

Abstract
We studied the effect of microtubule-associated tau protein on trafficking of vesicles and organelles in primary cortical neurons, retinal ganglion cells, and neuroblastoma cells. Tau inhibits kinesin-dependent transport of peroxisomes, neurofilaments, and Golgi-derived vesicles into neurites. Loss of peroxisomes makes cells vulnerable to oxidative stress and leads to degeneration. In particular, tau inhibits transport of amyloid precursor protein (APP) into axons and dendrites, causing its accumulation in the cell body. APP tagged with yellow fluorescent protein and transfected by adenovirus associates with vesicles moving rapidly forward in the axon (∼80%) and slowly back (∼20%). Both movements are strongly inhibited by cotransfection with fluorescently tagged tau (cyan fluorescent protein–tau) as seen by two-color confocal microscopy. The data suggests a linkage between tau and APP trafficking, which may be significant in Alzheimer's disease.
Keywords: axonal traffic; microtubules; tau protein; amyloid precursor protein; oxidative stress
Introduction
Tau protein, a neuronal microtubule-associated protein (MAP),* is known for its role in the stabilization of microtubules, which is important for the generation and maintenance of neurites (Cleveland et al., 1977; Binder et al., 1985; Drubin and Kirschner, 1986; Caceres and Kosik, 1990). In Alzheimer's disease, tau accumulates in neurons, aggregates into paired helical filaments, and loses its microtubule-binding and stabilizing function, leading to the degeneration of neurons (Garcia and Cleveland, 2001). Besides the role in microtubule stabilization, tau has other functions, such as membrane interactions or anchoring of enzymes (Brandt et al., 1995; Lee et al., 1998; Liao et al., 1998; Sontag et al., 1999). A further recently discovered function of tau is its ability to inhibit the transport of cell components in cultured fibroblasts. This may lead to the gradual retraction of mitochondria toward the cell center where the minus ends of microtubules are anchored. A similar behavior is observed for the ER and intermediate filaments. These effects can be explained by an interference between tau and motors, leading to a preferential inhibition of kinesin-like motors, which are responsible for transport towards the cell periphery (Ebneth et al., 1998). If such effects of tau on intracellular transport also took place in Alzheimer's disease neurons, one might expect a shortage of mitochondria and peroxisomes in cell processes with a concomitant loss of energy production and accumulation of reactive oxygen species. In addition, the anterograde transport of vesicles necessary for growth cones and synapse function might be slowed down. With these questions in mind, we studied neuron-like cell models (N2a neuroblastoma), cultured primary cortical neurons from rats or mice, and chicken retinal ganglion cells (RGCs) for the effects of tau overexpression. We show that these cells have an intact microtubule network, but their processes are deficient in mitochondria or peroxisomes, their growth is retarded, and they are highly sensitive to oxidative stress. Neurofilament protein accumulates in the cell body rather than distributing along the processes, reminiscent of ALS-like symptoms in tau-expressing transgenic mice (Ishihara et al., 1999, 2001; Götz et al., 2001; Lewis et al., 2001). In addition, the transport of Golgi-derived vesicles into axons is inhibited. Notably, vesicles carrying the amyloid precursor protein (APP) are retained in the cell body, which would allow an enhanced production of toxic Aβ peptides (Xu et al., 1997). Since tau may be increased in Alzheimer's disease neurons (Khatoon et al., 1992), these observations could provide a link to pathological features observed in Alzheimer's disease, such as oxidative stress, impaired energy supply, loss of intracellular transport, or altered processing of APP.
Results
Tau inhibits the transport of peroxisomes, neurofilaments, and vesicles along neurites
Upon differentiation with retinoic acid, N2a cells develop neurites, which grow to typical lengths of 30 μm after 2 d. The neurites contain the components necessary for sustained growth, such as mitochondria for the generation of chemical energy, peroxisomes for detoxification of H2O2, neurofilaments and microtubules for structural stability and intracellular transport, and transport vesicles carrying supplies for the growth cone (Figs. 1 and 2). N2a cells contain only a low concentration of endogenous tau hardly visible by immunofluorescence and are therefore useful models for testing the consequences of enhanced tau expression. Its effect on peroxisomes is illustrated in Fig. 1 A; these organelles are normally distributed throughout the cell body and neurites, but in cells with elevated tau they are virtually absent from the neurites, become clustered in the cell center around the microtubule organizing center (MTOC), and the neurites are inhibited in their growth (Fig. 1 A). In contrast, microtubules, the tracks of transport, are present throughout the neurites. An analogous behavior is observed with Golgi-derived vesicles visualized by WGA (Fig. 1 B): in normal N2a cells, they are visible in the cell body (mostly around the Golgi apparatus) and along the extended neurites (Fig. 1 B), but in cells with elevated tau they are strongly reduced in neurites and instead are concentrated in the Golgi area. Likewise, neurofilaments and mitochondria, which are normally present in neurites (Fig. 1 D), become clustered around the MTOC and Golgi area where microtubules originate. In all examples, microtubules and associated proteins such as tau are still capable of spreading throughout the neurite, both in control cells and at elevated tau concentrations. This means that the tracks for intracellular transport are laid out in the neurite, but the transport along them is severely impaired.
Figure 1.
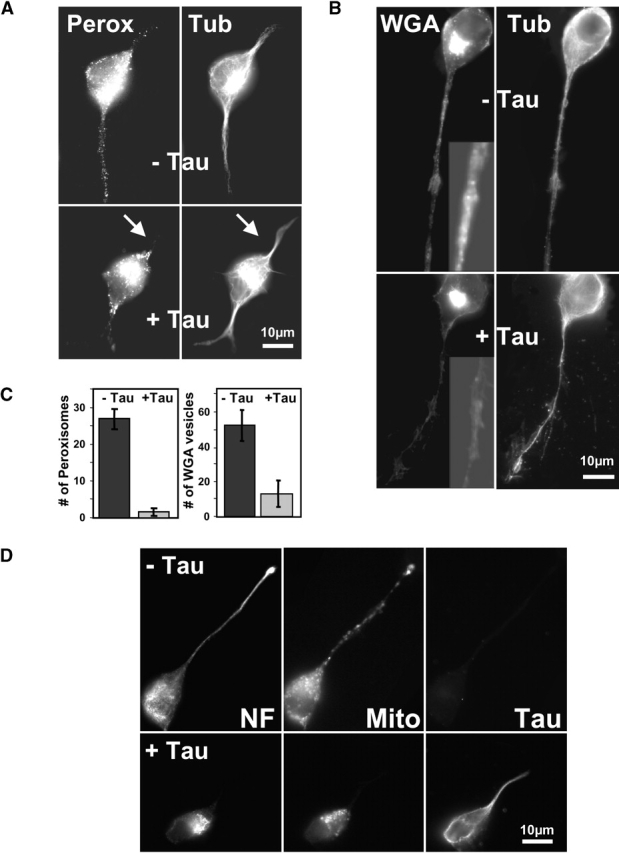
Tau causes the disappearance of vesicles and organelles from neurites. (A) Peroxisomes: N2a cells were differentiated for 2 d by 1 μM retinoic acid, fixed in methanol, and immunostained for peroxisomes (left) and tubulin (right). In mock-transfected controls (top), peroxisomes and MTs are visible in the cell body and throughout the neurite. In cells stably expressing tau (bottom), peroxisomes are mostly absent from the neurites and clustered around the MTOC in the cell body. These cells also have shorter neurites than the controls, but microtubules are still present throughout the neurites. (B) WGA-labeled vesicles: similar experiment as in A but staining with rhodamine-labeled WGA (left). In mock-transfected cells (top), WGA-stained vesicles (presumably Golgi-derived [Gonatas and Avrameas, 1977]) are visible in the cell body and neurite (inset, proximal neurite). In tau stable cells (bottom), the vesicles are strongly reduced in the neurites, and the trans-Golgi network is more contracted in the cell body. (C) Quantitation of the number of peroxisomes (left) and Golgi-derived vesicles (right) in neurites of N2a cells derived from 20-μm stretches of proximal neurites. In tau-stable cells, the density of peroxisomes decreases to ∼6% of the control cells, and the density of vesicles decreases to ∼25% of the control. (D) Disappearance of neurofilaments and mitochondria from neurites of tau-expressing N2a cells. In control N2a cells (top), neurofilaments (left, stained by antibody SMI32) and mitochondria (center, stained by MitoTracker Red) both extend along the entire length of the neurite, whereas tau (right, stained by antibody K9JA) is not visible in the cells because its endogenous concentration is too low. In tau-stable cells (bottom), neurofilaments and mitochondria are strongly reduced in the neurite and clustered in the cell body, whereas tau is visible in both compartments.
Figure 2.

Axons of primary rat neurons transfected by HSV-tau become depleted of mitochondria. (A, top) Rat primary hippocampal neurons contain mitochondria (left, stained with MitoTracker Red) and endogenous tau (right, stained with antibody KJ9A) in the cell body and neurites. (A, bottom) When primary neurons are treated with an HSV vector containing htau40 (transfection rate 75%), the positive cells are strongly stained with the tau antibody (bottom right), and mitochondria largely disappear from the neurites (bottom left, arrowhead). (B) Quantitation of mitochondria (left) and peroxisomes (right) measured in the proximal 50 μm of the neurite. The number of mitochondria decreases approximately ∼3-fold in tau-transfected cells compared with controls, and peroxisomes decrease ∼2.5-fold. The decrease of organelles is even more pronounced in the more distal parts of the neurites. Error bars show SEM; n = 10.
The quantitation of the density of organelles in neurites revealed a dramatic 17-fold decrease of peroxisomes in tau-transfected cells compared with controls (Fig. 1 C, left). The concentration of Golgi-derived vesicles in neurites also decreased strongly (about fourfold) (Fig. 1 C, right); in this case, the decrease was not as pronounced as for peroxisomes, presumably because of the higher diffusional mobility of vesicles, which makes them less dependent on active transport. For mitochondria, the results were comparable to peroxisomes, that is, nearly complete disappearance from neurites (Fig. 1 D, bottom). It implies that neurites lack the energy of ATP (because of the missing mitochondria) and lack protection against oxidative stress (because of the missing peroxisomes), both of which would explain why tau-expressing cells have fewer and shorter neurites (see below).
