

Abstract
We have previously shown that the neurotrophic effect of glial cell line–derived neurotrophic factor (GDNF) in vitro and in vivo requires the presence of transforming growth factor (TGF)β. Using primary neurons (chick E8 ciliary) we show that the combination of GDNF plus TGFβ promotes survival, whereas the single factors do not. This cooperative effect is inhibited by blocking the extracellular signal-regulated kinase (ERK)/MAPK pathway, but not by interfering with the PI3 kinase signaling cascade. Although there is no functional GDNF signaling in the absence of TGFβ, pretreatment with TGFβ confers GDNF responsiveness to the cells. This is not due to upregulation of GDNF receptors mRNA and protein, but to TGFβ-induced recruitment of the glycosyl-phosphatidylinositol-anchored GDNF receptor (GFR)α1 to the plasma membrane. This is supported by the fact that GDNF in the presence of a soluble GFRα1 can promote survival in the absence of TGFβ. Our data suggest that TGFβ is involved in GFRα1 membrane translocation, thereby permitting GDNF signaling and neurotrophic effects.
Keywords: neurotrophic factors; lipid raft; GFRα1; tyrosine kinases; MAPK pathway
Introduction
Neurotrophic factors are a broad set of peptide growth factors that tightly regulate development and survival of neurons of the central nervous system (CNS)* and the peripheral nervous system (for review see Huang and Reichardt, 2001; Neet and Campenot, 2001). Among the expanding list of neurotrophic factors, the discovery of glial cell line–derived neurotrophic factor (GDNF) as a survival and differentiation factor for midbrain dopaminergic neurons was a hallmark in the search for novel molecules that may have relevance in the treatment of neurodegenerative diseases, e.g., Parkinson's disease (PD) (Lin et al., 1993). The significance of GDNF is underscored by its efficacy in several animal models of PD, including: nonhuman primates (Hoffer et al., 1994; Beck et al., 1995; Sauer et al., 1995; Tomac et al., 1995; Gash et al., 1996); ubiquitous expression in neurons of the CNS (Pochon et al., 1997) and its widening spectrum of responsive neuron populations (Henderson et al., 1994; Arenas et al., 1995; Buj-Bello et al., 1995; Trupp et al., 1995; Farkas et al., 1997). GDNF is a member of the transforming growth factor (TGF)β superfamily (Kingsley, 1994), its closest relative being neurturin (Kotzbauer et al., 1996). Targeted mutations of GDNF (Moore et al., 1996; Pichel et al., 1996; Sanchez et al., 1996) or its receptors, c-Ret (Schuchardt et al., 1994) and GDNF receptor (GFR)-α1 (Cacalano et al., 1998; Enomoto et al., 1998), have indicated that GDNF is essentially required for the development of the kidney, major portions of the enteric nervous system, and the sympathetic superior cervical ganglion. Recently, Ret has been described as a key regulator of migration of neuronal precursors of the sympathetic nervous system (Enomoto et al., 2001). However, most strikingly, it has been shown that GDNF is not trophically active unless supplemented with TGFβ in vitro and in vivo (Krieglstein et al., 1998; Schober et al., 1999).
TGFβs are widely distributed cytokines with prominent roles in development and cell cycle control (Roberts and Sporn, 1990; Alexandrow and Moses, 1995; Krieglstein et al., 1995). TGFβs have been implicated in the regulation of neuronal survival of motoneurons (Martinou et al., 1990), sensory (Chalazonitis et al., 1992), and midbrain dopaminergic neurons (Krieglstein and Unsicker, 1994; Poulsen et al., 1994), for example. However, TGFβs show no or marginal effects on highly enriched, serum-free neuron cultures, such as sensory neurons (Krieglstein and Unsicker, 1996), suggesting that TGFβ may require cooperating factors for eliciting its trophic effects. Along this line, TGFβ actions are always referred to as being contextually dependent, suggesting that other molecules present significantly determine TGFβ actions and vice versa (Nathan and Sporn, 1991; Unsicker and Krieglstein, 2000). Putative contextual actions of TGFβs and additional molecules may also underlie the seemingly conflicting effects of TGFβ in the regulation of both survival and death of ciliary ganglionic (CG) neurons (Krieglstein et al., 2000). With regard to the regulation of survival, TGFβ cooperates with GDNF, whereas TGFβ-induced cell death may be the consequence of a lack of the appropriate neurotrophic factor, like GDNF, or may be the result of cooperativity with other yet unidentified cytokines.
A fundamental question regarding GDNF-TGFβ cooperativity concerns the underlying molecular mechanism. GDNF signals via binding as a homodimer to a heterotetrameric complex of two glycosyl-phosphatidylinositol (GPI)-anchored GFRα1 (Jing et al., 1996; Treanor et al., 1996) and two molecules of the transmembrane tyrosine kinase Ret (Durbec et al., 1996; Trupp et al., 1996). Upon autophosphorylation of Ret, two signaling pathways are activated: one is the Ras/Raf/MAPK pathway, and the other involves activation of PI3 kinase and its downstream target PKB/Akt (Chiariello et al., 1998; Trupp et al., 1999).
The present study identifies the Ras/MAPK pathway as the survival promoting signaling cascade activated by treatment of CG neurons with GDNF and TGFβ. We show that TGFβ mediates recruitment of the GPI-linked GFRα1 to the site of consecutive signaling, probably lipid rafts.
Results
Survival of chick E8 CG neurons requires cooperativity of GDNF and TGFβ
Glial cell line–derived neurotrophic factor (GDNF) has been described as a potent neurotrophic molecule for various neurons of the peripheral and CNS (Lin et al., 1993; Arenas et al., 1995; Buj-Bello et al., 1995; Farkas et al., 1997). We have previously shown that the survival promoting effect of GDNF in vivo and in vitro requires the presence of TGFβ (Krieglstein et al., 1998).
In this study we have asked how the cooperation of these two factors is mediated on the molecular level using primary chick embryonic (E8) CG neurons as a model system. Survival of CG neurons is strictly dependent on the presence of neurotrophic support. Fig. 1 A demonstrates that GDNF plus TGFβ promotes survival of the cells, whereas the single factors do not. This effect was dose dependent with concentrations of GDNF ranging from 2 to 10 ng/ml. However, TGFβ was almost as effective at 0.125 ng/ml as at 2 ng/ml (Fig. 1 B). As a positive control, ciliary neurotrophic factor (CNTF) was used, which is known to promote survival of CG neurons independent of TGFβ (Krieglstein et al., 1998).
Figure 1.
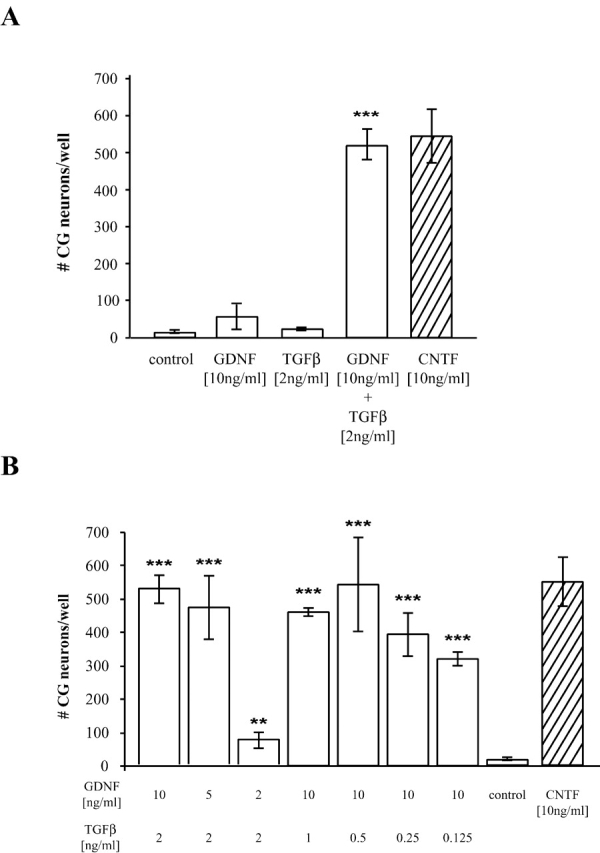
GDNF-induced survival of CG neurons is strictly dependent on the presence of TGFβ. CG neurons were seeded onto PORN/laminine-coated microtiter plates at a density of 3,000 cells per well. (A) Cells were treated with 10 ng/ml GDNF or 2 ng/ml TGFβ3 or both factors. As a control the neurons were left untreated or CNTF (10 ng/ml) was added. (B) Cells were treated with combinations of the indicated concentrations of GDNF (0.5 ng/ml to 10 ng/ml) and TGFβ (0.125 ng/ml to 2 ng/ml). The cultures were fixed after 24 h and the numbers of surviving neurons were counted. Each bar represents the mean and standard deviation of at least three independent experiments with every condition tested in triplicate per assay (***, P ≤ 0.01, **, P ≤ 0.05; determined in a Student's t test).
