

Abstract
Oxysterol binding proteins (OSBPs) comprise a large conserved family of proteins in eukaryotes. Their ubiquity notwithstanding, the functional activities of these proteins remain unknown. Kes1p, one of seven members of the yeast OSBP family, negatively regulates Golgi complex secretory functions that are dependent on the action of the major yeast phosphatidylinositol/phosphatidylcholine Sec14p. We now demonstrate that Kes1p is a peripheral membrane protein of the yeast Golgi complex, that localization to the Golgi complex is required for Kes1p function in vivo, and that targeting of Kes1p to the Golgi complex requires binding to a phosphoinositide pool generated via the action of the Pik1p, but not the Stt4p, PtdIns 4-kinase. Localization of Kes1p to yeast Golgi region also requires function of a conserved motif found in all members of the OSBP family. Finally, we present evidence to suggest that Kes1p may regulate adenosine diphosphate-ribosylation factor (ARF) function in yeast, and that it may be through altered regulation of ARF that Kes1p interfaces with Sec14p in controlling Golgi region secretory function.
Keywords: Kes1p; phosphoinositides; yeast Golgi; Sec14p; ARF
Introduction
Oxysterol binding proteins (OSBPs)* define a large protein family whose members are found in mammals (Dawson et al., 1989), Drosophila (Alphey et al., 1998), C. elegans ( Caenorhabditis elegans Sequencing Consortium, 1998), and yeast (Jiang et al., 1994; Fang et al., 1996; Beh et al., 2001). Initial studies suggested that OSBPs mediate the potent downregulation of sterol biosynthesis by oxysterols (Taylor et al., 1984; Dawson et al., 1989). Although evidence consistent with this hypothesis was culled from genetic experiments in yeast and mammalian cells (Jiang et al., 1994; Lagace et al., 1997; Beh et al., 2001), other evidence indicates that OSBPs are not direct mediators of oxysterol signaling (Brown and Goldstein, 1999). Rather, these proteins have other functions that interface with membrane trafficking from the Golgi complex (Fang et al., 1996), progression of cells through the cell cycle (Alphey et al., 1998), and tumor metastasis (Fournier et al., 1999).
Yeast express seven OSBPs. Three (Osh1p, Osh2p, and Osh3p) are classified as long OSBPs, typified by the mammalian OXYB, where a large extension is found NH2-terminal to the 430 amino acid oxysterol binding homology domain (Fig. 1 A). The remaining four are homologous to the oxysterol binding domain of OXYB and are classified as short OSBPs (Fig. 1 A). None of the yeast OSBPs individually plays an essential function, but deletion of all seven OSBP genes results in inviability (Jiang et al., 1994; Fang et al., 1996; Levine and Munro, 1998; Beh et al., 2001).
Figure 1.


Kes1p function and the Sec14p pathway. (A) The yeast OSBP family. The general organization of the seven yeast OSBP family members is given and compared with the human OSBP (hOSBP). Yeast proteins are identified at left by their common designations (see Beh et al., 2001). Regions homologous to the oxysterol binding domain of hOSBP are shown in black, and the primary sequence identities within that region are given. The three long yeast OSBPs (Osh1p, Osh2p and Osh3p) share homologies at their COOH-terminal domains with hOSBP (stippled areas), whereas these particular homologies are present in the four short yeast OSBPs (Kes1p, Hes1p, Osh6, and Osh7). Ultimate residues for each protein are numbered at right. (B) Pathway for Sec14p-dependent Golgi secretory function. Roles for phosphatidylcholine (PC), DAG, PA, PI(4)P, and PI(4,5)P2 in stimulating yeast Golgi complex secretory function have been proposed. PC, DAG, and PA are proposed to mediate combinatorial regulation of a pair of imperfectly redundant ARFGAPs (Gcs1p and Age2p) whose activity is required for Sec14p pathway function (Yanagisawa, L., and V.A. Bankaitus, unpublished data). The known execution points for relevant genes whose inactivation effects “bypass Sec14p” (i.e., CKI1, PCT1, CPT1, and SAC1) are shown. The execution point of the KES1 gene product in the Sec14p pathway is unknown.
Sec14p is the major yeast PI/PC-transfer protein (PITP) (Bankaitis et al., 1990; Kearns et al., 1998a; Li et al., 2000b). Analyses of mutations that allow yeast to grow in the absence of the essential Sec14p (i.e., ‘bypass Sec14p’ mutations) demonstrate that Sec14p regulates lipid metabolism so that a permissive membrane environment for Golgi complex secretory function is maintained (Fig. 1 B; Cleves et al., 1991a,b; McGee et al., 1994; Huijbregts et al., 2000; Li et al., 2000b; Xie et al., 2001). An important component of this Sec14p-mediated regulation of lipid metabolism appears to involve stimulation of the adenosine diphosphate-ribosylation factor guanosine triphosphatase activating protein (ARFGAP) activities of the yeast GCS1 and AGE2 gene products (Yanagisawa, L., and V.A. Bankaitus, unpublished data). In this fashion, Sec14p may impose a trans-Golgi region–specific regulation of the adenosine diphosphate-ribosylation factor (ARF) GTPase cycle in yeast. A functional relationship between Sec14p and OSBP function in yeast became apparent when it was discovered that inactivation of the KES1 gene, which encodes one of the short OSBP family members, uniquely elicits a “bypass Sec14p” phenotype (Fang et al., 1996). No other individual yeast OSBP defect elicits “bypass Sec14p” (Fang et al., 1996; Beh et al., 2001). This “bypass Sec14p” phenotype makes it possible to dissect the various functional properties of Kes1p.
Herein, we demonstrate that yeast Kes1p is a phosphoinositide (PIP) binding protein whose lipid binding activities are necessary, but not sufficient, for Kes1p localization to yeast Golgi membranes. We also show that Golgi complex localization is critical for Kes1p function. Finally, we present evidence to suggest that Kes1p activity influences ARF function in yeast. The collective data suggest that OSBPs are compartment specific regulators (or effectors) of ARF function in eukaryotes.
Results
Kes1p functional domains
We independently recovered 51 kes1 loss-of-function mutations in a “bypass Sec14p” mutant screen (Cleves et al., 1991b; Fang et al., 1996). Of these, nine sustain wild-type levels of Kes1p. Gap repair experiments, coupled with DNA sequence determinations, revealed these nine mutations define six alleles (Fig. 2 A). Four represent a GAA → AA mutation that substitutes lysine for glutamate at Kes1p residue 312 (E312K). One represents a double mutation that converts codons 201 and 202 from TTGCAT to TTCAAT, and substitutes phenylalanine-asparagine for leucine-histidine (LH20/202FN).
Figure 2.



Kes1p functional domains. (A) Kes1p domain structure. The 434-residue Kes1p sequence is represented with the OSBP and PH domains identified by stippled boxes as indicated at top. The six kes1 inactivating mutations of interest are identified at bottom, and the individual sites of missense substitution relative to the Kes1p primary sequence representation are indicated by the black lines. (B) Kes1p PH domain. The primary sequence of the Kes1p PH domain is aligned other known PH domains (from Lemmon, 1999). A secondary structure diagram derived from the PLC-δ1 PH domain crystal structure is given at top. The signature tryptophan of PH domains is boxed (W317 for Kes1p). Basic residues (i.e., R236, K242, and K243) in the predicted variable loop 2 (VL-2) of Kes1p that likely form the inositide binding motif are identified (solid circles). (C) OSBP domains from the yeast OSBPs and hOSBP are aligned. The canonical OSBP domain consists of two conserved motifs (identified by black bars at top) separated by linkers of 13–20 amino acids.
The sequences surrounding P30T, S43Y, and LH201/202FN revealed no informative homologies. However, clues were gleaned from regions surrounding the E312K, E107K, and K109A substitutions. E312K lies within a domain that exhibits features of Pleckstrin homology (PH) domains. Although this region is not scored as a PH domain by various search programs, it includes the signature invariant tryptophan (W317) that is positioned ∼100 residues downstream of a GIL motif (Kes1p residues 205–207). PH domains are often bounded by a GXL motif at the NH2 terminus, and the signature tryptophan is positioned ∼110 residues downstream of that motif (Lemmon, 1999). Alignment of the putative Kes1p PH domain (residues 180–330) with known PH domains is shown (Fig. 2 B). Although proof that this domain conforms to the PH domain fold awaits structural analyses, for purposes of convenience we will refer to this domain as the Kes1p PH domain.
BLAST analyses also identify a motif (Kes1p residues 108–149) that is present only in OSBPs and is highly conserved among them (Fig. 2 C). This OSBP domain consists of two 11-residue motifs separated by a linker of ∼20 divergent residues. Two of the kes1 alleles alter residues adjacent to (E107K), or within (H143Y), this domain.
Kes1p is a PIP binding protein
As PI(4,5)P2 and other PIPs are common ligands for PH domains, we assessed binding of purified Kes1p to PI(4,5)P2 or its soluble headgroup inositol-1,4,5-trisphosphate (IP3). To this end, [3H]-BZDC-PI(4,5)P2 and [3H]-BZDC-IP3 were used as photo-affinity ligands (Dorman and Prestwich, 1994; Prestwich, 1996; Prestwich et al., 1997; Chaudhary et al., 1998a,b; Feng et al., 2001). The [3H]-BZDC-PI(4,5)P2 binding experiments were performed in a mixed micelle system where the photo-affinity ligand was a trace component. Thus, the photolabeling assay requires Kes1p to interact with photo probe monomers in the face of large detergent excess. Although Sec14p fails to bind either probe (unpublished data), Kes1p binds both [3H]-BZDC-PI(4,5)P2 and [3H]-BZDC-IP3 avidly, as indicated by recovery of appropriate covalent adducts (Fig. 3 A). The intensities of Kes1p photolabeling were similar to those scored for the PIP binding protein gelsolin.
Figure 3.