Elevation of tau causes transport defects in primary neuronal cells
Since N2a is a neuroblastoma cell line, we wanted to test if primary neurons would show a similar behavior when tau is increased. Hippocampal neurons were prepared from the cortices of rats or mice (following Banker and Goslin, 1998) and transfected with tau by three different methods, by herpes simplex virus (HSV) or adenovirus (AV) vectors containing the longest human tau isoform (htau40) or by the calcium phosphate method (Fig. 2). The transfected tau was detected by the enhanced immunofluorescence of an antibody against human tau (Fig. 2 A, bottom right) or by an antibody against the HA tag on tau. In control rat primary neurons, mitochondria were spread throughout the cell body and the cell processes (Fig. 2 A, top left). However, when cells were transfected with tau by either method mitochondria disappeared from the neurites and became concentrated in the cell body (Fig. 2 A, bottom left). Cells transfected by the vector only did not show an effect (unpublished data). At 16 h after transfection, the average number of mitochondria found in the initial 50 μm of the cell process decreased threefold (Fig. 2 B, left), and the decrease was even more marked further toward the growth cone. Similar observations were made with peroxisomes (Fig. 2 B, right). This means that the elevation of tau has similar effects on intracellular transport in primary neurons, neuron-like cell models, or nonneuronal cells, that is, the preferential inhibition of plus-end–directed transport by kinesin motors along microtubules so that minus-end–directed transport by a dynein-like motor becomes dominant.
Tau expression makes neurites vulnerable against stress
Since the depletion of organelles from the neurites in cells with elevated tau would be expected to make them more vulnerable, we tested their susceptibility against oxidative stress. Fig. 3 illustrates the reaction of differentiated N2a cells against 250 μM H2O2. This leads to a gradual loss of cell processes and eventually to cell death (Fig. 3 A). However, in tau-transfected cells the degradation of neurites is much more rapid (Fig. 3 A, bottom), and long neurites are more vulnerable than short ones. This was quantitated separately for long (>30 μm) neurites where the decay was noticeable already at lower H2O2 and shorter times (Fig. 3 B). Starting from a cell population differentiated for 2 d where 12.3% of the cells contained a long neurite, 150 μM H2O2 caused a loss of half the long neurites (down to 5.9%) (Fig. 3 B, third bar) in the control cells within 40 min. Tau-expressing cells had somewhat fewer long neurites to begin with (9.9%), but the loss upon exposure to H2O2 was much more dramatic (33-fold, down to 0.3%) (Fig. 3 B, seventh bar). This could be explained by the depletion of peroxisomes containing catalase for the detoxification of H2O2. We added catalase (0.02 U/μl) to the medium to check that the degeneration effect was due to the extracellular added hydrogen peroxide. Catalase alone had no effect on the survival of neurites (Fig. 3 B, second and sixth bar), but when challenging the cells with H2O2 and then immediately adding the catalase it provided an efficient protection (Fig. 3 B, fourth and eighth bars).
Figure 3.

Neurites of N2a cells overexpressing tau are highly susceptible to oxidative stress. (A, top) Control N2a cells (mock-transfected with vector) differentiated for 2 d and exposed to 250 μM H2O2 for up to 2 h. At 60 min, long neurites (>30 μm) stained for microtubules are less frequent but still clearly visible, and even at 120 min some thin neurites can be discerned. (A, bottom) Tau-stable N2a cells treated with H2O2. Neurites (>30 μm) disappear quickly and are no longer visible at 60 min. (B) Quantitation of oxidative stress by H2O2 and protection by catalase. Differentiated N2a cells (2 d) were treated with 30 μM H2O2, and neurites longer than 30 μm (twice the cell body diameter) were scored. In mock-transfected cells (left group of bars), the fraction of cells with long neurites drops from an initial value of 12.3 to 5.9% (∼ 2-fold) after 40 min of H2O2. In tau-transfected cells, (right group of bars) this fraction drops from an initial 9.9% down to 0.3% (∼33-fold). Adding catalase (0.02 U/μl) but no H2O2 to the medium does not change the number of long neurites (black bars). When catalase is added immediately after H2O2 exposure in control cells, it protects the cells so that 8.7% still have long neurites after 40 min H2O2 treatment compared with only 5.9% without catalase. In the case of tau-overexpressing cells, the fraction of cells with long neurites drops from 9.9 to 0.3% after H2O2 treatment but only to 5.6% with catalase. Thus, the higher vulnerability of neurites in tau-expressing cells can be explained by the lack of peroxisomes carrying catalase. Experiments were done in triplicate, 3 × 150 cells were counted for each condition. Error bars show SEM. (C) Inhibition of catalase exacerbates vulnerability of neurites against H2O2 in controls but not in tau-transfected cells. (Left) Mock-transfected cells differentiated for 2 d: the fraction of cells with long neurites (>30 μm) drops from 11.5% (untreated) to 5.2% after H2O2 treatment (30 μM, 40 min). Inhibition of catalase with 10 mM 3-AT for 60 min causes some degeneration (to 9.0%), but treatment first with 10 mM 3-AT for 60 min and then 30 μm H2O2 for 40 min causes a drop to 2.7%. Thus, the inhibition of catalase enhances the toxic effect of H2O2. (Right) N2a cells stably transfected with tau have a lower initial number of long neurites (8.4%). They are diminished somewhat by 3-AT treatment (to 6.6%) but almost disappear after H2O2 treatment (0.7%). In these conditions, a similar value (0.9%) is observed with 3-AT + H2O2 treatment because the peroxisomes are already excluded from the neurites (which is equivalent to catalase inhibition). Error bars show SEM; n = 300.
To corroborate these findings, we next tested the effect of inhibiting directly the endogenous catalase contained in the peroxisomes using the inhibitor 3-aminotriazole (3-AT). Among the population of mock-transfected N2a cells shown in Fig. 3 C, initially 11.5% contained long neurites. This value dropped to about half (5.2%) after exposure to 150 μM H2O2 for 40 min (Fig. 3 C, third bar). The decay became even more pronounced after pretreatment with 10 mM 3-AT (down to 2.7%) (Fig. 3 C, fourth bar), whereas 3-AT alone without H2O2 had only a moderate effect (decay to 9.0%) (Fig. 3 C, second bar). In tau-overexpressing N2a cells, H2O2 strongly reduced the number of neurites (from 8.4 to 0.7%) (Fig. 3 C, fifth compared with seventh bar), but in this case there was no further reduction by 3-AT (Fig. 3 C, eighth bar). The experiment illustrates that the inhibition of catalase exacerbates the oxidative damage induced by H2O2 but only if peroxisomes are present in the neurites. However, if they are depleted from the neurites by overexpression of tau the catalase inhibitor 3-AT has no target and thus no further effect. (Note that we disregard here, in first approximation, the diffusion of H2O2 between the neurites and the cell body where peroxisomes are concentrated in the tau-transfected cells because we are scoring only long neurites, and we also disregard the small contribution of cytosolic catalase.)
One concern in interpreting these results is that tau expression might have a negative effect on cell viability or the cell's capacity for detoxification, independently of tau's effect on transport after differentiation. In other words, elevated tau might somehow be toxic for the cell independently of its state of differentiation. Therefore, as a control the survival of undifferentiated N2a cells was checked by the 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT) test (which senses the reducing capacity of mitochondria [Mattson et al., 1995]) or the trypan blue test (which senses the leakiness of the plasma membrane). By both assays, survival rates were the same in tau-expressing and control cells (unpublished data). Furthermore, we compared the metabolic pathways for the detoxification of peroxides in N2a mock and tau-transfected cells. After treatment of the cells with 100 μM H2O2, the concentration of the peroxide decreased with comparable rates in both cell lines. The inhibitory effect of 3-AT was also similar (∼30%) and indicates that catalase is mainly responsible for H2O2 detoxification in both cell clones (unpublished data). Various concentrations (1 and 10 mM) of hydrogen peroxide were used to probe for an involvement of the glutathione system in peroxide detoxification. No immediate rise in glutathione disulfide was detectable after the addition of the peroxide (Dringen et al., 1999); thus, both cell clones make little use of glutathione peroxidase in detoxification of peroxide. Therefore, we conclude that the expression of tau is not toxic by itself, it has no negative effect on the overall biochemical pathways in the transfected cells as such, so that the decrease in neurite growth and viability must be ascribed to the inhibition of transport processes.
The trafficking of APP is inhibited by tau
APP is a membrane protein implicated in Alzheimer's disease by improper proteolysis and accumulation of its Aβ peptide fragment. After synthesis in neurons, it is initially transported to the axon by Golgi-derived vesicles, and later a small fraction travels back to the dendrites by transcytosis (Simons et al., 1995). Since tau inhibits the transport of vesicles down the axon, we investigated whether tau would affect the anterograde transport of APP as well using N2a cells stably transfected with the human isoform APP695 (Thinakaran et al., 1996) and detecting it by immunofluorescence. The inhibition is indeed observed. In the absence of tau, APP-containing vesicles can be visualized in the cell body around the Golgi area and throughout the neurite (Fig. 4 A). When the cells are additionally transfected with tau, it appears throughout the neurites (Fig. 4 A, right), but APP vesicles are strongly reduced in the neurites after 12 h. Quantification shows that the level of APP-carrying vesicles in neurites is decreased about threefold (Fig. 4 B) after tau transfection comparable to the observations with Golgi-derived vesicles in general (Fig. 1 B). The results demonstrate a direct connection between the elevation of tau and the inhibition of anterograde APP trafficking.
Figure 4.
