

Abstract
14-3-3 proteins regulate the cell cycle and prevent apoptosis by controlling the nuclear and cytoplasmic distribution of signaling molecules with which they interact. Although the majority of 14-3-3 molecules are present in the cytoplasm, we show here that in the absence of bound ligands 14-3-3 homes to the nucleus. We demonstrate that phosphorylation of one important 14-3-3 binding molecule, the transcription factor FKHRL1, at the 14-3-3 binding site occurs within the nucleus immediately before FKHRL1 relocalization to the cytoplasm. We show that the leucine-rich region within the COOH-terminal α-helix of 14-3-3, which had been proposed to function as a nuclear export signal (NES), instead functions globally in ligand binding and does not directly mediate nuclear transport. Efficient nuclear export of FKHRL1 requires both intrinsic NES sequences within FKHRL1 and phosphorylation/14-3-3 binding. Finally, we present evidence that phosphorylation/14-3-3 binding may also prevent FKHRL1 nuclear reimport. These results indicate that 14-3-3 can mediate the relocalization of nuclear ligands by several mechanisms that ensure complete sequestration of the bound 14-3-3 complex in the cytoplasm.
Keywords: 14-3-3; FKHRL1; phosphoserine; nuclear export; Crm1
Introduction
14-3-3 proteins are a highly conserved family of molecules that regulate intracellular signal transduction events within all eukaryotic cells. These ∼30 kD α-helical molecules include 9 proteins in mammals and 2 to 12 proteins in yeast, fungi, and plants (Fu et al., 2000; Tzivion et al., 2001; van Hemert et al., 2001). 14-3-3 proteins form homo- and heterodimeric U-shaped structures (Liu et al., 1995; Xiao et al., 1995) that bind to a large number of phosphorylated signaling molecules through the large central channel in the center of the U. For example, 14-3-3 proteins specifically recognize one or more short phosphoserine/threonine-containing sequence motifs (Muslin et al., 1996; Yaffe et al., 1997) on target proteins such as Raf to control mitogen-activated protein kinase activation, Cdc25 to control G2/M cell cycle progression, and BAD and FKHRL1 to control apoptosis. The molecular details of how 14-3-3 functions in many of these interactions remains obscure, though it appears that for at least some of its substrates 14-3-3 participates in regulating their subcellular localization. Several 14-3-3 binding molecules perform critical functions within the nucleus. These include the mitotic phosphatase Cdc25 (Peng et al., 1997; Dalal et al., 1999; Kumagai and Dunphy, 1999; Lopez-Girona et al., 1999; Yang et al., 1999; Zeng and Piwnica-Worms, 1999; Davezac et al., 2000), the proapoptotic transcription factor FKHRL1 (Brunet et al., 1999), the yeast transcriptional activators MSN2 and MSN4 (Beck and Hall, 1999), the mammalian transcriptional coactivator TAZ and YAP (Kanai et al., 2000), and the histone deacetylases HDAC4, -5, and -7 (Grozinger and Schreiber, 2000; Wang et al., 2000; Kao et al., 2001). When complexed to 14-3-3, these molecules become sequestered in the cytoplasm, impairing their nuclear function. Curiously, 14-3-3 proteins are found primary within the cytoplasm, raising the question of whether phosphorylation and 14-3-3 binding occurs in the cytoplasm or in the nucleus. In addition, there is controversy as to the mechanisms through which 14-3-3 causes the relocalization of these substrates from the nucleus into the cytoplasm.
Insight into the mechanism of 14-3-3–dependent cytosolic sequestration has emerged from a series of studies on the interaction between 14-3-3 and Cdc25. Initial studies by Lopez-Girona et al. (1999) identified a leucine-rich sequence within the COOH-terminal helix (helix αI) of 14-3-3 that was highly homologous to known nuclear export signals (NESs).* These authors proposed that 14-3-3 functioned essentially as an “attachable NES” to transport nuclear Cdc25 to the cytoplasmic compartment by a piggy-back mechanism. This view was contradicted by detailed studies by both Kumagai and Dunphy (1999) and Zeng and Piwnica-Worms (1999). These authors showed that point mutations in this leucine-rich sequence of 14-3-3 prevented Cdc25 from being sequestered in the cytoplasm by disrupting its binding to 14-3-3. Furthermore, our x-ray crystallographic studies of 14-3-3–peptide complexes of two canonical 14-3-3 binding motifs R[S/Ar]XpSXP and RX[Ar/S]XpSXP, (pS denotes phosphoserine and Ar denotes aromatic residues) showed that the critical leucine and isoleucine residues within helix αI were directly involved in binding to the phosphopeptide (Rittinger et al., 1999). Taken together, these studies raise the possibility that the primary function of helix αI may be to mediate interactions between 14-3-3 and many phosphoprotein ligands but do not prove that this sequence cannot function as an NES.
The current prevailing mechanism for how 14-3-3 proteins mediate changes in the subcellular localization of Cdc25 comes from elegant studies by Kumagai and Dunphy (1999) and Yang et al. (1999). Cdc25 was found to be exported from the nucleus at a constant rate. This process involved intrinsic NES sequences in Cdc25 but was not modified by phosphorylation/14-3-3 binding. In contrast, 14-3-3–bound Cdc25 within the cytoplasm showed a decreased rate of nuclear import as a result of masking a Cdc25 nuclear localization signal (NLS) sequence, thereby inhibiting the interaction of Cdc25 with importin-α. Whether this mechanism operates for all 14-3-3–bound ligands or whether a subset of ligands can undergo an increase in nuclear export upon phosphorylation/14-3-3 binding is not known.
In the present study, we sought to clarify where 14-3-3 binding to its ligands occurred, determine whether the leucine-rich helix αI truly functions as an NES, and identify features of both 14-3-3 and one of its nuclear ligands, the transcription factor FKHRL1, that are critical for the movement of this nuclear ligand to the cytoplasm. Surprisingly, we find that in the absence of bound ligands the majority of 14-3-3 is localized to the nucleus, where a large number of 14-3-3 ligands are present. For FKHRL1, growth-factor stimulation, which activates AKT, first leads to the phosphorylation of FKHRL1 at its 14-3-3 binding site within the nucleus immediately before FKHRL1 nuclear export. Helix αI of 14-3-3 does not function as an attachable NES but is involved in a general manner in the binding of phosphorylated ligands. Instead, rapid nuclear export of FKHRL1 requires both NES sequences that are intrinsic to FKHRL1 and phosphorylation/14-3-3 binding. Once in the cytoplasm, FKHRL1 remains phosphorylated and bound to 14-3-3, thereby preventing nuclear reimport. Together with the studies on Cdc25 cited above, our study suggests that 14-3-3 can function through multiple mechanisms that may cooperate to ensure complete sequestration of the phosphorylated 14-3-3 ligand in the cytoplasm.
Results
Different intracellular localization of 14-3-3 and its ligands
To investigate the normal intracellular distribution of 14-3-3 proteins and their ligands, U2OS cells were immunostained with a polyclonal antibody that recognizes all 14-3-3 isotypes (pan α–14-3-3) or with a monoclonal antibody generated against the 14-3-3σ isotype (Fig. 1 A). Identical immunostaining results were obtained with both antibodies, though the performance of the monoclonal anti–14-3-3σ antibody was superior, and this antibody was therefore used preferentially. Both antibodies showed that the vast majority of 14-3-3 had a diffuse cytoplasmic and perinuclear distribution in unsychronized cells, consistent with findings reported by others (Hermeking et al., 1997; Kumagai and Dunphy, 1999; Lopez-Girona et al., 1999; Grozinger and Schreiber, 2000). A similar cytoplasmic distribution of 14-3-3 was observed after biochemical separation of the cells into cytoplasmic and nuclear fractions followed by immunoblotting with the pan α–14-3-3 antibody (Fig. 1 B). To investigate the intracellular localization of all ligands capable of 14-3-3 binding, cells were metabolically labeled with [35S]methionine, subjected to nuclear and cytoplasmic fractionation, and incubated with 14-3-3–GST fusion proteins in pull-down assays. Surprisingly, these experiments revealed that many 14-3-3 binding ligands were in the nuclear fraction (Fig. 1 C) despite the predominantly cytoplasmic location of endogenous 14-3-3.
Figure 1.