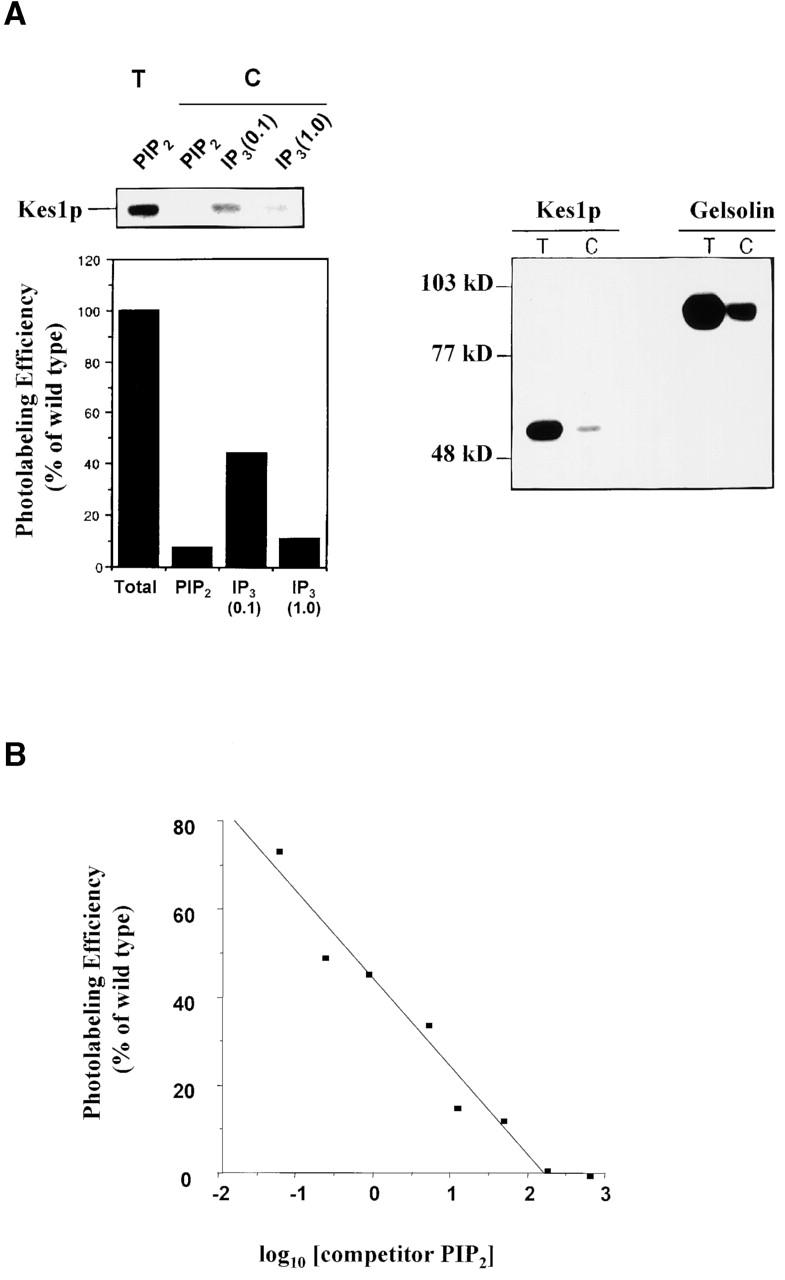
Kes1p binds PIPs. (A) Photo-cross-linking of Kes1p to [3H]BZDC-PI(4,5)P2 and [3H]BZDC-IP3. Purified recombinant Kes1p was incubated with [3H]BZDC-PI(4,5)P2 photo-probe in a mixed micelle system, the reaction was irradiated under long wave UV light, and covalent adducts were resolved by SDS-PAGE and autoradiography. In the left panels, a representative autoradiogram is shown with a corresponding densitometric quantification of photo-cross-linking provided below. A reaction of Kes1p and [3H]BZDC-PI(4,5)P2 with no competitor yields the total photolabeling lane (T). Addition of excess competitor PI(4,5)P2 (10 μM) or 0.1 mM or 1 mM IP3, as indicated in the competition (C) lanes, displaces photo-probe from Kes1p and reduces photolabeling. In the right panel, a representative autoradiogram is shown from a Kes1p/[3H]BZDC-IP3 photolabeling reaction with no competitor IP3 (T) or with excess competitor IP3 (C). Purified gelsolin (gift from Paul Janmey, Harvard Medical School, Cambridge, MA) was employed as a positive control. Purified Sec14p fails to bind photo-probe and served as negative control (unpublished data). (B) Concentration-dependent displacement of photo-probe from Kes1p by PI(4,5)P2. Purified Kes1p was incubated in a series of mixed micelle photolabeling reactions supplemented with increasing concentrations of PI(4,5)P2. Efficiency of photolabeling was determined by densitometric scanning of autoradiograms, and is plotted as a function of the log10 of the competitor PI(4,5)P2 concentration. (C) Kes1p PH domain is sufficient for PIP binding. Purified recombinant Kes1p and Kes1p PH domain were incorporated into reactions using [3H]BZDC-PI(4,5)P2 as photo-probe. Total (T) and competition (C) reactions are as above. (D) Kes1p has weak affinity for other acidic phospholipids. Kes1p was incorporated into a standard mixed micelle photolabeling reaction using [3H]BZDC-PI(4,5)P2 as photo-probe. Reactions with no competitor (Total), and with competitor lipids (10 μM final concentration; indicated at top), are shown. PIP, phosphatidylinositol-4-P; PI, phosphatidylinositol; PE; phosphatidylethanolamine; PS, phosphatidylserine; DiC8, di-C8:0 diacylglycerol.
Kes1p-[3H]-BZDC-IP3 solution binding experiments also impose rigorous constraints on binding. Both photo-probes are displaced from Kes1p by challenge with excess unlabeled PI(4,5)P2 or IP3 competitor. However, an 83-fold molar excess of unlabeled PI(4,5)P2 displaces both photo-probes, whereas a 103-fold molar excess of IP3 is required for displacement of [3H]-BZDC-PI(4,5)P2 (Fig. 3 A). These data suggest that Kes1p has a 10-fold higher affinity for PI(4,5)P2 than it does for IP3.
To quantify Kes1p affinity for PI(4,5)P2, we determined the concentration of competitor required for 50% displacement of [3H]BZDC-PI(4,5)P2 from Kes1p (IC50). Although photoaffinity labeling is a nonequilibrium process (Dorman and Prestwich, 1994) and cannot directly give equilibrium dissociation constants, displacement by competing ligands yields a rank order of relative affinities. A dose-dependent reduction in photolabeling efficiency was observed when PI(4,5)P2 was employed as competitor (Fig. 3 B). The IC50 for PI(4,5)P2 is 1.9 μM with an estimated K D = 2.5 μM for Kes1p-[3H]BZDC-PI(4,5)P2 binding. These values resemble those measured for PI(4,5)P2 and [3H]BZDC-PI(4,5)P2 binding by PLC-δ1 (Tall et al., 1997; Lemmon, 1999).
Kes1p PH domain is sufficient for PIP binding
We tested whether the Kes1p PH domain represents the PI(4,5)P2 and IP3 binding module. Indeed, a 144 amino acid Kes1p fragment that consists largely of the PH domain (residues 171–314) is sufficient for PI(4,5)P2 and IP3 binding (Fig. 3 C). A 110 amino acid region of Kes1p (residues 205–314) that defines the putative Kes1p PH domain (Fig. 2 B) also binds IP3 photo-probe (unpublished data). Neither a smaller region of Kes1p (residues 205–309) that fully overlaps this domain, nor a Kes1p domain (residues 220–330) that incompletely overlaps this domain exhibits any inositide binding activity (unpublished data). Interestingly, the minimum Kes1p inositide binding module does not include W317, the signature residue of PH domains (Fig. 2 B; see below).
Specificity of PIP binding
To characterize specificity of Kes1p binding to PIPs, we employed a competitive displacement strategy using a variety of binding substrates in a Kes1p-[3H]BZDC-PI(4,5)P2 photolabeling assay. At 400-fold molar excess, all PIPs tested and other acidic phospholipids (phosphatidylserine, phosphatidic acid, and cardiolipin) displaced [3H]BZDC-PI(4,5)P2 (Fig. 3 D). Other lipids (phosphatidylethanolamine, phosphatidylcholine, ceramide, and diacylglycerol) were ineffective. Soluble inositol-polyphosphates (IP3, IP4, IP6) also failed to displace [3H]BZDC-PI(4,5)P2 from Kes1p (unpublished data). When unlabeled competitor lipids were reduced to a 10-fold molar excess relative to [3H]BZDC-PI(4,5)P2, only PI(4,5)P2 displaced photo-probe. PI, PI(4)P, phosphatidic acid (PA), and other acidic phospholipids were ineffective (unpublished data). A 10-fold molar excess of PI(3,4,5)P3, PI(3,4)P2, or PI(3,5)P2 also failed to displace [3H]BZDC-PI(4,5)P2 (Fig. 3 D). Thus, PI(4,5)P2 is the preferred Kes1p ligand in vitro.
Mutations in the Kes1p PH domain influence PIP binding
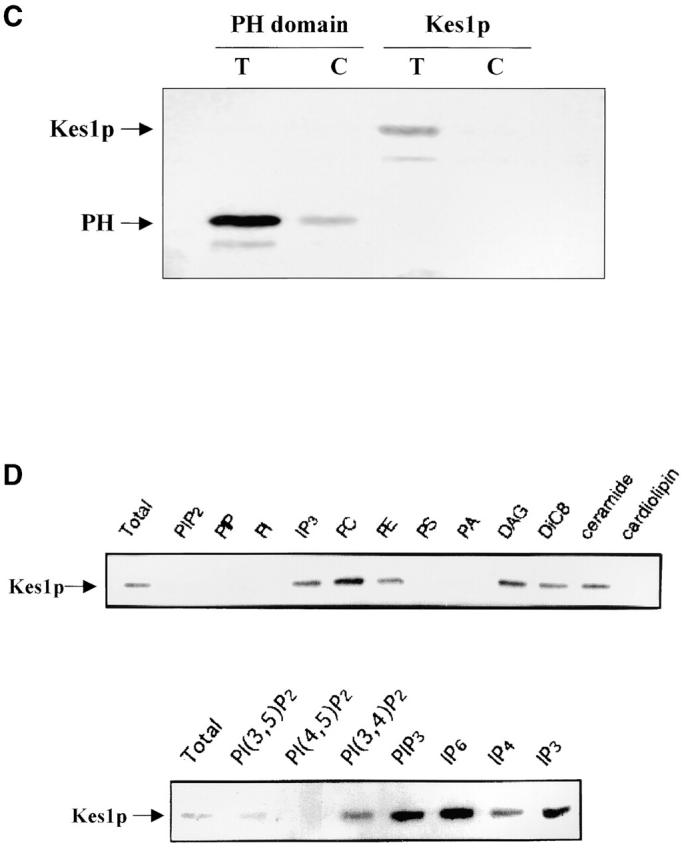
Since the Kes1p PH domain binds PI(4,5)P2, we investigated whether integrity of this domain is required for PIP binding. To this end, we characterized the binding properties of Kes1pE312K and Kes1p W317A (Kes1pW317A) using the photolabeling assay described above. In neither case could we detect binding of full-length mutant protein to [3H]BZDC-PI(4,5)P2 (Fig. 4 A) or to [3H]BZDC-IP3 (unpublished data). In vivo complementation experiments reveal that Kes1pE312K and Kes1pW317A, while stable proteins in yeast, are nonfunctional (unpublished data).
Figure 4.


Kes1p PH domain mutations and PIP binding. (A) Mutations in Kes1p PH domain compromise PIP binding. Photoaffinity-labeling assays were performed with mutant Kes1ps using [3H]BZDC-PI(4,5)P2 as photo-probe. Covalent Kes1p–photo-probe adducts were resolved by SDS-PAGE and visualized by autoradiography. Kes1pE312K and full-length Kes1pW317A fail to bind [3H]BZDC-PI(4,5)P2. A proteolytic breakdown product of Kes1pW317A (Kes1pW317A*) regains PIP binding competence. (B) MALDI-TOF mass spectrometry of Kes1pW317A and Kes1pW317A*. The mass to charge ratios of full-length Kes1pW317A and the Kes1pW317A* truncation product are shown and the corresponding mass values for each are indicated. Below is an illustration of the Kes1p domain structure where the OSBP and PH domains are designated by the black and gray boxes, respectively. The location of the W317A missense substitution within the PH domain is shown by a black bar, and the surrounding wild-type sequence is displayed at bottom (W corresponds to W317). The R314K315 motif that defines the site of proteolysis that generates Kes1pW317A* is underlined. (C) COOH-terminal Kes1p truncation and PIP binding. Wild-type and mutant forms of Kes1p were genetically truncated after residue K314 to generate protein fragments consisting of otherwise wild-type primary sequence (Kes1p1–314). GST-tagged versions of Kes1p1–314 and Kes1pE312K* were purified and photolabeled with [3H]BZDC-IP3 in a mixed micelle system in the absence (T) or presence (C) of competitor PI(4,5)P2. Both full-length protein and fragments associate with [3H]BZDC-IP3 in a manner that is subject to displacement by PI(4,5)P2. (D) COOH-terminal truncation of Kes1p3E fails to rescue PIP binding. GST-tagged Kes1p and Kes1p3E, and the corresponding COOH-terminal truncation fragments Kes1p1–314 and Kes1p3E* were purified were photolabeled with [3H]BZDC-IP3 in a mixed micelle system without (T) and with (C) unlabeled PI(4,5)P2 competitor. Full-length Kes1p3E fails to bind photo-probe, and this failure is not rescued by Kes1p truncation. (E) PIP binding is essential for Kes1p function in vivo. Centromeric plasmids (YCp) that drive physiological levels of KES1 or kes1 3E expression were transformed into the kes1 strain CTY159. Transformants were streaked for isolation onto selective minimal media and incubated for 3 d at 26°C or 37°C. Complementation of kes1 defects is scored by failure of transformants to grow at 37°C.
