

Abstract
Salmonella enterica, the causative agent of food poisoning and typhoid fever, induces programmed cell death in macrophages, a process found to be dependent on a type III protein secretion system, and SipB, a protein with membrane fusion activity that is delivered into host cells by this system. When expressed in cultured cells, SipB caused the formation of and localized to unusual multimembrane structures. These structures resembled autophagosomes and contained both mitochondrial and endoplasmic reticulum markers. A mutant form of SipB devoid of membrane fusion activity localized to mitochondria, but did not induce the formation of membrane structures. Upon Salmonella infection of macrophages, SipB was found in mitochondria, which appeared swollen and devoid of christae. Salmonella-infected macrophages exhibited marked accumulation of autophagic vesicles. We propose that Salmonella, through the action of SipB, kills macrophages by disrupting mitochondria, thereby inducing autophagy and cell death.
Keywords: apoptosis; bacterial pathogenesis; membrane fusion; host–pathogen interactionl; type III protein secretion
Introduction
Central to the pathogenicity of Salmonella enterica is the function of a type III secretion system (TTSS) encoded within a discrete region of its chromosome known as Salmonella pathogenicity island 1 (SPI-1; Galán, 2001). This system injects into host cells a battery of effector proteins that have the capacity to precisely modulate cellular functions. In intestinal epithelial cells, Salmonella induces profuse actin cytoskeleton rearrangements and nuclear responses that ultimately lead to bacterial uptake and the production of proinflammatory cytokines. These responses are the consequence of the activation of the Rho family GTPases Cdc42 and Rac by the bacterially encoded exchange factors SopE and SopE2, and the phosphoinositide phosphatase SopB, which are delivered into host cells by the SPI-1 TTSS (Galán, 2001).
In contrast, in macrophages, Salmonella induces programmed cell death, a process that is also dependent on the function of the SPI-1 TTSS (Chen et al., 1996; Monack et al., 1996). The mechanisms by which Salmonella kills macrophages are poorly understood. Macrophages undergoing Salmonella-induced cell death exhibit heterogeneous morphological features indicative of both necrotic and apoptotic cell death (Chen et al., 1996; Monack et al., 1996; Brennan and Cookson, 2000; Boise and Collins, 2001). It is clear that more than one mechanism is involved in the Salmonella-induced killing of macrophages. One type of death, which occurs within 1 h of infection, exhibits “necrotic-like” features, such as organelle degeneration and lack of chromatin condensation. The other type of death, which occurs after ∼5 h of infection, exhibits more typical features of apoptosis, such as chromatin condensation and DNA breakage. Experiments have shown that the rapid death, which has been referred to as “programmed necrosis,” is dependent on caspase-1 because Salmonella-induced cell death is significantly delayed in caspase-1–defective macrophages (Hersh et al., 1999; Brennan and Cookson, 2000; Jesenberger et al., 2000). Very little is known about the mechanisms that lead to Salmonella-induced caspase-1–independent macrophage cell death. Both forms of macrophage cell death triggered by Salmonella are strictly dependent on the SPI-1 TTSS. Previous work has shown that the caspase-1–dependent macrophage cell death is triggered by SipB, a Salmonella protein that is delivered into host cells by the SPI-1 TTSS (Hersh et al., 1999). SipB apparently binds and activates caspase-1, resulting in the stimulation of an unconventional form of programmed cell death with features of necrosis.
Nothing is known about the Salmonella SPI-1 TTSS effector protein(s) that may be responsible for the stimulation of caspase-1–independent programmed cell death. Dissecting the different mechanisms by which Salmonella triggers programmed cells death is essential to understand their biological significance and contribution to pathogenesis. In these analyses, we have focused on the caspase-1–independent pathway of Salmonella-induced macrophage cell death. We found that this pathway is the consequence of the activity of the SPI-1 TTSS effector protein translocase SipB. During infection, SipB localized to mitochondria, and when transiently expressed in cultured cells, it formed multilamellar structures that resembled autophagic vesicles and contained ER and mitochondrial markers. Furthermore, macrophages infected with Salmonella accumulated numerous autophagosomes. We propose that Salmonella induces autophagy-mediated programmed cell death in macrophages by disrupting mitochondria.
