SNAP-25 in the t-cyt preparation).
Next, we ascertained whether the complex of vN-pep, vC-pep, and the t-SNARE is fusogenic. Donor liposomes containing lipid-anchored vC-pep were prepared, and we determined whether they could fuse with t-SNARE acceptor liposomes when vN-pep is added. For this purpose, a variant of vC-pep with a COOH-terminal cysteine was synthesized to allow covalent attachment to a lipid via the peptide's -SH group (Fig. 1, vC-pep-cys). The vC-pep-cys was then covalently joined to a C45-polyisoprenoid using a maleimide isoprenoid derivative synthesized for this purpose (McNew et al., 2000b). This C45 lipid anchor has previously been shown to function in place of the natural transmembrane protein domain of VAMP2 (McNew et al., 2000b).
Donor liposomes bearing vC-pep-cys-C45 do not themselves fuse with t-liposomes, but they fuse efficiently when free vN-pep is added (Fig. 2 B). Fusion is vN-pep dose-dependent, and is inhibited by either the complete cytoplasmic domain of VAMP2 (v-cyt) or free vC-pep (unpublished data). Free vC-pep inhibits rather than activates, because free vC-pep + vN-pep results in irreversible occupation of the VAMP2 binding site on the t-SNARE as observed in Fig. 1 A. Evidently, continuity through the length of the v-SNARE coil is not an essential feature for fusion. Fusion does not occur when vN-pep (whether joined to C45 maleimide via a COOH- or NH-terminal Cys-SH) is anchored to the donor liposomes and free vC-pep is added (unpublished data).
Fusion when one coil is separated into two pieces, forming a discontinuous helix, is not unprecedented, but is surprising for VAMP in light of some published results. For example, BoNT/E toxin treatment of cracked PC12 cells specifically removes the last 26 amino acids from SNAP-25, rendering it incapable of forming an SDS-resistant complex (Hayashi et al., 1994) and incapable of participating in neuroendocrine exocytosis (Chen et al., 1999). However, exocytosis can be recovered if a peptide correlating to the cleaved fragment is added back at high concentration (Chen et al., 2001). Cleavage of VAMP2 by BoNT/D releases the NH2-terminal 59 amino acids from the protein and eliminates exocytosis. However, in this case, exocytosis cannot be recovered by addition of the cleaved fragment (Chen et al., 2001). Peptides that exactly correspond to the BoNT/D cleavage site (VAMP2 aa 25–59 and 60–94-cys) were equally efficient at mediating liposome fusion (unpublished data). The difference in the two studies perhaps indicates that the cleaved fragment is not stable in permeabilized cells.
The core SNARE complex migrates as an SDS-resistant band on SDS–polyacrylamide gels (Fasshauer et al., 1998; Poirier et al., 1998a; Fig. 2 C, lane 2; 65-kD band). This complex consists of the cytoplasmic domain of VAMP2 (v-cyt) bound to the cytoplasmic domain of the exocytic t-SNARE (t-cyt), a complex of the cytoplasmic domains of syntaxin and SNAP-25. No such SDS-resistant complex is formed between t-cyt and one or both peptides (Fig. 2 C, lanes 3–5). Preassembled v-cyt/t-cyt SDS-resistant complexes are unaffected by the addition of one or even both peptides (Fig. 2 C, lane 6). Also, when the t-SNARE is incubated with either vN-pep or vC-pep alone and then v-cyt is added, SDS-resistant complex formation is unaffected (Fig. 2 C, lanes 8 and 9). However, when the t-SNARE is preincubated with both vN-pep and vC-pep before the addition of v-cyt, SDS-resistant complex formation is now largely prevented (Fig. 2 C, lane 7). This establishes that vN-pep and vC-pep can bind simultaneously and stably to the t-SNARE, and when they are both bound, neither can be competed off by the v-SNARE.
Two conformations in the coiled-coil core of the t-SNARE detected by limited proteolysis
Two structural states of the coiled-coil core region of exocytic t-SNAREs have previously been characterized using proteins from different organisms. In the yeast Sso1–Sec9 complex, the SNARE motif within the cytoplasmic domain of the t-SNARE is a three-stranded coiled coil in its approximately NH2-terminal half, but is less ordered in its approximately COOH-terminal half (Fiebig et al., 1999). The only structures available for the mammalian complex are limited to the coil-forming SNARE domains of syntaxin 1A and SNAP-25, which give rise to a four-helix bundle composed of the two coils from SNAP-25 and two syntaxin H3 coils (Margittai et al., 2001; Xiao et al., 2001). However, it is likely that the mammalian t-SNARE also goes through a three-stranded intermediate to allow for VAMP2 binding, and that such an intermediate would be structurally analogous to the yeast complex. In fact, EPR studies on the SNARE motifs of the mammalian t-SNARE complexed with an NH2-terminal fragment of the VAMP2 SNARE domain display significant disorder over the COOH-terminal half of the t-SNARE (Margittai et al., 2001). Furthermore, work in our lab suggests that the NRD, which is not present in the studies cited above, may be essential to maintain a 1:1 syntaxin/SNAP-25 stoichiometry (unpublished observations). When the core domains of the mammalian t-SNARE are bound to the cytoplasmic domain of the v-SNARE VAMP2, the t-SNARE is fully structured throughout as three strands of the resulting four-helix bundle (Sutton et al., 1998). So, although the partly bundled conformation in the isolated t-SNARE was initially identified by nuclear magnetic resonance and only with the yeast homologues, we will illustrate that a similar change from partly bundled to more completely structured can also conveniently be monitored in the mammalian t-SNARE using a limited proteolysis assay that is better suited for routine biochemical studies.
The SNARE complex yields a protease-resistant core limited to the four-SNARE motif coil regions (Fasshauer et al., 1998; Poirier et al., 1998a). Proteolysis of t-cyt alone yields a 14-kD band that originates from the Habc portion of the NRD (Fasshauer et al., 1998), and a distinct set of smaller (∼6 kD) peptide fragments (Fig. 3 A, lane 2), whose origin was established by a combination of Western blot analysis and NH2-terminal sequencing.
Figure 3.