Suppression of PIP binding defects by COOH-terminal truncation of Kes1p
During the course of the photolabeling experiments, we noted that Kes1pW317A purifies as two species. Both species bind polyclonal anti-Kes1p immunoglobulin, suggesting that the smaller species represents a Kes1p fragment. Surprisingly, this truncated Kes1p (Kes1pW317A*) binds PI(4,5)P2, whereas full-length Kes1pW317A does not (Fig. 4 A). Thus, removal of COOH-terminal Kes1p sequences rescues PIP binding by Kes1pW317A. Although Kes1pW317A was most prominent in exhibiting this degradation product, preparations of wild-type Kes1p also contain detectable amounts of such a degradation product. This fragment also retains PI(4,5)P2 binding capability (unpublished data). Eleven cycles of Edman degradation indicate a sequence of MRGSH6M for the Kes1pW317A* NH2 terminus, and this sequence corresponds to the NH2-terminal sequence of the His6-tagged Kes1p produced in Escherichia coli (see Materials and methods). Consequently, the proteolytic event that generates Kes1pW317A* must remove a COOH-terminal Kes1p domain.
The site of Kes1p proteolysis that generates Kes1pW317A* was determined. Matrix-assisted laser ionization coupled with time-of-flight mass analysis (MALDI-TOF) analyses yield a mass of 50.663 kD for Kes1p and 36.838 kD for Kes1pW317A* (Fig. 4 B). These data assigned the Kes1pW317A* cleavage site to the amide bond of a dibasic motif comprised of Kes1p residues R314 and K315, indicating that Kes1pW317A* is devoid of the COOH-terminal 120 residues (Fig. 4 B). Interestingly, this cleavage site lies upstream of the W317A substitution. Thus, Kes1pW317A* represents a proteolytic fragment consisting solely of wild-type Kes1p primary sequence.
To confirm this result, we purified GST-tagged versions of full-length Kes1p and of a protein fragment generated by engineering a Kes1p truncation immediately following residue R314. This truncation product is designated Kes1p(1–314). Consistent with the Kes1pW317A* data, Kes1p(1–314) binds [3H]BZDC-IP3 with an affinity that is similar to that exhibited by Kes1p itself (Fig. 4 C). We also tested whether the COOH-terminal truncation restores PIP binding to the mutant Kes1pE312K. This experiment was of interest for two reasons. First, it tests the allele specificity of the suppression effect elicited by the COOH-terminal truncation mutation. Second, because E312K is retained in the truncated product (unlike the W317A case where the fragment consists of wild-type Kes1p sequence), this experiment tests whether E312K imposes an intrinsic PIP binding defect on Kes1p. Photolabeling data demonstrated that Kes1pE312K* binds PIPs, whereas full-length Kes1pE312K cannot (Fig. 4 C), indicating deletion of the COOH-terminal 120 Kes1p residues rescues both Kes1pE312K and Kes1pW317A PIP binding defects. Thus, neither E312 nor W317 are intrinsically essential for PIP binding. Rather, COOH-terminal Kes1p sequences inhibit PIP binding by the Kes1p PH domain.
Kes1p mutants intrinsically defective in PIP binding are nonfunctional
The data indicate that Kes1pE312K and Kes1pW317A exhibit PIP binding defects of a regulatory nature. This complicates conclusions that can be drawn regarding the significance of PIP binding for Kes1p activity from functional analyses of Kes1pE312K and Kes1pW317A alone. To more directly assess the functional significance of PIP binding by Kes1p, we generated Kes1p derivatives with intrinsic defects in PIP binding. Our criteria for such mutants was that these will fail to bind inositide photo-probe, and that these binding failures will not be rescued by COOH-terminal Kes1p truncation.
To generate mutant Kes1p intrinsically defective in PIP binding, we were guided by structural analyses indicating that basic residues in the PH domain variable loop 1 and 2 regions engage inositide headgroups (Lemmon, 1999). We generated a triple mutant Kes1p (Kes1p3E) where residues R236, K242, and K243 in the presumed loop 2 region of the Kes1p PH domain were replaced with glutamate (see Fig. 2 B). Kes1p3E fails to bind photo-probe in vitro, and truncation of Kes1p3E distal to residue K314 fails to restore PIP binding (Fig. 4 D). Thus, Kes1p3E is intrinsically defective in PIP binding.
The biochemical PIP binding defect translates to loss of Kes1p activity in yeast. Complementation analyses demonstrate that Kes1p3E is nonfunctional in vivo (Fig. 4 D). This defect is manifested even though Kes1p3E is a stable cellular protein that accumulates to steady-state levels that are several-fold greater than those of wild-type Kes1p. Thus, PIP binding is required for Kes1p function in vivo.
Kes1p OSBP domain is nonessential for PIP binding
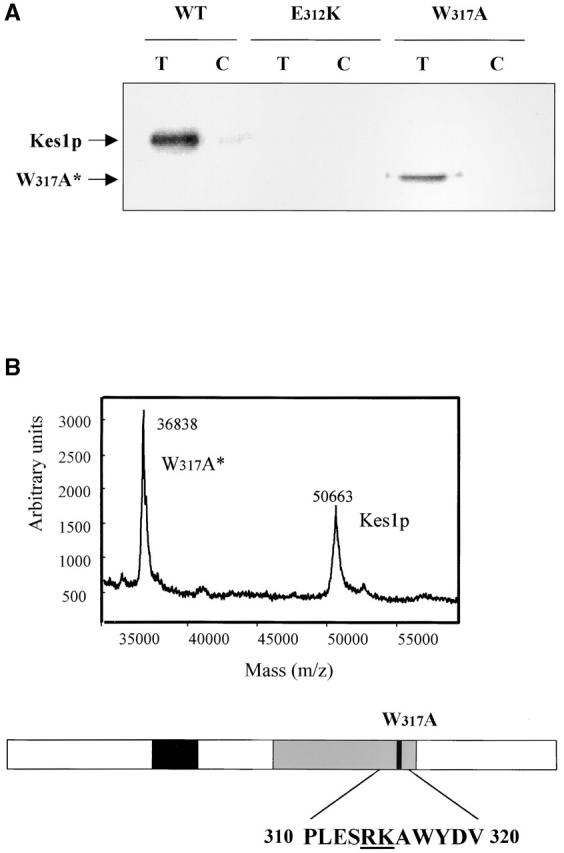
To investigate the OSBP motif in detail, we performed alanine-scanning mutagenesis throughout the OSBP domain (Fig. 5 A). Complementation experiments show that Kes1pHH143/144AA, Kes1pK109A, and Kes1pE117A are nonfunctional in vivo (Fig. 5 B), as are Kes1pLG115/116AA and Kes1pEQ139/140AA. All of these proteins are expressed as stable polypeptides in cells (unpublished data). Thus, the OSBP domain is critical for Kes1p function in yeast. Photolabeling experiments demonstrate that OSBP domain mutants avidly bind photo-probe in a manner that is subject to competitive displacement by unlabeled PI(4,5)P2 competitor (Fig. 5 A). These data show that the OSBP domain is not required for PI(4,5)P2 binding as scored by the photolabeling assay.
Figure 5.
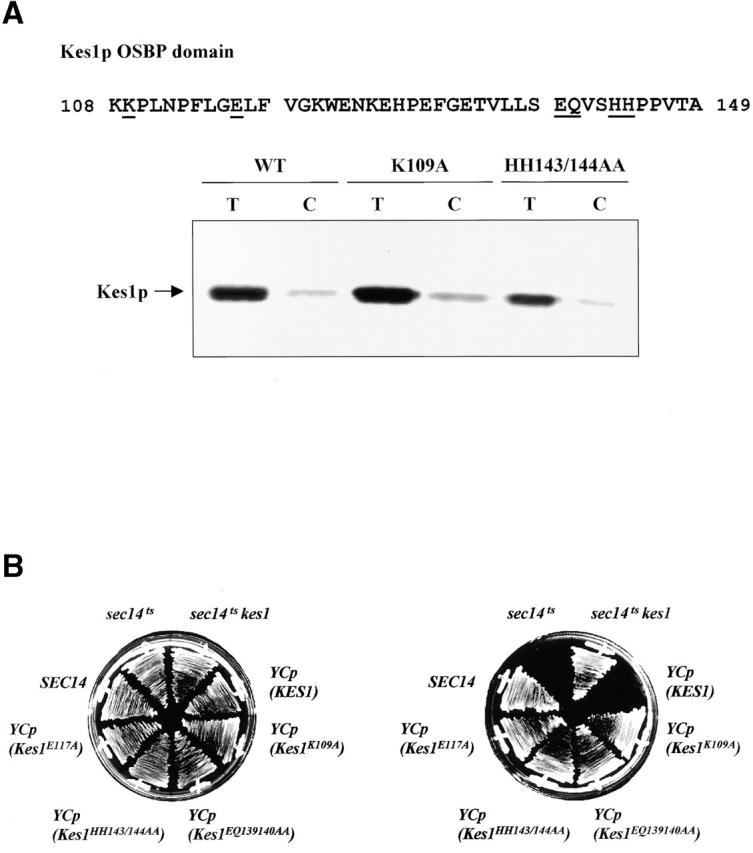

Analysis of Kes1p OSBP domain mutants. (A) OSBP domain is not required for PIP binding. The four missense substitutions, as underlined, were engineered into the Kes1p OSBP domain. Two representative proteins, Kes1pK109A and Kes1pHH143/144AA, were purified and employed in photolabeling assays using [3H]BZDC-IP3 as photo-probe. These mutant proteins, like Kes1p, retain PIP binding capability. (B) OSBP domain mutants are nonfunctional. Four low copy mutant plasmids were transformed into strain CTY159. Transformants were restreaked onto appropriate media and grow for 2–3 d at 26°C or 37°C. Complementation was assessed as before. (C) PIP binding by Kes1pS43Y, Kes1pE107A, and Kes1pLH201/202FN. The proteins were purified and photolabeled with [3H]BZDC-IP3. These mutant proteins retain PIP binding capability.