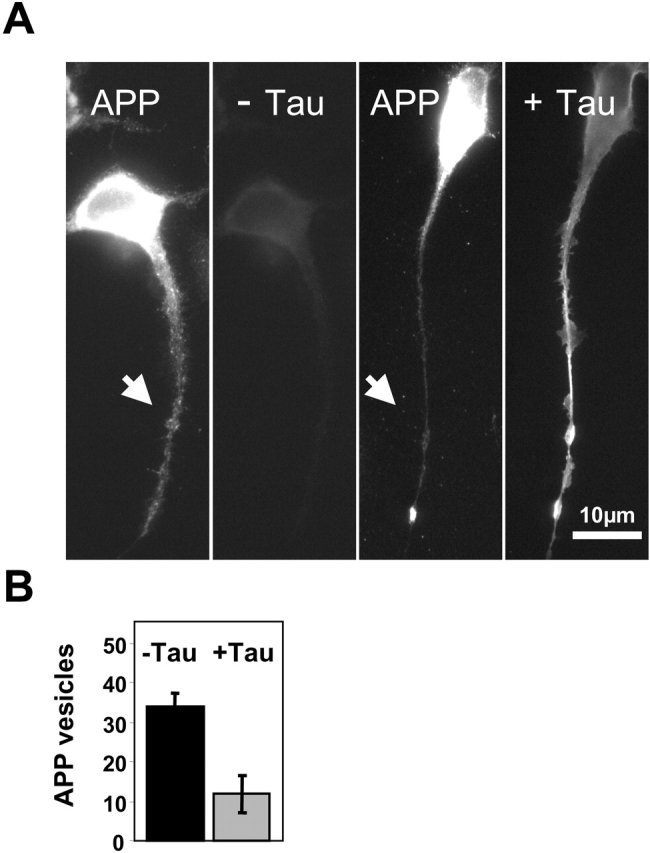
Reduction of APP-containing vesicles in neurites of N2a cells by tau. (A) N2a cells stably overexpressing APP695 were differentiated for 1 d by serum deprivation and 1 μM retinoic acid, and then transiently transfected with htau40 for 12 h. Cells were fixed by methanol and immunostained with antibodies against APP (myc tag) and tau (HA tag). (Left) Control cells without tau transfection show APP in the cell body and along neurites but no staining of tau. (Right) Cells transfected with tau show APP vesicles clustered in the Golgi area but not in the neurite (arrow), whereas tau is present throughout the neurite. (B) Quantitation: the number of APP vesicles stained with an antibody against human APP (B5 5313) in a stretch of 30-μm length drops approximately threefold after expression of tau.
Time-lapse imaging of mitochondria, APP-derived, and Golgi-derived vesicles and effect of tau in cortical and retinal ganglion neurons
Thus far we have described the effect of tau on the distribution of cell structures at fixed time points. The next step was to observe the movements of particles in real time in the presence or absence of tau. To analyze the transport of fluorescent APP vesicles in living cultured cortical neurons in the presence and absence of overexpressed htau40, APP and htau40 were tagged with yellow fluorescent protein (YFP) and cyan fluorescent protein (CFP), two spectral variants of green fluorescent protein (GFP), which allow simultaneous detection of two different fusion proteins in a living cell (Ellenberg et al., 1999; Lu and Kosik, 2001). For transfection of the cultured hippocampal neurons, the fusion constructs were each inserted into the genome of an E1/E3-deleted replication-deficient AV (serotype Ad5). Western Blots showed that the CFP-htau40 and APP-YFP proteins were properly expressed (unpublished data). Anterograde and retrograde movements of the YFP fluorescent structures were observed in transfected hippocampal neurons. The example of Fig. 5 A shows two particles moving toward each other at mean velocities of 0.60 μm/s (anterograde) and 0.36 μm/s (retrograde). Rapidly moving vesicles (instantaneous velocities up to 10 μm/s) often had an elongated morphology, whereas slowly or nonmoving fluorescent structures were globular. The mobility of axonal particles was generally more pronounced in the axons of cells with a low level of APP expression, whereas higher APP concentrations tended to induce clusters of immobile vesicles in “traffic jams.”
Figure 5.
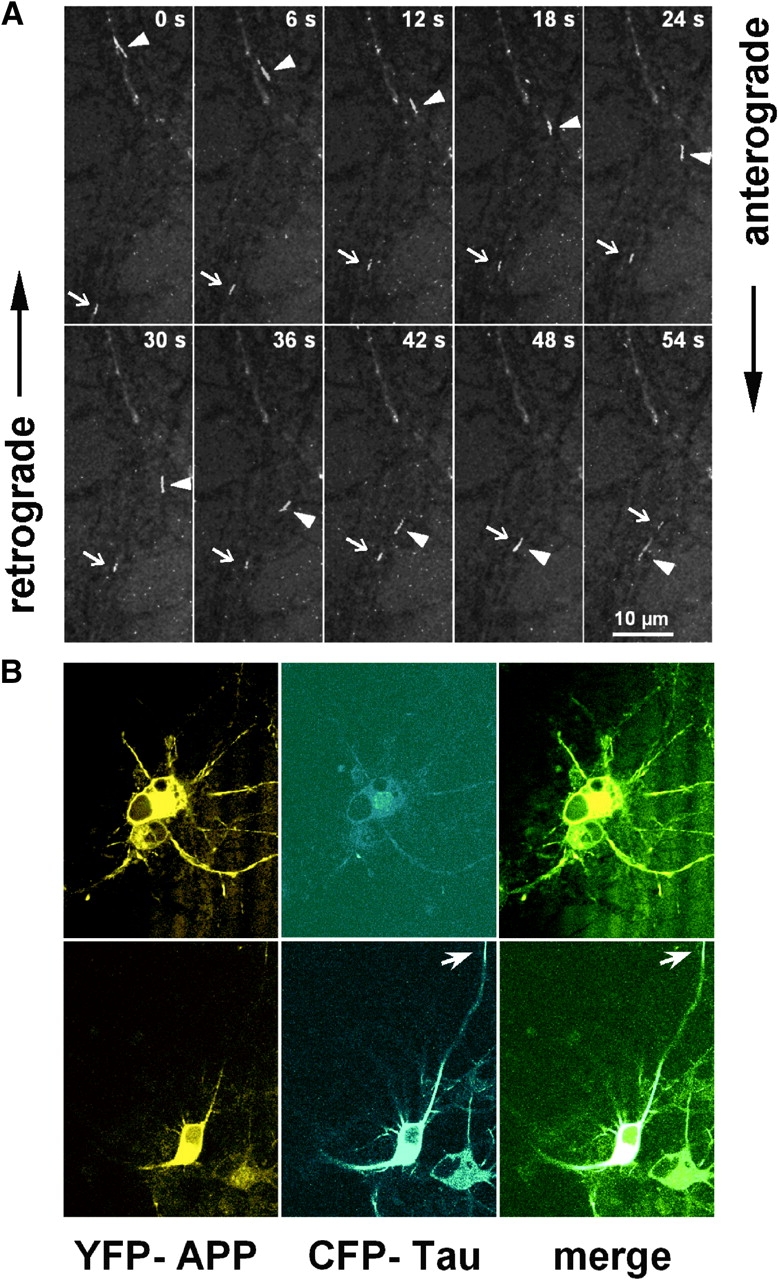
APP-YFP transport in cultured cortical neurons. (A) After 8 d in culture rat, cortical neurons were transfected with APP-YFP AV and analyzed 24 h later by live cell light microscopy. Confocal images were collected in 6-s time frames. The arrowheads and arrows point at two tubular vesicles carrying APP-YFP that move toward each other, crossover at t = 48 s, and continue without interference (velocities: top particle, 0.60 μm/s and bottom particle, 0.36 μm/s). (B) Dual color live imaging of APP and tau in cultured cortical neurons cotransfected with APP-YFP and CFP-htau40 AV and analyzed 24 h later by live cell confocal microscopy. Transfected neuron expressing APP-YFP (top) and doubly transfected neuron expressing APP-YFP and CFP-htau40 (bottom). Note that APP alone distributes throughout the cell body and neurites (top), but with elevated tau it is largely restricted to the cell body (bottom left), whereas tau is visible along the axons (bottom middle, arrow).
Although fluorescent particles were very visible in live cortical neurons, their direction of movement was often ambiguous, since the polarity of the cell processes and their identity (axon or dendrite) was not easily determined in the absence of markers. Therefore, we turned to the model system of chick RGCs where axons grow from a retinal explant with a well-defined polarity on a laminin-coated surface and show transport of APP vesicles (Walter et al., 1987; Morin et al., 1993; Amaratunga and Fine, 1995). This system allows the evaluation of parameters, such as anterograde and retrograde direction of movement, the fraction of moving particles, and the instantaneous and average velocities in both directions. Fig. 6 A shows the tip and growth cone of an axon of a retinal ganglion neuron after 50 h in culture, 24 h after transfection with APP-YFP (efficiency 40–80%). The highlighted elongated APP particle in Fig. 6 A moves with a speed of 4.5 μm/s toward the growth cone, remains stationary there, and eventually disappears. Instantaneous speeds of this particle reaches up to 7 μm/s. During the typical observation time of 6 min, ∼30% of the fluorescent vesicles were moving; the rest of the vesicles were stationary and clustered in immobile groups (these clusters were not included in the analysis) (Fig. 6 C). Movement was recorded in 3.9-s time intervals and occurred in both directions, predominantly (80%) in the anterograde direction. Velocities were remarkably heterogeneous among different particles, and even a given particle showed considerable variations in instantaneous speeds. 83 vesicles were scored in 13 axons in four experiments (Fig. 6, A and C). The same analysis was applied to cells labeled with fluorescent WGA (Fig. 7 A). In live cells, lectins added to the medium are taken up by endocytosis primarily in the neuronal cell body, accumulate in the trans-Golgi network, and can then be found in vesicles derived from the TGN (Gonatas and Avrameas, 1977). Thus, the labeled structures observed after 15 h in the axon are Golgi-derived vesicles. In this case, the fraction of moving particles was smaller (20%) than with APP vesicles, and velocities were slower (Fig. 7 C).
Figure 6.