vC-pep binding changes the t-SNARE conformation. t-cyt was incubated in the presence or absence of VAMP2 and VAMP2 peptides for 20 min at RT, and then was digested with proteinase K, as described in Materials and methods. Concentrations of the components in the digest were as follows: 2.2 mg/ml t-cyt, 2 mg/ml vC-pep, 2 mg/ml vN-pep, 1.2 mg/ml v-cyt, and 50 μg/ml proteinase K. (A) t-cyt– and t-cyt + v-cyt–digested samples were boiled, resolved by SDS-PAGE on a 12% gel (Novex) with bis-Tris buffer, and stained with Coomassie brilliant blue. The bands were identified by microsequencing and mass spectrometry. The lines between lanes 2 and 3 indicate coils that have gotten smaller (SNAP-25N) or larger (H3 and SNAP-25C). The low molecular mass band originating from v-cyt is a proteinase K–resistant fragment from the highly charged COOH terminus of v-cyt, and is observed when either v-cyt or vC-pep are proteolyzed alone. (B and C) Proteolytic fragments of SNAP-25 were detected by Western blot analysis. Samples were digested and resolved as above and transferred to nitrocellulose. The Western blot was probed, stripped, and reprobed sequentially with antibodies against the SNAP-25 NH2 terminus (B; Cl 71.1) and the SNAP-25 COOH terminus (C; anti–amino acids 207–218). (D) t-liposomes were incubated with or without peptides and proteolyzed as in A–C. Protection of the COOH terminus of SNAP-25 was detected by Western blotting with the same peptide antibody as in C. Note that the large amount of full-length SNAP-25 remaining after proteolysis reflects the fully protected protein in the vesicle lumen.
The addition of v-cyt dramatically changes the proteinase K fragmentation pattern (Fig. 3 A, lane 3), resulting in a larger number of 6–11-kD peptide bands that correspond to the previously reported protease-resistant fragment pattern derived from the 7S core complex (Fasshauer et al., 1998). As expected, this VAMP2-induced conformational change of the t-SNARE results in complete protection up to the COOH terminus of the helical domain of both syntaxin (H3) and one of the two SNAP-25 coils (the COOH-terminal helix). Surprisingly, the proteolytically resistant portion of the coil originating from the SNAP-25 NH2 terminus becomes smaller after addition of VAMP2. This appears to reflect an increase in the proteolytic sensitivity of the loop connecting the two helices that remains associated with SNAP-25N in the t-SNARE alone, but is more exposed after VAMP2 addition. Thus, all three coils show changes in their proteolytic sensitivity after addition of VAMP2.
vC-pep structures the membrane-proximal portion of the t-SNARE
To monitor the effects of peptide addition on each SNAP-25 coil, we used antibodies directed against the extreme NH2 terminus or COOH terminus of SNAP-25. The addition of vC-pep (Fig. 3, B and C) shifts the proteolytic sensitivity of each coil in a manner identical to that observed with the complete v-cyt; SNAP-25N becomes shorter due to increased proteolysis of the adjoining loop (Fig. 3 B), and SNAP-25C becomes protected at its COOH terminus (the site of the antibody; Fig. 3 C), which is apparent in the Coomassie gel as an increase in the molecular mass of the band (Fig. 3 A). This implies that binding of vC-pep by itself shifts the conformational equilibrium of the COOH-terminal half of the t-SNARE to a more structured state: a tC fusion switch. vN-pep does not significantly change the protease digestion pattern of t-cyt.
We also performed the same analysis with the full-length t-SNARE inserted into lipid bilayer vesicles (Fig. 3 D). As a result of membrane insertion, now both H3 of syntaxin and SNAP-25N are protected from proteolysis to a much greater degree. No change in the pattern of the protection of SNAP-25N or H3 is now caused by adding either peptide or even v-cyt (unpublished data). The greater resistance to proteolytic cleavage of H3 of syntaxin and SNAP-25N when they are attached to the membrane has any of several explanations. Access to the t-SNARE could be sterically restricted by the close proximity of the lipid bilayer. Alternatively, this portion of the t-SNARE could be occluded within oligomers organizing a fusion “ring” on the bilayer surface. However, just as the specificity of SNARE–SNARE interactions is greatly influenced by membrane association (McNew et al., 2000a; Scales et al., 2000), it may well be that the range of conformations that the SNAREs can sample is altered in the context of membrane insertion. Interestingly, a dramatic increase in the protection of the COOH terminus of SNAP-25C is still observed when vC-pep is added (Fig. 3 D). Taken together, these results suggest that membrane-proximal portions of SNAP-25N and syntaxin H3 t-SNAREs have a more ordered structure when associated with a membrane than they do as a soluble complex. However, the COOH terminus of the COOH-terminal helix of SNAP-25 remains unstructured and proteolytically accessible in both the soluble and the bilayer-anchored t-SNAREs, and vC-pep binding thus protects this region of the t-SNARE.
Polar effects of N versus C v-SNARE peptides imply N→C zippering during fusion
Fig. 4 A shows that vN-pep reduces the initial rate of fusion between t- and v-liposomes to nearly zero. Inhibition is dose dependent and complete at close to 1 mol vN-pep/mol t-SNARE (Fig. 4 C). However, fusion eventually begins after several minutes, even when it had initially been completely inhibited by a saturating concentration (5.4 μM) of vN-pep (Fig. 4 B). This is expected because the higher affinity, irreversibly bound, full-length v-SNARE will eventually displace reversibly bound vN-pep at a rate determined by the rate of dissociation of vN-pep from the t-SNARE (Fig. 2 C). This is consistent with the N→C zippering model and inconsistent with random (nonpolarized) zippering models.
Figure 4.

vN-pep reversibly inhibits fusion. t-liposomes (20 mg protein) were incubated with vN-pep for 7 min at RT before initiating the fusion assay by addition of v-liposomes. (A) vN-pep reduces the initial rate of liposome fusion, almost to zero. Data are plotted in rounds of fusion (Parlati et al., 1999). No peptide (○), 0.54 μM vN-pep (▪), 1.6 μM vN-pep (▵), 5.4 μM vN-pep (•), 5.4 μM vN-pep + 38 μM V-cyt (□). (B) After a prolonged lag-phase, fusion recovers. The initial kinetics in A correspond to the black-boxed region. (C) vN-pep–induced lag is dose dependent and maximal at concentrations that are approximately stoichiometric with accessible t-SNARE. The rate of fusion over the first 6 min of the fusion assay is plotted at varying peptide to t-SNARE ratios in terms of rounds of fusion per minute. Accessible t-SNARE is defined as t-SNAREs in which the coiled-coil domain is facing out on the liposome and is estimated to be 70% of the total t-SNARE (Weber et al., 1998).
In marked contrast, vC-pep dramatically enhances the initial rate and extent of fusion in a saturable and dose-dependent manner (Fig. 5 A). The lack of inhibition by vC-pep does not support the simplest alternative, a C→N zippering model. Maximal activation occurs at ∼1 mol vC-pep/mol t-SNARE (Fig. 5 B). Fusion completes at least 8.3 of the theoretical limit of 9.7 maximal rounds (determined by the relative amounts of SNARE and lipid in donor membranes vs. acceptor membranes) within 2 h. Fig. 5 C shows that vC-pep will activate fusion when added at any time of incubation, even after the first round of fusion has taken place.
Figure 5.