Other mutant forms of Kes1p (S43Y, E107K, LH201/202FNo) harbor unadulterated PH and OSBP domains, yet are nonfunctional in vivo (Fig. 2 A). We find that Kes1pS43Y, Kes1pE107K, and Kes1pLH201/202FN are all proficient in [3H]BZDC-PI(4,5)P2 binding. Moreover, bound photo-probe is efficiently displaced by the incorporation of excess unlabeled PI(4,5)P2 into the photolabeling assay (Fig. 5 C). Competitive displacement assays using purified Kes1pLH201/202FN revealed an IC50 of photo-probe displacement of 6.1 μM when PI(4,5)P2 was employed as competitor (unpublished data), a value that resembles the IC50 of 1.9 μM recorded for wild-type Kes1p (Fig. 3 B). Thus, these mutant Kes1ps bind PI(4,5)P2 with affinities similar to that measured for Kes1p (K D = 8.0 μM vs. 2.5 μM, respectively). We conclude that PIP binding is necessary, but insufficient, for Kes1p function in vivo.
Kes1p localizes to Golgi membranes
Fractionation studies revealed that Kes1p resides in both soluble and membrane-bound pools (Fang et al., 1996). To localize Kes1p more precisely, a YCp(KES1-YFP) plasmid was constructed that drives physiological expression levels of a chimera consisting of Kes1p fused via its COOH terminus to the green fluorescent protein (GFP) NH2 terminus. This chimera is both stable and functional (unpublished data). To assess Kes1p-GFP distribution, appropriate yeast strains were cultured to midlogarithmic growth phase in minimal medium at 26°C and cells were imaged. As control, we also monitored localization of GFP expressed from an isogenic YCp plasmid carrying a KES1 promoter cassette. Whereas GFP is distributed diffusely throughout the cytosol, Kes1p-GFP adopts both a diffuse cytosolic location and localization to 3–10 punctate structures dispersed throughout the cytoplasm (Fig. 6).
Figure 6.

Kes1p localization. YCp (KES1-YFP) and YEp (KEX2-RFP) plasmids were cotransformed into the kes1 yeast strain CTY159. Liquid cultures of the plasmid-bearing derivative strain were incubated overnight at 26°C with shaking in the appropriate minimal medium. The cultures were then diluted to OD600 = 0.2∼0.3, and incubated for an additional 2 to 3 h at 26°C. Cells were gently pelleted and resuspended in ∼50 μl residual medium, 5–8 μl of the suspension was mounted onto glass slides, and the samples were visualized by fluorescence microscopy. The excitatory filters employed for visualization of GFP/YFP and RFP fluorescence were the quinacrine mustard filter (excitation wavelength 495 nm) and rhodamine filter (excitation wavelength 545 nm), respectively. Images were collected by IP Lab 2.0 software and processed with Adobe Photoshop® 5.0. Arrows identify some examples of colocalization.
The punctate component of Kes1p-GFP staining primarily corresponds to a Golgi membrane–associated Kes1-GFP pool. We observed that ∼85% of the Kes1-GFP–positive structures costain with the yeast Golgi complex marker Kex2p-RFP (Fig. 6). The remaining 15% of the Kes1-GFP–positive structures costain with the fluorescent dye FM4-64 under conditions that trap this lipophilic molecule in early endosomes.
Localization of Kes1p to Golgi membranes requires PIP binding
Previous reports had documented that PIP binding is necessary and sufficient for localization of long OSBPs to target membranes (Levine and Munro, 1998). To determine whether PIP binding plays the same role in targeting of short OSBPs, representative mutations in the Kes1p PH domain were introduced into a YCp(KES1-GFP) expression cassette. Localization of mutant Kes1-GFP proteins was then monitored in living cells by fluorescence microscopy.
As expected, the Kes1p3E-GFP chimera exhibit a diffuse intracellular distribution that contrasts with the punctate Golgi region staining recorded for wild-type Kes1p-GFP (Fig. 7 A). Thus, Kes1p3E-GFP fails to localize to Golgi membranes in vivo. We also assessed localization of Kes1pE312K and Kes1pW317A. These species fail to bind PIPs in vitro as full-length polypeptides, but regain PIP binding activity when the COOH-terminal 120 Kes1p residues are removed. Interestingly, Kes1pE312K-GFP and Kes1pW317A-GFP exhibit localization profiles similar to those recorded for wild-type Kes1p-GFP, suggesting Kes1pE312K and Kes1pW317A target to Golgi membranes (Fig. 7 A). Thus, unlike the PIP-binding defective Kes1p3E, Kes1pE312K and Kes1pW317A are apparently subject to regulatory compensation that exposes their PIP binding activities in the appropriate in vivo context. Kes1pE312K and Kes1pW317A remain nonfunctional, however.
Figure 7.
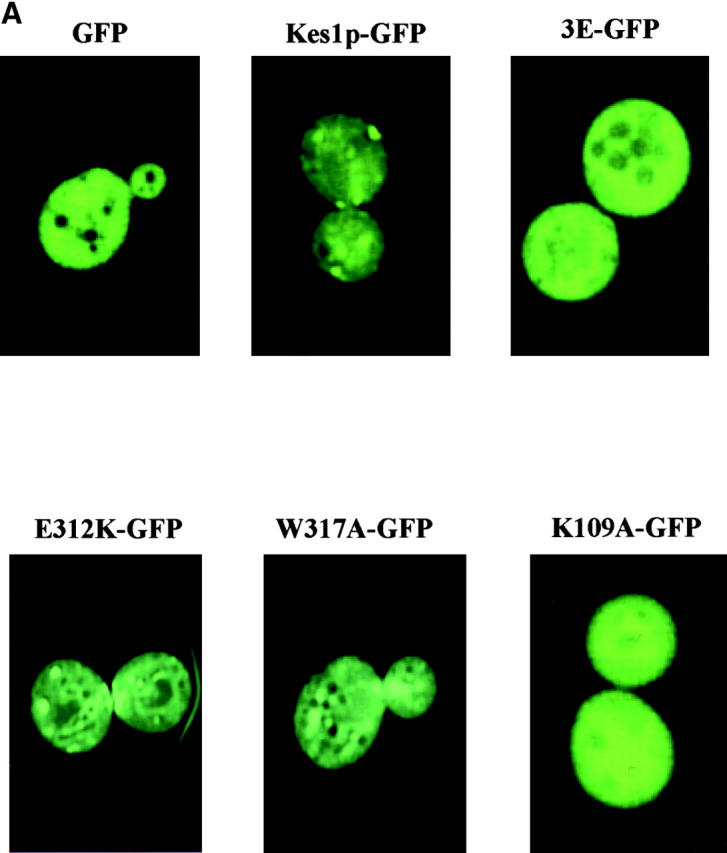
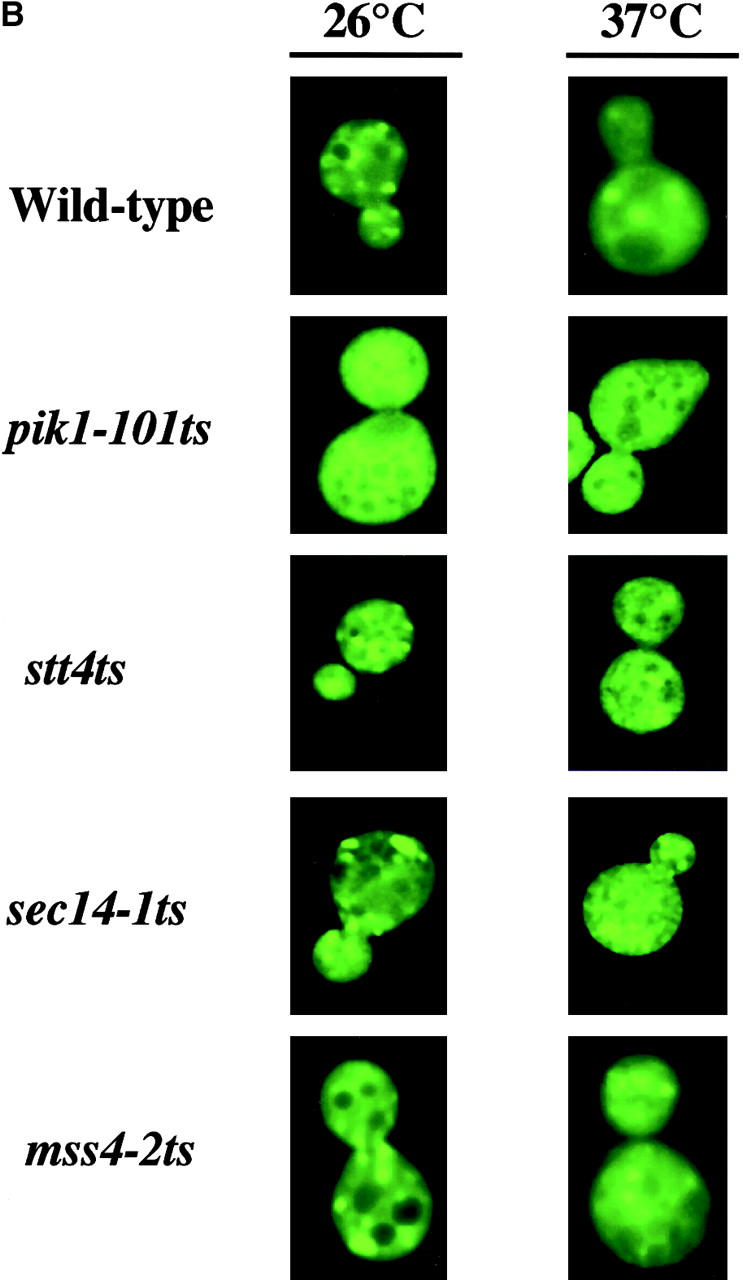
Kes1p targeting to yeast Golgi membranes. (A) Kes1p localization domains. YCp(GFP), YCp(KES1-GFP) and mutant YCp(kes1 3E-GFP), YCp(kes1 E312K-GFP), YCp(kes1 W317A-GFP), and YCp(kes1 K109A-GFP) plasmids were introduced into a kes1 yeast strain. Fluorescence images were captured at 26°C. The kes1p3E-GFP and chimeras exhibit a diffuse cytosolic distribution that mimics that of GFP alone. In these three cases, signal is excluded from vacuoles (the dark circular structures). The wild-type (WT) control Kes1p-GFP and the mutant Kes1pE312K-GFP and Kes1pW317A-GFP chimeras exhibit typical punctate staining patterns. (B) PI(4)P synthesis and Kes1p localization. A YCp(KES1::GFP) plasmid was introduced into wild-type, pik1-101 ts, stt4 ts, sec14-1 ts, and mss4-2 ts strains as indicated at right. Fluorescence images were captured at 26°C and after shift to 37°C for 2 h (indicated at top) at an excitation wavelength of 495nm. Pik1p defects exert major defects in Kes1p localization to Golgi membranes, whereas inactivation of Stt4p and Sec14p has more modest effects.