Different subcellular distributions of 14-3-3 and its ligands. (A) U2OS cells immunostained for 14-3-3σ (left) and corresponding DAPI staining (right). Bar, 10 μm. (B) Biochemical fractionation of U2OS cells followed by immunoblotting using an antibody recognizing all 14-3-3 isotypes (N, nuclear fraction; C, cytoplasmic fraction). Blots were probed with anti–α-tubulin (cytoplasmic) or anti–lamin B (nuclear) as controls. A very small amount of 14-3-3 is present in the nuclear fraction but cannot be seen in this exposure. (C) U2OS cells were metabolically labeled with [35S]methionine, fractionated, and nuclear (N) and cytoplasmic (C) fractions containing equal amounts of protein and radioactivity were incubated with beads containing GST–14-3-3 or GST alone. Bound proteins were visualized by SDS-PAGE and autoradiography. A small portion of the input sample is shown on the left (Lysate lanes). Positions of molecular weight markers, in kD, are indicated. The prominent band at ∼28 kD in the cytoplasmic/14-3-3–GST lane (arrow) is endogenous U2OS 14-3-3 (Rittinger et al., 1999).
14-3-3 normally transits into and out of the nucleus
The findings in Fig. 1 suggest that free 14-3-3 might dynamically transit into the nucleus to bind ligands followed by export of the ligand-bound 14-3-3 complex out of the nucleus. To investigate if 14-3-3 was actively exported out of the nucleus, U2OS cells were incubated with leptomycin B (LMB), a specific inhibitor of Crm1- and NES-dependent nuclear export (Fornerod et al., 1997; Fukuda et al., 1997; Ossareh-Nazari et al., 1997), and the distribution of 14-3-3 was analyzed by immunofluorescence. As shown in Fig. 2, A and B , incubation of U2OS cells with LMB caused the progressive accumulation of endogenous 14-3-3 in the nucleus, causing a 2.5-fold increase in nuclear 14-3-3 staining within 1.5 h and a nearly 15-fold increase in nuclear 14-3-3 accumulation by 18 h with the majority of 14-3-3 being clearly localized within the nuclear compartment (Fig. 2). These experiments demonstrate that nuclear export of 14-3-3 is an NES/Crm1-dependent process but do not reveal whether the NES is present on 14-3-3 or on the bound ligand.
Figure 2.
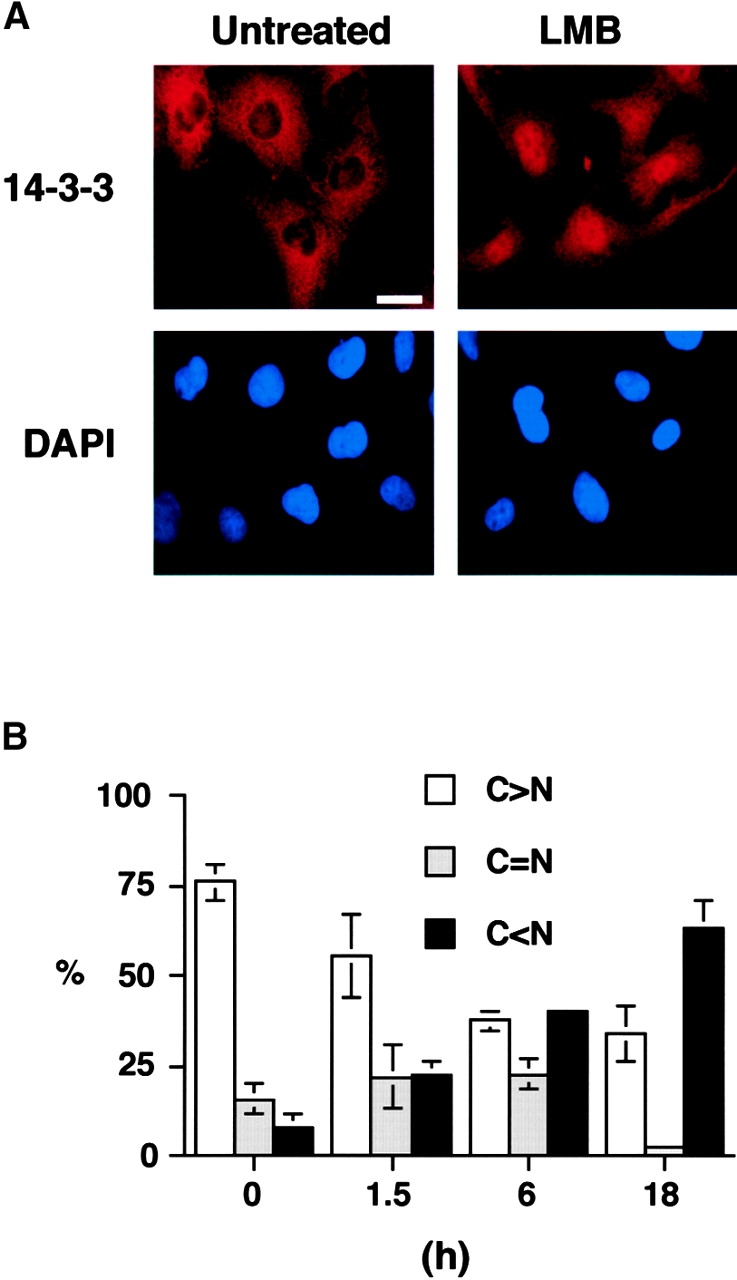
LMB sequesters endogenous 14-3-3 in the nucleus. (A) Immunostaining of U2OS cells for endogenous 14-3-3σ with (right) or without (left) treatment with LMB for 18 h. Corresponding DAPI staining is shown on the bottom. Bar, 10 μm. (B) Quantitative analysis of 14-3-3σ location. Subcellular localization of 14-3-3σ was scored according to whether it was higher in the nucleus (N > C), evenly distributed between nucleus and cytoplasm (C = N), or higher in cytoplasm (C < N) for 300 cells at each time point. Results are the mean ± SD from three separate experiments.
The leucine-rich sequence in 14-3-3 functions in ligand binding rather than nuclear export
14-3-3 proteins contain a leucine-rich sequence in helix αI that matches the consensus sequence for nuclear export, and Lopez-Girona et al. (1999) have argued that this sequence functions as an “attachable NES” for 14-3-3–dependent nuclear export of Cdc25. However, several lines of evidence suggest that this leucine-rich sequence in helix αI functions primarily in binding to phosphorylated ligands, including crystallographic studies of two 14-3-3–phosphopeptide complexes (Yaffe et al., 1997; Rittinger et al., 1999) and mutation and deletion studies involving interactions between this region of 14-3-3 with two specific phosphorylated ligands, Cdc25C (Kumagai and Dunphy, 1999; Zeng and Piwnica-Worms, 1999) and Raf (Thorson et al., 1998).
To determine if helix αI of 14-3-3 has a very general function in binding to many serine-phosphorylated 14-3-3 binding proteins, we used commercial antibodies that had been generated against one of the major 14-3-3 phosphoserine binding motifs. This motif is present in many 14-3-3 ligands (Muslin et al., 1996; Yaffe et al., 1997), and this antibody detects a large number of immunoreactive bands in 293T total cell lysates (Fig. 3 A, TCL lane), which are specifically pulled down by wild-type (WT) 14-3-3–GST (14-3-3ɛ WT lane) but not by a 14-3-3–GST point mutant that is unable to bind to ligands (14-3-3ɛ MT lane, discussed in more detail below). Deletion of residues 215–255 in 14-3-3ɛ, which includes both helix αI and the extreme COOH-terminal tail (Fig. 3 A, ΔC lanes, and Fig. 4 A), completely abrogated binding of 14-3-3 to all of the detected proteins. The extreme COOH-terminal tail of 14-3-3 (residues 233–255 in ɛ) cannot be visualized in 14-3-3 x-ray structures (Liu et al., 1995; Xiao et al., 1995) (Fig. 4 A), and its contribution to ligand binding is unknown. Removal of this region while maintaining helix αI reduced but did not eliminate 14-3-3 binding (Fig. 3 A, ΔT lanes), demonstrating that helix αI plays an essential role in binding to many phosphorylated protein ligands.
Figure 3.