Inhibition of MAPK activity decreases neuronal survival mediated by the synergistic action of GDNF and TGFβ
To further elucidate the intracellular mechanisms underlying the survival effect of GDNF in the presence of TGFβ, we first asked which intracellular pathway transmits the survival signal. The PI3 kinase pathway and the Ras/MAPK signaling cascade activated by binding of GDNF to its receptors at the cell surface of responsive cells are schematically depicted in Fig. 2 A. To clarify the involvement of these two pathways in the cooperative survival effect of GDNF and TGFβ, specific inhibitors were used to block the two signaling cascades. Wortmannin (Ward et al., 1996) and LY294002 (Vlahos et al., 1994) were used to specifically block the PI3 kinase downstream signaling, and the MEK inhibitors PD98059 (Dudley et al., 1995) and U0126 (Favata et al., 1998) were employed for blocking the Ras/MAPK pathway. Cell counts after 24 h revealed that the neurotrophic effect of GDNF plus TGFβ is significantly reduced by blocking the ERK/MAPK pathway, but not (or only marginally) by interfering with the PI3 kinase signaling cascade (Fig. 2 B). To exclude a toxic effect of the inhibitors and demonstrate their specificity, CNTF and FGF-2 were used as controls. As shown in Fig. 2 B, none of the inhibitors was able to block the neurotrophic effect of CNTF. Many functions of CNTF have been shown to be mediated through the JAK/STAT and ERK/MAPK pathway, without activating PI3 kinase (Boulton et al., 1994; Stahl and Yancopoulos, 1994; Segal and Greenberg, 1996). Apparently, the survival signal in CG neurons does not involve ERK/MAPK. In agreement with previous findings (Besser et al., 1995; Piiper et al., 1996), blocking both pathways diminished FGF-2 mediated survival of CG neurons demonstrating that the inhibitors used were working properly. The finding that the PI3 kinase pathway is not crucial for the cooperative survival promoting effect of GDNF and TGFβ is in contrast to our previous observations (Krieglstein et al., 1998). To test whether this may be due to the fact that we excluded insulin from the CG culture medium in the experiments presented here while it was present in previous studies, we examined the effect of the kinase inhibitors on CG-survival in the presence of insulin. In corroboration of our previous findings, in the presence of insulin the application of both the PI3 kinase and the MEK inhibitors decreased the numbers of surviving neurons in the presence of GDNF and TGFβ (Fig. 2 C).
Figure 2.
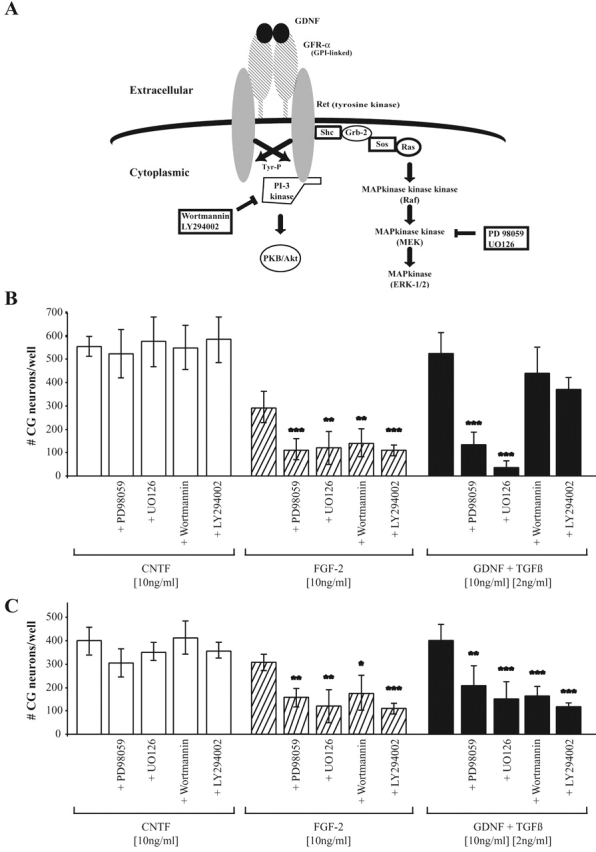
Survival of CG neurons in the presence of GDNF and TGFβ is mediated by activation of the ERK/MAPK pathway. (A) Simplified general scheme of GDNF signaling. (B) Survival of CG neurons in the presence of GDNF (10 ng/ml) and TGFβ3 (2 ng/ml), FGF-2 (10 ng/ml) or CNTF (10 ng/ml), respectively, in the absence or presence of the kinase inhibitors Wortmannin (100 nM), LY294002 (5 μM), U0126 (10 μM), and PD98059 (50 μM). The surviving neurons were counted after 24 h, each bar represents the mean and standard deviation of at least three independent experiments with every condition tested in triplicate per assay (***, P ≤ 0.01; **, P ≤ 0.05; *P ≤ 0.1). (C) Survival assay as described in (B) except that insulin (5 μg/ml) was added.
GDNF responsiveness essentially requires pretreatment with TGFβ
We next tested whether TGFβ is required to achieve downstream signaling of GDNF by investigating the phosphorylation status of ERK and Akt in total lysates from CG neurons using antibodies directed against the activated forms of both kinases (Fig. 3 A). At 30 min there was no GDNF-stimulated phosphorylation of ERK or Akt in the absence of TGFβ or after simultaneous addition of the two factors. However, treatment of CG neurons for 3 h with TGFβ, and subsequent addition of GDNF for 30 min, lead to enhanced phosphorylation of ERK. Interestingly, phosphorylation of Akt was only seen if neurons were simultaneously treated with GDNF and TGFβ after TGFβ pretreatment. Levels of nonphosphorylated ERK were not affected by TGFβ pretreatment but remained unchanged throughout the experiment; hence, they were used as a control for equal loading. Applying the kinase inhibitors, which were used for the survival assay, proved that PD98059 could efficiently block GDNF-induced phosphorylation of ERK without changing Akt phosphorylation in the presence of GDNF and TGFβ, and Wortmannin inhibited Akt phosphorylation without affecting ERK phosphorylation (Fig. 3 B). This observation further confirmed the notion that although Wortmannin efficiently blocked Akt-phosphorylation induced by GDNF plus TGFβ, it did not have a major effect on survival promoted by the cooperative action of the two factors.
Figure 3.
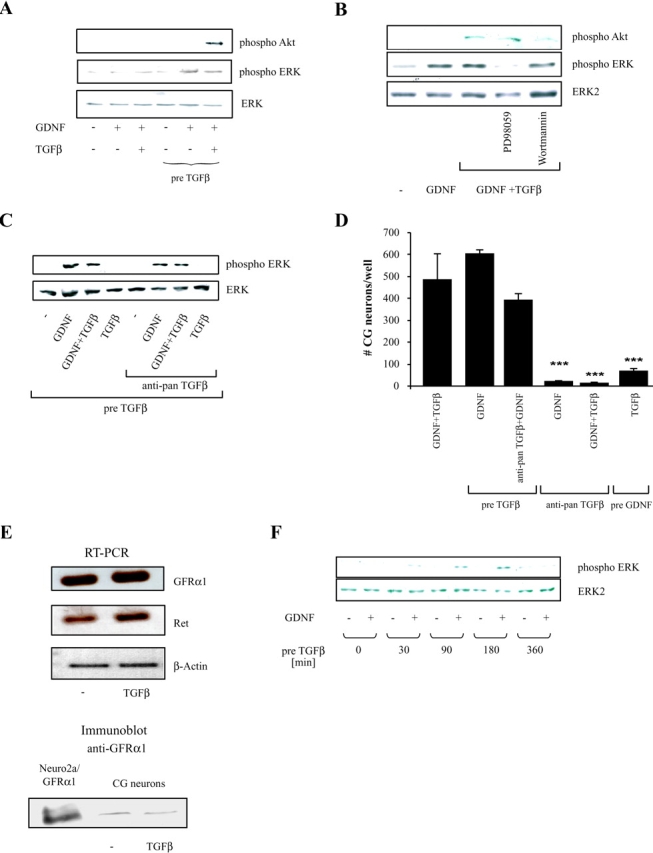
Pretreatment with TGFβ induces GDNF responsiveness, but does not affect expression of the GDNF receptors. (A) 100,000 CG neurons per well were plated and allowed to attach for 2 h. The cells were treated with 2 ng/ml TGFβ for 3 h or left untreated as indicated. Subsequently no factor or GDNF (10 ng/ml) or GDNF (10 ng/ml) plus TGFβ (2 ng/ml) were added for 30 min before harvesting of the cells in Laemmli sample buffer. (B) CG neurons were handled as in (A) except that PD98059 or Wortmannin was added to prove specific inhibition of ERK- and Akt-phosphorylation by the respective inhibitors. (C) Cells were pretreated for 3 h with TGFβ (2 ng/ml), thereafter an antibody blocking all three TGFβ-isoforms (anti-pan TGFβ) was added where indicated. 10 min later GDNF (10 ng/ml), TGFβ (2 ng/ml) or both factors were added for 30 min and the cells subsequently harvested in Laemmli sample buffer. Protein extracts (A–C) were separated by 10% SDS-PAGE, electroblotted onto nitrocellulose membranes and probed with anti– phospho ERK-, anti–phospho Akt- or anti–GFRα1-antibodies. To control for equal loading the membrane stripped and probed with an antibody against nonphosphorylated ERK. (D) CG neurons were differently treated to investigate the requirement of persistent TGFβ or GDNF treatment for survival. Cells were pretreated for 3 h with TGFβ3 (2 ng/ml). Thereafter, either TGFβ was blocked by addition of a pan TGFβ antibody and GDNF (10 ng/ml) was added or only GDNF was added in the absence of the antibody. Similarly the neurons were pretreated with GDNF (10 ng/ml) for 3 h and after a washout of GDNF, TGFβ3 (2 ng/ml) was added. As controls, neurons were simultaneously treated with GDNF and TGFβ in the presence or absence of the pan TGFβ antibody. The surviving neurons were counted after 24 h, each bar represents the mean and standard deviation of two independent experiments with every condition tested in triplicate per assay (***, P ≤ 0.01). (E) RT-PCR of the GDNF-receptors GFRα1 and Ret and immunoblot of GFRα1 using total RNA and protein extracts, respectively, from CG neurons treated for 3 h with TGFβ or untreated controls. For the immunoblot Neuro2a cells stably expressing GFRα1 were used as positive control. (F) Anti phospho-ERK immunoblot with extracts from CG neurons, pretreated with TGFβ for the indicated time points and subsequently treated with GDNF for 30 min or left untreated. As control for equal loading an immunoblot with an antibody detecting nonphosphorylated ERK was used.