Results
A Salmonella enterica serovar Typhimurium strain devoid of its SPI-1 TTSS effector proteins retains cytotoxicity for caspase-1–deficient macrophages
TTSSs secrete a set of proteins that are either effectors of cellular responses or are involved in the translocation of the effector proteins into host cells (Galán and Collmer, 1999). In the case of the Salmonella SPI-1 TTSS, the translocases SipB, SipC, and SipD mediate the transfer of a battery of effector proteins with diverse functions (Collazo and Galán, 1997). It is unclear whether Salmonella uses a distinct set of SPI-1 TTSS–secreted proteins to induce caspase-1–independent macrophage cell death. To investigate this issue, we constructed strains of Salmonella enterica serovar Typhimurium (S. typhimurium) carrying loss-of-function mutations in the genes encoding each of all known SPI-1 TTSS effector proteins, including the Cdc42 and Rac activators SopE (Hardt et al., 1998b) and SopE2 (Stender et al., 2000), the phosphoinositide phosphatase SopB (Norris et al., 1998), the actin-binding protein SipA (Zhou et al., 1999), the tyrosine phosphatase and GTPase-activating protein SptP (Fu and Galán, 1999), and AvrA (Hardt and Galán, 1997), a protein highly related to the ubiquitin-like protein proteinase YopJ from Yersinia spp., which has been implicated in the induction of apoptosis by these bacteria (Orth et al., 2000). All these mutant strains retained the ability to kill caspase-1–defective bone marrow–derived primary macrophages (BMDPM) in a manner that was indistinguishable from that of the wild type (unpublished data). To examine the possibility of redundancy in the function of effector proteins, we constructed an S. typhimurium strain simultaneously carrying loss-of-function mutations in all known effector proteins of the SPI-1 TTSS (hereafter refer to as “effectorless”). The effectorless Salmonella mutant was able to induce cell death in caspase-1–defective BMDPM in a manner that was indistinguishable from that of the wild type (Fig. 1, A and B). Because this mutant strain presumably can only deliver SipB, SipD, and SipC, these results indicated that any of these proteins (or a combination thereof) could be directly responsible for macrophage cytotoxicity.
Figure 1.
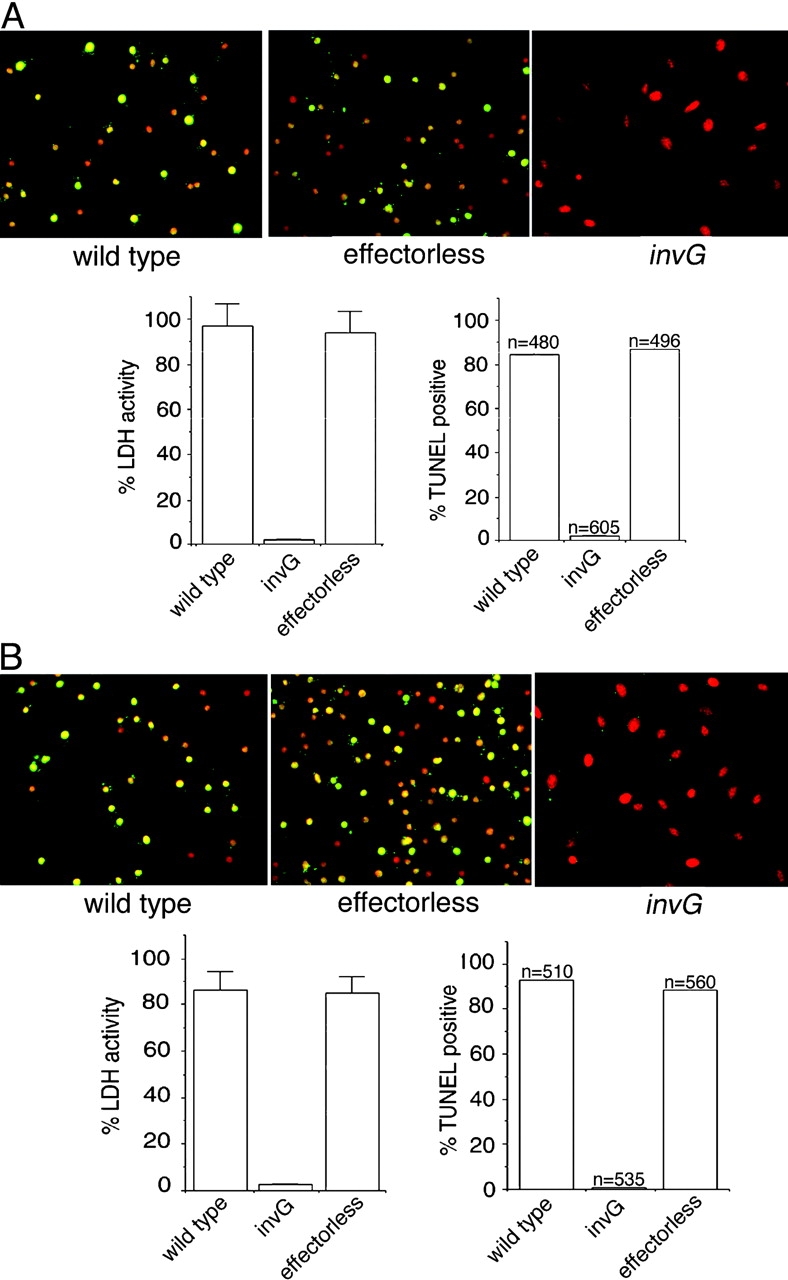
An S. typhimurium strain devoid of its SPI-1 TTSS effector proteins is able to induce programmed cell death in macrophages. BMDPM from wild-type (A) or caspase-1−/− (B) mice (Kuida et al., 1995) were infected with wild-type S. typhimurium or mutant derivative either lacking all TTSS effector proteins (effectorless) or defective for TTSS secretion by virtue a having a mutation on an essential component of this system (invG). 6 h after infection, cytotoxicity was evaluated by lactate dehydrogenase release or by terminal deoxynucleatide transferase (TUNEL) staining. Values for the lactate dehydrogenase release represent the average of at least three independent measurements. Equivalent results were obtained in several repetitions of these experiments.