The Leu-rich sequence in helix αI of 14-3-3 is responsible for ligand binding. (A) In the context of the entire 14-3-3 molecule, the Leu-rich helix αI functions primarily in ligand binding. Total cell lysates from 293T cells were incubated with beads containing the indicated GST–14-3-3 constructs or GST alone and bound proteins analyzed by SDS-PAGE and immunoblotting using an antibody against the 14-3-3 phosphoserine-binding epitope. TCL, total cell lysate; WT, WT 14-3-3; MT, K49E mutant; ΔC, GST–14-3-3 (1–214); ΔT, GST–14-3-3 (1–232). The mobility of molecular mass standards in kD is indicated. (B and C) Direct contribution of leucine and isoleucine residues in helix αI to ligand binding. 293T cell lysates as in A were incubated with beads containing immobilized WT GST–14-3-3ɛ or GST–14-3-3ɛ containing sequential Ala substitutions for Leu/Ile residues in helix αI. Bound proteins were analyzed by SDS-PAGE followed by immunoblotting using the antibody against the 14-3-3 phosphoserine-binding epitope (top) or stained with Coomassie blue to quantitate the amount of bead-bound GST–14-3-3 protein and GST alone (bottom, arrows). The bottom part of C shows the contributions from each Leu/Ile residue to phosphopeptide binding based on the x-ray structure of a 14-3-3–phosphopeptide complex (Rittinger et al., 1999).
Figure 4.

A mutant 14-3-3, which cannot bind to ligands, accumulates in the nucleus. (A) Structural cartoon of the 14-3-3 dimer. Monomers (red and green) consists of nine α-helices. Helix αI containing the leucine-rich sequence is purple with leucine and isoleucine residues shown in ball and stick. Lys-49 in the phosphoserine-binding pocket is indicated. Only three residues in the extreme COOH-terminal tail (cyan) are visible in the x-ray structures. The figure was generated with Molscript and Raster3D. (B) Expression of Xpress-tagged 14-3-3 WT and K49E mutant protein in different stable U2OS cell lines. Total cell lysates corresponding to equivalent amounts of total protein from stable cell lines expressing empty vector (lane 1), WT 14-3-3 (lanes 2–4), or K49E mutant (lanes 5–7) were analyzed by SDS-PAGE and immunoblotting using an anti-Xpress epitope antibody. (C) Comparison of Xpress-tagged and endogenous 14-3-3 levels in several U2OS stable cell lines. Total cell lysates from stable cell lines containing empty vector (lane 1), WT 14-3-3ɛ (clone #15) (lane 2) and 14-3-3ɛ K49E mutant (clone #21) (lane 3) were immunoblotted using a pan anti–14-3-3 antibody. Arrowhead indicates endogenous 14-3-3 in U2OS cells; arrow indicates Xpress-tagged 14-3-3. (D) Immunostaining of stable U2OS cell lines expressing WT 14-3-3ɛ (clone #15, left) or 14-3-3ɛ K49E mutant (clone #21, right) with the anti-Xpress antibody. Corresponding DAPI staining was shown in the middle, and the merged image is shown on the bottom. Bar, 10 μm. Each of three stable cell lines showed identical results.
To specifically address the role of the leucine and isoleucine residues within helix αI, we progressively mutated these residues to alanines in a systematic manner and determined if the 14-3-3 mutants could bind to ligands (Fig. 3, B and C). Mutation of a single leucine, Leu-216, resulted in an ∼50% global reduction in ligand binding (Fig. 3 B, LA1 lane). A double Ala substitution for Leu-216/Ile-217 (LA2 lane) or a triple Ala substitution for Leu-216/Ile-217/Leu-220 (LA3 lane) further reduced ligand binding to ∼5% or less of that seen with WT 14-3-3. Additional Ala substitutions of Leu-221 (LA4 lane), Leu-225 (LA5 lane), and Leu-227 (LA6 lane) eliminated binding altogether. Thus, the major role of the leucine and isoleucine residues within helix αI appears to be binding phosphoproteins.
A 14-3-3 mutant, which cannot bind to ligands, accumulates in the nucleus
The experiments described above strongly argue that helix αI functions primarily in ligand binding but cannot completely exclude the possibility that helix αI might also somehow mediate nuclear export. To directly address whether helix αI can function as an NES in vivo, we took advantage of a 14-3-3 point mutant in which Lys-49 is replaced by Glu (K49E) in the presence of an intact WT helix αI. This mutant, developed by Fu and colleagues, has the same conformation as WT 14-3-3 and is able to form homo- and heterodimers in vitro and in vivo (Zhang et al., 1997; unpublished data) but is unable to bind to Raf (Zhang et al., 1997). X-ray crystallographic studies revealed that Lys-49 forms part of a positively charged substrate-binding pocket in the central binding groove of 14-3-3 (Fig. 4 A), where it directly coordinates the phosphoserine phosphate of 14-3-3 binding peptides (Yaffe et al., 1997; Petosa et al., 1998; Rittinger et al., 1999). These structural observations on peptides suggested that the K49E mutation would be generally deficient in binding to phosphorylated protein ligands, while preserving its leucine-rich NES-like sequence. Indeed, the 14-3-3ɛ K49E mutant is unable to bind to numerous 14-3-3 binding proteins in cell extracts (Fig. 3 A, MT lanes), allowing the intrinsic function of the leucine-rich sequence in helix αI to be directly investigated without complexities arising from 14-3-3 ligand interactions.
If the leucine-rich sequence in helix αI functions as a true NES for 14-3-3, then the unliganded 14-3-3ɛ K49E protein should localize exclusively in the cytoplasm. Stable U2OS cell lines expressing either WT or K49E mutant Xpress-tagged 14-3-3ɛ proteins were generated, and three different stable cell lines for each construct were studied in detail (Fig. 4, B–D). The ligand-binding defective 14-3-3ɛ K49E mutant was found predominantly in the nucleus in 100% of the stable transfected cells in all cell lines investigated (Fig. 4 D, left). In contrast, the tagged WT 14-3-3 protein was found almost exclusively in the cytoplasm (Fig. 4 D, right), showing the identical staining pattern as that seen with endogenous 14-3-3 (Fig. 1 A). Expression levels of tagged WT 14-3-3ɛ in all three cell lines were ∼3.5- to 5-fold lower than those seen with the K49E mutant (Fig. 4, B and C), perhaps due to deleterious effects of 14-3-3 on cell growth (Hermeking et al., 1997; Kumagai et al., 1998). However, in all cases the expression levels of the stably transfected 14-3-3 proteins were less than or equal to those of endogenous 14-3-3 (Fig. 4 C). The possibility that localization of the K49E mutant compared with WT was due to differences in expression levels was ruled out by transient transfection experiments. Under these conditions, both WT and mutant proteins are expressed at equivalent levels (albeit at significantly higher concentrations than the endogenous 14-3-3 proteins) and resulted in the same nuclear localization for the K49E mutant (unpublished data). Together, these findings show conclusively that the leucine-rich sequence within 14-3-3 does not function as an NES in the absence of bound ligands.
The nuclear localization of the K49E 14-3-3 mutant strongly argues that nuclear export of 14-3-3 requires ligand binding. In addition, the LMB experiments indicate that the 14-3-3 is exported in an NES-dependent manner. Therefore, we hypothesized that it is NES sequences on the bound ligand which drive the export of 14-3-3–ligand complexes out of the nucleus and that this process is facilitated by 14-3-3 binding. To directly investigate the contribution of the 14-3-3–bound cargo to nuclear export, we focused the remainder of this study on one particular 14-3-3 binding ligand, the human Forkhead transcription factor FKHRL1 (Brunet et al., 1999).
The export of the 14-3-3 ligand FKHRL1 requires Crm1, intrinsic FKHLR1 NES sequences, and 14-3-3 binding
The subcellular localization of FKHRL1 is dependent on its phosphorylation by the protein kinase Akt. In the absence of growth factor, Akt is inactive and FKHRL1 is present in the nucleus in an unphosphorylated form. Upon growth factor stimulation, Akt is activated, directly phosphorylating FKHRL1 at T32 and S253, and to a lesser extent at S315, and resulting in FKHRL1 relocalization from the nucleus to the cytoplasm (Brunet et al., 1999). Similar AKT-dependent changes in subcellular localization have also been observed for the additional FKHRL1 human homologs AFX and FKHR (Brownawell et al., 2001; Rena et al., 2001) and for the Caenorhabditis elegans homologue daf-16 (Cahill et al., 2001). Phosphorylation at T32 and S253 causes FKHRL1 to bind to 14-3-3, suggesting that the interaction between FKHRL1 and 14-3-3 may play an important role in regulating the subcellular localization of the complex. COOH-terminal to the two 14-3-3 binding sites, FKHRL1 contains two sequences that closely match a consensus for NES sequences (Fig. 5 A), suggesting that nuclear export of 14-3-3–FKHRL1 complexes might involve a cooperative process involving both the FKHRL1 NES and 14-3-3 binding.