When TGFβ was removed from the culture medium after a 3-h pretreatment period by changing the medium and adding a blocking pan-TGFβ antibody (blocking all three TGFβ isoforms), increased phosphorylation of ERK was still observed in the presence of GDNF (Fig. 3 C).
Corroborating this result survival of CG neurons was also promoted when TGFβ, initially added without GDNF, was neutralized after 3 h and GDNF was subsequently added. In contrast, neutralization of TGFβ at the beginning of the experiment and administration of GDNF alone or GDNF and TGFβ after 3 h did not promote survival. This proved that the TGFβ antibody was present in excess and completely blocked TGFβ activity throughout the culture period. Please note that a consecutive treatment of first 3 h with TGFβ followed by GDNF was sufficient to attain GDNF-induced cell survival. Further along this line pretreatment with GDNF, followed by a change of medium and addition of TGFβ did not elicit a survival promoting effect (Fig. 3 D). Together, these data suggest that GDNF responsiveness essentially requires pretreatment with TGFβ.
One possible explanation for this TGFβ requirement may be an effect on the expression of Ret and GFRα1. Therefore, we investigated the mRNA level of both receptors using semi-quantitative RT-PCR with primers specific for chicken Ret and GFRα1 in the presence and absence of TGFβ. As shown in Fig. 3 E, there was no change in the amount of the two receptor mRNAs. Consistent with the unchanged mRNA level, we found no effect of TGFβ pretreatment on the GFRα1 protein level in a Western blot analysis using a GFRα1-specific antibody. We next investigated the time dependence for pretreatment with TGFβ. We found increased ERK phosphorylation in GDNF-treated neurons after 30 min, which peaked after 3 h and disappeared after 6 h (Fig. 3 F). This kinetics is also indicative of an effect independent of protein synthesis. One possible explanation could be that the availability of the GDNF receptors for interaction with their ligand at the plasma membrane might be regulated by TGFβ.
In the presence of a soluble GFRα1, TGFβ is not required to promote survival and activation of ERK by GDNF
GFRα1 is attached to the outer cell membrane by a GPI modification (Jing et al., 1996). We have previously shown that cleavage of this GPI anchor from the membrane by phosphoinositide-specific phospholipase C disrupts the survival promoting effect of GDNF and TGFβ, and that this effect can be partly reversed by adding TGFβ before cleavage by phosphoinositide-specific phospholipase C (Krieglstein et al., 1998). This observation strongly argued for an effect of TGFβ on the recruitment of GFRα1 to or its stabilization on the plasma membrane. Therefore, we asked whether an exogenously added receptor may confer GDNF responsiveness to CG neurons in the absence of TGFβ.
To test this hypothesis we added a GFRα1-IgG chimeric protein to the supernatant of the primary neuron cultures. Cell counts after 24 h showed that GDNF fully promoted CG neuron survival in the presence of soluble GFRα1, indicating that addition of TGFβ is not required under these conditions (Fig. 4 A). Thus, TGFβ may function by recruiting the GFRα1 by yet unknown mechanisms. Interestingly, additional treatment with TGFβ further increased numbers of surviving cells, suggesting that TGFβ may also affect components of the GDNF signaling pathway other than GFRα1. Neutralization of endogenous TGFβ resulted in a minor decrease in survival. Along this line, phosphorylation of ERK in the presence of soluble GFRα1 also does not require TGFβ. However, Akt phosphorylation was only observed if the cells were treated simultaneously with GDNF and TGFβ (Fig. 4 B).
Figure 4.
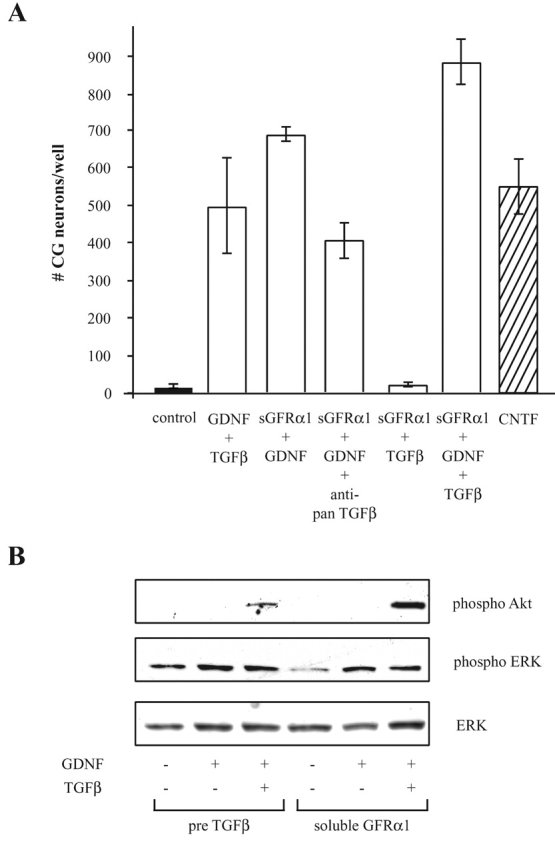
In the presence of a soluble GFRα1 TGFβ is not required to induce GDNF responsiveness. (A) CG neurons were seeded onto PORN/laminine-coated microtiter plates at a density of 3,000 cells per well. Cells were treated with combinations of 10 ng/ml GDNF, 2 ng/ml TGFβ3, soluble GFRα1 (sGFRα1, 100 nM), or anti-pan TGFβ antibody (10 μg/ml) as indicated. As a control the neurons were left untreated or CNTF (10 ng/ml) was added. (B) Immunoblot with a phospho-specific antibodies to Akt and ERK with extracts from CG neurons that have been either pretreated with TGFβ or a soluble GFRα1 for 3 h and where subsequently GDNF or GDNF plus TGFβ was added for 30 min.
Treatment of CG neurons with Brefeldin A blocks activation of the MAPK pathway by GDNF and TGFβ
It has been recently shown that GPI-linked molecules constantly cycle between the Golgi apparatus and the plasma membrane (Nichols et al., 2001). To test whether the shuttle of GFRα1 between Golgi and plasma membrane might be influenced by TGFβ, we treated CG neurons with Brefeldin A, a fungal metabolite that causes disassembly of the Golgi complex and retainment of proteins in the endoplasmic reticulum (Nuchtern et al., 1990; Oda et al., 1990). After pretreatment with TGFβ or soluble GFRα1 for 3 h, neurons were exposed to Brefeldin A (2.5 μM) for 10 min, and then GDNF was added for another 30 min and the amount of phosphorylated ERK protein was detected by Western blot analysis. Brefeldin A completely blocked GDNF-stimulated phosphorylation of ERK after TGFβ pretreatment. However, in the presence of soluble GFRα1, GDNF-induced ERK phosphorylation was only slightly affected by Brefeldin A (Fig. 5). This adds to the notion that TGFβ is required for presenting the GFRα1 to the ligand on the cell membrane.
Figure 5.

Treatment with Brefeldin A abolishes ERK phosphorylation in TGFβ pretreated cells. Immunoblot with a phospho-specific antibody to ERK with protein extracts from CG neurons. Cells were incubated with Brefeldin A (2.5 μM) or vehicle alone for 10 min before treatment with TGFβ or soluble GFRα1 for 3 h, followed by addition of GDNF for 30 min. A quantification of the bands representing phosphorylated ERK normalized to total ERK is shown in the bar diagram.