Transient expression of the SPI-1 translocase/effector protein SipB in caspase-1–deficient macrophages results in cell death
Salmonella strains carrying loss-of-function mutations in sipB, sipC, or sipD are unable to induce macrophage cell death (Chen et al., 1996). SipB, SipC, and SipD are referred to as translocases because they mediate the passage of all effector proteins through the host cell plasma membrane (Collazo and Galán, 1997). For this reason, it is unclear whether the inability of these mutants to kill macrophages is due to their role in the translocation of each other or to their direct ability to induce cell death. To investigate the potential role of the TTSS protein translocases in the stimulation of caspase-1–independent programmed cell death, we expressed SipB, SipC, or SipD in BMDPM from caspase-1−/− mice and examined their cytotoxicity. Expression of either SipC or SipD did not lead to cytotoxicity (Fig. 2; unpublished data). Expression of full-length SipB was not detectable, presumably due to the rapid death of the transfected macrophages combined to the intrinsic low level of expression characteristic of most TTSS effector proteins containing their secretion and translocation domain (Hardt et al., 1998a; unpublished data). In an attempt to improve the level of expression of SipB, we removed its putative secretion and translocation domain located at the amino terminus. Expression of this SipB deletion could be detected in BMDPM from caspase-1−/− mice only early after transfection (Fig. 2; unpublished data), presumably because transfected macrophages detached from the dish. These macrophages exhibited obvious signs of cytotoxicity and marked chromatin condensation (Fig. 2; unpublished data). Together, these results indicate that SipB is cytotoxic for macrophages, and is most likely directly responsible for the induction of caspase-1–independent macrophage cell death.
Figure 2.

Transient expression of SipB in BMDPM from caspase-1 −/− mice results in cell death. Plasmids expressing either SipB, SipC, or SipD fused to YFP were introduced into BMDPM from caspase-1−/− mice as indicated in the Materials and methods section. Macrophages were then fixed, stained with DAPI (to visualize chromatin), and observed under a fluorescence microscope. Cells expressing SipC or SipD do not show any signs of cytotoxicity. In contrast, cells expressing SipB show clear signs of cytotoxicity and chromatin condensation. Arrows indicate the nucleus of transfected cells.