Figure 5.

Both FKHRL1 nuclear export sequences and interaction with 14-3-3 are necessary for efficient nucleocytoplasmic transport. (A) Schematic illustration of FKHRL1 showing the T32, S253, and S315 phosphorylation sites and the 14-3-3 binding sites. The two NES sequences in FKHRL1 are shown (NES1, amino acids 369–378; NES2, amino acids 386–396). (B) LMB treatment prevents FKHRL1 nuclear export even in the presence of growth factors. CCL39 cells were transfected with a WT HA-FKHRL1 construct. Cells were starved for 20 h, incubated with LMB for 2 h, and then stimulated with 10% serum (FCS) or 100 ng/ml IGF-I for 15 min. Localization of FKHRL1 WT or mutants was monitored by immunolocalization with the anti-HA antibody. Quantitative analysis of a representative experiment is shown. C, cytoplasm; C + N, cytoplasm and nucleus; N, nucleus. (C) Mutation of FKHRL1 NES or 14-3-3 binding site prevents FKHRL1 relocalization from the nucleus to the cytoplasm in the presence of growth factors. CCL39 cells were transfected with a WT HA-FKHRL1 construct or a mutant of both NES mutant of FKHRL1 (NESm) or a mutant in which all three phosphorylation sites of FKHRL1 have been replaced by alanine (TM). Cells were incubated in the presence of 10% serum (FCS). Localization of FKHRL1 WT or mutants was monitored by immunolocalization with the anti-HA antibody. (D) Quantitative analysis of FKHRL1 localization. Results are the mean ± SD from three separate experiments. C, cytoplasm; C + N, cytoplasm and nucleus; N, nucleus. (E) Mutation of FKHRL1 NES does not abolish binding to 14-3-3, whereas mutation of the phosphorylation sites of FKHRL1 does. 293T cells were cotransfected with a construct encoding M2–14-3-3ζ and either empty vector (CTL) or vectors encoding HA-FKHRL1 WT, T32A/S253A/S315A (TM), or a mutant of the putative NES1 and 2 (NESm). 14-3-3 was immunoprecipitated with the anti-M2 antibody, and the immune complex was analyzed by SDS-PAGE and immunoblotted with the anti-HA antibody (top). Total cell lysates (TCL) were also analyzed by direct immunoblotting with the anti-HA antibody (middle) or the anti-M2 antibody (bottom).
To first determine if FKHRL1 nuclear export is a Crm1- and NES-dependent process, we followed FKHLR1 subcellular localization in fibroblasts in the presence or absence of LMB upon stimulation by growth factors (Fig. 5 B). We found that treatment of cells with LMB prevented the relocalization of FKHRL1 from the nucleus to the cytoplasm in response to serum or insulin-like growth factor (IGF)-I, indicating that FKHRL1 was exported from the nucleus in an NES-dependent manner.
To investigate whether the intrinsic FKHRL1 NES sequences were responsible for the NES-dependent FKHRL1 nuclear export, we generated mutants of FKHRL1 in which the critical leucine or methionine residues in NES1 or NES2 were replaced by alanines either individually or in combination (Fig. 5, C and D). We found that an FKHRL1 mutant in which both NES1 and -2 were mutated remains localized in the nucleus even in the presence of growth factors, indicating that the NES sequences of FKHRL1 are required for the nuclear export of this protein. Importantly, we verified that the FKHRL1 NES mutant protein, which localizes to the nucleus, still binds to 14-3-3 (Fig. 5 E). This indicates that in the absence of a functional NES in FKHRL1, 14-3-3 binding alone is not sufficient to promote the efficient localization of FKHRL1 from the nucleus to the cytoplasm.
Consistent with our previous results, we found that a triple point mutant of FKHRL1, which does not bind to 14-3-3 (Fig. 5 E) because the key 14-3-3 binding residues T32 and S253 has been mutated to alanine, remains localized within the nucleus even in the presence of growth factors (Fig. 5, C and D). As this FKHRL1 mutant contains the intact NESs, this result further indicates that the NESs of FKHRL1 are not sufficient to lead to the net export of FKHRL1 out of the nucleus in the absence of 14-3-3 binding. Therefore, both the NES of FKHRL1 and FKHRL1's ability to bind 14-3-3 appear to be necessary to promote efficient FKHRL1 nuclear export. In addition, we found in transient transfection studies that FKHRL1 bound to Crm1 in a manner that is dependent on FKHRL1 NESs, further confirming that the NES sequences of FKHRL1 play a role in a Crm1-dependent nuclear export of this transcription factor (unpublished data). However, in these overexpression studies we have been unable to show that the interaction between FKHRL1 and Crm1 is facilitated by 14-3-3. Nevertheless, 14-3-3 may enhance the binding of FKHRL1 to Crm1 in a context where both proteins are expressed at physiological levels, or alternatively 14-3-3 may enhance the export of FKHRL1 without directly enhancing its binding to Crm1.
FKHLR1 undergoes phosphorylation within the nucleus upon growth factor stimulation before nuclear export—roles of intrinsic NES and NLS sequences and the 14-3-3 binding site in FKHRL1 localization
The experiments described above reflect the overall subcellular distribution of FKHRL1 at a given time but do not address the dynamic sequence of events involving FKHRL1 relocalization from the nucleus to the cytoplasm. Based on the results from Figs. 1–4, we hypothesized that 14-3-3 binding to FKHRL1 might occur in the nucleus, exposing the FKHRL1 NES and facilitating export of the 14-3-3–FKHRL1 complex to the cytoplasm. Once in the cytoplasm, 14-3-3 may have additional function in masking potential NLS, thereby preventing reentry of the ligands into the nucleus.
This sequence of events would require the phosphorylation of FKHRL1 in the nucleus to precede its cytoplasmic translocation. To investigate this, WT HA-tagged FKHRL1 was expressed in CCL39 fibroblasts, and the kinetics of phosphorylation and FKHRL1 relocalization were examined using immunofluorescence experiments with an antibody to HA and an antibody specific to phospho-Thr32 (Fig. 6 , WT panels). When cells were deprived of survival factors, WT FKHRL1 was localized in the nucleus and was not phosphorylated at Thr32. At short times after stimulation by IGF-1 phosphorylation of WT FKHRL1 at the 14-3-3 consensus binding site, Thr32 was detected, whereas FKHRL1 remained in the nucleus (Fig. 6, WT, 2 min). Upon longer stimulation times, the phosphorylated form of FKHRL1 began to appear predominantly in the cytoplasm (Fig. 6, WT, 5 and 15 min). In contrast, the NES1/2 mutant (NESm) was efficiently phosphorylated by IGF-1 within the nucleus, but unlike WT FKHRL1 the FKHRL1 NESm remained within the nucleus even after relatively long incubation times (Fig. 6, NESm 2, 5, and 15 min panels). Thus, phosphorylation of FKHRL1 at Thr32, which generates a 14-3-3 binding site, occurs within the nucleus before FKHRL1 cytoplasmic relocalization. The finding that phosphorylation of FKHRL1 to generate 14-3-3 binding sites occurs within the nucleus together with the nuclear localization of ligand-free 14-3-3 proteins further suggests that 14-3-3 binding to nuclear FKHRL1 may facilitate its NES-dependent export.
Figure 6.

Phosphorylation of FKHRL1 at the 14-3-3 binding site occurs within the nucleus immediately before nuclear export. CCL39 fibroblasts were transfected with WT HA-tagged FKHRL1 or a mutant of NES1 and -2 (NESm). Cells were serum starved for 12 h and incubated in the presence of 20 μM LY294002 for the last hour and then incubated in the presence of 100 ng/ml IGF-I for the indicated periods of time. Cells were rapidly fixed with paraformaldehyde, and FKHRL1 was detected by immunolocalization with an anti-HA antibody. Phosphorylation of FKHRL1 was assessed by immunolocalization with an antibody to phospho-Thr32. Representative pictures are shown. NESm, mutant of NES1 and NES2 of FKHRL1.