Defects in PI-4-P synthesis compromise Kes1p localization to Golgi membranes
To test whether defects in PIP synthesis release of Kes1p from Golgi membranes, we expressed Kes1p-GFP in strains carrying ts alleles of each of the two known yeast phosphatidylinositol (PI) 4-kinases (Pik1p and Stt4p). As described above, Kes1p-GFP adopts a characteristic punctate localization pattern in wild-type cells incubated at 26°C (Fig. 7 B). By contrast, Kes1p-GFP redistributes from Golgi membranes to the cytosol in the pik1-101 ts mutant incubated at either 26°C or 37°C (Fig. 7 B). Thus, Pik1p, which localizes to Golgi membranes (Walch-Solimena and Novick, 1999), catalyzes synthesis of a PIP pool, PI(4)P and/or PI(4,5)P2, that is required for localization of Kes1p to yeast Golgi membranes. Kes1p mislocalization in the pik1-101 ts strain at 26°C indicates that this pik1 ts allele is quite defective even at permissive temperatures, a result in accord with the potent synthetic lethalities exhibited by pik1-101 ts when combined with a number of other mutations that compromise secretory pathway function (Walch-Solimena and Novick, 1999). By contrast, inactivation of Stt4p at 37°C results in some mislocalization of Kes1p to cytosol at 26°C (Fig. 7 B). Relative to pik1-101 ts, these effects are modest. Thus, Stt4p-derived PIPs play more minor roles in Kes1p targeting to Golgi membranes than do Pik1p-derived PIPs. To distinguish whether PI(4)P or PI(4,5)P2 is a Golgi region ligand for Kes1p, we assessed Kes1p-GFP localization in mutants inactivated for Mss4p, the yeast PI(4)P 5-kinase. Even long shift of mss4-2 ts mutants to restrictive temperatures (37°C) fails to compromise Kes1p-GFP association with Golgi membranes (Fig. 7 B).
We also tested whether inactivation of the Golgi complex–associated Sec14p affects Kes1p localization. Sec14p stimulates PIP synthesis in a variety of in vitro systems (Hay and Martin, 1993; Cunningham et al., 1996; Jones et al., 1998). Moreover, Sec14p stimulates PI(4)P synthesis in yeast (Hama et al., 1999; Phillips et al., 1999). Kes1p localization is not compromised in sec14-1 ts strains at 26°C. Shift of the sec14-1 ts strain to 37°C for 30 min reduces, but does not abolish, Kes1p association with Golgi membranes (Fig. 7 B).
Localization of Kes1p to Golgi membranes requires a functional OSBP domain
Analyses of mutants compromised for OSBP domain function demonstrate that an intact OSBP domain is also essential for proper localization of Kes1p-GFP in living cells. Imaging experiments show that the Kes1pK109A-GFP chimera adopts a diffuse cytosolic distribution with no visible concentration in punctate structures (Fig. 7 C). Kes1p-GFP chimeras with mutant OSBP domains (i.e., Kes1pHH143/144AA-GFP, Kes1pLG115/116AA-GFP, and Kes1pVS141/142AA-GFP) also exhibit exclusively cytosolic staining profiles (unpublished data). Of interest is the allele specificity that underlies these localization defects. E107K represents a missense substitution at a position only two residues upstream of K109A. Yet, Kes1pE107K does not mistarget to the cytosol (unpublished data), whereas Kes1pK109A does. Finally, mutations that release Kes1p-GFP to the cytosol are not limited to the OSBP domain. For example, LH201/202FN also has this effect (unpublished data). We conclude the region bounded by Kes1p residues 109–202 is critical for the Kes1p localization to membranes, and that the OSBP domain is an essential component of this membrane targeting information.
Because Kes1ps with defective OSBP domains bind PIPs, we tested whether this domain contributes to Golgi region targeting by binding another lipid. To this end, we determined whether the phospholipase D (PLD)-driven accumulation of PA that accompanies Sec14p inactivation is required for Kes1p localization to the yeast Golgi region. This effort was motivated by the weak binding of Kes1p to PA monomers (see above). PLD deficiency markedly reduces Golgi region/endosomal PA levels in Sec14p-deficient yeast (Li et al., 2000a), but has no effect on Kes1p localization in Sec14p-deficient strains (unpublished data). In accord with our previous findings that the involvement of Kes1p in yeast Golgi region function is independent of metabolic flux through the yeast sterol biosynthetic pathway (Fang et al., 1996), we also find that mutations compromising sterol biosynthesis (e.g., erg6) do not result in obvious mislocalization of Kes1p from Golgi membranes (unpublished data).
Relationship between magnitude of Kes1p overexpression required for compromise of “bypass Sec14p” and PI(4)P levels
Since genetic data indicate Kes1p antagonizes activity of the Sec14p pathway for Golgi region secretory function, we overexpressed Kes1p in “bypass Sec14p” mutant strains. In agreement with previous demonstrations (Fang et al., 1996), all mechanisms for “bypass Sec14p” are sensitive to increased KES1 gene dosage (Fig. 8 A). Kes1p is unique in this respect. Increased dosage of structural genes for enzymes of the CDP-choline pathway (i.e., CKI1, PCT1, and CPT1) or the Sac1p PIP phosphatase (SAC1) have no effect on nonallelic mechanisms for “bypass Sec14p” (unpublished data). Although modest overproduction of Kes1p compromises all other mechanisms of “bypass Sec14p”, robust overexpression of Kes1p is required for this effect in sac1 mutants (Fig. 8 A).
Figure 8.



Kes1p dosage and “bypass Sec14p”. (A) Distinct sensitivities of different mechanisms for “bypass Sec14p” to increased Kes1p dosages. Kes1p was modestly overexpressed by introduction of YCp (KES1), or highly overexpressed by introduction of YEp(PPGK:: KES1), which drives Kes1p expression from a powerful PGK promoter (see Materials and methods). All mechanisms of “bypass Sec14p” except that mediated by sac1 are sensitive to modest increases (two- to threefold) in Kes1p levels. A high degree (∼20-fold) of Kes1p is required for compromise of “bypass Sec14p” in sac1 mutants. (B) Structure of Sac1pΔTM. The first 462 codons of Sac1p, which include the catalytic domain but not the transmembrane domain, were fused to a protein A tag. This construct is designated Sac1pΔTM and its expression is under control of the native SAC1 promoter. Details of the construction are available from the authors by request. (C) PI(4)P levels as a function of Sac1pΔTM expression. A wild-type SAC1 strain, a sac1 mutant strain, and isogenic sac1 derivatives transformed either with YCp(sac1 Δ TM) or YEp(sac1 Δ TM) were pulse radiolabeled for 20 min at 26°C with [32P]-orthophosphate (10 μCi/ml), phospholipids were extracted under acidic conditions, lipids were resolved by two-dimensional paper chromatography, and PI(4)P was identified and quantified by phosphorimaging (Rivas et al., 1999). PI(4)P is expressed as a percentage of total extractable phospholipid. Relevant strain genotypes are indicated. The various strains incorporated similar amounts of [32P]-radiolabel into total lipid. Data are expressed as the mean ± standard deviation from at least three independent experiments. (D) Sac1pΔTM expression sensitizes sac1-mediated “bypass Sec14p” to Kes1p. Isogenic sac1 and sac1/YEp(sac1 Δ TM) yeast strains were transformed with YCp(KES1). Transformants were subsequently isolated and streaked for isolated colonies on selective medium at 37°C. Growth was scored after 48 h of incubation. Relevant genotypes are given.
Because sac1 mutants are unique among “bypass Sec14p” mutants in their massive overproduction of PI(4)P (Guo et al., 1999; Rivas et al., 1999; Stock et al., 1999), there is a correlation between PI(4)P levels and the magnitude of Kes1p overproduction required to abolish “bypass Sec14p”. To investigate whether the Kes1p levels required to abolish “bypass Sec14p” in sac1 mutants are proportional to PI(4)P levels, we expressed in sac1 mutants a truncated Sac1p. This truncated Sac1pΔTM contains the PIP phosphatase catalytic domain but lacks the COOH-terminal transmembrane domain (Fig. 8 B). Sac1pΔTM is a partially functional protein as it partially alleviates the derangement of PIP metabolism that characterizes sac1 mutants (Fig. 8 C). As shown previously, PI(4)P constitutes 17.8 ± 0.8% and 3.6 ± 0.6% of extractable phospholipid in sac1 mutants and wild-type yeast, respectively (Guo et al., 1999; Rivas et al., 1999; Stock et al., 1999). By comparison, PI(4)P levels are markedly reduced to 8.1 ± 1.2% in the sac1 mutant that expresses Sac1pΔTM from YEp(sac1 Δ TM) (Fig. 8 C). Expression of Sac1pΔTM from a YCp(sac1 Δ TM) vector effects only minor reductions in PI(4)P levels in a sac1 genetic background (15.3 ± 0.5%).
The reduction in PI(4)P recorded in the YEp(sac1ΔTM) strain corrects the inositol auxotrophy and cs growth phenotypes of sac1 mutants, but fails to ablate sac1-mediated “bypass Sec14p” (unpublished data). However, Sac1pΔTM-mediated reduction in PI(4)P levels strongly sensitizes the “bypass Sec14p” phenotype of sac1 mutants to elevated Kes1p. Whereas large increases in KES1 dosage are required for compromise of “bypass Sec14p” in sac1 mutants, modest increases in KES1 dosage ablate “bypass Sec14p” in sac1 YEp(sac1ΔTM) strains (Fig. 8 D). Thus, the magnitude of Kes1p overproduction required for compromise of “bypass Sec14p” is proportional to cellular PI(4)P levels.
Kes1p is mislocalized in sac1 mutants
Based on analyses of PI(4)P levels in sac1 mutants bearing pik1 ts and stt4 ts mutations, it is clear that the accumulated PI(4)P is predominantly synthesized via the Stt4p PI 4-kinase and not the Pik1p kinase (Nemoto et al., 2000; Foti et al., 2001). Because Pik1p is the Golgi region–localized PI 4-kinase (Walch-Solimena and Novick, 1999) and PI(4)P is likely the physiological ligand for Kes1p, we assessed the localization of Kes1p-GFP in sac1 mutants. As shown in Fig. 9 A, the intracellular profile of Kes1p-GFP distribution in sac1 mutants is dramatically different from its normal punctate distribution in wild-type strains. Rather, Kes1p-GFP localizes to a few large patches in sac1 cells that are frequently located in a juxtanuclear position. This altered Kes1p localization pattern does not reflect abnormal sac1 Golgi region morphology. Distribution of the Golgi complex marker Kex2p is not altered in sac1 mutants (Fig. 9 B). Rather, consistent with the significant (if not exclusive) localization of a Sac1p to ER compartments in yeast and mammalian cells (Whitters et al., 1993; Nemoto et al., 2000; Foti et al., 2001), we conclude that Kes1p is mistargeted to what are likely ER membranes in sac1 mutants. We suggest that Kes1p is mistargeted to ER membrane domains enriched in the PI(4)P that accumulates in these mutants. In support of this idea, the Kes1p PH domain is required for this mislocalization as Kes1p3E-GFP retains its cytosolic localization in sac1 mutants, and fusion of the Sac1p PIP phosphatase catalytic domain to the ER-resident protein Sec61p yields a fully functional Sac1p whose expression complements all sac1Δ phenotypes (including the “bypass Sec14p” phenotype [unpublished data]).
Figure 9.