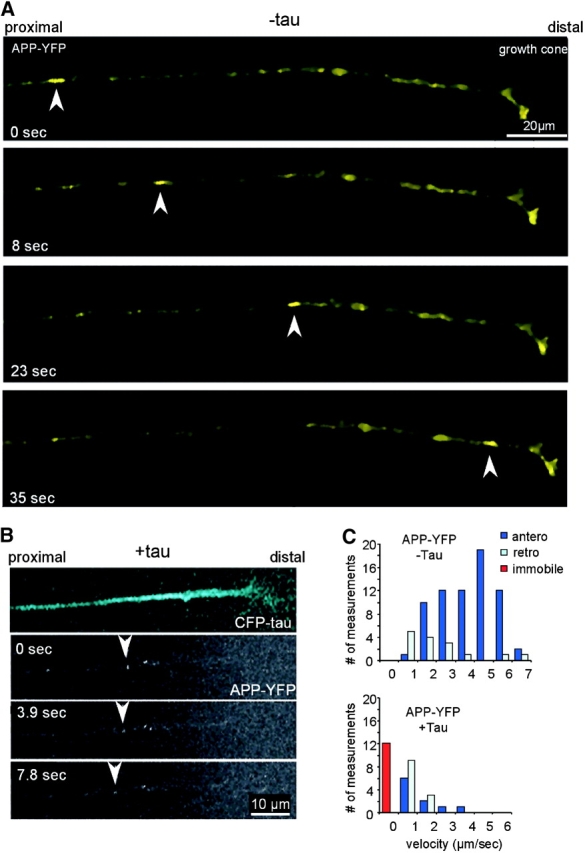
Time-lapse imaging of APP-YFP transport in cultured chick retinal ganglion neurons and inhibition by tau. (A) After 1 d in culture, the RGCs were transfected with APP-YFP AV and analyzed 48 h later (transfection rate 80%). Image frames were collected every 3.9 s. The arrowheads point at an elongated vesicle carrying APP-YFP moving rapidly toward the growth cone on the right (average velocity, 4.5 μm/s; instantaneous velocity, up to 7 μm/s). The particle resides in the growth cone and eventually disappears. (B) Double transfection of RGCs with CFP-htau40 and APP-YFP (30% double transfection). The top shows the blue fluorescence of tau throughout the axon and growth cone, and the bottom shows yellow fluorescent APP vesicles. Very few particles are visible in the presence of tau, and they slowly move in the retrograde direction (0.4 μm/s; arrowheads). (C) Quantification: fractions of particles moving in RGCs transfected with APP only or with APP and tau. Particle movement was classified by direction (anterograde and retrograde) and speed (bins of 1 μm/s). Anterograde vesicles predominate, and speeds can be up to 7 μm/s (clustered particles forming immobile traffic jams were not scored here). After transfection with tau, very few vesicles move, many are immobile (red bar), and slow speeds predominate in both directions.
Figure 7.
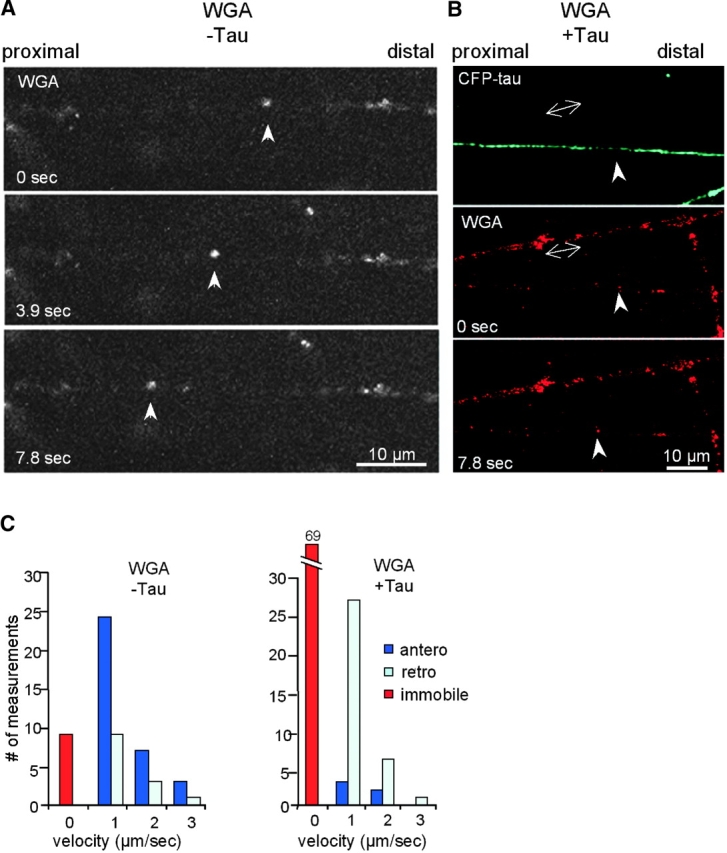
Transport of Golgi-derived vesicles in chick retinal ganglion neurons. The growth cone is on the right (outside the picture frame). (A) After 2 d in culture, RGCs were stained with WGA, and movements of vesicles were monitored by confocal microscopy. The arrowheads point to an example of a retrogradely moving particle at 1.2 μm/s. (B) After 2 d in culture, RGCs were transfected with CFP-tau, and 2 d later they were stained with WGA. The tau-expressing axon (top, blue) contains almost no WGA-stained vesicles; one of the rare retrograde movements is indicated by arrows (0.7 μm/s; middle and bottom). By contrast, the axon not overexpressing tau (double arrows) contain numerous mobile particles. (C) Quantification of WGA vesicle transport without or with tau transfection. WGA particles generally move more slowly than APP vesicles. The distribution between slow and medium velocities (shown in 1 μm/s bins) is roughly comparable without or with tau, but the absolute numbers of moving particles are much smaller after transfection with tau.
We next investigated the effect of tau on transport in RGCs by cotransfecting CFP-htau40 and APP-YFP (efficiency 20–40%). After 24 h, tau was expressed and distributed over the length of the axon and growth cone (Fig. 6 B, top). At the bottom of Fig. 6 B, an APP particle is seen, which slowly moves retrogradely with a speed of 0.4 μm/s. The overall effect on movement was dramatic: the axons became essentially devoid of vesicles, and there were almost no forward moving particles. The few remaining visible particles moved slowly (<1 μm/s) in the retrograde direction and had a globular shape. A similar picture was seen with WGA vesicle movements after transfection of RGCs with CFP-htau40: very few particles were visible in the axon expressing tau (Fig. 7 B, top, blue and bottom, arrowhead), and these were mostly stationary or moved only retrogradely at slow or medium speeds (Fig. 7 B, middle and bottom, arrowheads). By contrast, axons that do not express tau (Fig. 7 B, double arrows) have numerous vesicles, many of which are mobile in both directions.
The same effect of overexpressed tau on the transport of APP was seen in cultured hippocampal neurons, which were simultaneously transfected with APP-YFP and CFP-htau40 by using the recombinant adenoviral vectors. A sufficient amount of doubly transfected neurons was obtained (∼50% of cells). Transfection of APP alone resulted in APP vesicles distributed throughout the cell body and neurites (Fig. 5 B, top left), but cells containing both transfected APP and tau showed that APP was restricted mostly to the cell body, and dendrites and axons were almost completely devoid of YFP fluorescent vesicles (Fig. 5 B, bottom left). As a consequence, only very few moving YFP fluorescent structures could be observed, moving mainly retrogradely with velocities <0.4 μm/s (in this case the axons were clearly defined by the fluorescence of tau) (Fig. 5 B, arrow).
Similar experiments were performed to observe the movement of mitochondria (stained with Mitotracker red) in RGCs (Fig. 8). Without tau, most particles were mobile with instantaneous velocities ∼1 μm/s, and anterograde traffic dominated (Fig. 8 A). After AV-mediated tau transfection, about half of the mitochondria became immobile over extended periods, and the rest moved mainly retrogradely (Fig. 8 B). The quantification (Fig. 8 C) shows that tau biases the particles toward the immobile and retrograde fractions without significant changes in instantaneous velocities. These results explain the gradual disappearance of mitochondria from the cell processes discussed above (Fig. 1 D).
Figure 8.
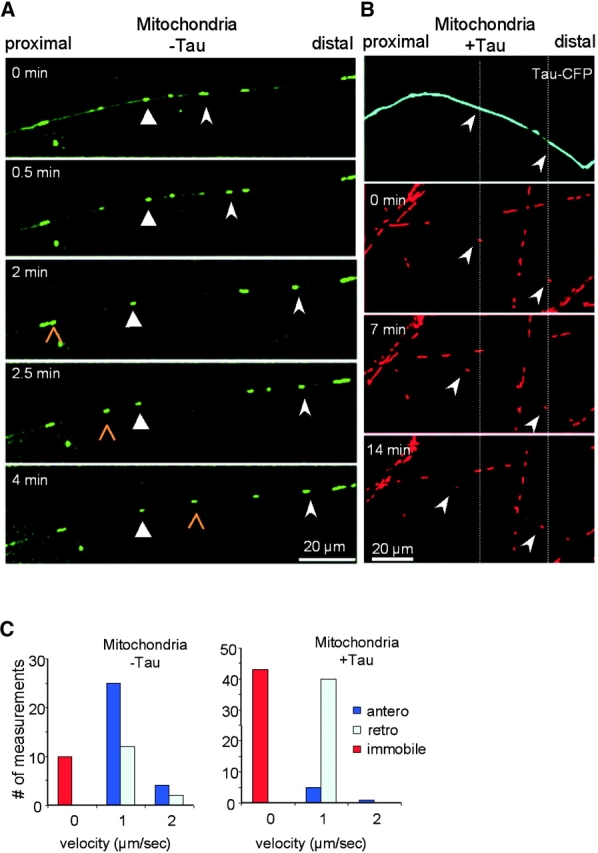
Transport of mitochondria in chick retinal ganglion neurons. The growth cone is on the right (outside the picture frame). (A) After 2 d in culture, RGCs were stained with MitoTracker green, and movements of mitochondria were monitored by confocal microscopy. The triangular arrowhead (middle) points at a nearly immobile particle (with only small excursion in either direction). The slender arrowhead (right) and the open arrowhead show particles with anterograde movement. Average velocities are ∼0.5 μm/s, but instantaneous velocities are ∼1 μm/s. (B) After 2 d in culture, RGCs were transfected with CFP-tau by AV (5 h) and incubated overnight with MitoTracker red and observed 24 h later by confocal microscopy. The tau-expressing axon (top, blue) contains very few mitochondria, which are mostly stationary or gradually move retrogradely (arrowheads). By contrast, the axons not overexpressing tau contain numerous mobile particles. (C) Quantification of mitochondria transport in 1 μm/s bins without or with tau transfection (left and right). Instantaneous velocities are similar in both direction (mostly ∼1 μm/s). Without tau, 20% of the mitochondria are immobile, 55% move anterogradely, and 25% move retrogradely. With tau, this distribution shifts to ∼50% immobile, ∼50% retrograde, and only a small fraction shows anterograde movement.