It is possible that 14-3-3 binding to FKHRL1 may also function in the cytoplasm to prevent FKHRL1 reentry into the nucleus, perhaps by masking NLS. Two regions in the primary sequence of FKHRL1 represent putative NLS sequences in the vicinity of the second phosphorylation site (Ser 253). To determine whether these regions indeed played a role in the subcellular localization of FKHRL1, we mutated the arginines 248, 249, and 250 into alanines (RRR → AAA) and the lysines 269, 270, and 271 to alanines (KKK → AAA). Expression of these FKHRL1 mutants within cells followed by immunofluorescence experiments revealed that the RRR → AAA mutant of FKHRL1 no longer localized in the nucleus, even in the absence of survival factors (Fig. 7, A and B , − lanes) or after complete inhibition of AKT using the phosphatidylinositol 3-kinase inhibitor LY294002 (Fig. 7, A and B, LY lanes). The KKK → AAA mutant of FKHRL1 also showed less nuclear localization in the absence of survival factors compared with WT FKHRL1 (Fig. 7 B) but did accumulate in the nucleus when the basal activity of Akt in serum-starved cells was completely eliminated by LY294002 (Fig. 7, A and B). Thus, the RRR sequence plays a major role, whereas the KKK region plays a minor role, in the nuclear localization of FKHRL1. These results indicate that the region surrounding the second 14-3-3 phosphorylation site is an NLS and further suggests that phosphorylation of this site, either by introducing negatively charged residue or by masking due to 14-3-3 binding, may participate in the retention of FKHRL1 in the cytoplasm. These results are in excellent agreement with the role of similar RRR and KKK sequences in AFX (Brownawell et al., 2001) and an RRR sequence in FKHR (Rena et al., 2001).
Figure 7.

Role of NLS and NES sequences in FKHRL1 subcellular localization. (A) CCL39 fibroblasts were transfected with a WT HA-tagged FKHRL1 construct or with FKHRL1 mutant constructs. Cells were either incubated in the presence of 10% serum (FCS) or 100 ng/ml IGF-I (IGF-I), or serum-starved for 12 h (−), or incubated in the presence of 20 μM LY294002 for 1 h (LY). FKHRL1 was detected by immunolocalization with an anti-HA antibody. RRR, RRR → AAA mutant of FKHRL1; KKK, KKK → AAA mutant of FKHRL1; NESm, mutant of NES1 and NES2 of FKHRL1; TM, triple mutant of FKHRL1 (T32A/S253A/S315A). (B) Quantification of a representative experiment is shown.
To experimentally determine the relative contributions of the NES, NLS, and 14-3-3 in promoting the export and/or preventing the reimport of FKHRL1, we analyzed the kinetics of cytoplasmic relocalization of a series of FKHRL1 mutants (Fig. 8) . Cells were first treated with the phosphatidylinositol 3-kinase inhibitor LY294002 to drive localization of all FKHRL1 mutants into the nucleus. LY294002 was then washed out, and cells were stimulated by IGF-I for short periods of time. FKHRL1 subcellular localization was determined by immunolocalization, and the number of cells in which FKHRL1 is present in the cytoplasm was quantified. We found that WT FKHRL1 was exported rapidly to the cytoplasm, and the percentage of cells showing exclusively cytoplasmic FKHRL1 remained constant at ∼40% (Fig. 8, WT) with the remaining 60% of cells displaying both cytoplasmic and nuclear localization. In contrast, the NES mutant of FKHRL1 did not relocalize to the cytoplasm even after longer stimulation with IGF-I. Next, we tested the cytoplasmic relocalization of the FKHRL1 RRR → AAA NLS mutant. We found that this NLS mutant was also exported from the nucleus with similar kinetics at early times to those of WT FKHRL1 (Fig. 8, RRR). However, after longer times a larger number of cells containing this mutant displayed exclusively cytoplasmic FKHRL1 staining, suggesting that the NLS of FKHRL1 plays a role after longer times of stimulation to promote the reentry of WT FKHRL1 into the nucleus. Phosphorylation of Ser253 could interfere with the basic residues of the NLS or 14-3-3 binding may mask this stretch of basic residues, thereby preventing FKHRL1 from being reimported to the nucleus at longer time of stimulation by growth factors.
Figure 8.

Kinetics of FKHRL1 cytoplasmic relocalization after IGF-I stimulation shows a requirement for both 14-3-3 binding and NES sequences for rapid nuclear export and NLS for inhibition of nuclear reimport. CCL39 fibroblasts were transfected with a WT HA-tagged FKHRL1 construct or with various FKHRL1 mutant constructs. Cells were treated as in the legend to Fig. 6, and FKHRL1 was detected by immunolocalization using an anti-HA antibody. The data presented correspond to the mean and variation of two experiments. Error bars are present for all time points, though in some cases they are obscured by the plot symbol. RRR, RRR → AAA mutant of FKHRL1; NESm, mutant of NES1 and NES2 of FKHRL1; TM, triple mutant of FKHRL1 (T32A/S253A/S315A).
To specifically address the role of 14-3-3 binding in the export of FKHRL1 from the nucleus, we next tested the export kinetics of FKHRL1 mutants in which the canonical 14-3-3 binding site at T32 or both 14-3-3 binding sites at T32 and S253 were replaced by alanines (Fig. 8, T32A and T32A/S253A). These experiments conclusively show that neither T32A nor T32A/S253A are exported efficiently from the nucleus even at short times of stimulation by growth factors. These results indicate that 14-3-3 binding to FKHRL1 is critical for the early nuclear events leading to export of FKHRL1 to the cytoplasm in a manner that requires FKHRL1 NES.
Together, these findings suggest that 14-3-3 binding to ligands can occur within the nucleus and may facilitate the nuclear export of the 14-3-3–ligand complex through the NES of the bound ligand. In the cytoplasm, 14-3-3 may have an additional role in preventing the reimport of ligands such as FKHRL1.
Discussion
In this study, we have shown that (a) although 14-3-3 proteins are predominantly localized in the cytoplasm, a large number of ligands are localized within the nucleus; (b) endogenous 14-3-3, fully competent to bind ligands, can be trapped in the nucleus by inhibiting Crm1-dependent nuclear export with LMB; (c) the leucine-rich putative NES sequence in 14-3-3, despite its structural homology to known NES sequences (Rittinger et al., 1999) and its ability to function as an NES in isolation when fused to GFP (unpublished data), functions primarily in phosphoprotein binding and not as an NES in the context of the intact 14-3-3 molecule; (d) a mutant 14-3-3, which cannot bind to ligands, homes to the nucleus; (e) the nuclear 14-3-3 ligand FKHRL1 is phosphorylated at its major 14-3-3 binding site within the nucleus before its export into the cytoplasm; and (f) at least for FKHRL1, rapid export from the nucleus to the cytoplasm requires both phosphorylation/14-3-3 binding and NES sequences within the bound ligand. In addition, we have identified NLS sequences within FKHRL1 that are responsible for its nuclear localization in the absence of phosphorylation. These data suggest that for nuclear FKHRL1, the sequence of events after growth factor stimulation involves phosphorylation at the 14-3-3 binding site within the nucleus, where ligand-free 14-3-3 molecules are located, followed by rapid nuclear export that requires both phosphorylation/14-3-3 binding and intrinsic NES sequences in FKHRL1. Once FKHRL1 has been exported to the cytoplasm, phosphorylation/14-3-3 binding may play an additional role in preventing nuclear reimport possibly by masking the FKHRL1 NLS.