TGFβ causes clustering of GFRα1 immunoreactivity in lipid rafts
We next studied the effect of TGFβ on the localization of the GFRα1 in the plasma membrane of CG neurons. CG neurons were treated for 3 h with TGFβ or left untreated; subsequently, the medium was removed and the neurons were incubated with an anti-GFRα1 antibody on ice before fixation to stain for the receptor at the cell surface of the living cells. As shown in Fig. 6 A, in the absence of TGFβ, GFRα1 immunoreactivity is dispersed all over the cellular surface. After pretreatment with TGFβ, clusters of receptor immunoreactivity become visible. As a control for the specificity of the antibody, the murine neuroblastoma cell line Neuro2a, which is devoid of GFRα1 (Jing et al., 1996), and a Neuro2a clone stably transfected with GFRα1 (Neuro2a/GFRα1), were subject to the same staining procedure. Fig. 6 B demonstrates that wild-type Neuro2a cells display no GFRα1 immunostaining, whereas Neuro2a/GFRα1 cells reveal staining of clustered receptors. This is in accordance with the finding that in these stable transfectants the GFRα1 protein is predominantly located in lipid rafts at the plasma membrane (Tansey et al., 2000). The localization of GFRα1 in lipid rafts can be visualized by double labeling of the cells with the anti-GFRα1 antibody and an FITC-coupled cholera toxin B which specifically binds to the lipid raft marker GM1, a cell surface ganglioside (Van Heyningen et al., 1976; Iwamori et al., 1985). We used Triton extraction of the cells to visualize detergent-insoluble membrane compartments as described by Paratcha et al. (2001). In Neuro2a/GFRα1 cells, GFRα1 is highly colocalized with GM1 (Fig. 7 A). Using the same labeling procedure for the CG neurons revealed that there is a markedly enhanced colabeling of GFRα1 and GM1 after pretreatment of the cells with TGFβ, compared with untreated controls (Fig. 7 B). This can also be observed if incubation with the antibody for GFRα1 was performed on the neurons in culture, and FITC-cholera toxin B was added after fixation (Fig. 7 C).
Figure 6.
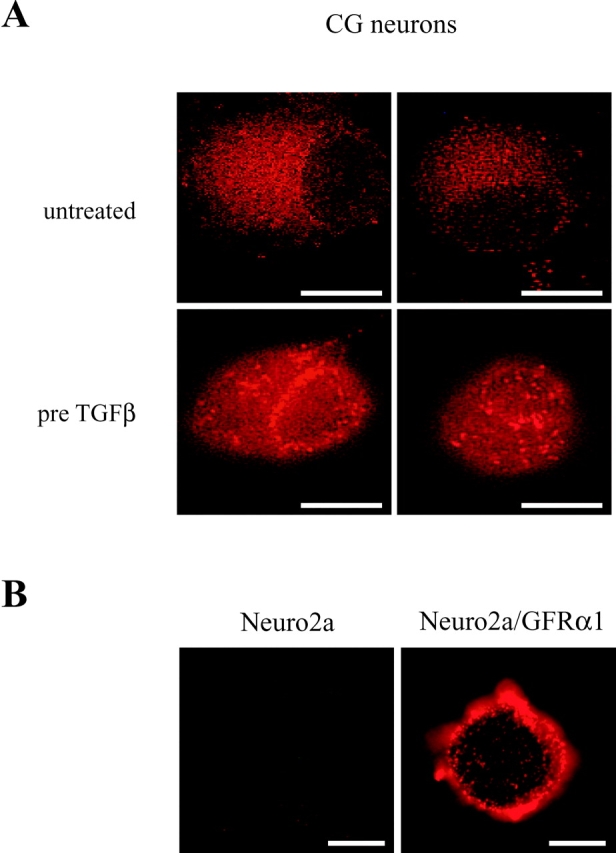
Pretreatment of CG neurons with TGFβ induces clustering of GFRα1 immunoreactivity. (A) Living cells were incubated with a GFRα1 antibody for 1 h on ice and then fixed with 4% PFA before secondary antibody treatment. In the absence of TGFβ GFRα1 immunoreactivity is evenly distributed on the cell surface of two representative cells. After incubation with TGFβ (2 ng/ml) for 3 h GFRα1 immunoreactivity is clustered in a patch-wise fashion. Bars, 5 μM. (B) Specificity of GFRα1 antibodies was ascertained by staining wild-type Neuro2a cells, which lack GFRα1 receptors, and Neuro2a cells transfected with a GFRα1 construct. Bars, 10 μM.
Figure 7.
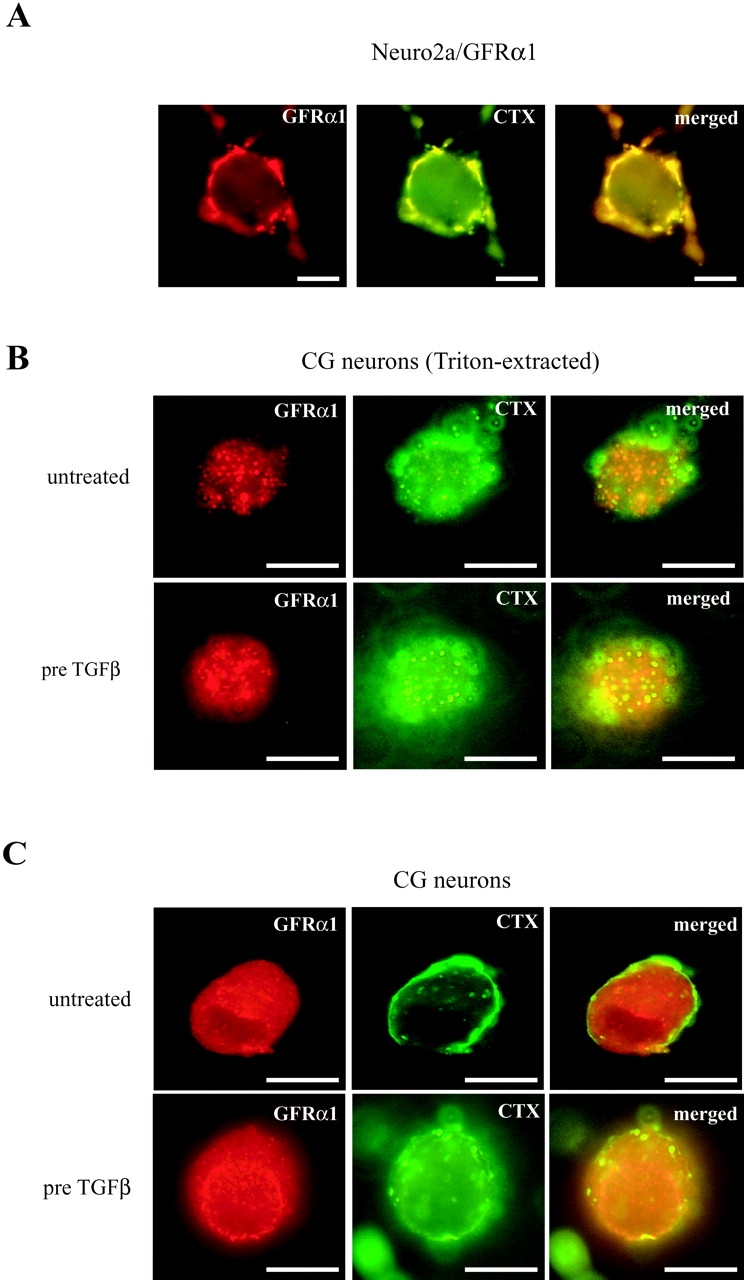
GFRα1 colocalizes with a marker for lipid rafts in the presence of TGFβ. (A) Neuro2a cells transfected with a GFRα1 construct were Triton-extracted and fixed with 4% PFA before staining with an antibody to GFRα1 and cholera toxin (CTX), which specifically binds to the lipid raft ganglioside GM1. The merged images shows colocalization of GFRα1 immunoreactivity and CTX. Bars, 10 μM. (B) CG neurons were either treated with TGFβ (2 ng/ml) for 3 h or left untreated. Cells were processed and stained as described in (A). In the presence of TGFβ colocalization of GFRα1 and CTX is increased. Bars, 5 μM. (C) CG neurons were either treated with TGFβ (2 ng/ml) for 3 h or left untreated. Living cells were incubated with GFRα1 antibody for 1 h on ice and fixed with 4% PFA before incubation with a secondary antibody to GFRα1 and CTX. Note copatching of GFRα1 and CTX after treatment with TGFβ. Bars, 5 μM.
Taken together, we propose a cooperation of GDNF and TGFβ on the recruitment to, and activation of, the GFRα1 at the plasma membrane. Our data strongly indicate that the availability of GFRα1 on the cell surface may limit neuronal survival promoted by GDNF, and that TGFβ is involved in GFRα1 membrane translocation.
Discussion
Response of CG neurons to GDNF requires pretreatment with TGFβ
In the present study we have analyzed the molecular basis of GDNF-TGFβ cooperativity in primary chick E8 CG neurons. We investigated different putative explanations for the GDNF-TGFβ synergism at the following levels of GDNF signaling: (a) with regard to signaling cascade components; (b) at the receptor expression level; and (c) at the level of translocation of GFRα1 to lipid rafts. Using specific inhibitors proved that the major portion of the survival promoting effect of GDNF in our system is mediated via the ERK/MAP kinase pathway. A prerequisite for GDNF activity is pretreatment of the cells with TGFβ, both with regard to ERK phosphorylation and to survival. After the initial pretreatment, continuous presence of TGFβ is no longer required. Concerning activity of PI3 kinase as measured by phosphorylation of its target Akt, the requirement for TGFβ is different in that a simultaneous addition of both GDNF and TGFβ is necessary. However, inhibition of PI3 kinase activity only has a minor effect on survival. The difference between our results presented here and previous observations on the effect of PI3 kinase inhibition (Krieglstein et al., 1998) can be explained by the fact that we excluded insulin from the culture medium. The rationale for this exclusion was that insulin also promotes survival of CG neurons and is known to signal via the PI3 kinase pathway. However, as we show here, the GDNF-TGFβ cooperativity is not dependent on the presence of insulin. The physiological relevance of Akt phosphorylation and the underlying molecular mechanisms of the TGFβ-GDNF synergism remain to be clarified.