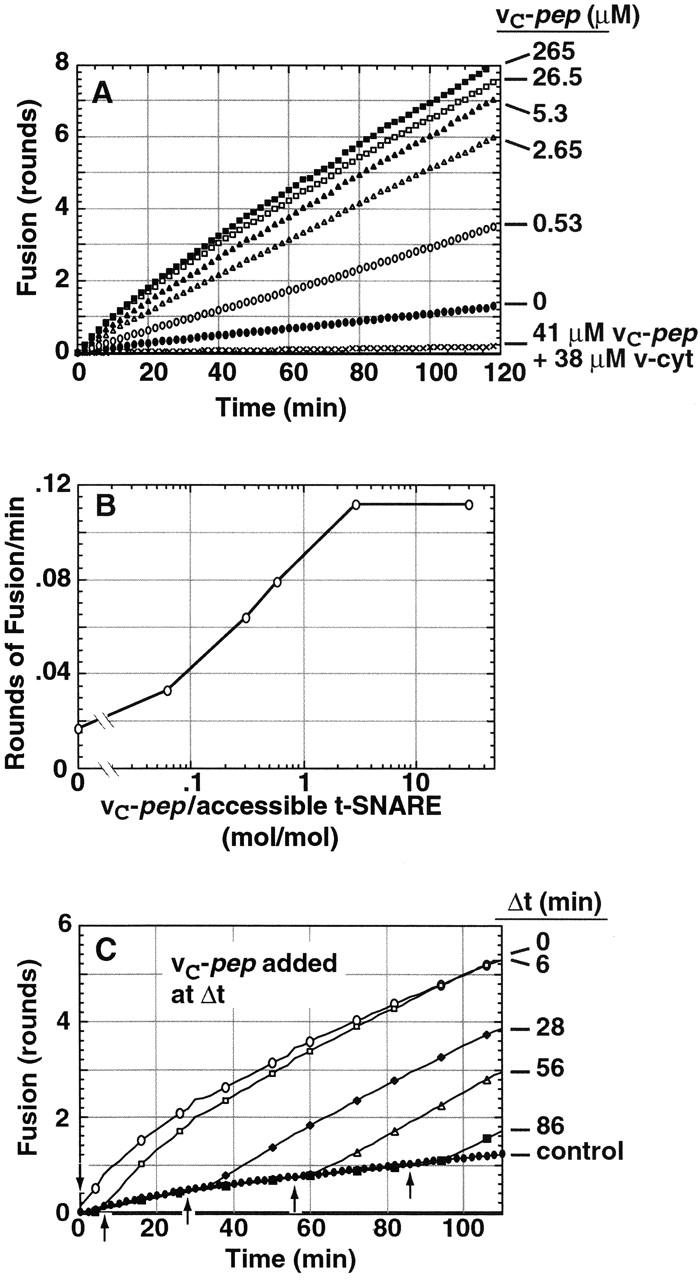
vC-pep stimulates liposome fusion. t-liposomes (36 μg protein) were incubated with vC-pep for 7 min before initiating the fusion assay by addition of v-liposomes. (A) vC-pep promotes a dose-dependent increase in liposome fusion. No peptide (•), 0.53 μM vC-pep (○), 2.65 μM vC-pep (▵), 5.3 μM vC-pep (▴), 26.5 μM vC-pep (□), 265 μM vC-pep (▪), 41 μM vC-pep + 38 μM v-cyt (X). (B) vC-pep stimulation reaches a maximum at about a threefold molar excess. Initial rates (in rounds of fusion per minute) are plotted, and accessible t-SNARE is defined as in Fig. 4. (C) Addition of 10 μM vC-pep at any point (arrows) during the assay stimulates fusion.
The fact that v-cyt completely overcomes the activation by vC-pep and then completely blocks fusion, even in the presence of saturating concentrations of vC-pep (Fig. 5 A), rules out the alternative formal explanation of its “activating” property, that vC-pep is itself fusogenic. The specificity of vC-pep is further underscored by the fact that corresponding peptides representing a variety of other v-SNAREs (yeast Snc1, Snc2, Bet1, and Sft1) are without significant effect on fusion mediated by VAMP, syntaxin, and SNAP-25 (Fig. 5, not depicted).
At saturating concentrations of vC-pep, the initial rate of fusion corresponds to a half-time for one round of ∼4 min (t 1/2 = 4.1 ± 1.5 min). This rate is faster than that observed when the syntaxin NRD is removed (t 1/2 = 10 min; Parlati et al., 1999; Fig. 7 A) or when donor and acceptor liposomes are allowed to dock at 0–4°C before permitting fusion at 37°C (t 1/2 = 7 min; Parlati et al., 1999). The t 1/2 of fusion in the presence of vC-pep of 4.1 ± 1.5 min should be considered an upper limit because it must include the unknown time required for vC-pep to dissociate.
Figure 7.

Docking and fusing liposomes rapidly develop resistance to vN-pep. t-liposomes and v-liposomes were prewarmed to 37°C and then mixed to start a fusion assay. At the indicated times, 7.6 μM vN-pep was added and the fusion kinetics monitored. The data is plotted as rounds of fusion.
The finding that significantly less time is required for the completion of fusion from the vC-pep–sensitive step than from the NRD-sensitive step establishes that the first step inhibited by NRD (the NRD docking switch) occurs upstream of the last vC-pep–sensitive step in the fusion pathway. Keeping this in mind, the fact that the rate and final extent of fusion in the presence of vC-pep is the same with or without the NRD (Fig. 6 A) implies that vC-pep binding can simultaneously release autoinhibition of docking by the NRD (i.e., the NRD docking switch) and overcome the later kinetic roadblock to the completion of zippering imposed by the poorly structured membrane-proximal portion of the t-SNARE (i.e., the tC fusion switch).
Figure 6.

vN-pep and vC-pep act directly upon the coiled-coil region of the t-SNARE. The NRD of syntaxin was removed from t-liposomes (32 μg protein before digestion) by proteolytic digestion at a thrombin site introduced at residue 181 of syntaxin (Parlati et al., 1999). The resulting-t-SNARE liposomes (TΔNRD) were incubated with vC-pep (A) or vN-pep (B) for 4 min before initiating the fusion assay by addition of v-liposomes (V). The control uncleaved t-liposomes (T) were incubated with preinactivated thrombin. The symbols in all panels are as follows: T + V (○), T + V + peptide (•), TΔNRD + V (□), TΔNRD + V + peptide (▪), TΔNRD + V + vC-pep + vN-pep (▴). (C) Protein pattern of t-liposomes analyzed by SDS-PAGE and Coomassie blue staining. Thrombin cleavage generates the NRD and H3 regions of syntaxin. SNAP-25 and syntaxin in the lumen of the liposomes is protected from thrombin.
vN-pep still inhibits the initial rate of fusion when the NRD is removed from the t-SNARE (Fig. 6 B). This establishes that polarized N→C zippering is an intrinsic property of the t-SNARE's coil region, and is not a functional asymmetry imposed by asymmetrical binding of the NRD to the coil region. It also rules out the formal possibility that vN-pep inhibits fusion by stabilizing the auto-inhibited conformation of the NRD (i.e., the off state of the NRD docking switch).