Our observation that many potential 14-3-3 binding substrates are present in the nuclear fraction and that FKHRL1 is phosphorylated at its major 14-3-3 binding site within the nucleus suggests that kinases, such as AKT, Chk1, and Chk2, may act preferentially within this compartment, conferring 14-3-3 binding to some of their substrates. Furthermore, the observations that LMB treatment led to accumulation of endogenous 14-3-3 in the nucleus and that a K49E mutant of 14-3-3, which does not bind to ligands, localizes preferentially within the nucleus suggests that unliganded 14-3-3 normally transits to the nuclear compartment where binding to some ligands can occur. It would also be interesting to investigate the rate of import of 14-3-3 into the nucleus to address whether dissociation of ligands from preformed cytoplasmic complexes determines the rate of 14-3-3 nuclear import. We found that elimination or mutation of the leucine-rich sequence within helix αI disrupts binding to numerous phosphorylated protein ligands. This is in excellent agreement with the results of more focused mutational studies reported by others examining 14-3-3 binding to Raf and Cdc25 (Thorson et al., 1998; Wang et al., 1998; Kumagai and Dunphy, 1999; Zeng and Piwnica-Worms, 1999). Thus, the primary and general function of this α-helix is to directly participate in ligand binding. Furthermore, the observation that this leucine-rich sequence is incapable of maintaining unliganded 14-3-3 in the cytoplasm is incompatible with its proposed function as a true NES (Lopez-Girona et al., 1999). Instead, our present study of FKHRL1 and previous studies on Cdc25 (Kumagai and Dunphy, 1999; Yang et al., 1999; Davezac et al., 2000) indicate that export of the 14-3-3–bound complex involves NES sequences that are intrinsic to the ligands.
We reported previously an interaction between 14-3-3 and Crm1 within eukaryotic cell lysates that was disrupted by a 14-3-3 binding peptide (Rittinger et al., 1999). At that time, we interpreted this as evidence that the leucine-rich region within helix αI could interact with either ligands or Crm1 but not both simultaneously. In light of our current findings, the interaction that we observed between 14-3-3 and Crm1 was most likely mediated through the 14-3-3–bound ligands, which were competed off by the 14-3-3 binding peptide. In agreement with this, we have been unable to show a direct interaction between 14-3-3 and Crm1 when both are expressed in Escherichia coli (unpublished data).
Our findings are consistent with several different functions for AKT and 14-3-3 in regulating the activity and subcellular localization of FKHRL1. We have shown that AKT-mediated phosphorylation of FKHRL1 at the 14-3-3 binding site within the nucleus results in accelerated nuclear export of FKHRL1. This role for phosphorylation/14-3-3 binding in nuclear export is novel and complements recent results from other investigators who reported, while our paper was under revision, that AKT phosphorylation of the FKHRL1 homologs AFX and FKHR can inhibit nuclear reimport (Brownawell et al., 2001; Rena et al., 2001). In addition, Brownawell et al. (2001) proposed that phosphorylation at the 14-3-3 binding site did not affect nuclear export, since a triple phosphorylation site mutant of AFX that could not bind to 14-3-3 was able to shuttle between nuclei in heterokaryon fusion experiments. However, this study did not examine the time dependence of the import or export process, and our data in Fig. 8 indicates that phosphorylation at the 14-3-3 binding site is critical for rapid nuclear export of FKHRL1. Therefore, it is likely that AKT phosphorylation of Forkhead family members affects both nuclear export and import: phosphorylation/14-3-3 binding increases the rapid export of FKHRL1 from the nucleus in a manner that requires the intrinsic NES sequences on FKHRL1, whereas phosphorylation near the NLS site may disrupt FKHRL1 subsequent nuclear reimport. Furthermore, Cahill et al. (2001) showed that phosphorylation of the C. elegans FKHRL1 homologue daf-16 by AKT together with 14-3-3 binding directly inhibited its DNA binding ability. Thus, AKT- and 14-3-3–mediated inhibition of FKHRL1 function is likely to involve multiple events, first by blocking DNA binding and then by ensuring a complete sequestration of this transcription factor in the cytoplasm away from their nuclear target genes.
We find that a K49E mutant of 14-3-3 is unable to bind to ligands or be exported from the nucleus, despite its ability to form heterodimers with endogenous WT 14-3-3 in vivo and in vitro. This finding underscores the important role of ligand binding by each monomer within the 14-3-3 dimer for 14-3-3 function. A requirement for dimeric 14-3-3 has also been demonstrated for both 14-3-3–mediated Raf activation (Tzivion et al., 1998) and for 14-3-3–mediated disruption of daf-16 DNA binding after AKT phosphorylation (Cahill et al., 2001) using a mutant 14-3-3 in which the dimerization interface was disrupted.
Our functional data shown in Fig. 8, examining FKHRL1 export as a function of time, shows an increase in nuclear export upon FKHRL1 phosphorylation and 14-3-3 binding, which suggests that 14-3-3 may enhance the export process. Exactly how 14-3-3 accomplishes this remains to be defined. 14-3-3 binding might facilitate the formation of productive complexes between FKHRL1 and regulatory components of the nuclear export machinery, perhaps through inducing conformational changes in the 14-3-3–bound ligand. Recent data from Brownawell et al. (2001) on AFX and our unpublished observations on FKHRL1 suggest, however, that 14-3-3 binding does not cause an increase in the bulk association of these transcription factors with Crm1, though both proteins are grossly overexpressed in these assays. It has not been technically possible to measure the kinetics with which the association occurs between the endogenous proteins within cells. Alternatively, 14-3-3 binding could facilitate the ability of FKHRL1–Crm1 complexes to bind to endogenous Ran-GTP to trigger nuclear export. Another possibility is that 14-3-3 binding protects phosphorylation sites from the action of phosphatases and/or thereby permits additional modifications of FKHRL1, which are themselves involved in the export process.
We have focused this study specifically on one particular 14-3-3 ligand, FKHRL1, but studies on other 14-3-3 binding ligands such as HDAC4, -5, and -7 and Cdc25 are consistent with the idea that cytoplasmic relocalization involves both 14-3-3 proteins and ligand NES and NLS sequences (Kumagai and Dunphy, 1999; Yang et al., 1999; Davezac et al., 2000; Grozinger and Schreiber, 2000; Wang et al., 2000; Kao et al., 2001). Furthermore, not all 14-3-3–bound ligands are destined for cytoplasmic sequestration. For other molecules, 14-3-3 binding may preferentially sequester them within the nucleus as has been reported for the catalytic subunit of human telomerase (Seimiya et al., 2000) and the HOX11 homeobox transcription factor Tlx-2 (Tang et al., 1998). Thus, it appears that the ligand, rather than 14-3-3, dictates the resulting subcellular location (Muslin and Xing, 2000). In this manner, 14-3-3 functions as a type of “molecular chauffeur” where the destination of the 14-3-3–bound complex is determined by instructions contained within the sequence and structure of the bound cargo rather than through any intrinsic properties of 14-3-3. The ultimate fate of such cargo is then determined by 14-3-3–mediated alterations in the kinetics of dynamic nuclear-cytoplasmic transport.
Materials and methods
Antibodies
Monoclonal antibodies against 14-3-3σ were raised by immunizing DnJ mice with purified GST–14-3-3σ (Yaffe et al., 1997) and spleen fusions to NS1 myeloma cells performed as described (Dalal et al., 1999). Clones specifically recognizing 14-3-3σ but not the other 14-3-3 isoforms were identified by immunoprecipitation of in vitro–translated 14-3-3σ and immunoblotting using GST fusion proteins of all mammalian 14-3-3 isotypes. Additional details will be published elsewhere (unpublished data). An anti–phospho-14-3-3 binding motif antibody was obtained from New England Biolabs. An antibody recognizing all 14-3-3 isotypes (K-19) were purchased from Santa Cruz Biotechnology. Anti–α-tubulin antibody (DM1A) and anti-M2 antibody were purchased from Sigma-Aldrich. Anti-HA antibody (12CA5) and anti–lamin B (Ab-1) antibody were obtained from Roche-Boehringer Mannheim and Oncogene Research Products, respectively. Anti-Xpress antibody and Cy3-conjugated anti–mouse IgG secondary antibody were obtained from Invitrogen and Jackson ImmunoResearch Laboratories, respectively. The anti-phosphoThr32 FKHRL1 antibody has been described previously (Brunet et al., 1999).