Discussion
Tau expression in neurons and retrograde transport bias
Tau has a variety of functions, most prominently in microtubule stabilization or neurite outgrowth (for reviews see Gundersen and Cook, 1999; Garcia and Cleveland, 2001). Another emerging role is the effect of tau and related MAPs on transport along microtubules (Sato-Harada et al., 1996; Bulinski et al., 1997; Ebneth et al., 1998). This emphasizes the interplay between the cytoskeletal fibers and motors in integrating the physiological functions within cell processes (Bradke and Dotti, 1998; Rogers and Gelfand, 2000). Tau influences the rates of attachment and detachment of motors from microtubules with the net result that dynein-mediated movements (toward the cell center) become predominant (Trinczek et al., 1999). The effect is strictly dependent on the binding of tau to microtubules; as shown previously, variants of tau which lack the microtubule-binding domain do not have an effect on transport, and conversely the size and composition of the nonbinding domains is of little importance (Ebneth et al., 1998; Trinczek et al., 1999). Similarly, other proteins have an effect on transport as long as they bind tightly to microtubules, such as MAP4 (Bulinski et al., 1997) or MAP2 (unpublished data). The predominance of minus-directed transport leads to the retraction of slow moving cell components, such as the ER, mitochondria, or peroxisomes. The transport bias can be overcome by inhibiting the retrograde motor dynein or by diffusion, that is, our earlier results on CHO cells showed that small vesicles with high mobility could still distribute throughout the cytoplasm even though their run lengths became shorter. Given this background, we asked what the effect of tau would be on highly asymmetric cells such as neurons where diffusion is much more restricted. Tau is the predominant MAP in axons (Binder et al., 1985; Riederer and Binder, 1994); if the plus-end–directed transport were retarded by elevated tau, this would impair the supply of material and energy to the growth cone and make the cell processes vulnerable. This issue gains additional significance in the context of Alzheimer's disease and related tauopathies, which are characterized by the elevation and aggregation of tau (see below).
We have tested three types of cells: differentiated neuroblastoma cells, primary hippocampal neurons from rat or mouse brain, and chick RGCs. Tau was elevated either by stable or transient transfection, transfection with viral vectors (AV or HSV), or the calcium phosphate method. We studied the distribution of cytoskeletal components (microtubules, tau, and neurofilaments), organelles (mitochondria and peroxisomes), and vesicles derived from the Golgi complex, in particular vesicles carrying APP. In addition, we monitored the growth of neurites and their response to oxidative stress. All of the observed effects support the view that tau generally inhibits transport along microtubules, preferentially in the plus-end direction, that is, toward the growth cone. This applies to organelles, vesicles, and neurofilaments (Figs. 1–3). The inhibition of transport is so efficient that organelles are largely excluded from cell processes, and vesicles are dramatically reduced (Fig. 1 C). In this regard, neurons are more vulnerable than the CHO cells studied earlier because diffusion is too limited. The effects of tau are observed both in neuron-like cell models (N2a), primary neurons, and RGCs, confirming that the underlying interactions between microtubules, motors, and cargo are similar.
The inhibition of organelles, vesicles, and neurofilaments is consistent with the view that these components are carried down the axon by a kinesin-dependent transport along microtubules (Hirokawa et al., 1998; Baas, 1999; Chou et al., 2001; Goldstein, 2001; Shea and Flanagan, 2001; Zhou et al., 2001). The motor-cargo connection may be direct (Liao and Gundersen, 1998; Yabe et al., 1999; Kamal et al., 2000), or it could be mediated by adaptor proteins (Morris and Hollenbeck, 1995; Ratner et al., 1998; Setou et al., 2000; Verhey et al., 2001). By contrast, the inhibition does not apply to microtubules or their associated proteins (e.g., tau itself), presumably because their transport into the cell process is mediated by a different type of mechanism. This would be consistent with the assumption that microtubules and MAPs might be carried by a dynein-mediated transport along actin microfilaments (Sheetz et al., 1998; Baas, 1999). Thus, tau highlights the difference between microtubules and neurofilaments even though both are transported as “slow components” of axonal transport. Using GFP-tagged intermediate filament subunits, several groups showed recently that they are transported by a fast motor of the kinesin family except that the average speed in axons is slowed down by frequent long pauses (Roy et al., 2000; Wang et al., 2000).
It is interesting to compare the tau-induced transport effects with the accumulation of proteins into aggresomes (Kopito, 2000). There is a superficial similarity because in both cases there is a net transport toward the microtubule minus ends around the MTOC. Aggresomes are formed by improperly folded and aggregated proteins, which are transported toward the cell center by a dynein-mediated process. They selectively contain cellular factors engaged in protein folding or degradation, such as proteasomes, heat shock proteins, and enzymes of the ubiquitin pathway, but aggresome formation does not affect the distribution of organelles such as mitochondria, peroxisomes, or the ER. Moreover, aggresomes represent the cell's response to (nearly) irreversible protein aggregation (e.g., proteins with polyglutamine stretches [Muchowski et al., 2000]); this does not occur with tau protein in transfected cells, which remains either microtubule-bound or highly soluble in the cytosol (Preuss et al., 1997; Lu and Kosik 2001). Finally, aggresomes cannot be redispersed, in contrast to the clusters of cell components described here, which dissolve again when microtubules are destroyed or when dynein-mediated transport is impaired (e.g., by dynamitin expression [Ebneth et al., 1998]).
One can expect that transport inhibition has serious consequences for the growth and survival of cell processes. We have tested this for two cases, the growth of neurites and their vulnerability to oxidative stress. Indeed, tau-transfected cells have shorter neurites (Fig. 3 A, bottom), and they become highly sensitive to oxidative conditions (H2O2). This is due to the absence of peroxisomes, since exogenous catalase can provide substantial protection (Fig. 3 B, left). The alternative pathway of detoxification by glutathione peroxidase plays little role in neurons (Dringen et al., 1999), and we have verified this for our cell lines (unpublished data). The role of catalase can be probed specifically with the catalase inhibitor 3-AT. In control cells, it amplifies the toxic effect of H2O2 but not in tau-expressing cells (Fig. 3 C). Thus, roughly speaking the loss of peroxisomes from the neurites due to elevated tau is as damaging as the direct inhibition of catalase. By the same argument, the exclusion of mitochondria from the cell processes implies a local depletion of ATP. This might be bearable for a compact cell where ATP diffuses throughout the cytoplasm but becomes a problem in extended cell processes.
Implications of tau-induced transport defects for neurodegeneration
The results described here may be important for understanding neurodegenerative disorders such as Alzheimer's disease, frontotemporal dementias, and others, which are characterized by elevated and aggregated tau protein (for reviews see Buee et al., 2000; Hutton, 2001). Alzheimer's disease is traditionally characterized by two types of protein deposits in the brain, the extracellular amyloid plaques, consisting largely of the peptide Aβ, a derivative of the membrane protein APP, and the intracellular neurofibrillary tangles, consisting mostly of tau protein (Price et al., 1998). However, these visible signs of pathology must be preceded by more subtle changes. The progression of the disease correlates with the spreading of the neurofibrillary tangles, whereas the amyloid plaques have a more generalized distribution (Braak and Braak, 1991; Arriagada et al., 1992). One of the earliest detectable signs is the loss of synapses and retrograde degeneration (“dying back”) of neurons, which appears to be accompanied by a decay of intracellular transport (Terry, 1998). We suggest that the data presented here speak both to the amyloid and the tau dysfunction, linking them through the impairment of intracellular traffic.
APP appears to have neurotrophic functions and is carried by kinesin-driven vesicles along axons and dendrites (Ferreira et al., 1993; Amaratunga et al., 1995; Simons et al., 1995; Kaether et al., 2000). The anterograde movement is dependent on a kinesin motor; specifically, the cytosolic COOH-terminal tail of APP interacts directly with a kinesin light chain (Kamal et al., 2000). Despite their high mobility, the APP vesicles are effectively eliminated from the axon once tau becomes elevated, and APP is found concentrated in the cell body (Fig. 5 B, bottom). This is a prominent locus for the generation of the Aβ peptides; in particular, both Aβ40 and the more toxic variant Aβ42 are generated in the trans-Golgi network (Xu et al., 1997). Thus, if the dwell time of APP were increased by a tau-dependent retardation of traffic one would expect an increase in the production of Aβ with the known pathological consequences of aggregation and toxicity. This would be analogous to the increased accumulation of Aβ by other treatments, which inhibit vesicle budding or transport (Greenfield et al., 1999). We note that the inhibition of APP transport by tau is a robust phenomenon that can be achieved in different cell models, for example, by transient transfection of tau in N2a cells stably transfected with APP or transfecting primary cortical or retinal ganglion neurons with APP and tau using AV vectors. In the latter case, the transfected proteins were tagged with fluorescent markers (YFP for APP and CFP for tau), which allows one to monitor vesicle movements and tau distributions by two-color live cell imaging. Several features were notable. (a) APP vesicles can be remarkably fast: whereas the known kinesins typically move ∼0.6–0.8 μm/s in vitro, APP vesicles in cortical and RGCs can reach up to 10 μm/s. (b) Fast transport occurs mainly in the anterograde direction: retrograde movement is generally slower, corresponding to the activity of dynein (Fig. 6 C). These results are in good agreement with recent findings by Kaether et al. (2000). (c) The majority of mobile particles (80%) moves anterogradely. This is the main reason for the strong bias of transport toward the synapse, independently of other transport characteristics. (d) Transport is nearly halted in both directions when tau is increased, that is, the number of moving particles becomes minute and the few mobile ones are retrograde (Fig. 6, B and C). (e) Remarkably, the transport infrastructure is much more resistant than traffic itself: microtubule tracks survive for many hours after mitochondria, peroxisomes, neurofilaments, APP vesicles, and others have deserted the neurite and accumulated in the cell body (Fig. 3), but eventually microtubules also disappear when the cell processes degenerate. On the basis of these data, one could imagine two potential causes for neuronal damage due to transport inhibition of APP. One is the depletion of APP from the synapse where it would loose its neurotrophic function (loss of function), and the second is its retention in the cell body where increased levels of Aβ would be produced (gain of toxic function). It remains to be seen which of these is more important in the context of brain tissue.