Rapid, reversible assembly of SNAREpins with closed t-SNAREs
The initial rate of fusion is inhibited by vN-pep binding to t-SNAREs, which blocks the initiation of zippering (Fig. 4 C). This implies that a binding site for the NH2-terminal portion of the v-SNARE is open and available for binding and necessary to initiate zippering. If this is correct, then it follows that if the v-SNARE were allowed to bind to initiate its zippering with the t-SNARE in the absence of vN-pep, then subsequently, fusion should be resistant to vN-pep.
In fact, within the first 2 min of incubation, the initial rate of fusion (Fig. 7) becomes largely resistant to the inhibitory effects of vN-pep. Resistance to vN-pep requires coincubation of v- with t-liposomes (not depicted). Yet, at these same early time points the liposomes have progressed through <20% of the first round of fusion, and therefore are unlikely to have completed zippering. This rapid acquisition of resistance to vN-pep implies that the v-SNARE initiates N→C zippering with the t-SNARE, and that fusion is rate-limited by the slow completion of zippering at the membrane-proximal COOH termini of the SNAREs.
COOH-terminal deletions of SNAP-25 reduce SNARE-mediated fusion and abolish the stimulatory effect of vC-pep
The proteolysis results (Fig. 3) combined with the predicted binding site of vC-pep strongly suggest that the tC fusion switch is located near the COOH-terminal, membrane-proximal portion of the coiled-coil region on the t-SNARE, and specifically involves the COOH terminus of SNAP-25. To test this hypothesis, we deleted the COOH-terminal 9 and 26 amino acids of SNAP-25 to generate proteins that correspond to the well-characterized products of proteolysis by the botulinum neurotoxins A and E, respectively. Deletion of the nine COOH-terminal residues of SNAP-25 reduces the initial rate and final extent of liposome fusion (Fig. 8 A). Importantly, fusion by the SNAP-25Δ9 t-SNARE was still accelerated by removal of the NRD (from 0.5 to 1.0 rounds of fusion at 2 h; Fig. 8 B), indicating that the SNAP-25 deletion does not grossly change the structure of the t-SNARE, and confirming that the effects of the NRD switch are restricted to the NH2-terminal portion of the coiled coil. The BoNT/E-like SNAP-25Δ26 t-SNARE did not show any fusion under any conditions, including after removal of the NRD, despite the fact that the concentration of the t-SNAREs and the extent of proteolysis were comparable for all liposome preparations (Fig. 8 C). Addition of vC-pep had no effect on the fusogenic capacity of the SNAP-25Δ9 t-SNARE, even when present at a 10-fold excess and at concentrations in excess of 100 μM (Fig. 8 D). Therefore, in this t-SNARE that retains significant fusion activity and full control of the NRD switch, all regulation of the tC fusion switch has been lost, at least via the mechanism used by vC-pep.
Figure 8.

The tC fusion switch involves the COOH-terminal nine amino acids of SNAP-25. SNAP-25 deletion constructs were made to produce t-SNARE analogous to botulinum-neurotoxin A (SNAP-25Δ9) and E (SNAP-25Δ26) treatments. The deletion mutants and wild-type SNAP25 were each expressed with thrombin-cleavable syntaxin and reconstituted into liposomes. Fusion assays were performed at 37°C either with full-length syntaxin (A) or after thrombin cleavage to remove the NRD (B). The symbols in A and B are as follows: (♦) Syntaxin/SNAP-25, (□) Syntaxin/SNAP-25Δ, (▵) Syntaxin/SNAP-25Δ26. (C) Coomassie blue–stained protein profile of proteoliposomes used in A and B containing tcSyntaxin/SNAP-25, tcSyntaxin/SNAP-25Δ9, or tcSyntaxin/SNAP-25Δ26, before thrombin (lanes 1, 3, and 5, respectively) and after thrombin (lanes 2, 4, and 6, respectively). (D) SNAP-25Δ9/wild-type syntaxin proteoliposomes are insensitive to vC-pep. Initial rates (in rounds of fusion per minute) are plotted, and accessible t-SNARE is defined as in Fig. 4.
These results indicate that trans-SNARE pair assembly involves at least three stages (Fig. 9). (Unstable SNAREpins) Zippering is initiated at the NH2-terminal ends of the v- and t-SNARE, as the first SNAREs rapidly assemble into unstable, reversible SNAREpins. (Stable, partly zippered SNAREpins) The v-/t-SNARE complexes form a stable association once the NRD docking switch in the t-SNARE opens. Zippering progresses, but completion of zippering is still blocked by the unstructured tc-fusion switch. (Fully zippered SNAREpins) The tc portion of the t-SNARE becomes structured, turning on the tc switch. Zippering is completed and fusion occurs.
Figure 9.

Polarized assembly of SNAREpins and membrane fusion. SNAREpin assembly proceeds in at least three successive stages: unstable SNAREpins; stable, partly zippered SNAREpins; and fully zippered SNAREpins. (unstable SNAREpins) SNAREpins assemble between liposomes via their membrane distal NH2-terminal ends, but are only partly zipped. This can occur rapidly, even when the t-SNARE is closed by NRD autoinhibition. (stable, partly-zippered SNAREpins) When the t-SNARE opens, further zippering-up occurs to produce stable SNAREpins that indicate irreversible docking between bilayers that are not yet fused. (structured t-SNARE) vc-pep structures, removing the impediment to the completeion of zippering. (fully zippered SNAREpins) Zippering is completed and fusion occurs. This takes ∼4 min in the presence of bound vC-pep that needs to dissociate as part of this step, and could therefore be intrinsically faster than this.
SNAREpin assembly appears to proceed through at least three successive stages (Fig. 9): unstable SNAREpins; stable, partly zippered SNAREpins; and fully zippered SNAREpins. (Unstable SNAREpins) SNAREpins assemble between liposomes via their membrane distal NH2-terminal ends, but are only partly zipped. This can occur very rapidly. (Stable, partly zippered SNAREpins) When the NRD docking switch in the t-SNARE opens, further zippering occurs to produce stable SNAREpins that irreversibly dock the bilayers. This intermediate accumulates when the tC fusion switch is off. (Fully zippered SNAREpins) Zippering is completed and fusion occurs. This takes ∼5 min in the presence of bound vC-pep, which needs to dissociate as part of this step, and may limit the rate of fusion at this stage.
Discussion
In principle, fusion by SNARE proteins can be regulated at any stage: (a) before SNAREpin assembly, regulating the reactivity of v- and t-SNAREs toward each other, and therefore the number of vesicles competent to initiate fusion; (b) during SNAREpin assembly, controlling the rate of fusion; or (c) after assembly of SNARE complexes, controlling their rate of disassembly, controlling the rate of vesicle recycling, and potentially the rate of fusion pore opening.