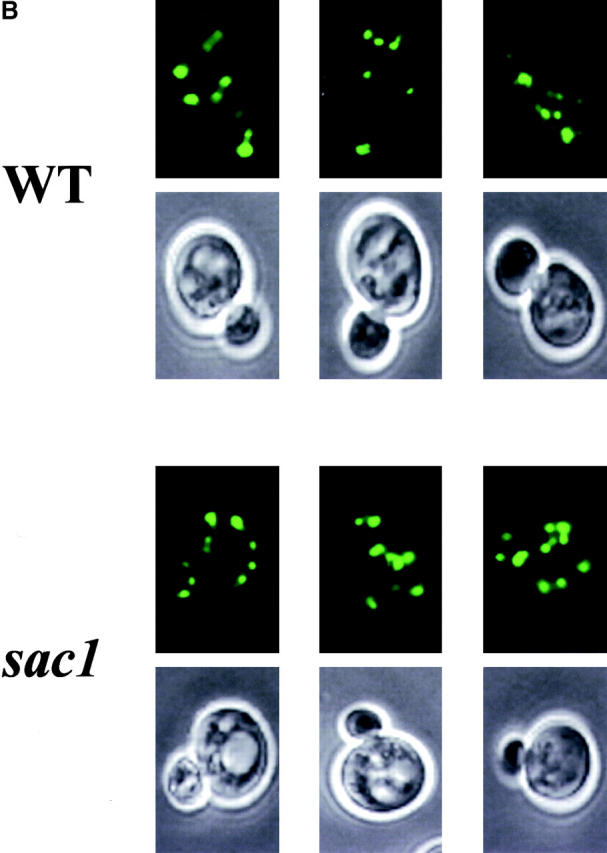
Kes1p mislocalization in sac1 mutants. YCp(KES1-GFP) and YEp(KEX2-RFP) plasmids were transformed into wild-type or isogenic sac1 yeast. Liquid cultures of the plasmid-bearing derivatives were incubated at 26°C in the appropriate minimal medium. Cells were mounted and visualized by fluorescence microscopy. Kes1p-GFP and Kex2p-RFP profiles are shown in A and B, respectively. Relevant genotypes are indicated at left. Because Kex2p is an integral membrane protein, Nomarski images are shown below the corresponding fluorescence images in B.
Genetic interactions of kes1Δ with arf1Δ, gcs1Δ, and pik1-101 ts
To further understand how Kes1p functions in yeast, we screened for instructive genetic interactions. The phenotypes associated with Kes1p overproduction in many ways mimic those associated with reduced PLD activity (Xie et al., 1998), suggesting that Kes1p is an inhibitor of PLD in yeast. However, our in vivo experiments indicate PLD activity is not sensitive to high levels of Kes1p (unpublished data). Rather, we observed a linkage between Kes1p function and the yeast ARF cycle. Both arf1Δ and gcs1Δ alleles abrogate kes1Δ-mediated “bypass Sec14p”. Although an sec14-1 ts mutant cannot grow at 37°C, the sec14-1 ts kes1Δ derivative grows at wild-type rates. However, the isogenic sec14-1 ts kes1Δ arf1Δ and sec14-1 ts kes1Δ gcs1Δ triple mutants are no longer viable at 37°C (Fig. 10 A). This is not the result of more complex genetic interactions between kes1Δ and arf1Δ or gcs1Δ as introduction of SEC14 into sec14-1 ts kes1Δ arf1Δ and sec14-1 ts kes1Δ gcs1Δ mutants restores viability at 37°C (unpublished data). Gcs1p is an ARFGAP, and it is this biochemical activity that is required for kes1-mediated “bypass Sec14p” (Yanagisawa, L., and V.A. Bankaitus, unpublished data).
Figure 10.


Genetic interactions between kes1 Δ , arf1 Δ , gcs1 Δ , and pik1–101ts alleles. (A) “Bypass Sec14p” in arf1Δ strains. Isogenic sets of wild-type (WT), sec14-1 ts, sec14-1 ts kes1Δ, sec14-1 ts kes1Δ arf1Δ, and sec14-1 ts kes1Δ gcs1Δ strains were incubated on YPD agar at 26°C or 37°C (a restrictive temperature for sec14-1 ts strains) as indicated for 72 h. WT and sec14-1 ts strains represent positive and negative growth controls, respectively. The kes1Δ allele rescues sec14-1 ts lethality at 37°C, and this rescue is abolished by arf1Δ and gcs1Δ. The arf1Δ and gcs1Δ alleles do not compromise growth of otherwise WT strains at 37°C. (B) Kes1p deficiency partially suppresses growth defects of the pik1 ts strain. Each strain was serially diluted from original culture (OD600 = 0.6) by 10-, 100-, and 1,000-fold. 3 μl of each dilution was inoculated onto YPD plates, and plates were incubated for 3 d at 26°C and 35°C, respectively.
Second, we recorded a genetic interaction between kes1Δ and a mutation that compromises activity of the Pik1p PI 4-kinase. Yeast strains carrying pik1-101 ts grow at 26°C, but not at 35°C or 37°C (Walch-Solimena and Novick, 1999; Fig. 10 B). A kes1Δ allele clearly improves the growth of pik1-101 ts strains at 35°C. In a serial dilution array incubated at 35°C, the pik1-101 ts mutant forms colonies only in the undiluted culture inoculum. By contrast, the wild-type control exhibits growth in undiluted and low dilution innocula; i.e., single colonies are scored in regions spotted with aliquots of 1,000-fold dilutions of culture sample. Thus, relative to wild-type, there is an ∼1,000-fold reduction in the viability of pik1-101 ts mutants cultured at 35°C. The kes1Δ pik1-101 ts mutant exhibits a growth profile intermediate between those of wild-type and pik1-101 ts strains (Fig. 10 B). Growth was recorded in the inoculum of a 10-fold culture dilution. Thus, kes1Δ increases the viability of pik1-101 ts mutants some 10-fold at 35°C. This effect is not observed at 37°C, indicating kes1Δ exerts only partial suppression of pik1-101 ts defects (unpublished data). Nonetheless, this effect is specific by two criteria. First, kes1Δ allele has no beneficial effect on the growth of yeast strains defective in Stt4p (unpublished data). Second, no other “bypass Sec14p” alleles (including sac1 alleles) suppress pik1-101 ts–associated growth or secretory defects at 35°C (unpublished data).
Genetic interactions of kes1Δ with ARF and ARFGAP deficiencies
Kes1p deficiency elicits a “bypass Sec14p” that, like all “bypass Sec14p” phenotypes, requires Gcs1p ARFGAP activity (Yanagisawa, L., and V.A. Bankaitus, unpublished data). Our finding that Kes1p overproduction mimics Gcs1p defects in “bypass Sec14p” mutants further supports the possibility that Kes1p influences regulation of the ARF cycle. Therefore, we tested whether kes1Δ suppresses phenotypes associated with Gcs1p or ARF dysfunction.
Sodium fluoride (NaF) sensitivity is associated with defects in the yeast ARF cycle (Zhang et al., 1998). Gcs1p-deficient mutants are sensitive to NaF, as are arf2Δ strains carrying the arf1-3 ts mutation (Zhang et al., 1998; Table I). Based on the results described above, we predicted that Kes1p defects might suppress NaF sensitivity in gcs1Δ and arf1-3 arf2Δ strains. Although both gcs1Δ and arf1-3 arf2Δ mutants fail to grow in the presence of 30 and 50 mM NaF, isogenic wild-type strains are NaF resistant (Table I). However, kes1Δ suppresses Gcs1p and ARF1p deficiency in this assay as isogenic kes1Δ gcs1Δ and kes1Δ arf1-3 arf2Δ mutants grow when challenged with either 30 or 50 mM NaF (Table I). Serial dilution experiments indicate that the viability of gcs1Δ strains is increased ∼1,000-fold relative by kes1Δ.
Table I. Interactions of kes1 mutations with perturbations in Arf function.
| Genotype | NaF (0 mM) | NaF (30 mM) | NaF (50 mM) |
|---|---|---|---|
| WT | +++ | +++ | +++ |
| sec14-1ts | +++ | +++ | +++ |
| arf1-3 arf2Δ | ++ | − | − |
| arf1-3 arf2Δ kes1Δ | +++ | − | − |
| sec14-1ts gcs1Δ | ++ | − | − |
| sec14-1ts gcs1Δ kes1Δ | +++ | +++ | +++ |
| pik1-101ts | +++ | − | − |
| pik1-101ts kes1Δ | +++ | +++ | +++ |
Genetic interactions between Kes1p and ARF cycle components. KES1 was deleted in gcs1D, arf1-3ts arf2D, and in pik1ts mutants, and strains were grown on YPD agar with indicated NaF concentrations. Wild-type colony size is indicated by +++, smaller colonies by ++, and − indicates no growth at all after 3 d at 26°C. In arf1, gcs1, and pik1 mutants challenged with 50 mM NaF, kes1D affects 103-fold increases in cell viability.
This is not a trivial result of Kes1p defects reducing either the permeability of cells to NaF or the capacity of yeast to accumulate NaF as kes1Δ does not increase the threshold of yeast tolerance to NaF. Moreover, kes1Δ only modestly influences other phenotypes associated with Gcs1p defects (e.g., sensitivity to hyperosmotic stress). The viability of gcs1Δ mutants is reduced ∼10-fold relative to that of isogenic gcs1Δ kes1Δ mutants (unpublished data). Zhang et al. (1998) reported that overproduction of any one of several ARFGAPs, including Gcs1p, suppresses NaF sensitivity in arf1-3 arf2Δ yeast strains. Thus, kes1Δ phenocopies the elevation of ARFGAP levels in arf1-3 arf2Δ yeast mutants. Finally, we note that the pik1-101 ts mutant resembles gcs1Δ and arf1-3 ts strains in its NaF sensitive growth, and that NaF sensitivity is relieved by kes1Δ (Table I).
Discussion
Herein, we show Kes1p binds PIPs and this PIP-binding property, in conjunction with its conserved OSBP domain, is essential for Kes1p localization to Golgi membranes. The PIP pool required for Kes1p targeting to Golgi membranes is driven by activity of the Pik1p PI 4-kinase, and Golgi region targeting of Kes1p is required for inhibition of Sec14p-dependent Golgi region function in vivo. We further demonstrate that inactivation of Kes1p not only affects “bypass Sec14p”, but that it also elicits a suppression of some phenotypes associated with ARF and ARFGAP dysfunction. We propose that it is through its effects on ARF that Kes1p regulates Sec14p-dependent Golgi membrane secretory function in yeast.
The PIP binding activity resides in what appears to be a Kes1p PH domain. Although the existence of PH domains has been documented for long OSBPs (e.g., OXYB yeast Osh1; Levine and Munro, 1998), our findings represent the first demonstration that short OSBPs also binds PIPs. Moreover, our data demonstrate that PIP binding, and its role in targeting an OSBP to Golgi membranes, is a functionally important activity in cells. With regard to the binding of Kes1p to other lipids, we failed to detect significant binding of Kes1p to 25-hydroxycholesterol (unpublished data).
One unanticipated result obtained from analysis of mutations in the Kes1p PH domain is our finding that the E312K and W317A mutations abolish PIP binding in the context of full-length Kes1p, but not when the COOH-terminal 120 Kes1p residues are removed. Thus, E312K and W317A do not affect PIP binding directly. Rather, these substitutions likely affect Kes1p conformation in such a way that the PH domain is not accessible to PIPs. Consistent with this model, E312 and W317 do not reside in the predicted PIP binding loops of the Kes1p PH domain, and truncation of the Kes1p COOH terminus does not rescue PIP binding defects associated with mutations in those loops. Because removal of the Kes1p COOH terminus suppresses E312K- and W317A-associated PIP binding defects, we speculate that the Kes1p COOH terminus interferes with PH domain function by occluding the PIP binding site. This proposal suggests that PIP binding by Kes1p is subject to conformational regulation by posttranslational modification, or by interaction of Kes1p with some other binding partner (either protein or lipid). This concept is supported by our demonstration that the E312K and W317A substitutions do not abrogate Kes1p targeting to the Golgi complex in vivo, whereas intrinsic Kes1p defects in PIP binding and defects in PIP synthesis by the Pik1p PI-kinase do.