TGFβ treatment induces clustering of GFRα1 in lipid microdomains on the cell surface
TGFβ induction of GDNF responsiveness occurred within 30 min, excluding an effect that is dependent on new protein synthesis. Consistently, TGFβ-induced GDNF responsiveness did not involve upregulation of expression of the receptors cRet and GFRα1. Our data suggest that TGFβ does affect the availability of GFRα1 for the ligand GDNF. This is shown by the following: (a) the inhibitory effect of BFA on ERK phosphorylation; (b) the clustering of GFRα1 immunoreactivity and the costaining of GFRα1 and the lipid raft marker GM1; and (c) preliminary observations that TGFβ pretreatment leads to a shift of GFRα1 from cytoplasmic to membrane fractions of CG neuron protein extracts (unpublished data).
It has been shown that GFRα1 is necessary to recruit Ret to lipid rafts, which attenuates downstream signaling pathways as well as neuronal differentiation and survival (Tansey et al., 2000; Paratcha et al., 2001). Lipid rafts are thought to represent specialized signaling organelles within the plasma membrane because of an enrichment of many adaptor and signaling molecules (Anderson, 1998). Tansey et al. (2000) have analyzed the functional importance of the GPI anchorage of GFRα1 in mediating GFRα1/Ret signalling. They were able to show that both the anchored and the soluble GFRα1 mediate Ret translocation to lipid rafts, and thereby attenuate downstream kinase activity. Paratcha et al. (2001) extended these studies by providing additional evidence, suggesting that Ret-translocation is best mediated by soluble GFRα1, which may be provided by ectopic sources. Most studies addressing GFRα1/Ret interaction have been performed using cell lines which were transfected with either GFRα1 alone or both GFRα1 and Ret. In this context, the overexpressed GPI-linked receptor is constitutively present in the lipid raft fraction on the cellular membrane. So far, it has not been addressed whether the presence of endogenous GFRα1 at the plasma membrane might be subject to regulation. It has been shown that GPI-linked proteins continuously shuttle between the Golgi complex and the plasma membrane without involvement of new synthesis of this proteins (Nichols et al., 2001). Regulation of this cycling might thus play an important role in lipid raft associated signaling cascades.
Mechanisms of TGFβ-induced recruitment of GFRα1 to its active site at the plasma membrane
There are several possible ways that TGFβ may be mediating the availability of GFRα1 at lipid rafts. First, it has to be clarified whether TGFβ is acting via its receptors, TβRI and TβRII, and whether downstream signaling of TGFβ is required. GDNF-TGFβ cooperativity might also involve a direct interaction of the corresponding receptors at the plasma membrane. Furthermore, cooperativity could be due to an indirect effect involving a protein for which the receptors compete. One example for such a competition has been shown for binding of caveolin-1 to the NGF receptors TrkA and p75 which determines the NGF response of PC12 cells (Bilderback et al., 1999). Caveolin-1 is a principal component of cholesterol-enriched plasma membrane microdomains, called caveolae, which are involved in vesicular transport and signal transduction (for review see Schlegel and Lisanti, 2001). It has been proposed that caveolin could function as a transmembrane adaptor molecule that couples GPI-linked proteins with signaling molecules during GPI-membrane trafficking or GPI-mediated signal transduction events (Sargiacomo et al., 1993).
It has been shown that TβRI interacts with caveolin-1, and that upon binding of TGFβ to its receptors at the membrane, this interaction is increased and leads to downregulation of the TGFβ response, possibly by internalization of TβRI (Razani et al., 2001). Whether a similar interaction of GFRα1 with caveolin-1 occurs and whether this is changed in the presence of TGFβ remains to be established.
Cooperative effects of GDNF with other factors on the survival of different neuronal populations
The requirement of a synergism between GDNF and another factor for the survival of specific neuronal populations has been described for BDNF (sensory neurons of the nodose-petrosal ganglion complex; Erickson et al., 2001), CNTF (mouse photoreceptors; Ogilvie et al., 2000), IGF-I (motoneurons; Bilak and Kuncl, 2001; Bilak et al., 2001), osteogenin-1/bone morphogenetic protein 7, a member of the TGFβ superfamily (explanted embryonic chicken ganglia and dissociated ganglionic neuron; Bengtsson et al., 1998), and cardiotrophin-1 (embryonic rat motoneurons; Arce et al., 1998). These findings underline the importance of modulation of the GDNF response by other (growth) factors, but the mechanistic explanation for any of these synergisms has yet to be provided.
Interference between TGFβ signaling and signal transduction through receptor tyrosine kinases
In contrast to GDNF synergisms, molecular mechanisms of interaction of TGFβ family members with other signaling cascades have been studied in various cellular systems. Interferon-γ inhibits TGFβ-induced phosphorylation of Smad3 and downstream TGFβ effects by an effect involving new protein synthesis, namely by induction of the expression of the antagonistic SMAD, Smad7, through Jak1 and Stat1 (Ulloa et al., 1999). Synergistic effects involving direct interaction of Stat proteins and Smads have been reported for Smad3 (activated by TGFβ) and STAT3 (activated by Interleukin-6) in the Hep3B hepatoma cell line (Yamamoto et al., 2001) and for Smad1 (activated by bone morphogenetic protein 2) and STAT3 (activated by leukemia inhibitory factor) on primary fetal neural progenitor cells during the induction of astrocyte differentiation (Nakashima et al., 1999). These activities were due to physical interactions between STAT3 and the Smads bridged by the transcriptional coactivator p300.
Evidence for inhibitory effects of receptor tyrosine kinase pathways on signaling of members of the TGFβ family concerns the level of signal transduction through phosphorylated Smads. It has been shown that EGF and hepatocyte growth factor exhibit an inhibitory effect on bone morphogenetic protein–induced Smad1 activation by phosphorylation of specific serines in the linker region of Smad1. This phosphorylation is catalysed by the MAPK ERK in mink lung epithelial cells (Kretzschmar et al., 1997). In contrast to our findings of a TGFβ-GDNF cooperativity, in these cases the cells receive opposing regulatory inputs through the receptor tyrosine kinases and the serine/threonine kinase. There are also reports about how receptor tyrosine kinases (RTKs) or downstream kinases can positively interfere with the TGFβ signaling cascade. For example, in contrast to their above described inhibitory effect, hepatocyte growth factor and EGF have been shown to induce Smad2 phosphorylation and nuclear translocation in epithelial cells in the absence of TGFβ and independent of TGFβ receptors (de Caestecker et al., 1998). These observations suggest that the quality of interference of RTK- and TGFβ-induced pathways is dependent on the context of the analyzed cellular system. In contrast to all these observations in which RTK signaling positively affects TGFβ signaling, in our neuronal model, TGFβ facilitates RTK signaling, primarily by exerting an effect on the GFRα1 receptor. Whether there also exists a physical interaction of downstream effectors of TGFβ and GDNF which could explain the differential TGFβ requirement for Akt phosphorylation needs to be examined.
Interaction of Smads with other transcription factors on the promoter level
Cooperativity of the TGFβ signaling pathway with other pathways has also been described to involve regulation of ligand-induced gene transcription at the promoter level. A nuclear Smad3/Smad4 can activate a TPA responsive gene promoter element directly or through interaction with both c-Jun and c-Fos (Zhang et al., 1998). Smad3 has also been reported to potentiate ligand-induced transactivation of the vitamin D receptor as a coactivator through binding of the steroid receptor coactivator-1 protein in the nucleus (Yanagi et al., 1999; Yanagisawa et al., 1999) or by binding to a Smad binding element adjacent to a vitamin D receptor binding element on the human osteocalcin promoter (Subramaniam et al., 2001). In human neuroblastoma cells, CNTF and TGFβ synergistically induce vasoactive intestinal peptide (VIP) mRNA expression and transcription through the cytokine response element in the VIP promoter. This synergy is dependent on binding of Smad, STAT, and AP-1 to distinct sites within the VIP promoter. Whether any physical interaction of the signaling components is necessary has not been analyzed in this report (Pitts et al., 2001).
As discussed above, crosstalks of TGFβ family members with other signaling pathways have been described to occur both in the cytosol and the nucleus (for review see Wrana, 2000). Our finding that TGFβ cooperativity also involves molecular interactions at the plasma membrane adds a new point of view to signaling interactions of RTKs and TGFβ signaling.
In conclusion, our data show for the first time that TGFβ signaling facilitates tyrosine kinase signaling by modulating the availability of the GDNF receptor on the cell surface. TGFβ-mediated recruitment of the GFRα1 is crucial for GDNF-dependent signaling and survival.
Materials and methods
Cell culture and preparation of protein extracts
Fertilized white Leghorn chicken eggs were incubated in a humidified egg chamber at 37.8°C. Embryonic chick (E8) CG were dissected, freed from nerve roots, and connective tissue, and collected in Ca2+/Mg2+-free HBSS (CMF; Sigma-Aldrich). Ganglia were trypsinized, washed in CMF, and triturated as described (Lachmund et al., 1994). Single cell suspensions were seeded onto poly-l-ornithine (PORN), laminine-coated plates or coverslips in DME supplemented with N1 additives w/o insulin (Bottenstein and Sato, 1980), 0.25% BSA (Sigma-Aldrich), and incubated at 37°C in a 5% CO2 and 95% air atmosphere (Lachmund et al., 1994).