Examples of conformational switches controlling these steps are now coming to light. Post-fusion cis-SNARE complexes are disassembled and their subunits are recycled by the ATPase NSF and SNAP proteins (Söllner et al., 1993) using an ATP/ADP conformational switch that provides mechanical force to rip open the SNARE complex (Vale, 2000; Dalal and Hanson, 2001). Fusion can be regulated before SNAREpin assembly by switching the assembly of t-SNAREs from their subunits on and off. When the NRD assembly switch is off, the assembly of Sso1 and Sec9 to form the yeast exocytic t-SNARE is auto-inhibited by the NRD of the syntaxin heavy chain (Munson et al., 2000).
The rate of SNAREpin formation between v-SNARES and already assembled t-SNAREs on separate membranes is regulated through the distinct and later-acting NRD docking switch in the assembled t-SNARE. In the absence of other regulatory factors, this switch is off and fusion is slow. Removal of the NRD leaves the t-SNARE in a constitutive “on” state, and fusion is correspondingly accelerated (Parlati et al., 1999). The degree of structural similarity between the NRD assembly switch in isolated syntaxin and the NRD docking switch in the assembled syntaxin–SNAP-25 exocytic t-SNARE is presently unknown, because the structure of the syntaxin–SNAP-25 heterodimer has not been established. However, there must be some differences between the way the NRD interacts with the coil portion of the t-SNARE in the off state of the NRD assembly and docking switches because the linker portion of NRD binds the coil in an isolated yeast syntaxin at the same site at which a portion of SNAP-25 binds in the mammalian t-SNARE (Munson et al., 2000). Other proteins such as munc-13 may “open” the NRD assembly and/or docking switches in syntaxin by binding the NRD (Betz et al., 1997; Richmond et al., 2001).
The molecular mechanisms by which fusion is regulated during SNAREpin assembly are less clear. Certainly, it is clear that regulation of zippering is physiologically important (Hua and Charlton, 1999; Xu et al., 1999), and proteins such as synaptotagmin and complexin may act at this stage (Fernandez-Chacon et al., 2001; Reim et al., 2001; Tokumaru et al., 2001). In fact, recent structural studies suggest that complexin functions to stabilize contacts in the COOH-terminal half of the heterotrimeric core complex (Chen et al., 2002). The results presented in this paper argue that the SNARE complex obligatorily zippers in the N→C direction (toward the membranes) as an intrinsic property of its coil regions. And, as importantly, there is an inherent molecular switch (the tC fusion switch) built into the zippering mechanism that controls the rate of zippering, and thus, fusion.
Recent structural studies have suggested that the SNARE motif of the exocytic t-SNARE is separated into distinct tN and tC domains, most likely by the highly polar zero layer located about halfway along its length (Hua and Charlton, 1999; Xu et al., 1999). The membrane-distal tN domain forms a stable three-stranded helical bundle, but in the membrane-proximal tC domain this bundle is inherently unstable (Fiebig et al., 1999). The data presented here establish that the tN and tC domains are functionally distinct with respect to bilayer fusion, and that the v-SNARE is similarly divided into corresponding cognate vN (membrane-distal) and vC (membrane-proximal) domains. Like their cognate tN and tC domains, the vN and vC domains can independently switch into and out of the helical bundle conformation, and independently pair with tN and tC. Polarized zippering results because vN pairs with tN before vC pairs with tC. The isolated vN and vC domains of the v-SNARE (vN-pep and vC-pep) can even drive bilayer fusion when vC-pep is lipid-anchored.
The simplest structural interpretation of our functional data is that in the NRD-docking off state, the NRD covers a portion of the coil domain, sterically inhibiting further zippering, but leaves at least part of the tN domain uncovered. vC-pep likely sterically competes with NRD binding to a portion of tC. vC-pep binding would then release NRD and throw this switch for docking. Simultaneously, vC-pep binding will also stabilize tC as a helical bundle, turning on this switch for fusion. We have already used the information gleaned here to design peptide activators of two other SNARE-dependent fusion pathways (Paumet et al., 2001; Parlati et al., 2002), which suggests that the structural changes and the opportunity for regulation are not limited to the neuronal secretory pathway.
Why is vC-pep more efficient at carrying out NRD displacement and tC structuring than the COOH-terminal part of the full-length v-SNARE? After the initial interaction between the v- and t-SNARE liposomes has occurred, which takes much less than 5 min (Fig. 7), one could suppose that the COOH terminus of VAMP2 is now present in a very high local concentration and would then be the more efficient activator. However, this is precisely the point at which fusion becomes energetically challenging, when two membranes are brought into close apposition, and water must be displaced before the bilayers will get any closer. The steric penalties for allowing the COOH termini of the v- and t-SNAREs to zip up become enormous. The free peptide does not invoke these penalties and therefore could bind (or remain bound) and activate the t-SNARE tC fusion switch more efficiently.
Once the t-SNARE is activated, zippering is an intrinsically more efficient process. Of course once the peptide dissociates, the already NH2-terminally associated VAMP2 would have a significant advantage in the completion of zippering due to its proximity and much higher affinity, provided that the t-SNARE remains activated for some finite period of time. It is perhaps a bit surprising that the peptide, which binds so tightly and overcomes two independent regulatory mechanisms, would then freely dissociate and not instead act as an impediment to the completion of fusion. Consistent with this view, we suspect that the rate of peptide dissociation (t 1/2 = 3–5 min, as measured by protease sensitivity; unpublished data) is the limiting factor in our accelerated fusion, which has a half-time to one round of fusion on the order of 4 min.
One can easily imagine how cellular regulatory proteins could stabilize the helical bundle conformation of tC (analogous to the synthetic peptide, vC-pep), and in so doing, trigger fusion. Like vC-pep, they could do so at any time physiology demands (Fig. 5 C). It can also be imagined that cellular regulatory proteins might stabilize a noncoil conformation of tC, or permanently occupy or cover the v-SNARE binding site of tC. These proteins would block fusion, whereas reversal of their binding activity would activate fusion.