With regard to tau protein, the most visible change in Alzheimer's disease and related tauopathies is the aggregation into neurofibrillary tangles, which is accompanied by an increase in the level of tau, hyperphosphorylation, and loss of microtubule binding in the affected neurons. These observations are usually interpreted within a hypothesis where the physiological function of tau (stabilization of microtubules) is disrupted due to excess phosphorylation; the unbound tau then is thought to aggregate in a pathological manner and obstructs the cell interior, and the microtubules disassemble so that axonal transport is disrupted (for review see Buee et al., 2000). This hypothesis is based on the view that tau's role is to promote neurite outgrowth by stabilizing microtubule bundles. Indeed, tau is upregulated during neuronal differentiation, adult isoforms are generated by alternative splicing, and phosphorylation then gradually decreases, all of which favor tighter binding of tau to microtubules (Mandell and Banker, 1996). Thus, the presence of tau would appear to be beneficial for the neuron. However, several observations do not fit into this scheme: tau-deficient transgenic mice show no major phenotype, since tau can be substituted by other cofactors (MAP1b) (Takei et al., 2000), mice overexpressing tau show transport defects even though microtubules are intact and tau aggregates are absent or minor (Ishihara et al., 1999; Götz et al., 2001; Lewis et al., 2001), and tau-overexpressing flies also show defects in neuronal traffic without evidence of tau aggregation (Wittmann et al., 2001). These observations argue that even “normal” tau may be detrimental when it becomes elevated.
How can we reconcile the requirement for tau in neurite outgrowth with the damage inflicted on transport? A hint comes from the fact that the tau to tubulin ratio is normally quite low, in the range of a few percent (Cleveland et al., 1977). This means that relatively few tau molecules may suffice to initiate a growing neurite, and the tau concentration required for microtubule stabilization along the axon may be low in the presence of other stabilizing factors (consistent with the tau knockout results [Harada et al., 1994]). Thus, in a physiological environment tau's effect on transport is negligible. However, it can become noticeable if tau becomes elevated in degenerating neurons as reported by Khatoon et al. (1992). The reason for the increase in tau is unclear, but one possibility is the neuron's tendency for reactive sprouting to counteract toxic challenges in an aging brain (Savaskan and Nitsch, 2001). Once traffic jams are initiated, they are exacerbated by the loss of mitochondria and peroxisomes from the axons, eventually speeding up degeneration. This could be the situation mimicked by the cell models described here. In conclusion, tau may be beneficial for the neuron at physiologically low concentrations but becomes detrimental if the concentration is elevated. This may simply happen in response to an adverse cellular environment or a consequence of a possible transcription factor activity of the cleaved COOH-terminal stub of APP (as proposed recently [Cao and Sudhof, 2001]). In addition, the cell could make use of the “friction” of tau to actively regulate intracellular transport. This could be achieved by regulating the concentration of tau or more flexibly by the kinases and phosphatases that determine tau's affinity for microtubules. Thus, pathological “hyperphosphorylation” could be the neuron's response to a traffic jam.
Materials and methods
Cell lines
N2a neuroblastoma cells were grown in MEM with Earle's salts supplemented with 10% FCS, 1 mM glutamine, and 1% nonessential amino acids (Sigma-Aldrich) at 37°C with 5% CO2. Differentiation was induced by 1 μM retinoic acid, 0.1% FCS at a density of 2 × 104 cells per cm2 for 24–48 h. N2a cells were stably transfected with tau as described (Ebneth et al., 1998). An N2a neuroblastoma cell line stably transfected with APP695 was provided by Drs. G. Thinakaran and S. Sisodia (University of Chicago, Chicago, IL) (Thinakaran et al., 1996). Wistar rats and C57black mice for the preparation of primary neurons were obtained from the animal facility of Hamburg University Medical School.
Fusion proteins
APP-EYFP.
EYFP cDNA (CLONTECH Laboratories, Inc.) without start codon was fused to the 3′ end of human APP695 cDNA omitting the stop codon. The COOH-terminal part of APP was amplified from pSG5-hAPP695 (provided by B. De Strooper, Katholieke Universiteit Leuven, Leuven, Belgium). The fragment with HA tag was subcloned into a pCR Blunt II TOPO vector using the Zero Blunt TOPO cloning kit (Invitrogen). The resulting plasmid pCR Blunt II APP-HA was used for the insertion of EYFP DNA. The obtained fragment was subcloned into pCR Blunt II TOPO vector. The EYFP fragment was purified and cloned into pCR Blunt II APP-HA vector to obtain pCR Blunt II APP-HA-EYFP. The sequences generated by PCR were checked by sequencing.
ECFP-htau40.
The plasmid pECFP-htau40 was provided by J. Kupper, Max-Plank-Institute for Biochemistry, Martinsried, Germany. To allow the cloning of ECFP-htau40 into an AV vector, the SalI restriction site was introduced at the 5′ end of the ECFP-htau40 cDNA using QuikChange site-directed mutagenesis (Stratagene).
AV vectors encoding fluorescent fusion proteins
Recombinant AVs were generated following He et al. (1998). Briefly, the SalI-XbaI fragment that contained the CFP-htau40 cDNA and the HindIII-XbaI fragment that contained the APP-YFP cDNA were subcloned into the respective restriction sites of the pShuttle-CMV vector. The resulting plasmids were linearized with PmeI and cotransfected with the pAdEasy 1 vector into Escherichia coli BJ 5183 for homologous recombination. Plasmid DNA was amplified in E. coli DH10B and digested with PacI to cut out the entire recombinant adenoviral DNA, which was transfected into 911 cells, using Lipofectamine transfection (Life Technologies). The generation of the viruses in the 911 cells was monitored by fluorescence microscopy using an Axioplan fluorescence microscope (ZEISS). The cells were harvested, resuspended in PBS, and lysed by three freeze-thaw cycles. Cellular debris and nuclei were removed by centrifugation, and the virus suspension was purified by two CsCl gradient centrifugations. CsCl was removed by gel filtration and equilibrated in storage buffer (10 mM Tris/Cl, 135 mM NaCl, 3 mM KCl, 1 mM MgCl2, 10% glycerol, pH 8.0).
Transfection of primary cortex and retinal ganglion neurons
Cultures of E18 (rat) or E15 (mouse) hippocampal neurons were prepared according to Banker and Goslin (1998). Cells were plated in HBSS buffer (Biochrom) at a density of 7 × 104 cells per cm2 on a glass surface coated with poly-l-lysine (0.01% in 100 mM borate buffer, pH 8.5) and fibronectin (0.001% in HBSS; Life Technologies). To cultivate the neurons for live observation, 4.3 cm2 Lab-Tek chambers were used (Nunc) and coated as described above. Cells were transfected with tau either using a HSV vector or an AV vector. Cells were transfected between days 4 and 8 in culture. The HSV virus carrying the gene of htau40 was provided by Dr. R. Brandt, University of Heidelberg, Heidelberg, Germany. 10 μl virus suspension was added per 1.5 × 105 cells and incubated for 48 h. Then, the cells were fixed for immunofluorescence. For adenoviral transfection of APP-YFP or CFP-htau40, a 100-fold multiplicity of infection (multiplicity of infection of 100, 3 × 107 infectious particles) was applied to primary neurons. In the case of double transfections, 3 × 107 infectious particles of each recombinant AV were added. After 4 h incubation, the viral suspensions were removed. Vesicle movement was observed by confocal microscopy 24–48 h after transfection.
RGCs were prepared from white leghorn chicken eyes at embryonic day 7. Glass bottom dishes were coated overnight with 4 μg/ml laminin (Sigma-Aldrich), washed with sterile H2O, and dried. Retinae were mounted on nitrocellulose filter as described previously (Walter et al., 1987) and cut with a scalpel into 1–2-mm-wide stripes. Retinal explant was placed into the dish, and DME-F12 media (GIBCO BRL) with 10% FCS and 0.4% methyl cellulose was added. Explants were then cultured for 24 h at 37°C, 5% CO2 and 100% relative humidity prior viral transfection. Transfection and observation was done as above.
For staining of mitochondria in RGCs, the medium was removed 24–48 h after explantation of the retina and replaced with medium containing MitoTracker red 589 (Molecular Probes) at a final concentration of 12 nM or MitoTracker green FM (Molecular Probes) at a final concentration of 100 nM. Cells were incubated overnight under growth conditions. Then, the MitoTracker solution was replaced with fresh prewarmed medium, and movement of mitochondria was observed. For double labeling with CFP-tau AV, virus was added and removed after 5 h of incubation. MitoTracker red solution was added overnight. The expression of tau and the movement of mitochondria were observed by confocal microscopy 24–48 h after transfection.
Antibodies and dyes
Rat monoclonal antitubulin antibody YL1/2 and mouse monoclonal antibody DM1A were purchased from Serotec and Sigma-Aldrich. Polyclonal rabbit antitau antibody K9JA was from Dako, polyclonal rabbit anti-PMP69 antibody for peroxisomes was a gift from Dr. W. Just (University of Heidelberg, Heidelberg, Germany). All fluorescently (TRITC, FITC, and AMCA) labeled secondary antibodies were from DIANOVA. Fluorescent dyes MitoTracker red and rhodamine-labeled WGA were purchased from Molecular Probes. The monoclonal mouse antibody SMI32 (Chemicon) was used for the detection of unphosphorylated neurofilaments. The monoclonal tag antibodies from mouse against HA tag (12CA5) and myc tag were obtained from Roche Diagnostics and Invitrogen. Polyclonal antibody B5 (5313) against human APP (residues 444–592) was a gift from Dr. C. Haass (University of München, München, Germany), and monoclonal antibody 6E10 was from Senetek.