Unlike the case for long OSBPs, PIP binding is insufficient for Kes1p targeting to Golgi membranes. An intact OSBP domain is also required. These data provide the first indication of a function for this signature motif of OSBPs. It remains unclear how the OSBP domain is involved in localization of Kes1p to Golgi membranes. One possibility is that the Kes1p OSBP motif interacts with the Kes1p COOH terminus to expose the PH domain for PIP binding. We do not find compelling evidence in favor of this model. Inactivating mutations in the OSBP domain neither abolish PIP binding by Kes1p, nor do these strongly compromise the affinity of Kes1p binding of PIP monomers in a mixed micelle system. Alternatively, the OSBP domain may increase the avidity of Kes1p for Golgi membranes by binding another Golgi complex–localized ligand. Finally, the OSBP domain may represent an interface for homotypic binding of Kes1p molecules to each other so that the PH domains are configured in a dimeric arrangement where the PIP binding loops of each PH domain unit are exposed on one face of the dimer. This would generate a PIP binding module with a higher affinity for PIP than that exhibited by monomeric Kes1p. However, an increased binding affinity of dimeric Kes1p would not be apparent in the mixed micelle system we have employed to measure Kes1p–PIP interaction.
Although Kes1p plays an important role in Golgi function in yeast (Fang et al., 1996), the in vivo functions of OSBPs in eukaryotic cells remain unresolved. We suggest that Kes1p function somehow interfaces with activity of the yeast ARF cycle. We find that arf1Δ and gcs1Δ both abrogate kes1Δ-mediated “bypass Sec14p”, and several other lines of genetic evidence indicate that Kes1p dysfunction mimics elevated ARFGAP function, whereas elevated Kes1p phenocopies reduced ARFGAP function. Thus, Kes1p may act as: (a) an inhibitor of the Gcs1p ARFGAP activity that is required for yeast Sec14p-dependent Golgi complex function (Yanagisawa, L., and V.A. Bankaitus, unpublished data), (b) a novel ARF nucleotide exchange factor, or (c) an activator of an ARF nucleotide exchange factor. In our hands, Kes1p fails to inhibit the ARFGAP activity of Gcs1p in vitro and we have not detected intrinsic ARF nucleotide exchange activity associated with Kes1p (unpublished data).
Our data suggest that Kes1p may regulate ARF function through its effects on PIP synthesis via the Golgi complex–associated Pik1p as evidenced by the demonstration Kes1p defects partially suppress growth defects associated with Pik1p dysfunction. Kes1p may function, directly or indirectly, as a Pik1p inhibitor in vivo. Alternatively, effects of Kes1p on the ARF cycle may influence Pik1p activity. Linkage of Kes1p localization and function to Pik1p-mediated PI(4)P synthesis suggests that PI(4)P is the physiological Kes1p ligand. Although PI(4,5)P2 is bound by Kes1p with a 10-fold higher affinity, PI(4)P mass is fourfold greater than that of PI(4,5)P2 in yeast. Moreover, defects in PI(4,5)P2 synthesis do not compromise Kes1p localization to Golgi membranes, and massive accumulation of PI(4)P in a non-Golgi compartment entices Kes1p from Golgi membranes. The latter result suggests that a component of the mechanism by which sac1-mediated PI(4)P accumulation contributes to “bypass Sec14p” is an indirect one that involves Kes1p mistargeting to what is likely the ER. Finally, the data identify Kes1p as a reporter for Pik1p-driven PIP synthesis on yeast Golgi membranes. Thus, Pik1p facilitates the paradoxical recruitment to Golgi membranes of a protein whose dysfunction permits Sec14p-independent secretory function of Golgi membranes.
Materials and methods
Strains, media, and reagents
Genotypes for yeast strains are listed in Table II. Media are described elsewhere (Sherman et al., 1983), as are E. coli and yeast plasmid transformation techniques (Ito et al., 1983). Vector pQE30 was from QIAGEN. HiTrap Ni+ chelating columns and glutathione-Sepharose 4B beads were from Amersham Pharmacia Biotech. PI(4,5)P2, PI(4)P, and IP3 were from CalBiochem. PI(3,4)P2 and PI(3,5)P2 were from Echelon Research Laboratories. Other lipids were from Avanti Polar Lipids, Inc. Fine chemicals were from Sigma-Aldrich unless specified. Restriction enzymes were purchased from Promega. [α-35S]-dATP was obtained from Amersham Pharmacia Biotech.
Table II. Yeast strains and plasmids.
| Genotypes | Source | |
|---|---|---|
| Strain number | ||
| CTY1-1A | MATa ura3-52 his3Δ-200 lys2-801 sec14-1 ts | Bankaitis et al., 1989 |
| CTY100 | MATa ura3-52 his3Δ-200 lys2-801 sec14-1 ts sac1-26 cs | Cleves et al., 1989 |
| CTY159 | MATa ura3-52 his3Δ-200 lys2-801 sec14-1 ts kes1-1 | This study |
| CTY160 | MATa ura3-52 his3Δ-200 lys2-801 sec14-1 ts cki1-1 | Cleves et al., 1989 |
| CTY182 | MATa ura3-52 his3Δ-200 lys2-801 SEC14 | Bankaitis et al., 1989 |
| CTY1537 | MATa ura3-52 leu2 pik1-101 ts | Novick et al., 1980 |
| CTY1585 | MATa ura3-52 leu2 pik1-101 ts kes1Δ::URA3 | This study |
| Plasmid number | ||
| pCTY204 | YIp (KES1) | |
| pCTY244 | YCp (KES1) | |
| pCTY574 | YCp (KES1-GFP) | |
| YEp (KEX2-RFP) | ||
| pRE540 | His 6 -KES1 | |
| pRE682 | GST-KES1 | |
| pRE657 | GST-KES1(1–314) | |
| pRE665 | GST-GST-PH domain |
Recovery of genomic kes1 alleles
An integrative KES1 plasmid (pCTY204) was restricted at a unique site within the KES1 promoter with AflII. Linearized plasmid was transformed into kes1 strains and recombinants at the KES1 locus were identified. Genomic DNA was prepared from transformants, restricted with ClaI, and ligated at 15°C. Following transformation into E. coli DH5α, plasmid DNA was purified and used as template for nucleotide sequencing.
Site-directed mutagenesis
Mutageneses employed QuickChange™ (Stratagene) and confirmed by DNA sequencing analysis (Sanger et al., 1977) using the Sequenase version 2.0 kit (Amersham Pharmacia Biotech).
Plasmid construction
Details of the various plasmid constructions are included in supplemental materials or are available from the authors by request.
Expression and purification of His6-tagged and GST-tagged proteins from E. coli
One liter of Superbroth (12 g tryptone, 24 g yeast extract, 4 g glycerol, 0.17 M KH2PO4, 0.72 M K2HPO4, 50 μg/ml ampicillin) was inoculated with 10 ml of an overnight culture of E. coli strain KK2186 harboring plasmids expressing His6- or GST-tagged proteins. Protein production was induced with isopropyl β-D-thiogalactopyranoside (0.5 mM) and, after 3–5 h, cells were harvested by centrifugation. Cells were disrupted by sonication, and Triton X-100 was added (final concentration 0.1% vol/vol). After addition of DNase I (10 μg/ml) and MgCl2 (10 mM), lysate was clarified and filtered.
His6-tagged proteins were purified by HiTrap Ni-chelating column chromatography (Amersham Pharmacia Biotech) as per the manufacturer's instructions.
Peptide sequencing
Purified proteins were transferred to Immuno-Blot PVDF membranes (Bio-Rad Laboratories) using the semidry Transblot apparatus (Bio-Rad Laboratories). Blotted PVDF membrane were washed with distilled water and stained with Ponceau S. Kes1p species were excised from membranes and subjected to automated NH2-terminal Edman degradation.
Mass spectrometry
MALDI-TOF mass spectrometry was performed using positive mode on a Voyager Elite unit with delayed extraction technology (PerSeptive Biosystems). Samples were diluted 1:10 with matrix, and 1 μl of the resulting mix was deposited onto a smooth plate. Acceleration voltage was set at 25 kV and 10–50 laser shots were summed. Sinapinic acid (D13, 460-0; Sigma-Aldrich) dissolved in acetonitrile: 0.1% TFA (1:1) was used as matrix. The mass spectrometer was calibrated with bovine serum albumin.
Online supplemental materials
PIP photolabeling assay. Photolabeling and displacement assays were performed as described previously (Kearns et al., 1998b). Details are available at http://www.jcb.org/cgi/content/full/jcb.200201037/DC1 or from the authors by request.
Supplemental Material
Acknowledgments
This work was supported by National Institutes of Health grant GM44530 awarded to V.A. Bankaitis. G.D. Prestwich thanks the National Institutes of Health for grant NS29632 for support, and Drs. Jian Chen, Gyorgy Dorman, and Qu-Ming Gu for synthetic intermediates and procedures.
The online version of this article contains supplemental material.
Footnotes
Abbreviations used in paper: ARF, adenosine diphosphate-ribosylation factor; ARFGAP, ARF guanosine triphosphatase activating protein; BZDC, ([p-benzoyldihydrocinnamidyl]-amino)propyl; GFP, green fluorescent protein; IP3, inositol-1,4,5-trisphosphate; MALDI-TOF, matrix-assisted laser ionization coupled with time-of-flight mass analysis; OSBP, oxysterol binding protein; PA, phosphatidic acid; PC, phosphatidylcholine; PH, Pleckstrin homology, PI, phosphatidylinositol; PIP, phosphoinositide; PLD, phospholipase D.