The cell line Neuro2a was cultured in DME medium containing 10% bovine calf serum, 100 U/ml penicillin, 0.50 μg/ml streptomycin, 100 μg/ml neomycin (Invitrogen) and 1% nonessential amino acids. The stably transfected Neuro2a-GFRα1 clone, provided by M. Saarma (University of Helsinki, Helsinki, Finland) was cultured in RPMI containing 10% bovine calf serum, 100 U/ml penicillin, 0.50 μg/ml streptomycin and 250 μg/ml G418. Total cell lysates were obtained by solubilizing the cells in Laemmli sample buffer (80 mM Tris, pH 6.8, 2% SDS, 10% glycerol, 2% β-mercaptoethanol, 0.01% bromophenol blue) and sonication.
Evaluation of cell survival
CG neurons were seeded onto PORN/laminine-coated microtiter plates (A/2; Costar) at a density of 3,000 cells per well with or without different recombinant neurotrophic factors CNTF, GDNF, FGF2, and TGFβ3. In the experiments where 5 μg/ml insulin was added to the culture medium, 1,000 cells were seeded per well. The recombinant GFRα1/Fc chimera (soluble GFRα1) was obtained from R&D and applied in a concentration of 100 nM. The concentrations of the kinase inhibitors were as follows: Wortmannin (Calbiochem) 100 nM; PD98059 (Calbiochem) 50 μM; LY294002 (Calbiochem) 5 μM; and U0126 (Promega) 10 μM. After 24 h, cultures were fixed by addition of 2.5% glutaraldehyde in PBS. Numbers of surviving neurons were determined by direct counting using phase contrast microscopy.
Immunoblotting
The cells were allowed to attach to the PORN/laminine-coated dishes for at least 2 h and treated as indicated. Total cell lysates were subjected to a 8–10% SDS polyacrylamide gel electrophoresis and electroblotted onto nitrocellulose membranes. For detection of proteins the membranes were incubated with the specific antibodies, prior to the addition of the corresponding secondary antibody conjugated to horseradish peroxidase and detection by the enhanced chemoluminescence method (ECL; Amersham Biosciences). The antibodies were obtained from different sources as follows: anti–phospho-ERK and anti-ERK2 from Santa Cruz Biotechnology; anti–phospho-Akt (Ser-437) from New England Biolabs; biotinylated anti-GFRα1 antibody from R&D Systems; and streptavidine-coupled horseradish peroxidase from Amersham Biosciences. Quantification of immunoreactive bands was performed with Scion Image software and statistics performed with Excel.
Measurement of GFRα1 and cRet mRNA levels
Total RNA was extracted from neurons that had been cultured in the presence and absence of TGFβ using Total RNA reagent (Biomol) according to the manufacturer's protocol. Three micrograms of RNA were reverse transcribed with MMLV-RT (Promega) in a 50 μl reaction containing the manufacturer's buffer supplemented with 0.8 mM dNTPs and 0.02 μg/μl random hexanucleotides. Aliquots of 4 μl of the reverse transcription reaction were used for amplification in 30 μl PCR reactions with the following specific forward and reverse primers: GFRα1 forward: 5′-TTGACAAAGTTCCCCCAAAG-3′; GFRα1 reverse: 5′-GTTCGGTGTCATCACTGTGC-3′; Ret forward: 5′-GATGCTGTCGTGGAGTTCAA-3′; Ret reverse: 5′-TCGTTCACCAAAACATCCAA-3′; β-actin forward: 5′-CCAGCCATCTTTCTTGGGTA-3′; and β-actin reverse: 5′-GCGCATTTATGGGTTTTGTT-3′.
Immunocytochemistry
Staining for GFRα1 was done with a biotinylated anti-rat GFRα1 antibody (R&D) for either 2 h on ice prior to fixation of the cells with 4% PFA or after extraction with Triton X-100. In the latter case cells were washed in PBS, incubated on ice in buffer (2 mM MgCl2, 10 mM EGTA, 60 mM Pipes, pH 7) and extracted for 8 to 9 min on ice with 1% Triton X-100 diluted in MSB buffer (Ledesma et al., 1998). The cells were washed with cold MSB and fixed with 4% PFA. After washing a secondary biotinylated antibody was added and immunoreactivity was detected by adding streptavidin-Cy3. For visualization of the lipid raft marker GM1 the cells were then labeled for 45 min with 0.1 μg/ml of fluorescein-conjugated cholera toxin B fragment (Sigma-Aldrich) in PBS with 0.1% BSA, washed with PBS, and mounted in mounting solution.
Acknowledgments
We thank A. Brüntgens, M. Bussacker-Scharpff, J. Fey, and U. Hinz for expert technical assistance. Neuro2a cells stably transfected with full-length GFRα1 were provided by M. Saarma, Helsinki.
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (SFB 530, C8).
Footnotes
Abbreviations used in this paper: CG, ciliary ganglionic; CNS, central nervous system; CNTF, ciliary neurotrophic factor; ERK, extracellular signal-regulated kinase; GDNF, glial cell line–derived neurotrophic factor; GFR, GDNF receptor; GPI, glycosyl-phosphatidylinositol; PD, Parkinson's disease; RTK, receptor tyrosine kinase; PORN, poly-l-ornithine; TGF, transforming growth factor; VIP, vasoactive intestinal peptide.
References
- Alexandrow, M.G., and H.L. Moses. 1995. Transforming growth factor beta and cell cycle regulation. Cancer Res. 55:1452–1457. [PubMed] [Google Scholar]
- Anderson, R.G. 1998. The caveolae membrane system. Annu. Rev. Biochem. 67:199–225. [DOI] [PubMed] [Google Scholar]
- Arce, V., R.A. Pollock, J.M. Philippe, D. Pennica, C.E. Henderson, and O. deLapeyriere. 1998. J. Neurosci. 18:1440–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas, E., M. Trupp, P. Akerud, and C.F. Ibanez. 1995. GDNF prevents degeneration and promotes the phenotype of brain noradrenergic neurons in vivo. Neuron. 15:1465–1473. [DOI] [PubMed] [Google Scholar]
- Beck, K.D., J. Valverde, T. Alexi, K. Poulsen, B. Moffat, R.A. Vandlen, A. Rosenthal, and F. Hefti. 1995. Mesencephalic dopaminergic neurons protected by GDNF from axotomy-induced degeneration in the adult brain. Nature. 373:339–341. [DOI] [PubMed] [Google Scholar]
- Bengtsson, H., S. Soderstrom, A. Kylberg, M.F. Charette, and T. Ebendal. 1998. Potentiating interactions between morphogenetic protein and neurotrophic factors in developing neurons. J. Neurosci. Res. 53:559–568. [DOI] [PubMed] [Google Scholar]
- Besser, D., M. Presta, and Y. Nagamine. 1995. Elucidation of a signaling pathway induced by FGF-2 leading to uPA gene expression in NIH 3T3 fibroblasts. Cell Growth Differ. 6:1009–1017. [PubMed] [Google Scholar]
- Bilak, M.M., and R.W. Kuncl. 2001. Delayed application of IGF-I and GDNF can rescue already injured postnatal motor neurons. Neuroreport. 12:2531–2535. [DOI] [PubMed] [Google Scholar]
- Bilak, M.M., A.M. Corse, and R.W. Kuncl. 2001. Additivity and potentiation of IGF-I and GDNF in the complete rescue of postnatal motor neurons. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2:83–91. [DOI] [PubMed]
- Bilderback, T.R., V.R. Gazula, M.P. Lisanti, and R.T. Dobrowsky. 1999. Caveolin interacts with Trk A and p75(NTR) and regulates neurotrophin signaling pathways. J. Biol. Chem. 274:257–263. [DOI] [PubMed] [Google Scholar]
- Bottenstein, J.E., and G.H. Sato. 1980. Fibronectin and polylysine requirement for proliferation of neuroblastoma cells in defined medium. Exp. Cell Res. 129:361–366. [DOI] [PubMed] [Google Scholar]
- Boulton, T.G., N. Stahl, and G.D. Yancopoulos. 1994. Ciliary neurotrophic factor/leukemia inhibitory factor/interleukin 6/oncostatin M family of cytokines induces tyrosine phosphorylation of a common set of proteins overlapping those induced by other cytokines and growth factors. J. Biol. Chem. 269:11648–11655. [PubMed] [Google Scholar]
- Buj-Bello, A., V.L. Buchman, A. Horton, A. Rosenthal, and A.M. Davies. 1995. GDNF is an age-specific survival factor for sensory and autonomic neurons. Neuron. 15:821–828. [DOI] [PubMed] [Google Scholar]
- Cacalano, G., I. Farinas, L.C. Wang, K. Hagler, A. Forgie, M. Moore, M. Armanini, H. Phillips, A.M. Ryan, L.F. Reichardt, et al. 1998. GFRalpha1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron. 21:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalazonitis, A., J. Kalberg, D.R. Twardzik, R.S. Morrison, and J.A. Kessler. 1992. Transforming growth factor beta has neurotrophic actions on sensory neurons in vitro and is synergistic with nerve growth factor. Dev. Biol. 152:121–132. [DOI] [PubMed] [Google Scholar]
- Chiariello, M., R. Visconti, F. Carlomagno, R.M. Melillo, C. Bucci, V. de Franciscis, G.M. Fox, S. Jing, O.A. Coso, J.S. Gutkind, et al. 1998. Signaling of the Ret receptor tyrosine kinase through the c-Jun NH2-terminal protein kinases (JNKS): evidence for a divergence of the ERKs and JNKs pathways induced by Ret. Oncogene. 16:2435–2445. [DOI] [PubMed] [Google Scholar]
- de Caestecker, M.P., W.T. Parks, C.J. Frank, P. Castagnino, D.P. Bottaro, A.B. Roberts, and R.J. Lechleider. 1998. Smad2 transduces common signals from receptor serine-threonine and tyrosine kinases. Genes Dev. 12:1587–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley, D.T., L. Pang, S.J. Decker, S.J. Bridges, and A.R. Saltiel. 1995. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. USA. 92:7686–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbec, P., C.V. Marcos-Gutierrez, C. Kilkenny, M. Grigoriou, K. Wartiowaara, P. Suvanto, D. Smith, B. Ponder, F. Costantini, M. Saarma, et al. 1996. GDNF signalling through the Ret receptor tyrosine kinase. Nature. 381:789–793. [DOI] [PubMed] [Google Scholar]