Expression of SipB in cells resistant to Salmonella-induced death results in the formation of multilamellar structures that contain mitochondrial and endothelial reticulum markers
Because expression of SipB in macrophages results in their rapid death, we examined the localization of SipB tagged with YFP in transiently transfected COS-2 cells, which are resistant to Salmonella-induced cell death. SipB-YFP localized almost exclusively to round vesicular structures of varying sizes that did not resemble typical organelles (Fig. 3 A). This localization pattern was not due to excessive expression of SipB because it was observed even when very low amounts of DNA were used for transfection, although in this case the structures were somewhat smaller in size and significantly fewer in number (Fig. 3 C; unpublished data). Mitochondria were frequently observed in close association with these unusual vesicular structures, or even contained within them (Fig. 3 B). In addition, the SipB-induced vesicular structures were labeled with the mitochondrial marker MitoTracker® Red (Fig. 3 B) and antibodies directed to the mitochondrial protein CoxIV (unpublished data). In transient cotransfection experiments, the vesicular structures also colocalized with the Sec61-CFP fusion protein, a marker for the ER (Fig. 3 C). Similar results were obtained when SipB was expressed in 293T, Henle-407, or Ref52 cells (unpublished data). These observations indicate that both mitochondria and ER-derived membranes are components of the SipB-induced vesicular structure.
Figure 3.
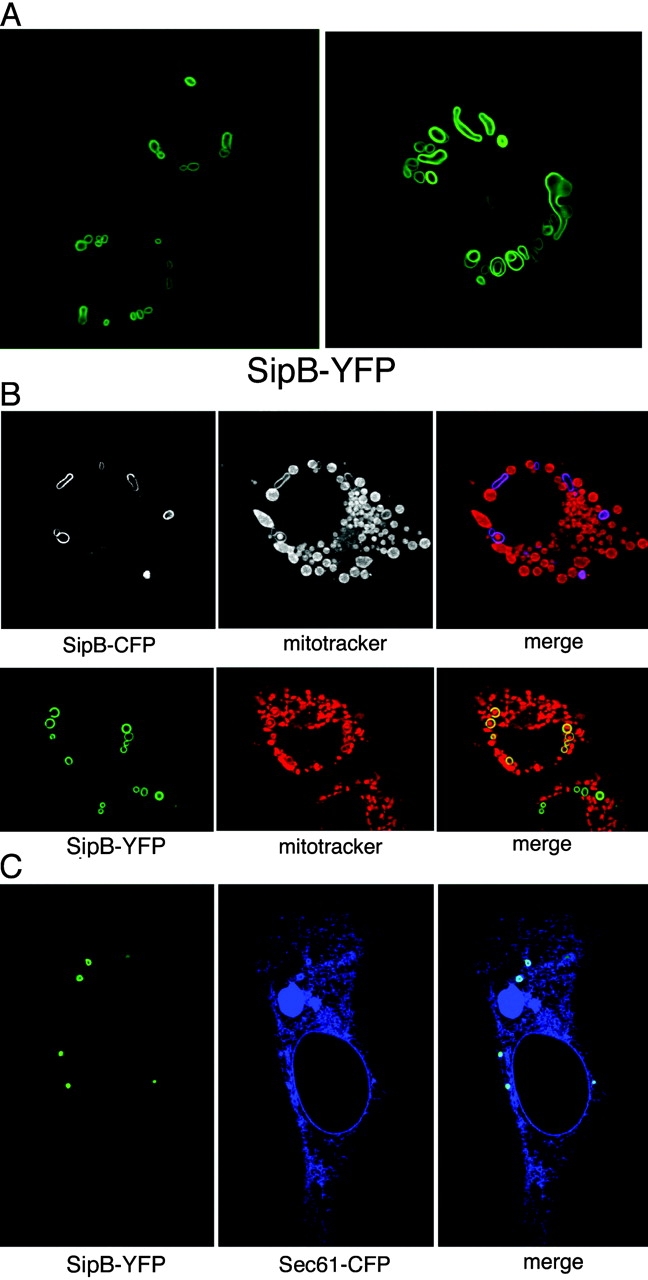
Transient expression of SipB in COS-2 cells. (A) A plasmid expressing YFP-tagged SipB was transfected into COS-2 cells, and its localization was examined under a confocal microscope. (B) SipB colocalized with mitochondrial markers in transfected cells. COS-2 cells were transfected with a plasmid encoding either SipB-CFP (top) or SipB-YFP (bottom). Transfected cells were stained with MitoTracker® Red (Molecular Probes, Inc.) to visualize mitochondria and were examined by confocal microscopy. The SipB-induced structures were readily stained with mitochondrial markers (top). Mitochondria were also seen contained within the SipB-induced structures (bottom). (C) SipB-induced structures were labeled with the ER marker Sec-61. COS-2 cells were cotransfected with plasmids encoding SipB-YFP and Sec61-CFP, and transfected cells were examined by confocal microscopy.