References
- Alphey, L., J. Jimenez, and D. Glover. 1998. A Drosophila homologue of oxysterol binding protein (OSBP) – implications for the role of OSBP. Biochim. Biophys. Acta. 1395:159–164. [DOI] [PubMed] [Google Scholar]
- Bankaitis, V.A., D.E. Malehorn, S.D. Emr, and R. Greene. 1989. The Saccharomyces cerevisiae SEC14 gene encodes a cytosolic factor that is required for transport of secretory proteins from the yeast Golgi complex. J. Cell Biol. 108:1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankaitis, V.A., J.R. Aitken, A.E. Cleves, and W. Dowhan. 1990. An essential role for a phospholipid transfer protein in yeast Golgi function. Nature. 347:561–562. [DOI] [PubMed] [Google Scholar]
- Beh, C.T., L. Cool, J. Phillips, and J. Rine. 2001. Overlapping functions of the yeast oxysterol binding protein homologues. Genetics. 157:1117–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, M.S., and J.L. Goldstein. 1999. A proteolytic pathway that controls the cholesterol content of membranes, cells and blood. Proc. Natl. Acad. Sci. USA. 96:11041–11048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caenorhabditis elegans Sequencing Consortium. 1998. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 282:2012–2018. [DOI] [PubMed] [Google Scholar]
- Chaudhary, A., J. Chen, Q.-M. Gu, W. Witke, D.J. Kwiatkowski, and G.D. Prestwich. 1998. a. Probing the phosphoinositide 4,5-bisphosphate binding site of human profilin I. Chem. Biol. 5:273–281. [DOI] [PubMed] [Google Scholar]
- Chaudhary, A., Q.-M. Gu, O. Thum, A.A. Profit, Y. Qing, L. Jeyakumar, S. Fleischer, and G.D. Prestwich. 1998. b. Specific interaction of Golgi coatomer α-COP with phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273:8344–8350. [DOI] [PubMed] [Google Scholar]
- Cleves, A.E., P.J. Novick, and V.A. Bankaitis. 1989. Mutations in the SAC1 gene suppress defects in yeast Golgi and yeast actin function. J. Cell Biol. 109:2939–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleves, A.E., T.P. McGee, and V.A. Bankaitis. 1991. a. Phospholipid transfer proteins: a biological debut. Trends Cell Biol. 1:30–34. [DOI] [PubMed] [Google Scholar]
- Cleves, A.E., T.P. McGee, E.A. Whitters, K.M. Champion, J.R. Aitken, W. Dowhan, M. Goebl, and V.A. Bankaitis. 1991. b. Mutations in the CDP-choline pathway for phospholipid biosynthesis bypass the requirement for an essential phospholipid transfer protein. Cell. 64:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham, E., S.K. Tan, P. Swigart, J. Hsuan, V.A. Bankaitis, and S. Cockcroft. 1996. The yeast and mammalian isoforms of phosphatidylinositol transfer protein can all restore phospholipase C-mediated inositol lipid signaling in cytosol-depleted RBL-2H3 and HL-60 cells. Proc. Natl. Acad. Sci. USA. 93:6589–6593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson, P.A., J.L. Van der Westhuyzen, J.L. Goldstein, and M.S. Brown. 1989. Purification of oxysterol binding protein from hamster liver cytosol. J. Biol. Chem. 264:9046–9052. [PubMed] [Google Scholar]
- Dorman, G., and G.D. Prestwich. 1994. Benzophenone photo-probes in biochemistry. Biochemistry. 33:5661–5673. [DOI] [PubMed] [Google Scholar]
- Fang, M., B.G. Kearns, A. Gedvilaite, S. Kagiwada, M. Kearns, M.K.Y. Fung, and V.A. Bankaitis. 1996. Kes1p shares homology with human oxysterol binding protein and participates in a novel regulatory pathway for yeast Golgi-derived transport vesicle biogenesis. EMBO J. 15:6447–6459. [PMC free article] [PubMed] [Google Scholar]
- Feng, L., M. Mejillano, H.L. Yin, J. Chen, and G.D. Prestwich. 2001. Full-contact domain labeling: identification of a novel phosphoinositide binding site on gelsolin that requires the complete protein. Biochemistry. 40: 904–913. [DOI] [PubMed] [Google Scholar]
- Foti, M., A. Audhya, and S.D. Emr. 2001. Sac1 lipid phosphates and Stt4 phosphatidylinositol 4-kinase regulate a pool of phosphatidylinositol 4-phosphate that functions in the control of the actin cytoskeleton and vacuole morphology. Mol. Biol. Cell. 12:2396–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier, M.V., F.C. Guimaraes, M.E. Paschoal, L.V. Ronco, M.G. Carvalho, and A.B. Pardee. 1999. Identification of a gene encoding a human oxysterol-binding protein-homologue: a potential genetic molecular marker for blood dissemination of solid tumors. Cancer Res. 59:3748–3753. [PubMed] [Google Scholar]
- Guo, S., L.E. Stolz, S. Lemrow, and J.D. York. 1999. SAC1-like domains of yeast SAC1, INP52 and INP53, and human synaptojanin encode polyPIP phosphatases. J. Biol. Chem. 274:12990–12995. [DOI] [PubMed] [Google Scholar]
- Hama, H., E.A. Schnieders, J. Thorner, J.Y. Takemoto, and D. DeWald. 1999. Direct involvement of phosphatidylinositol-4-phosphate in secretion in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 274:34294–34301. [DOI] [PubMed] [Google Scholar]
- Hay, J.C., and T.F.J. Martin. 1993. Phosphatidylinositol transfer protein is required for ATP-dependent priming of Ca2+-activated secretion. Nature. 366:572–575. [DOI] [PubMed] [Google Scholar]
- Huijbregts, R.P.H., L.L. Topalof, and V.A. Bankaitis. 2000. Lipid metabolism and membrane dynamics. Traffic. 1:195–202. [DOI] [PubMed] [Google Scholar]
- Ito, H., Y. Fukuda, K. Murata, and A. Kimura. 1983.Transformation of intact yeast cells treated with alkaline cations. J. Bacteriol. 153:163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, B., J.L. Brown, J. Sheraton, N. Fortin, and H. Bussey. 1994. A new family of yeast genes implicated in ergosterol synthesis is related to the human oxysterol binding protein. Yeast. 10:341–353. [DOI] [PubMed] [Google Scholar]
- Jones, S.M., J.G . Alb, Jr., S.E. Phillips, V.A. Bankaitis, and K.E. Howell. 1998. A phosphatidylinositol 3-kinase and phosphatidylinositol transfer protein act synergistically in formation of constitutive transport vesicles from the trans-Golgi network. J. Biol. Chem. 273:10349–10354. [DOI] [PubMed] [Google Scholar]
- Kearns, B.G., J.G. Alb, Jr., and V.A. Bankaitis. 1998. a. Phosphatidylinositol transfer proteins: the long and winding road to function. Trends Cell Biol. 8:276–282. [DOI] [PubMed] [Google Scholar]
- Kearns, M.A., D.E. Monks, M. Fang, M.P. Rivas, P.D. Courtney, J. Chen, G.D. Prestwich, A.B. Theibert, R.E. Dewey, and V.A. Bankaitis. 1998. b. Novel developmentally regulated PIP binding proteins from soybean whose expression bypasses the requirement for an essential phosphatidylinositol transfer protein in yeast. EMBO J. 17:4004–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagace, T.A., D.M. Byers, H.W. Cook, and N.D. Ridgway. 1997. Altered regulation of cholesterol and cholesteroyl ester synthesis in Chinese hamster ovary cells overexpressing the oxysterol binding protein is dependent on the pleckstrin homology domain. Biochem. J. 326:205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon, M.A. 1999. Structural basis for high-affinity PIP binding by pleckstrin homology domains. Biochem. Soc. Trans. 27:617–624. [DOI] [PubMed] [Google Scholar]
- Levine, T.P., and S. Munro. 1998. The pleckstrin homology domain of oxysterol-binding protein recognizes a determinant specific to Golgi membranes. Curr. Biol. 8:729–739. [DOI] [PubMed] [Google Scholar]
- Li, X., S. Routt, Z. Xie, X. Cui, M. Fang, M.A. Kearns, M. Bard, D. Kirsch, and V.A. Bankaitis. 2000. a. Identification of a novel family of nonclassical yeast PITPs whose function modulates activation of phospholipase D and Sec14p-independent cell growth. Mol. Biol. Cell. 11:1989–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X., Z. Xie, and V.A. Bankaitis. 2000. b. Phosphatidylinositol/phosphatidylcholine transfer proteins in yeast. Biochim. Biophys. Acta. 55626:1–17. [DOI] [PubMed] [Google Scholar]
- McGee, T.P., H.B. Skinner, E.A. Whitters, S.A. Henry, and V.A. Bankaitis. 1994. A phosphatidylinositol transfer protein controls the phosphatidylcholine content of yeast Golgi membranes. J. Cell Biol. 124:273–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto, Y., B.G. Kearns, M.R. Wenk, H. Chen, K. Mori, J.G. Alb, Jr., P. De Camilli, and V.A. Bankaitis. 2000. Functional characterization of a mammalian Sac1 and mutants exhibiting substrate specific defects in phosphoinositide phosphatase activity. J. Biol. Chem. 275:14446–14456. [DOI] [PubMed] [Google Scholar]
- Novick, P., C. Field, and R. Schekman. 1980. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 21:205–215. [DOI] [PubMed] [Google Scholar]
- Phillips, S., B. Sha, L. Topalof, Z. Xie, J. Alb, V. Klenchin, P. Swigart, S. Cockcroft, M. Luo, T. Martin, and V. Bankaitis. 1999. Yeast Sec14p deficient in phosphatidylinositol transfer activity is functional in vivo. Mol. Cell. 4:187–197. [DOI] [PubMed] [Google Scholar]
- Prestwich, G.D. 1996. Touching all the bases: inositol polyphosphate and phosphoinositide affinity probes from glucose. Acc. Chem. Res. 29:503–513. [Google Scholar]
- Prestwich, G.D., G. Dorman, J.T. Elliot, D.M. Marecak, and A. Chaudhary. 1997. Benzophenone photo-probes for PIPs, peptides and drugs. Photochem. Photobiol. 65:222–234. [DOI] [PubMed] [Google Scholar]
- Rivas, M.P., B.G. Kearns, S. Guo, Z. Xie, M.C. Sekar, K. Hosaka, S. Kagiwada, J.D. York, and V.A. Bankaitis. 1999. Relationship between altered phospholipid metabolism, diacylglycerol, ‘bypass Sec14p’, and the inositol auxotrophy of yeast sac1 mutants. Mol. Biol. Cell. 10:2235–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger, F., S. Nicklen, and A.R. Coulson. 1977. DNA sequencing with chain terminating inhibitors. Proc. Natl. Acad. Sci. USA. 74:5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, F., G.R. Fink, and J.B. Hicks. 1983. Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 1–113.
- Stock, S.D., H. Hama, D.B. DeWald, and J.Y. Takemoto. 1999. SEC14-dependent secretion in Saccharomyces cerevisiae: non-dependence on sphingolipid-coupled diacylglycerol synthesis. J. Biol. Chem. 274:12979–12983. [DOI] [PubMed] [Google Scholar]
- Tall, E., G. Dorman, P. Garcia, L. Runnels, S. Shah, J. Chen, A. Profit, Q.M. Gu, A. Chaudhary, G.D. Prestwich, and M.J. Rebecchi. 1997. PIP binding specificity among phospholipase C isozymes as determined by photo-crosslinking to novel substrate and product analogs. Biochemistry. 36:7239–7248. [DOI] [PubMed] [Google Scholar]
- Taylor, F.R., S.E. Saucier, E.P. Shown, E.J. Parish, and A.A. Kandutsch. 1984. Correlation between oxysterol binding to a cytosolic binding protein and potency in the repression of hydroxymethylglutaryl coenzyme A reductase. J. Biol. Chem. 259:12382–12387. [PubMed] [Google Scholar]
- Walch-Solimena, C., and P. Novick. 1999. The yeast phosphatidylinositol-4-OH kinase pik1 regulates secretion at the Golgi. Nat. Cell Biol. 1:523–525. [DOI] [PubMed] [Google Scholar]
- Whitters, E.A., A.E. Cleves, T.P. McGee, H.B. Skinner, and V.A. Bankaitis. 1993. SAC1p is an integral membrane protein that influences the cellular requirement for phospholipid transfer protein function and inositol in yeast. J. Cell Biol. 122:79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, Z., M. Fang, and V.A. Bankaitis. 2001. Evidence for an intrinsic toxicity of phosphatidylcholine to Sec14p-dependent protein transport from the yeast Golgi complex. Mol. Biol. Cell. 12:1117–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, Z., M. Fang, M.P. Rivas, A. Faulkner, P.C. Sternweis, J. Engebrecht, and V.A. Bankaitis. 1998. Phospholipase D activity is required for suppression of yeast phosphatidylinositol transfer protein defects. Proc. Natl. Acad. Sci. USA. 95:12346–12351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C.-J., M.M. Cavenagh, and R.A. Kahn. 1998. A family of ARF effectors defined as suppressors of the loss of Arf function in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 273:19792–19796. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.