Immunofluorescence
Neurons and neuroblastoma cells were fixed in methanol or 2% paraformaldehyde and incubated with antibodies. Cells were examined with an Axioplan fluorescence microscope (ZEISS) equipped with an 100× oil-immersion objective and filters optimized for triple label experiments (FITC, TRITC, and AMCA fluorescence). Pictures were taken with a cooled CCD camera (Visitron) and analyzed using the MetaMorph software package.
Quantification of vesicles, organelles, and neurofilament protein in N2a cells
Peroxisomes were stained with polyclonal anti-PMP69 antibody (45 min, 1:200). Golgi-derived vesicles were stained with 10 μM rhodamine-labeled WGA (Molecular Probes) for 5 min after methanol fixation. Mitochondria were visualized by adding 400 nM MitoTracker red (Molecular Probes) for 30 min in media at 37°C before fixation. Vesicles carrying APP were stained either with monoclonal anti-myc antibody (1:200; Invitrogen) or polyclonal anti-APP antibody (1:300). After fixation and immunofluorescence labeling, the pictures were recorded and a defined area (20–30 μm in length for N2a cells and 50 μm for neurons) beginning at the proximal neurite was circumscribed manually with the MetaMorph drawing tools. Usually >100 cells were recorded per experiment. Afterwards, signals of vesicles and organelles above the background threshold were visualized and counted using the threshold function of MetaMorph. Both in tau- and mock-transfected cells we quantified and compared commensurate areas.
Live cell light microscopy
For visualizing tau, primary cortex neurons (E18) were transfected at day 4 in culture with CFP-tau40 AV (or control virus) at multiplicity of infection of 30 for 24 h. For tracking APP vesicles, transfection with APP-YFP AV at multiplicity of infection of 100 was done at day 8 in culture for 4 h, and analysis was done 24 h later. For observing vesicles labeled with fluorescent lectins, cells at day 4 in culture were incubated with rhodamine-WGA (Molecular Probes) at a final concentration of 4 μg/ml for 15 min. Vesicles were tracked with an Axioplan fluorescence microscope (ZEISS). To visualize CFP fluorescence of CFP-htau40, the FITC filter set was employed; WGA-rhodamine–labeled vesicles were observed by using the TRITC filter set. The object plane was kept at 37°C by air heating. APP-YFP vesicles were tracked with a LSM 510 confocal microscope (ZEISS), equipped with an 63× oil-immersion objective, beam path, and laser settings for YFP and CFP fluorescence and a 37°C air-heated object plane. Image analysis was performed with LSM 510 software.
Response of cells to oxidative stress
N2a cells differentiated on coverslips were incubated in 150 μM H2O2 in MEM medium for 40 min or more. Cells were fixed in methanol, stained for immunofluorescence with antibodies K9JA (for tau) and YL1/2 (for tubulin), and the number and length of neurites was recorded. Protection against H2O2 was done by adding catalase (0.02 U/μl; Sigma-Aldrich) immediately after H2O2. Catalase was inhibited by 3-AT (Sigma-Aldrich) in concentrations of 0.1, 1, and 10 mM and incubating for 30 or 60 min before the addition of H2O2. The integrity of mitochondria was measured by the 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide test, and the viability of cells was measured by the trypan blue assay (Mattson et al., 1995). The capacity of the glutathione system for peroxide detoxification was determined following Dringen et al. (1999).
Acknowledgments
We thank G. Thinakaran and S. Sisodia (University of Chicago) for APP-expressing N2a cells, R. Brandt (University of Heidelberg) for the HSV vector containing htau40, and B. De Strooper (Katholieke Universiteit Leuven) for the APP plasmid. Antibodies against peroxisomes were kindly provided by W. Just (University of Heidelberg). We are grateful to J. Biernat, A. Marx (Hamburg) and T. Shea (University of Massachusetts, Lowell, Massachusetts) for help with clones, data processing, and stimulating discussions, and to K. Neumann and N. Habbe for excellent technical assistance.
This research was supported in part by the Deutsche Forschungsgemeinschaft.
Footnotes
Abbreviations used in this paper: APP, amyloid precursor protein; 3-AT, 3-aminotriazole; AV, adenovirus; CFP, cyan fluorescent protein; GFP, green fluorescent protein; HSV, herpes simplex virus; MAP, microtubule-associated protein; MTOC, microtubule organizing center; RGC, retinal ganglion cell; YFP, yellow fluorescent protein.
References
- Amaratunga, A., and R.E. Fine. 1995. Generation of amyloidogenic C-terminal fragments during rapid axonal transport in vivo of beta-amyloid precursor protein in the optic nerve. J. Biol. Chem. 270:17268–17272. [DOI] [PubMed] [Google Scholar]
- Amaratunga, A., S.E. Leeman, K.S. Kosik, and R.E. Fine. 1995. Inhibition of kinesin synthesis in vivo inhibits the rapid transport of representative proteins for three transport vesicle classes into the axon. J. Neurochem. 64:2374–2376. [DOI] [PubMed] [Google Scholar]
- Arriagada, P.V., J.H. Growdon, E.T. Hedley-Whyte, and B.T. Hyman. 1992. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 42:631–639. [DOI] [PubMed] [Google Scholar]
- Baas, P.W. 1999. Microtubules and neuronal polarity: lessons from mitosis. Neuron. 22:23–31. [DOI] [PubMed] [Google Scholar]
- Banker, G.A., and K. Goslin. 1998. Culturing Nerve Cells. MIT Press, Cambridge, Massachusetts. 666 pp.
- Binder, L.I., A. Frankfurter, and L.I. Rebhun. 1985. The distribution of tau in the mammalian central nervous system. J. Cell Biol. 101:1371–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak, H., and E. Braak. 1991. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 82:239–259. [DOI] [PubMed] [Google Scholar]
- Bradke, F., and C.G. Dotti. 1998. Membrane traffic in polarized neurons. Biochim. Biophys. Acta. 1404:245–258. [DOI] [PubMed] [Google Scholar]
- Brandt, R., J. Leger, and G. Lee. 1995. Interaction of tau with the neural plasma membrane mediated by tau's amino-terminal projection domain. J. Cell Biol. 131:1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buee, L., T. Bussiere, V. Buee-Scherrer, A. Delacourte, and P.R. Hof. 2000. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 33:95–130. [DOI] [PubMed] [Google Scholar]
- Bulinski, J.C., T.E. McGraw, D. Gruber, H.L. Nguyen, and M.P. Sheetz. 1997. Overexpression of MAP4 inhibits organelle motility and trafficking in vivo. J. Cell Sci. 110:3055–3064. [DOI] [PubMed] [Google Scholar]
- Caceres, A., and K.S. Kosik. 1990. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature. 343:461–463. [DOI] [PubMed] [Google Scholar]
- Cao, X., and T.C. Sudhof. 2001. A transcriptionally active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 293:115–120. [DOI] [PubMed] [Google Scholar]
- Chou, Y.H., B.T. Helfand, and R.D. Goldman. 2001. New horizons in cytoskeletal dynamics: transport of intermediate filaments along microtubule tracks. Curr. Opin. Cell Biol. 13:106–109. [DOI] [PubMed] [Google Scholar]
- Cleveland, D.W., S.Y. Hwo, and M.W. Kirschner. 1977. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 116:227–247. [DOI] [PubMed] [Google Scholar]
- Dringen, R., L. Kussmaul, J.M. Gutterer, J. Hirrlinger, and B. Hamprecht. 1999. The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. J. Neurochem. 72:2523–2530. [DOI] [PubMed] [Google Scholar]
- Drubin, D.G., and M.W. Kirschner. 1986. Tau protein function in living cells. J. Cell Biol. 103:2739–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebneth, A., R. Godemann, K. Stamer, S. Illenberger, B. Trinczek, and E. Mandelkow. 1998. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J. Cell Biol. 143:777–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenberg, J., J. Lippincott-Schwartz, and J.F. Presley. 1999. Dual-colour imaging with GFP variants. Trends Cell Biol. 9:52–56. [DOI] [PubMed] [Google Scholar]
- Ferreira, A., A. Caceres, and K.S. Kosik. 1993. Intraneuronal compartments of the amyloid precursor protein. J. Neurosci. 13:3112–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, M.L., and D.W. Cleveland. 2001. Going new places using an old MAP: tau, microtubules and human neurodegenerative disease. Curr. Opin. Cell Biol. 13:41–48. [DOI] [PubMed] [Google Scholar]
- Goldstein, L.S. 2001. Kinesin molecular motors: transport pathways, receptors, and human disease. Proc. Natl. Acad. Sci. USA. 98:6999–7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonatas, N.K., and S. Avrameas. 1977. Detection of carbohydrates with lectin-peroxidase conjugates. Methods Cell Biol. 15:387–406. [PubMed] [Google Scholar]
- Götz, J., F. Chen, J. van Dorpe, and R.M. Nitsch. 2001. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by AB42 fibrils. Science. 293:1491–1495. [DOI] [PubMed] [Google Scholar]
- Greenfield, J.P., J. Tsai, G.K. Gouras, B. Hai, G. Thinakaran, F. Checler, S.S. Sisodia, P. Greengard, and H. Xu. 1999. Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc. Natl. Acad. Sci. USA. 96:742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen, G.G., and T.A. Cook. 1999. Microtubules and signal transduction. Curr. Opin. Cell Biol. 11:81–94. [DOI] [PubMed] [Google Scholar]
- Harada, A., K. Oguchi, S. Okabe, J. Kuno, S. Terada, T. Ohshima, R. Sato-Yoshitake, Y. Takei, T. Noda, and N. Hirokawa. 1994. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 369:488–491. [DOI] [PubMed] [Google Scholar]