By EM, the vesicular structures were seen to present a striking “onion-like” appearance composed of a series of closely apposed membranes, most frequently (although not always) arranged in a circular fashion (Fig. 4). These structures stained readily with a gold-labeled antibody directed to SipB or GFP (Fig. 4). Mitochondria were often seen in close association with the onion-like structures (Fig. 4 A), or even within them (Fig. 4 D). On occasions, mitochondria appeared as if they were undergoing fusion to the SipB-induced structures (Fig. 4, B and C). Indeed, the SipB-induced membranous structures closely resemble the appearance of mitochondria in Drosophila spermatids during spermatogenesis. During this process, which is controlled by the fuzzy onion gene, mitochondria aggregate next to the spermatid nuclei and fuse into giant organelles that wrap around one another and are known as Nebenkern (Hales and Fuller, 1997). The SipB-induced structures also resemble the appearance of mitochondria in cells lacking the epsilon subunit of the ATP synthase (Paumard et al., 2002), which is involved in the formation of mitochondria cristae, and in cells overexpressing mitofusin 2 (Santel and Fuller, 2001), the mammalian homologue of fuzzy onion.
Figure 4.
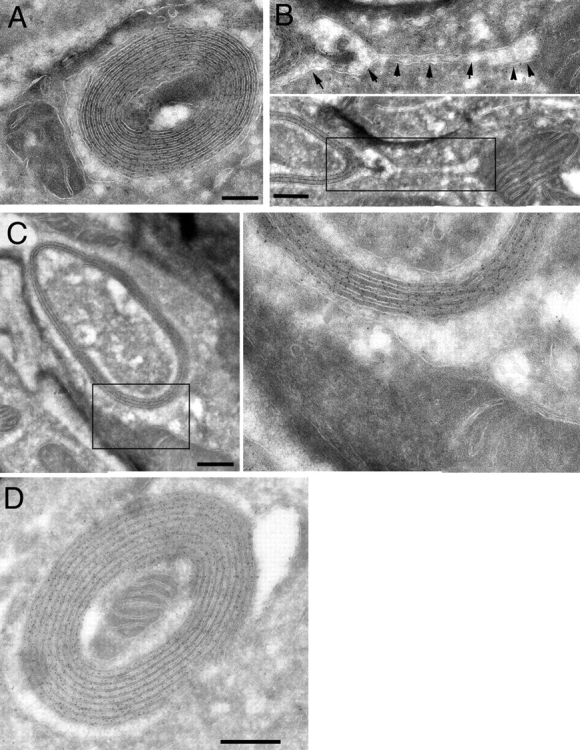
Immuno-EM of SipB-transfected COS-2 cells. COS-2 cells were transfected with a plasmid expressing SipB-YFP, and 24 h after transfection, cells were fixed and prepared for immuno-EM using an antibody specific for GFP and a gold-labeled anti–rabbit antibody. SipB was seen localized to structures made up of closely apposed membranes that were often in close proximity to mitochondria (A). Mitochondria were often seen apparently fusing to the SipB-induced membranous structure (B, see arrows in inset on top; C, inset to the right). Mitochondria were often seen contained with the SipB-induced membrane structures (D). Bars, 250 nm.