In summary, exocytic t-SNAREs possess distinct autoinhibitory docking and fusion switches that cellular regulatory proteins could control sequentially or independently (Fig. 9). When both the NRD docking switch and the tC fusion switch are off, fusion is extremely slow. When the NRD switch is thrown on, vesicles rapidly associate as vN fully zips up with tN. However, fusion is still slow because the SNAREpins cannot complete their zippering until tC is switched to its bundle, i.e., “on” conformation. When this happens, vC can then bind tC, zippering can be completed, and fusion can occur. Even though vC-pep binding turns both switches on, it is clear that the NRD and tC fusion switches are not obligatorily coupled. Thus, when NRD is physically removed, tC can still be switched on by vC-pep (Fig. 6), and when the SNAP-25 COOH terminus is distorted to inactivate the tC fusion switch, the NRD switch remains rate-limiting (Fig. 8). In fact, the ability to adopt a closed conformation need not be a universal feature of all syntaxins or t-SNAREs (Dulubova et al., 2001). Yet, all known syntaxins have a domain analogous to the NRD, and all known syntaxins and t-SNARE light chains have a central polar residue in the coil region that would divide other secretory t-SNAREs into potentially separately folding tN and tC domains, suggesting that one or both switches are likely present in most syntaxins. Furthermore, the steady-state accumulation of partially zipped SNAREpins in the docked, readily releasable pool of exocytic vesicles (Hua and Charlton, 1999; Xu et al., 1999) implies the two switches are in fact uncoupled physiologically.
Materials and methods
Peptides
The peptides in Fig. 1, derived from sequences in mouse VAMP2, were synthesized by The Microchemistry Core Facility of Memorial Sloan-Kettering Cancer Center.
Lipid mixtures
Lipid mixtures were made at the indicated concentrations and then stored in sealed ampules at −80°C. The donor lipid mix included the following: 82.5 mol% 1-palmitoyl-2-oleolyl-sn-glycero-3-phosphatidylcholine (POPC), 15 mol% 1,2-dioleolyl-sn-glycero-3-phosphatidylserine (DOPS), 1.5 mol% lissamine rhodamine B (rhodamine) 1,2-dipalmitoyl-sn-glycero-3-phosphatidylethanolamine, 1.5 mol% NBD 1,2-dipalmitoyl-sn-glycero-3-phosphatidylethanolamine, and 3 mM total lipid. The acceptor lipid mix included the following: 85 mol% POPC, 15 mol% DOPS, and 15 mM total lipid.
Plasmids
his6-SNAP-25Δ9 (a COOH-terminal deletion analogous to the product of botulinum-neurotoxin A proteolysis; amino acids 1–197) was PCR-amplified from SNAP25b (pFP247) using oligonucleotides OFP 82(5′-TCCATGGCCCATCATCATCATCATCATGCCGAGGACGCAGACATGC-3′) and oBJ4 (5′-GAATTCCTCGAGATTATTTATTTATTGGTTGGCTTCATCAATTCT-3′). This PCR product was digested with XhoI and NcoI restriction enzymes and inserted into plasmid pFP247 to create plasmid pBJ8.
his6-SNAP-25Δ26 (a COOH-terminal deletion analogous to the product of botulinum-neurotoxin E proteolysis; amino acids 1–180) was PCR-amplified from SNAP25b (pFP247) using oligonucleotides OFP82 and OBJ3 (5′-GAATTCCTGAGATTATTTATTTACCTGTCGATCTGGCG-3′). This PCR product was digested with XhoI and NcoI and inserted into plasmid pFP247 to create plasmid pBJ7.
Protein expression and purification
Full-length t-SNARE complex containing mouse his6-SNAP-25 and rat syntaxin 1A was expressed and purified from the polycistronic vector pTW34 (Weber et al., 2000). t-SNARE complex containing syntaxin, with a thrombin cleavage site at residue 181 and his6-SNAP-25, was expressed and purified from the polycistronic vector, pTW69 (Parlati et al., 1999). t-SNARE containing thrombin-cleavable syntaxin and either SNAP-25Δ9 or SNAP-25Δ26 was produced by the coexpression of pFP226 (Parlati et al., 1999) and either pBJ8 or pBJ7, respectively. The cytosolic domain of the t-SNARE complex with no internal cysteines (t-cyt) containing mouse his6-SNAP-25 (C→S) and rat syntaxin 1A (residues 1–265-L-C, C145S) was expressed from BL21 (DE3) Escherichia coli cotransfected with pJM72 and pJM57–2 as described previously (McNew et al., 2000b). Full-length mouse VAMP2-his6 was expressed and purified from pTW2 as described previously (Weber et al., 1998). The cytosolic domain of mouse VAMP2 (v-cyt) with a COOH-terminal his6 tag was expressed from pET-rVAMP2CD and purified as described previously (Weber et al., 1998). In some experiments, v-cyt with a COOH-terminal cysteine and an NH2-terminal, his6 tag (McNew et al., 2000b), was also used as an inhibitor and gave identical results.
Protein reconstitution into liposomes
VAMP2 and t-SNARE complexes were reconstituted into liposomes by detergent dialysis and isolated on a Nycodenz flotation gradient as described previously (Weber et al., 1998). In all experiments, VAMP2 was reconstituted with the donor lipid mix, and t-SNARE was reconstituted with the acceptor lipid mix. To remove the NRD from thrombin-cleavable syntaxin, thrombin-cleavable t-SNARE liposomes were incubated with 0.02 U/μl thrombin (Sigma-Aldrich) for 1 h at RT. The proteolysis was stopped by the addition of 2 mM AEBSF, and the liposomes were immediately used in fusion experiments.
Proteinase K protection assay
Limited proteolysis by proteinase K (Boehringer) was used to identify protected components of the core assembly, or of t-cyt in the presence of VAMP peptides, as described with some modifications (Fasshauer et al., 1998). In brief, t-cyt or t-liposomes were incubated with proteinase K at a 40:1 protein/proteinase molar ratio for 10 min at RT. Proteolysis was stopped by the addition of 5 mM PMSF and 5 mM AEBSF, and the samples were immediately boiled for 5 min. Novex LDS sample buffer was added and the samples were boiled again. To test for VAMP or VAMP peptide–dependent protection, v-cyt or VAMP peptides were added to t-cyt or to t-liposomes and preincubated for 20 min at RT before addition of proteinase K.
Electrophoresis and Western blotting
Electrophoresis was performed in precast 10 or 12% bis-Tris gels (Novex) running in MES buffer. For Western blot analysis, the protein fragments were transferred onto nitrocellulose and incubated with primary antibody for 30 min. Primary antibodies were used at the following dilutions: for HPC-1, 1:1,500 (syntaxin monoclonal), for 939-3, 1:1,000 (rabbit antibody raised against COOH-terminal peptide of SNAP-25; residues 207–218), for Cl 71.1, 1:5,000 (SNAP-25 NH2-terminal monoclonal [Cat. No. 111001; Synaptic Systems]). Goat anti–mouse and goat anti–rabbit secondary antibodies conjugated to HRP (Bio-Rad Laboratories) were incubated with the blot for 20 min at a 1:2,000 dilution, and the blots were developed with the ECL Western blotting detection system.