- Erickson, J.T., T.A. Brosenitsch, and D.M. Katz. 2001. Brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor are required simultaneously for survival of dopaminergic primary sensory neurons in vivo. J. Neurosci. 21:581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto, H., T. Araki, A. Jackman, R.O. Heuckeroth, W.D. Snider, E.M. Johnson, Jr., and J. Milbrandt. 1998. GFR alpha1-deficient mice have deficits in the enteric nervous system and kidneys. Neuron 21:317–324. [DOI] [PubMed] [Google Scholar]
- Enomoto, H., P.A. Crawford, A. Gorodinsky, R.O. Heuckeroth, E.M. Johnson, Jr., and J. Milbrandt. 2001. RET signaling is essential for migration, axonal growth and axon guidance of developing sympathetic neurons. Development 128:3963–3974. [DOI] [PubMed] [Google Scholar]
- Farkas, L., C. Suter-Crazzolara, and K. Unsicker. 1997. GDNF induces the calretinin phenotype in cultures of embryonic striatal neurons. J. Neurosci. Res. 50:361–372. [DOI] [PubMed] [Google Scholar]
- Favata, M.F., K.Y. Horiuchi, E.J. Manos, A.J. Daulerio, D.A. Stradley, W.S. Feeser, D.E. Van Dyk, W.J. Pitts, R.A. Earl, F. Hobbs, et al. 1998. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 273:18623–18632. [DOI] [PubMed] [Google Scholar]
- Gash, D.M., Z. Zhang, A. Ovadia, W.A. Cass, A. Yi, L. Simmerman, D. Russell, D. Martin, P.A. Lapchak, F. Collins, et al. 1996. Functional recovery in parkinsonian monkeys treated with GDNF. Nature. 380:252–255. [DOI] [PubMed] [Google Scholar]
- Henderson, C.E., H.S. Phillips, R.A. Pollock, A.M. Davies, C. Lemeulle, M. Armanini, L. Simmons, B. Moffet, R.A. Vandlen, and L.C. Simpson. 1994. GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science. 266:1062–1064. [DOI] [PubMed] [Google Scholar]
- Hoffer, B.J., A. Hoffman, K. Bowenkamp, P. Huettl, J. Hudson, D. Martin, L.F. Lin, and G.A. Gerhardt. 1994. Glial cell line-derived neurotrophic factor reverses toxin-induced injury to midbrain dopaminergic neurons in vivo. Neurosci. Lett. 182:107–111. [DOI] [PubMed] [Google Scholar]
- Huang, E.J., and L.F. Reichardt. 2001. Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24:677–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamori, M., J. Shimomura, and Y. Nagai. 1985. Specific binding of cholera toxin to rat erythrocytes revealed by analysis with a fluorescence-activated cell sorter. J. Biochem. 97:729–735. [DOI] [PubMed] [Google Scholar]
- Jing, S., D. Wen, Y. Yu, P.L. Holst, Y. Luo, M. Fang, R. Tamir, L. Antonio, Z. Hu, R. Cupples, J.C. Louis, S. Hu, B.W. Altrock, and G.M. Fox. 1996. GDNF-induced activation of the ret protein tyrosine kinase is mediated by GDNFR-alpha, a novel receptor for GDNF. Cell. 85:1113–1124. [DOI] [PubMed] [Google Scholar]
- Kingsley, D.M. 1994. The TGF-beta superfamily: new members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 8:133–146. [DOI] [PubMed] [Google Scholar]
- Kotzbauer, P.T., P.A. Lampe, R.O. Heuckeroth, J.P. Golden, D.J. Creedon, E.M. Johnson, Jr., and J. Milbrandt. 1996. Neurturin, a relative of glial-cell-line-derived neurotrophic factor. Nature. 384:467–470. [DOI] [PubMed] [Google Scholar]
- Kretzschmar, M., J. Doody, and J. Massague. 1997. Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature. 389:618–622. [DOI] [PubMed] [Google Scholar]
- Krieglstein, K., and K. Unsicker. 1994. Transforming growth factor-β promotes survival of midbrain dopaminergic neurons and protects them against N-methyl-4-phenylpyridinium ion toxicity. Neuroscience. 63:1189–1196. [DOI] [PubMed] [Google Scholar]
- Krieglstein, K., and K. Unsicker. 1996. Distinct modulatory actions of TGF-beta and LIF on neurotrophin-mediated survival of developing sensory neurons. Neurochem. Res. 21:843–850. [DOI] [PubMed] [Google Scholar]
- Krieglstein, K., C. Suter-Crazzolara, and K. Unsicker. 1995. Development of mesencephalic dopaminergic neurons and the transforming growth factor-beta superfamily. J. Neural Transm. Suppl. 46:209–216. [PubMed] [Google Scholar]
- Krieglstein, K., P. Henheik, L. Farkas, J. Jaszai, D. Galter, K. Krohn, and K. Unsicker. 1998. Glial cell line-derived neurotrophic factor requires transforming growth factor-β for exerting its full neurotrophic potential on peripheral and CNS neurons. J. Neurosci. 18:9822–9834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieglstein, K., S. Richter, L. Farkas, N. Schuster, N. Dünker, R.W. Oppenheim, and K. Unsicker. 2000. Reduction of endogenous transforming growth factors beta prevents ontogenetic neuron death. Nat. Neurosci. 3:1085–1090. [DOI] [PubMed] [Google Scholar]
- Lachmund, A., D. Gehrke, K. Krieglstein, and K. Unsicker. 1994. Trophic factors from chromaffin granules promote survival of peripheral and central nervous system neurons. Neuroscience. 62:361–370. [DOI] [PubMed] [Google Scholar]
- Ledesma, M.D., K. Simons, and C.G. Dotti. 1998. Neuronal polarity: essential role of protein-lipid complexes in axonal sorting. Proc. Natl. Acad. Sci. USA. 95:3966–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, L.F.H., D.H. Doherty, J.D. Lile, S. Bektesh, and F. Collins. 1993. GDNF – a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 260:1130–1132. [DOI] [PubMed] [Google Scholar]
- Martinou, J.C., A. Le Van Thai, A. Valette, and M.J. Weber. 1990. Transforming growth factor beta 1 is a potent survival factor for rat embryo motoneurons in culture. Brain Res. Dev. Brain Res. 52:175–181. [DOI] [PubMed] [Google Scholar]
- Moore, M.W., R.D. Klein, I. Farinas, H. Sauer, M. Armanini, H. Phillips, L.F. Reichardt, A.M. Ryan, K. Carver-Moore, and A. Rosenthal. 1996. Renal and neuronal abnormalities in mice lacking GDNF. Nature. 382:76–79. [DOI] [PubMed] [Google Scholar]
- Nakashima, K., M. Yanagisawa, H. Arakawa, N. Kimura, T. Hisatsune, M. Kawabata, K. Miyazono, and T. Taga. 1999. Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science. 284:479–482. [DOI] [PubMed] [Google Scholar]
- Nathan, C., and M. Sporn. 1991. Cytokines in context. J. Cell Biol. 113:981–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neet, K.E., and R.B. Campenot. 2001. Receptor binding, internalization, and retrograde transport of neurotrophic factors. Cell. Mol. Life Sci. 58:1021–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols, B.J., A.K. Kenworthy, R.S. Polishchuk, R. Lodge, T.H. Roberts, K. Hirschberg, R.D. Phair, and J. Lippincott-Schwartz. 2001. Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J. Cell Biol. 153:529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuchtern, J.G., W.E. Biddison, and R.D. Klausner. 1990. Class II MHC molecules can use the endogenous pathway of antigen presentation. Nature. 343:74–76. [DOI] [PubMed] [Google Scholar]
- Oda, K., T. Fujiwara, and Y. Ikehara. 1990. Brefeldin A arrests the intracellular transport of viral envelope proteins in primary cultured rat hepatocytes and HepG2 cells. Biochem. J. 265:161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogilvie, J.M., J.D. Speck, and J.M. Lett. 2000. Growth factors in combination, but not individually, rescue rd mouse photoreceptors in organ culture. Exp. Neurol. 161:676–685. [DOI] [PubMed] [Google Scholar]
- Paratcha, G., F. Ledda, L. Baars, M. Coulpier, V. Besset, J. Anders, R. Scott, and C.F. Ibanez. 2001. Released GFRalpha1 potentiates downstream signaling, neuronal survival, and differentiation via a novel mechanism of recruitment of c-Ret to lipid rafts. Neuron. 29:171–184. [DOI] [PubMed] [Google Scholar]
- Pichel, J.G., L. Shen, H.Z. Sheng, A.C. Granholm, J. Drago, A. Grinberg, E.J. Lee, S.P. Huang, M. Saarma, B.J. Hoffer, et al. 1996. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 382:73–76. [DOI] [PubMed] [Google Scholar]
- Piiper, A., D. Stryjek-Kaminska, R. Gebhardt, and S. Zeuzem. 1996. Pertussis toxin-sensitive G-proteins inhibit fibroblast growth factor-induced signaling in pancreatic acini. J. Cell. Physiol. 167:52–59. [DOI] [PubMed] [Google Scholar]
- Pitts, R.L., S. Wang, E.A. Jones, and A.J. Symes. 2001. Transforming growth factor-beta and ciliary neurotrophic factor synergistically induce vasoactive intestinal peptide gene expression through the cooperation of Smad, STAT, and AP-1 sites. J. Biol. Chem. 276:19966–19973. [DOI] [PubMed] [Google Scholar]
- Pochon, N.A., A. Menoud, J.L. Tseng, A.D. Zurn, and P. Aebischer. 1997. Neuronal GDNF expression in the adult rat nervous system identified by in situ hybridization. Eur. J. Neurosci. 9:463–471. [DOI] [PubMed] [Google Scholar]
- Poulsen, K.T., M.P. Armanini, R.D. Klein, M.A. Hynes, H.S. Phillips, and A. Rosenthal. 1994. TGF beta 2 and TGF beta 3 are potent survival factors for midbrain dopaminergic neurons. Neuron. 13:1245–1252. [DOI] [PubMed] [Google Scholar]
- Razani, B., X.L. Zhang, M. Bitzer, G. von Gersdorff, E.P. Bottinger, and M.P. Lisanti. 2001. Caveolin-1 regulates transforming growth factor (TGF)-beta/SMAD signaling through an interaction with the TGF-beta type I receptor. J. Biol. Chem. 276:6727–6738. [DOI] [PubMed] [Google Scholar]
- Roberts, A.B., and M.B. Sporn. 1990. The transforming growth factor-βs. Handbook of Experimental Pharmacology. M.B. Sporn and A.B. Roberts, editors. Springer/Germany, Heidelberg. 419–172.