Plasmids
Plasmids encoding GST–14-3-3ɛ, GST–14-3-3ɛ K49E, HA-FKHRL1 WT, and FKHRL1 T32A/S253A/S315A have been described previously (Brunet et al., 1999; Rittinger et al., 1999). GST–14-3-3ɛ ([1–214]; ΔC) and GST–14-3-3ɛ ([1–232]; ΔT) were generated by PCR amplification and cDNA cloning into pGEX4T. cDNAs of 14-3-3ɛ WT and 14-3-3ɛ K49E were subcloned into mammalian expression vector pcDNA3.1/His C (Invitrogen) to express Xpress epitope-tagged 14-3-3. The vector encoding the M2-tagged form of 14-3-3ζ subcloned in the pcDNAneo3 expression vector was provided by Dr. Haian Fu (Emory University School of Medicine, Atlanta, GA). Sequential point mutations of individual leucine and isoleucine residues to alanines in helix αI of GST–14-3-3ɛ were generated using specific oligonucleotides and the Quickchange site-directed mutagenesis kit (Stratagene). A mutant of FKHRL1 NES1 (amino acids 369–378) and NES2 (amino acids 386–396) was obtained as follows: first, a mutant of FKHRL1 NES2 (LMDDAADNATA) was generated using the following set of primers: forward, 5′-AAAGCTGCAGATAACGCCACGGCCCCGCCATCCCAGCCATCGC-3′ and reverse, 5′-AAAGGCCGTGGCGTTATCTGCAGCGTCGTCCATGAGGTTTTCAG-3′. The PCR fragment was subcloned into the Acc I sites of HA-FKHRL1 in the pECE vector. FKHRL1 NES2 mutant was then used as a template to generate FKHRL1 NES1-NES2 mutant (LTDMAGTANANDGATENLMDDAADNATA) using the following set of primers: forward, 5′-AAAGCGAATGCGAATGATGGGGCGACTGAAAACCTCATGGACGAC-3′ and reverse, 5′-AAACGCCCCATCATTCGCATTCGCGGTGCCTGCCATATCAGTCAGC-3′. The triple point mutants of FKHRL1 NLS sequences were produced with the Quickchange site-directed mutagenesis kit (Stratagene) using the following primers: RRR → AAA forward, 5′-GGGAAGAGCGGAAAAGCCCCCGCGGCGGCGGCTGTCTCCATGGACAATAGC-3′ and RRR → AAA reverse, 5′-GCTATTGTCCATGGAGACAGCCGCCGCCGCGGGGGCTTTTCCGCTCTTCCC-3′ and KKK → AAA forward, 5′-GAGCCGTGGCCGCGCAGCCGCGGCGGCGGCAGCCCTGCAGACAGCCCCCG-3′ and KKK → AAA reverse, 5′-CGGGGGCTGTCTGCAGGGCTGCCGCCGCCGCGGCTGCGCGGCCACGGCTC-3′.
Cell lines and transfection
The 293T human epithelial kidney cell line, human osteosarcoma U2OS cell line, and Chinese hamster lung fibroblast cell line CCL39 were cultured in DME supplemented with 10% FBS, and NIH3T3 cells were maintained in DME with 10% calf serum. LMB was used at final concentration of 20 ng/ml. To establish stable cell lines expressing 14-3-3 proteins, U2OS cells were transfected with pcDNA3.1/His-14-3-3ɛ or pcDNA3.1/His-14-3-3ɛ (K49E) using Superfect (QIAGEN) and transfected cells selected in media containing 400 μg/ml G418 (GIBCO BRL). After 2 wk, resistant colonies were tested for expression of epitope-tagged 14-3-3 by immunoblotting and used to propagate stable cell lines.
Cellular fractionation, immunoblotting, and 14-3-3 binding assays
U2OS cells were suspended in buffer A (10 mM Hepes, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.15% NP-40, 1 mg/ml each of AEBSF, aprotinin, leupeptin, and pepstatin), swollen for 10 min on ice, and centrifuged at 12,000 g for 30 s. Supernatant was collected as a cytoplasmic extract. The pellet was washed, resuspend in buffer B (20 mM Hepes, pH 7.9, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.5% NP-40, 4 μg/ml each of AEBSF, aprotinin, leupeptin, and pepstatin), and rocked for 15 min at 4°C. The supernatant after centrifugation was used as nuclear extract. Equal amounts of proteins from cytosol and nuclear extract were subjected to immunoblotting as described (Kanai et al., 2000). To investigate the distribution of 14-3-3 ligands, cells were metabolically labeled in the presence of [35S]methionine for 8 h and then separated into nuclear or cytoplasmic fractions as above. Samples (240 μg total protein containing equal amounts of radioactivity) were adjusted to the same final buffer composition and pulled down with beads containing 150 μg of GST–14-3-3ɛ or GST as described (Rittinger et al., 1999). Proteins binding to the beads were eluted, separated by SDS-PAGE, and analyzed by autoradiography. To analyze the interaction between FKHRL1 and 14-3-3, 293T cells transfected with HA-FKHRL1 constructs and M2–14-3-3ζ were lysed and subjected to immunoprecipitation and immunoblotting as described previously (Brunet et al., 1999).
Immunofluorescence microscopy
Immunofluorescent microscopy was done as described (Brunet et al., 1999; Kanai et al., 2000). U2OS cells grown on glass coverslips were fixed with 3% formaldehyde, permeabilized with 0.2% Triton X-100, and stained with an anti–14-3-3 antibody (K-19) or an anti–14-3-3σ monoclonal antibody and Cy3-conjugated secondary antibodies. Cells were counterstained with 1 μg/ml DAPI (Sigma-Aldrich) to visualize the nucleus. The anti-Xpress antibody was used to stain 14-3-3–expressing stable cell lines. For immunolocalization studies of FKHRL1, CCL39 fibroblasts on glass coverslips in 12-well dishes were transfected with 4 μg of the indicated plasmids using calcium phosphate. After 24 h, the cells were fixed in 4% formaldehyde/2% sucrose, permeabilized with 0.1% Triton X-100, and stained with anti-HA antibody, anti-phosphoT32 antibody, and Cy3- or Cy2-conjugated anti–mouse IgG secondary antibody and Cy3-conjugated anti–rabbit IgG secondary antibody. For quantification experiments, 150–300 cells were scored according to whether the protein of interest was higher in the nucleus, evenly distributed between nucleus and cytoplasm, or higher in cytoplasm. Microscopic images were acquired using Nikon Diaphot 300 microscope mounted with a Photometrics SenSys CCD camera and processed using Vaytek Imaging Software.
Acknowledgments
We thank Dr. M. Yoshida (University of Tokyo) for LMB, Colleen Schweitzer and Jianmin Gan (DeCaprio lab) for assistance with antibody production, and members of the Yaffe and Greenberg lab and Lewis C. Cantley for discussions.
A. Brunet was supported by a Goldenson-Berenberg fellowship, F. Kanai was supported by fellowships from the Cell Science Research Foundation and Sankyo Foundation of Life Science, and J. Stehn was supported by a C.J. Martin Fellowship from the National Health and Medical Research Council of Australia. J.V. Frangioni is a postdoctoral fellow of the Howard Hughes Medical Institute. S.N. Dalal was a fellow of the Leukemia Society of America. This work was funded by National Institutes of Health grant HD24926 and HD18655 to M.E. Greenberg, National Institutes of Health grant GM60594, and a Burroughs-Wellcome Career Development award to M.B. Yaffe.
A. Brunet and F. Kanai contributed equally to this work.
F. Kani's present address is Dept. of Gastroenterology, University of Tokyo, 7-3-1 Bunkyo-ku, Tokyo 113-8655, Japan.
Footnotes
Abbreviations used in this paper: IGF, insulin-like growth factor; LMB, leptomycin B; NES, nuclear export signal; NLS, nuclear localization signal; WT, wild-type.