Fusion assay
Fusion between t-SNARE liposomes and v-SNARE liposomes was monitored by the increase in fluorescence resulting from the dilution of a quenched FRET pair of fluorophores in the v-SNARE liposomes as described previously (Weber et al., 1998). For each assay, 45 μl of t-SNARE liposomes were mixed in 96-well Fluoronunc™ polysorb plates (Nunc) at 37°C with reconstitution buffer or the indicated amounts of v-cyt or peptide for 2–10 min. Stock concentrations of peptide were adjusted so that 2–10 μl of peptide were added to each 50-μl reaction and the balance made up with buffer. The fusion reaction was initiated by adding 5 μl of RT v-liposomes. NBD fluorescence was monitored at 2-min intervals for 160 min (excitation = 460 nm; emission = 538 nm) in a Fluoroskan II plate reader (Thermo Labsystems) at 37°C. At 120 min, 10 μl of 2.5% wt/vol n-dodecyl-maltoside (Boehringer) was added to completely dissolve the lipids and measure the NBD fluorescence at infinite dilution. The data were normalized and calibrated as described previously in order to derive the rounds of fusion (Weber et al., 1998; Parlati et al., 1999).
Acknowledgments
We thank the Microchemistry Core Facility of the Memorial Sloan-Kettering Cancer Center for protein sequencing and peptide synthesis, and particularly Hedia Erdjument-Bromage and Lynne Lacomis. We thank Stéphanie Bourdelle and Willa Bellamy for help with the completion of the manuscript. We would also like to thank Dr. Fabienne Paumet for critical reading of the manuscript.
Research was supported by a National Institutes of Health (NIH) grant R01 DK 27044 (to J.E. Rothman) and an NIH postdoctoral fellowship (to J.A. McNew).
Thomas Weber's present address is Carl C. Icahn Institute for Gene Therapy and Molecular Medicine, Mount Sinai Medical Center, Box 1496, One Gustave L. Levy Place, New York, NY 10029.
James A. McNew's present address is Dept. of Biochemistry and Cell Biology, Rice University, MS140, 6100 Main St., P.O. Box 1892, Houston, TX 77251.
Robert J. Johnston's present address is Dept. of Biochemistry and Molecular Biophysics, Columbia University, College of Physicians and Surgeons, 701 West 168th St., HHSC 724, Box 140, New York, NY 10032.
Frank Parlati's present address is Rigel Pharmaceuticals, Inc., 240 East Grand Ave., South San Francisco, CA 94080.
Footnotes
Abbreviations used in this paper: NRD, NH2-terminal regulatory domain; SNAP-25, synaptosomal-associated protein of 25 kD; t-cyt, cytoplasmic domain of syntaxin–SNAP-25 complex; VAMP, vesicle-associated membrane protein; v-cyt, cytoplasmic domain of VAMP.
References
- Betz, A., M. Okamoto, F. Benseler, and N. Brose. 1997. Direct interaction of the rat unc-13 homologue Munc 13-1 with the N-terminus of syntaxin. J. Biol. Chem. 272:2520–2526. [DOI] [PubMed] [Google Scholar]
- Block, M.R., B.S. Glick, C.A. Wilcox, F.T. Wieland, and J.E. Rothman. 1988. Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proc. Natl. Acad. Sci. USA. 85:7852–7856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, D.C., and P.S. Kim. 1998. HIV entry and its inhibition. Cell. 93:681–684. [DOI] [PubMed] [Google Scholar]
- Chen, X., D.R. Tomchick, E. Kovrigin, D. Arac, M. Machius, T.C. Sudhof, and J. Rizo. 2002. Three-dimensional structure of the complexin/SNARE complex. Neuron. 33:397–409. [DOI] [PubMed] [Google Scholar]
- Chen, Y.A., S.J. Scales, S.M. Patel, Y.-C. Doung, and R.H. Scheller. 1999. SNARE complex formation is triggered by Ca2+ and drives membrane fusion. Cell. 97:165–174. [DOI] [PubMed] [Google Scholar]
- Chen, Y.A., S.J. Scales, J.R. Jagath, and R.H. Scheller. 2001. A discontinuous SNAP-25 C-terminal coil supports exocytosis. J. Biol. Chem. 276:28503–28508. [DOI] [PubMed] [Google Scholar]
- Dalal, S., and P.I. Hanson. 2001. Membrane traffic: what drives the AAA motor? Cell. 104:5–8. [DOI] [PubMed] [Google Scholar]
- Dulubova, I., S. Sugita, S. Hill, M. Hosaka, I. Fernandez, T.C. Südhof, and J. Rizo. 1999. A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J. 18:4372–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova, I., T. Yamaguchi, Y. Wang, T.C. Südhof, and J. Rizo. 2001. Vam3p structure reveals conserved and divergent properties of syntaxins. Nat. Struct. Biol. 8:258–264. [DOI] [PubMed] [Google Scholar]
- Fasshauer, D., W.K. Eliason, A.T. Brünger, and R. Jahn. 1998. Identification of a minimal core of the synaptic SNARE complex sufficient for reversible assembly and disassembly. Biochemistry. 37:10354–10362. [DOI] [PubMed] [Google Scholar]
- Fernandez, I., J. Ubach, I. Dulubova, X. Zhang, T.C. Südhof, and J. Rizo. 1998. Three-dimensional structure of an evolutionarily conserved N-terminal domain of syntaxin 1A. Cell. 94:841–849. [DOI] [PubMed] [Google Scholar]
- Fernandez-Chacon, R., A. Konigstorfer, S.H. Gerber, J. Garcia, M.F. Matos, C.F. Stevens, N. Brose, J. Rizo, C. Rosenmund, and T.C. Sudhof. 2001. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 410:41–49. [DOI] [PubMed] [Google Scholar]
- Fiebig, K.M., L.M. Rice, R. Pollock, and A.T. Brünger. 1999. Folding intermediates of SNARE complex assembly. Nat. Struct. Biol. 6:117–123. [DOI] [PubMed] [Google Scholar]
- Hayashi, T., H. McMahon, S. Yamasaki, T. Binz, Y. Hata, T.C. Südhof, and H. Niemann. 1994. Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J. 13:5051–5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazzard, J., T.C. Südhof, and J. Rizo. 1999. NMR analysis of the structure of synaptobrevin and of its interaction with syntaxin. J. Biomol. NMR. 14:203–207. [DOI] [PubMed] [Google Scholar]
- Hua, S.-Y., and M.P. Charlton. 1999. Activity-dependent changes in partial VAMP complexes during neurotransmitter release. Nat. Neurosci. 2:1078–1083. [DOI] [PubMed] [Google Scholar]
- Lin, R.C., and R.H. Scheller. 2000. Mechanisms of synaptic vesicle exocytosis. Annu. Rev. Cell Dev. Biol. 16:19–49. [DOI] [PubMed] [Google Scholar]