- Sanchez, M.P., I. Silos-Santiago, J. Frisen, B. He, S.A. Lira, and M. Barbacid. 1996. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature. 382:70–73. [DOI] [PubMed] [Google Scholar]
- Sargiacomo, M., M. Sudol, Z. Tang, and M.P. Lisanti. 1993. Signal transducing molecules and glycosyl-phosphatidylinositol-linked proteins form a caveolin-rich insoluble complex in MDCK cells. J. Cell Biol. 122:789–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer, H., C. Rosenblad, and A. Bjorklund. 1995. Glial cell line-derived neurotrophic factor but not transforming growth factor beta 3 prevents delayed degeneration of nigral dopaminergic neurons following striatal 6-hydroxydopamine lesion. Proc. Natl. Acad. Sci. USA. 92:8935–8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel, A., and M.P. Lisanti. 2001. The caveolin triad: caveolae biogenesis, cholesterol trafficking, and signal transduction. Cytokine Growth Factor Rev. 12:41–51. [DOI] [PubMed] [Google Scholar]
- Schober, A., R. Hertel, U. Arumae, L. Farkas, J. Jaszai, K. Krieglstein, M. Saarma, and K. Unsicker. 1999. Glial cell line-derived neurotrophic factor rescues target-deprived sympathetic spinal cord neurons but requires transforming growth factor-beta as cofactor in vivo. J. Neurosci. 19:2008–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuchardt, A., V. D'Agati, L. Larsson-Blomberg, F. Costantini, and V. Pachnis. 1994. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 367:380–383. [DOI] [PubMed] [Google Scholar]
- Segal, R.A., and M.E. Greenberg. 1996. Intracellular signaling pathways activated by neurotrophic factors. Annu. Rev. Neurosci. 19:463–489. [DOI] [PubMed] [Google Scholar]
- Stahl, N., and G.D. Yancopoulos. 1994. The tripartite CNTF receptor complex: activation and signaling involves components shared with other cytokines. J. Neurobiol. 25:1454–1466. [DOI] [PubMed] [Google Scholar]
- Subramaniam, N., G.M. Leong, T.A. Cock, J.L. Flanagan, C. Fong, J.A. Eisman, and A.P. Kouzmenko. 2001. Cross-talk between 1,25-dihydroxyvitamin D3 and transforming growth factor-beta signaling requires binding of VDR and Smad3 proteins to their cognate DNA recognition elements. J. Biol. Chem. 276:15741–15746. [DOI] [PubMed] [Google Scholar]
- Tansey, M.G., R.H. Baloh, J. Milbrandt, and E.M. Johnson, Jr. 2000. GFRalpha-mediated localization of RET to lipid rafts is required for effective downstream signaling, differentiation, and neuronal survival. Neuron. 25:611–623. [DOI] [PubMed] [Google Scholar]
- Tomac, A., E. Lindqvist, L.F. Lin, S.O. Ogren, D. Young, B.J. Hoffer, and L. Olson. 1995. Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature. 373:335–339. [DOI] [PubMed] [Google Scholar]
- Treanor, J.J., L. Goodman, F. de Sauvage, D.M. Stone, K.T. Poulsen, C.D. Beck, C. Gray, M.P. Armanini, R.A. Pollock, F. Hefti, et al. 1996. Characterization of a multicomponent receptor for GDNF. Nature. 382:80–83. [DOI] [PubMed] [Google Scholar]
- Trupp, M., M. Ryden, H. Jornvall, H. Funakoshi, T. Timmusk, E. Arenas, and C.F. Ibanez. 1995. Peripheral expression and biological activities of GDNF, a new neurotrophic factor for avian and mammalian peripheral neurons. J. Cell Biol. 130:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trupp, M., E. Arenas, M. Fainzilber, A.S. Nilsson, B.A. Sieber, M. Grigoriou, C. Kilkenny, E. Salazar-Grueso, V. Pachnis, and U. Arumae. 1996. Functional receptor for GDNF encoded by the c-ret proto-oncogene. Nature. 381:785–789. [DOI] [PubMed] [Google Scholar]
- Trupp, M., R. Scott, S.R. Whittemore, and C.F. Ibanez. 1999. Ret-dependent and -independent mechanisms of glial cell line-derived neurotrophic factor signaling in neuronal cells. J. Biol. Chem. 274:20885–20894. [DOI] [PubMed] [Google Scholar]
- Ulloa, L., J. Doody, and J. Massague. 1999. Inhibition of transforming growth factor beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature. 397:710–713. [DOI] [PubMed] [Google Scholar]
- Unsicker, K., and K. Krieglstein. 2000. Co-activation of TGF-βs and cytokine signaling pathways are required for neurotrophic functions. Cytokine Growth Factor Rev. 11:97–102. [DOI] [PubMed] [Google Scholar]
- Van Heyningen, W.E., S. Van Heyningen, and C.A. King. 1976. The nature and action of cholera toxin. Ciba Found. Symp. 42:73–88. [DOI] [PubMed] [Google Scholar]
- Vlahos, C.J., W.F. Matter, K.Y. Hui, and R.F. Brown. 1994. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269:5241–5248. [PubMed] [Google Scholar]
- Ward, S.G., C.H. June, and D. Olive. 1996. PI3-kinase: a pivotal pathway in T-cell activation? Immunol. Today. 17:187–197. [DOI] [PubMed] [Google Scholar]
- Wrana, J. 2000. Crossing smads. Sci. STKE. 2000:RE1. [DOI] [PubMed]
- Yamamoto, T., T. Matsuda, A. Muraguchi, K. Miyazono, and M. Kawabata. 2001. Cross-talk between IL-6 and TGF-beta signaling in hepatoma cells. FEBS Lett. 492:247–253. [DOI] [PubMed] [Google Scholar]
- Yanagi, Y., M. Suzawa, M. Kawabata, K. Miyazono, J. Yanagisawa, and S. Kato. 1999. Positive and negative modulation of vitamin D receptor function by transforming growth factor-beta signaling through smad proteins. J. Biol. Chem. 274:12971–12974. [DOI] [PubMed] [Google Scholar]
- Yanagisawa, J., Y. Yanagi, Y. Masuhiro, M. Suzawa, M. Watanabe, K. Kashiwagi, T. Toriyabe, M. Kawabata, K. Miyazono, and S. Kato. 1999. Convergence of transforming growth factor-beta and vitamin D signaling pathways on SMAD transcriptional coactivators. Science. 283:1317–1321. [DOI] [PubMed] [Google Scholar]
- Zhang, Y., X.H. Feng, and R. Derynck. 1998. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature. 394:909–913. [DOI] [PubMed] [Google Scholar]