References
- Beck, T., and M.N. Hall. 1999. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature. 402:689–692. [DOI] [PubMed] [Google Scholar]
- Brownawell, A.M., G.J. Kops, I.G. Macara, and B.M. Burgering. 2001. Inhibition of nuclear import by protein kinase B (Akt) regulates the subcellular distribution and activity of the forkhead transcription factor AFX. Mol. Cell. Biol. 21:3534–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet, A., A. Bonni, M.J. Zigmond, M.Z. Lin, P. Juo, L.S. Hu, M.J. Anderson, K.C. Arden, J. Blenis, and M.E. Greenberg. 1999. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 96:857–868. [DOI] [PubMed] [Google Scholar]
- Cahill, C.M., G. Tzivion, N. Nasrin, S. Ogg, J. Dore, G. Ruvkun, and M. Alexander-Bridges. 2001. Phosphatidylinositol 3-kinase signaling inhibits DAF-16 DNA binding and function via 14-3-3-dependent and 14-3-3-independent pathways. J. Biol. Chem. 276:13402–13410. [DOI] [PubMed] [Google Scholar]
- Dalal, S.N., C.M. Schweitzer, J. Gan, and J.A. DeCaprio. 1999. Cytoplasmic localization of human cdc25C during interphase requires an intact 14-3-3 binding site. Mol. Cell. Biol. 19:4465–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davezac, N., V. Baldin, B. Gabrielli, A. Forrest, N. Theis-Febvre, M. Yashida, and B. Ducommun. 2000. Regulation of CDC25B phosphatases subcellular localization. Oncogene. 19:2179–2185. [DOI] [PubMed] [Google Scholar]
- Fornerod, M., M. Ohno, M. Yoshida, and I.W. Mattaj. 1997. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 90:1051–1060. [DOI] [PubMed] [Google Scholar]
- Fu, H., R.R. Subramanian, and S.C. Masters. 2000. 14-3-3 proteins: structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 40:617–647. [DOI] [PubMed] [Google Scholar]
- Fukuda, M., S. Asano, T. Nakamura, M. Adachi, M. Yoshida, M. Yanagida, and E. Nishida. 1997. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature. 390:308–311. [DOI] [PubMed] [Google Scholar]
- Grozinger, C.M., and S.L. Schreiber. 2000. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc. Natl. Acad. Sci. USA. 97:7835–7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermeking, H., C. Lengauer, K. Polyak, T.C. He, L. Zhang, S. Thiagalingam, K.W. Kinzler, and B. Vogelstein. 1997. 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol. Cell. 1:3–11. [DOI] [PubMed] [Google Scholar]
- Kanai, F., P.A. Marignani, D. Sarbassova, R. Yagi, R.A. Hall, M. Donowitz, A. Hisaminato, T. Fujiwara, Y. Ito, L.C. Cantley, and M.B. Yaffe. 2000. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 19:6778–6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, H.Y., A. Verdel, C.C. Tsai, C. Simon, H. Juguilon, and S. Khochbin. 2001. Mechanism for nucleocytoplasmic shuttling of histone deacetylase 7. J. Biol. Chem. 276:47496–47507. [DOI] [PubMed] [Google Scholar]
- Kumagai, A., and W.G. Dunphy. 1999. Binding of 14-3-3 proteins and nuclear export control the intracellular localization of the mitotic inducer cdc25. Genes Dev. 13:1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai, A., P.S. Yakowec, and W.G. Dunphy. 1998. 14-3-3 proteins act as negative regulators of the mitotic inducer Cdc25 in Xenopus egg extracts. Mol. Biol. Cell. 9:345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, D., J. Bienkowska, C. Petosa, R.J. Collier, H. Fu, and R. Liddington. 1995. Crystal structure of the zeta isoform of the 14-3-3 protein. Nature. 376:191–194. [DOI] [PubMed] [Google Scholar]
- Lopez-Girona, A., B. Fumari, O. Mondesert, and P. Russel. 1999. Nuclear localization of cdc25 is regulated by DNA damage and a 14-3-3 protein. Nature. 397:172–175. [DOI] [PubMed] [Google Scholar]
- Muslin, A.J., and H. Xing. 2000. 14-3-3 proteins: regulation of subcellular localization by molecular interference. Cell. Signal. 12:703–709. [DOI] [PubMed] [Google Scholar]
- Muslin, A.J., J.W. Tanner, P.M. Allen, and A.S. Shaw. 1996. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 84:889–897. [DOI] [PubMed] [Google Scholar]
- Ossareh-Nazari, B., F. Bachelerie, and C. Dargemont. 1997. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science. 278:141–144. [DOI] [PubMed] [Google Scholar]
- Peng, C.Y., P.R. Graves, R.S. Thoma, Z. Wu, A.S. Shaw, and H. Piwnica-Worms. 1997. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 277:1501–1505. [DOI] [PubMed] [Google Scholar]
- Petosa, C., S.C. Masters, L.A. Bankston, J. Pohl, B. Wang, H. Fu, and R.C. Liddington. 1998. 14-3-3zeta binds a phosphorylated Raf peptide and an unphosphorylated peptide via its conserved amphipathic groove. J. Biol. Chem. 273:16305–16310. [DOI] [PubMed] [Google Scholar]
- Rena, G., A.R. Prescott, S. Guo, P. Cohen, and T.G. Unterman. 2001. Roles of the forkhead in rhabdomyosarcoma (FKHR) phosphorylation sites in regulating 14-3-3 binding, transactivation and nuclear targeting. Biochem. J. 354:605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittinger, K., J. Budman, J. Xu, S. Volinia, L.C. Cantley, S.J. Smerdon, S.J. Gamblin, and M.B. Yaffe. 1999. Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol. Cell. 4:153–166. [DOI] [PubMed] [Google Scholar]
- Seimiya, H., H. Sawada, Y. Muramatsu, M. Shimizu, K. Ohko, K. Yamane, and T. Tsuruo. 2000. Involvement of 14-3-3 proteins in nuclear localization of telomerase. EMBO J. 19:2652–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, S.J., T.C. Suen, R.R. McInnes, and M. Buchwald. 1998. Association of the TLX-2 homeodomain and 14-3-3zeta signaling proteins. J. Biol. Chem. 273:25356–25363. [DOI] [PubMed] [Google Scholar]
- Thorson, J.A., L.W. Yu, A.L. Hsu, N.Y. Shih, P.R. Graves, J.W. Tanner, P.M. Allen, H. Piwnica-Worms, and A.S. Shaw. 1998. 14-3-3 proteins are required for maintenance of Raf-1 phosphorylation and kinase activity. Mol. Cell. Biol. 18:5229–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzivion, G., Z. Luo, and J. Avruch. 1998. A dimeric 14-3-3 protein is an essential cofactor for Raf kinase activity. Nature. 394:88–92. [DOI] [PubMed] [Google Scholar]
- Tzivion, G., Y.H. Shen, and J. Zhu. 2001. 14-3-3 proteins; bringing new definitions to scaffolding. Oncogene. 20:6331–6338. [DOI] [PubMed] [Google Scholar]
- van Hemert, M.J., H.Y. Steensma, and G.P. van Heusden. 2001. 14-3-3 proteins: key regulators of cell division, signalling and apoptosis. Bioessays. 23:936–946. [DOI] [PubMed] [Google Scholar]
- Wang, A.H., M.J. Kruhlak, J. Wu, N.R. Bertos, M. Vezmar, B.I. Posner, D.P. Bazett-Jones, and X.J. Yang. 2000. Regulation of histone deacetylase 4 by binding of 14-3-3 proteins. Mol. Cell. Biol. 20:6904–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H., L. Zhang, R. Liddington, and H. Fu. 1998. Mutations in the hydrophobic surface of an amphipathic groove of 14-3-3zeta disrupt its interaction with Raf-1 kinase. J. Biol. Chem. 273:16297–16304. [DOI] [PubMed] [Google Scholar]
- Xiao, B., S.J. Smerdon, D.H. Jones, G.G. Dodson, Y. Soneji, A. Aitken, and S.J. Gamblin. 1995. Structure of a 14-3-3 protein and implications for coordination of multiple signalling pathways. Nature. 376:188–191. [DOI] [PubMed] [Google Scholar]
- Yaffe, M.B., K. Rittinger, S. Volinia, P.R. Caron, A. Aitken, H. Leffers, S.J. Gamblin, S.J. Smerdon, and L.C. Cantley. 1997. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell. 91:961–971. [DOI] [PubMed] [Google Scholar]
- Yang, J., K. Winkler, M. Yoshida, and S. Kornbluth. 1999. Maintenance of G2 arrest in the Xenopus oocyte: a role for 14-3-3-mediated inhibition of Cdc25 nuclear import. EMBO J. 18:2174–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Y., and H. Piwnica-Worms. 1999. DNA damage and replication checkpoints in fission yeast require nuclear exclusion of the Cdc25 phosphatase via 14-3-3 binding. Mol. Cell. Biol. 19:7410–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L., H. Wang, D. Liu, R. Liddington, and H. Fu. 1997. Raf-1 kinase and exoenzyme S interact with 14-3-3zeta through a common site involving lysine 49. J. Biol. Chem. 272:13717–13724. [DOI] [PubMed] [Google Scholar]