- Margittai, M., D. Fasshauer, S. Pabst, R. Jahn, and R. Langen. 2001. Homo- and heterooligomeric SNARE complexes studies by site-directed spin labeling. J. Biol. Chem. 276:13169–13177. [DOI] [PubMed] [Google Scholar]
- Mayer, A., W. Wickner, and A. Haas. 1996. Sec18p (NSF)-driven release of Sec17p (alpha-SNAP) can precede docking and fusion of yeast vacuoles. Cell. 85:83–94. [DOI] [PubMed] [Google Scholar]
- McNew, J.A., T. Weber, D.M. Engelman, T.H. Söllner, and J.E. Rothman. 1999. The length of the flexible SNAREpin juxtamembrane region is a critical determinant of SNARE-dependent fusion. Mol. Cell. 4:415–421. [DOI] [PubMed] [Google Scholar]
- McNew, J.A., F. Parlati, R. Fukuda, R.J. Johnston, K. Paz, F. Paumet, T.H. Söllner, and J.E. Rothman. 2000. a. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature. 407:153–159. [DOI] [PubMed] [Google Scholar]
- McNew, J.A., T. Weber, F. Parlati, R.J. Johnston, T.J. Melia, T.H. Söllner, and J.E. Rothman. 2000. b. Close is not enough: SNARE-dependent membrane fusion requires an active mechanism that transduces force to membrane anchors. J. Cell Biol. 150:105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misura, K.M.S., R.H. Scheller, and W.I. Weis. 2000. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature. 404:355–362. [DOI] [PubMed] [Google Scholar]
- Munson, M., X. Chen, A.E. Cocina, S.M. Schultz, and F.M. Hughson. 2000. Interactions within the yeast t-SNARE Sso1p that control SNARE complex assembly. Nat. Struct. Biol. 7:894–902. [DOI] [PubMed] [Google Scholar]
- Nickel, W., T. Weber, J.A. McNew, F. Parlati, T.H. Söllner, and J.E. Rothman. 1999. Content mixing and membrane integrity during membrane fusion driven by pairing of isolated v-SNAREs and t-SNAREs. Proc. Natl. Acad. Sci. USA. 96:12571–12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlati, F., T. Weber, J.A. McNew, B. Westermann, T.H. Söllner, and J.E. Rothman. 1999. Rapid and efficient fusion of phospholipid vesicles by the alpha-helical core of a SNARE complex in the absence of an N-terminal regulatory domain. Proc. Natl. Acad. Sci. USA. 96:12565–12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlati, F., J.A. McNew, R. Fukuda, R. Miller, T.H. Söllner, and J.E. Rothman. 2000. Topological restriction of SNARE-dependent membrane fusion. Nature. 407:194–198. [DOI] [PubMed] [Google Scholar]
- Parlati, F., O. Varmalov, K. Paz, J.A. McNew, D. Hurtado, T.H. Söllner, and J.E. Rothman. 2002. Distinct SNARE complexes mediating membrane fusion in Golgi transport based on combinatorial specificity. Proc. Natl. Acad. Sci. USA. 99:5424–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paumet, F., B. Brügger, F. Parlati, J.A. McNew, T.H. Söllner, and J.E. Rothman. 2001. A t-SNARE of the endocytic pathway must be activated for fusion. J. Cell Biol. 155:961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier, M.A., J.C. Hao, P.N. Malkus, C. Chan, M.F. Moore, D.S. King, and M.K. Bennett. 1998. a. Protease resistance of syntaxin-SNAP-25-VAMP complexes: Implications for assembly and structure. J. Biol. Chem. 273:11370–11377. [DOI] [PubMed] [Google Scholar]
- Poirier, M.A., W. Xiao, J.C. Macosko, C. Chan, Y.-K. Shin, and M.K. Bennett. 1998. b. The synaptic SNARE complex is a parallel four-stranded helical bundle. Nat. Struct. Biol. 5:765–769. [DOI] [PubMed] [Google Scholar]
- Reim, K., M. Mansour, F. Varogueaux, H.T. McMahon, T.C. Sudhof, N. Brose, and C. Rosenmund. 2001. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell. 104:71–81. [DOI] [PubMed] [Google Scholar]
- Richmond, J.E., R.M. Weimer, and E.M. Jorgensen. 2001. An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature. 412:338–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scales, S.J., Y.A. Chen, B.Y. Yoo, S.M. Patel, Y.-C. Doung, and R.H. Scheller. 2000. SNAREs contribute to the specificity of membrane fusion. Neuron. 26:457–464. [DOI] [PubMed] [Google Scholar]
- Söllner, T., S.W. Whiteheart, M. Brunner, H. Erdjument-Bromage, S. Geromanos, P. Tempst, and J.E. Rothman. 1993. SNAP receptors implicated in vesicle targeting and fusion. Nature. 362:318–324. [DOI] [PubMed] [Google Scholar]
- Sutton, R.B., D. Fasshauer, R. Jahn, and A.T. Brünger. 1998. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 395:347–353. [DOI] [PubMed] [Google Scholar]
- Tokumaru, H., K. Umayahara, L.L. Pellegrini, T. Ishizuka, H. Saisu, H. Betz, G.J. Augustine, and T. Abe. 2001. SNARE complex oligomerization by synaphin/complexin is essential for synaptic vesicle exocytosis. Cell. 104:421–432. [DOI] [PubMed] [Google Scholar]
- Vale, R.D. 2000. AAA proteins: lords of the ring. J. Cell Biol. 150:F13–F20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, T., B.V. Zemelman, J.A. McNew, B. Westermann, M. Gmachl, F. Parlati, T.H. Söllner, and J.E. Rothman. 1998. SNAREpins: minimal machinery for membrane fusion. Cell. 92:759–772. [DOI] [PubMed] [Google Scholar]
- Weber, T., F. Parlati, J.A. McNew, R.J. Johnston, B. Westermann, T.H. Söllner, and J.E. Rothman. 2000. SNAREpins are functionally resistant to disruption by NSF and αSNAP. J. Cell Biol. 149:1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimbs, T., K. Mostov, S.H. Low, and K. Hofmann. 1998. A model for structural similarity between different SNARE complexes based on sequence relationships. Trends Cell Biol. 8:260–262. [DOI] [PubMed] [Google Scholar]
- Wild, C.T., D.C. Shugars, T.K. Greenwell, C.B. McDanal, and T.J. Matthews. 1994. Peptides corresponding to a predictive α-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA. 91:9770–9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, W., M.A. Poirier, M.K. Bennett, and Y.-K. Shin. 2001. The neuronal t-SNARE complex is a parallel four-helix bundle. Nat. Struct. Biol. 8:308–311. [DOI] [PubMed] [Google Scholar]
- Xu, T., B. Rammner, M. Margittai, A.R. Artalejo, E. Neher, and R. Jahn. 1999. Inhibition of SNARE complex assembly differentially affects kinetic components of exocytosis. Cell. 99:713–722. [DOI] [PubMed] [Google Scholar]