- He, T.C., S. Zhou, L.T. da Costa, J. Yu, K.W. Kinzler, and B. Vogelstein. 1998. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA. 95:2509–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa, N., Y. Noda, and Y. Okada. 1998. Kinesin and dynein superfamily proteins in organelle transport and cell division. Curr. Opin. Cell Biol. 10:60–73. [DOI] [PubMed] [Google Scholar]
- Hutton, M. 2001. Missense and splice site mutations in tau associated with FTDP-17: multiple pathogenic mechanisms. Neurology. 56:S21–S25. [DOI] [PubMed] [Google Scholar]
- Ishihara, T., M. Hong, B. Zhang, Y. Nakagawa, M.K. Lee, J.Q. Trojanowski, and V.M. Lee. 1999. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 24:751–762. [DOI] [PubMed] [Google Scholar]
- Ishihara, T., M. Higuchi, B. Zhang, Y. Yoshiyama, M. Hong, J.Q. Trojanowski, and V.M. Lee. 2001. Attenuated neurodegenerative disease phenotype in tau transgenic mouse lacking neurofilaments. J. Neurosci. 21:6026–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaether, C., P. Skehel, and C.G. Dotti. 2000. Axonal membrane proteins are transported in distinct carriers: a two-color video microscopy study in cultured hippocampal neurons. Mol. Biol. Cell. 11:1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal, A., G.B. Stokin, Z. Yang, C.H. Xia, and L.S. Goldstein. 2000. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron. 28:449–459. [DOI] [PubMed] [Google Scholar]
- Khatoon, S., I. Grundke-Iqbal, and K. Iqbal. 1992. Brain levels of microtubule-associated protein tau are elevated in Alzheimer's disease: a radioimmuno-slot-blot assay for nanograms of the protein. J. Neurochem. 59:750–753. [DOI] [PubMed] [Google Scholar]
- Kopito, R.R. 2000. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10:524–530. [DOI] [PubMed] [Google Scholar]
- Lee, G., S.T. Newman, D.L. Gard, H. Band, and G. Panchamoorthy. 1998. Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 111:3167–3177. [DOI] [PubMed] [Google Scholar]
- Lewis, J., D.W. Dickson, W.L. Lin, L. Chisholm, A. Corral, G. Jones, S.H. Yen, N. Sahara, L. Skipper, D. Yager, et al. 2001. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 293:1487–1491. [DOI] [PubMed] [Google Scholar]
- Liao, G., and G.G. Gundersen. 1998. Kinesin is a candidate for cross-bridging microtubules and intermediate filaments. Selective binding of kinesin to detyrosinated tubulin and vimentin. J. Biol. Chem. 273:9797–9803. [DOI] [PubMed] [Google Scholar]
- Liao, H., Y. Li, D.L. Brautigan, and G.G. Gundersen. 1998. Protein phosphatase 1 is targeted to microtubules by the microtubule-associated protein Tau. J. Biol. Chem. 273:21901–21908. [DOI] [PubMed] [Google Scholar]
- Lu, M., and K.S. Kosik. 2001. Competition for microtubule-binding with dual expression of tau missense and splice isoforms. Mol. Biol. Cell. 12:171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell, J.W., and G.A. Banker. 1996. A spatial gradient of tau protein phosphorylation in nascent axons. J. Neurosci. 16:5727–5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson, M.P., S.W. Barger, J.G. Begley, and R.J. Mark. 1995. Calcium, free radicals, and excitotoxic neuronal death in primary cell culture. Methods Cell Biol. 46:187–216. [DOI] [PubMed] [Google Scholar]
- Morin, P.J., C.R. Abraham, A. Amaratunga, R.J. Johnson, G. Huber, J.H. Sandell, and R.E. Fine. 1993. Amyloid precursor protein is synthesized by retinal ganglion cells, rapidly transported to the optic nerve plasma membrane and nerve terminals, and metabolized. J. Neurochem. 61:464–473. [DOI] [PubMed] [Google Scholar]
- Morris, R.L., and P.J. Hollenbeck. 1995. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J. Cell Biol. 131:1315–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchowski, P.J., G. Schaffar, A. Sittler, E.E. Wanker, M.K. Hayer-Hartl, and F.U. Hartl. 2000. Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. USA. 97:7841–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss, U., J. Biernat, E.M. Mandelkow, and E. Mandelkow. 1997. The ‘jaws’ model of tau-microtubule interaction examined in CHO cells. J. Cell Sci. 110:789–800. [DOI] [PubMed] [Google Scholar]
- Price, D.L., S.S. Sisodia, and D.R. Borchelt. 1998. Genetic neurodegenerative diseases: the human illness and transgenic models. Science. 282:1079–1083. [DOI] [PubMed] [Google Scholar]
- Ratner, N., G.S. Bloom, and S.T. Brady. 1998. A role for cyclin-dependent kinase(s) in the modulation of fast anterograde axonal transport: effects defined by olomoucine and the APC tumor suppressor protein. J. Neurosci. 18:7717–7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer, B.M., and L.I. Binder. 1994. Differential distribution of tau proteins in developing cat cerebellum. Brain Res. Bull. 33:155–161. [DOI] [PubMed] [Google Scholar]
- Rogers, S.L., and V.I. Gelfand. 2000. Membrane trafficking, organelle transport, and the cytoskeleton. Curr. Opin. Cell Biol. 12:57–62. [DOI] [PubMed] [Google Scholar]
- Roy, S., P. Coffee, G. Smith, R.K. Liem, S.T. Brady, and M.M. Black. 2000. Neurofilaments are transported rapidly but intermittently in axons: implications for slow axonal transport. J. Neurosci. 20:6849–6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato-Harada, R., S. Okabe, T. Umeyama, Y. Kanai, and N. Hirokawa. 1996. Microtubule-associated proteins regulate microtubule function as the track for intracellular membrane organelle transports. Cell Struct. Funct. 21:283–295. [DOI] [PubMed] [Google Scholar]
- Savaskan, N.E., and R. Nitsch. 2001. Molecules involved in reactive sprouting in the hippocampus. Rev. Neurosci. 12:195–215. [DOI] [PubMed] [Google Scholar]
- Setou, M., T. Nakagawa, D.H. Seog, and N. Hirokawa. 2000. Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor-containing vesicle transport. Science. 288:1796–1802. [DOI] [PubMed] [Google Scholar]
- Shea, T.B., and L.A. Flanagan. 2001. Kinesin, dynein and neurofilament transport. Trends Neurosci. 24:644–648. [DOI] [PubMed] [Google Scholar]
- Sheetz, M.P., K.K. Pfister, J.C. Bulinski, and C.W. Cotman. 1998. Mechanisms of trafficking in axons and dendrites: implications for development and neurodegeneration. Prog. Neurobiol. 55:577–594. [DOI] [PubMed] [Google Scholar]
- Simons, M., E. Ikonen, P.J. Tienari, A. Cid-Arregui, U. Monning, K. Beyreuther, and C.G. Dotti. 1995. Intracellular routing of human amyloid protein precursor: axonal delivery followed by transport to the dendrites. J. Neurosci. Res. 41:121–128. [DOI] [PubMed] [Google Scholar]
- Sontag, E., V. Nunbhakdi-Craig, G. Lee, R. Brandt, C. Kamibayashi, J. Kuret, C.L. White III, M.C. Mumby, and G.S. Bloom. 1999. Molecular interactions among protein phosphatase 2A, tau, and microtubules. Implications for the regulation of tau phosphorylation and the development of tauopathies. J. Biol. Chem. 274:25490–25498. [DOI] [PubMed] [Google Scholar]
- Takei, Y., J. Teng, A. Harada, and N. Hirokawa. 2000. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J. Cell Biol. 150:989–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry, R.D. 1998. The cytoskeleton in Alzheimer disease. J. Neural Transm. Suppl. 53:141–145. [DOI] [PubMed] [Google Scholar]
- Thinakaran, G., D.B. Teplow, R. Siman, B. Greenberg, and S.S. Sisodia. 1996. Metabolism of the “Swedish” amyloid precursor protein variant in neuro2a (N2a) cells. Evidence that cleavage at the “beta-secretase” site occurs in the golgi apparatus. J. Biol. Chem. 271:9390–9397. [DOI] [PubMed] [Google Scholar]
- Trinczek, B., A. Ebneth, E.M. Mandelkow, and E. Mandelkow. 1999. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J. Cell Sci. 112:2355–2367. [DOI] [PubMed] [Google Scholar]
- Verhey, K.J., D. Meyer, R. Deehan, J. Blenis, B.J. Schnapp, T.A. Rapoport, and B. Margolis. 2001. Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J. Cell Biol. 152:959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, J., B. Kern-Veits, J. Huf, B. Stolze, and F. Bonhoeffer. 1987. Recognition of position-specific properties of tectal cell membranes by retinal axons in vitro. Development. 101:685–696. [DOI] [PubMed] [Google Scholar]
- Wang, L., C.L. Ho, D. Sun, R.K. Liem, and A. Brown. 2000. Rapid movement of axonal neurofilaments interrupted by prolonged pauses. Nat. Cell Biol. 2:137–141. [DOI] [PubMed] [Google Scholar]
- Wittmann, C.W., M.F. Wszolek, J.M. Shulman, P.M. Salvaterra, J. Lewis, M. Hutton, and M.B. Feany. 2001. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 293:711–714. [DOI] [PubMed] [Google Scholar]
- Xu, H., D. Sweeney, R. Wang, G. Thinakaran, A.C. Lo, S.S. Sisodia, P. Greengard, and S. Gandy. 1997. Generation of Alzheimer beta-amyloid protein in the trans-Golgi network in the apparent absence of vesicle formation. Proc. Natl. Acad. Sci. USA. 94:3748–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabe, J.T., A. Pimenta, and T.B. Shea. 1999. Kinesin-mediated transport of neurofilament protein oligomers in growing axons. J. Cell Sci. 112:3799–3814. [DOI] [PubMed] [Google Scholar]
- Zhou, H.M., I. Brust-Mascher, and J.M. Scholey. 2001. Direct visualization of the movement of the monomeric axonal transport motor UNC-104 along neuronal processes in living Caenorhabditis elegans. J. Neurosci. 21:3749–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]