The membrane fusion activity of SipB is required for the formation of multilamellar structures
Previous works have shown that purified SipB can integrate into phospholipid membranes and induce liposomal fusion at neutral pH (Hayward et al., 2000). To explore the potential contribution of the SipB membrane fusion activity to the formation of multivesicular structures in transiently transfected cells, we expressed a SipB mutant (SipB428–593), which has been shown to lack fusion activity while retaining its ability to bind to membranes (Hayward et al., 2000; McGhie et al., 2002). Expression of SipB428–593 failed to induce mitochondrial remodeling and macrophage cell death (Fig. 5 A). However, SipB428–593 retained its ability to localize to mitochondria, as indicated by its colocalization with the mitochondrial marker MitoTracker® Red (Fig. 5 A; unpublished data). This mutant form of SipB has been previously shown to exert a dominant-negative effect over wild-type SipB. When simultaneously present in a membrane fusion assay, SipB428–593 was able to inhibit the ability of the wild-type protein to induce membrane fusion (Hayward et al., 2000; McGhie et al., 2002). Consistent with the requirement of the fusion activity of SipB for the formation of the multivesicular structures, SipB428–593 abolished the ability of wild-type SipB to induce the formation of these unusual structures when simultaneously coexpressed in cultured cells (Fig. 5 B; unpublished data). These results indicate that the membrane fusion activity of SipB is required for its ability to induce the formation of multivesicular compartments, but not for its ability to localize to mitochondria.
Figure 5.

The nonfusogenic SipB428–593 mutant localizes to the mitochondria, but does not form multivesicular structures and interferes with the activity of the wild-type protein. (A) COS-2 cells were transfected with a plasmid expressing YFP-tagged SipB428–593, stained with an antibody to the mitochondrial protein CoxIV, subunit VIc (Molecular Probes, Inc.), and examined by confocal microscopy. SipB428–593-YFP localized to mitochondria and did not induce the formation of multivesicular structures seen after expression of the wild-type protein. (B) COS-2 cells were cotransfected with plasmids expressing either SipB428–593-YFP or SipB-CFP, and transfected cells were examined under a confocal microscope. Expression of SipB428–593-YFP prevented wild-type SipB-CFP from forming multivesicular structures (notice that vesicular structures are only apparent in the absence, but not in the presence, of SipB428–593-YFP; right).
SipB localizes to mitochondria during S. typhimurium infection of caspase-1–defective macrophages
The transient transfection experiments suggested that SipB induces macrophage cell death by disrupting mitochondria. Therefore, we examined whether SipB localizes to mitochondria during bacterial infection. BMDPM from caspase-1−/− mice were infected with wild-type S. typhimurium or the TTSS translocation-defective sipD mutant and examined for the presence of SipB in mitochondria by immuno-EM and biochemical fractionation. The S. typhimurium sipD mutant strain can serve as a negative control because it is taken up by macrophages in a manner similar to the wild type and is competent for the secretion of SipB, but completely defective for its translocation into the host cell cytosol (Kaniga et al., 1995). Immuno-EM of infected macrophages showed the presence of SipB in the mitochondria of macrophages infected with wild-type Salmonella, but not in mitochondria of macrophages infected with the TTSS translocation-deficient sipD mutant (Fig. 6 A). Even at earlier times after infection, mitochondria of macrophages infected with wild-type Salmonella displayed disrupted morphology characterized by swelling and the disappearance of cristae (Fig. 6 A). At later times, the appearance of mitochondria was so severely disrupted that it was difficult to recognize them under the electron microscope (unpublished data). The presence of SipB in mitochondria was verified by cellular fractionation analysis. BMDPM from caspase-1−/− mice were infected with wild-type S. typhimurium or the sipD mutant strain, mitochondria were isolated by sucrose gradient centrifugation, and the presence of SipB was determined by Western immunoblot analysis. SipB was readily detected in mitochondria from macrophages infected with wild-type S. typhimurium, but not in mitochondria from macrophages infected with an S. typhimurium strain carrying a mutation in the sipD gene (Fig. 6 B). Reprobing the blots with an antibody to the mitochondrial protein CoxIV verified that in both cases, equal amount of material had been loaded (Fig. 6 B). These results indicate that SipB is also capable of targeting mitochondria during bacterial infection.
Figure 6.
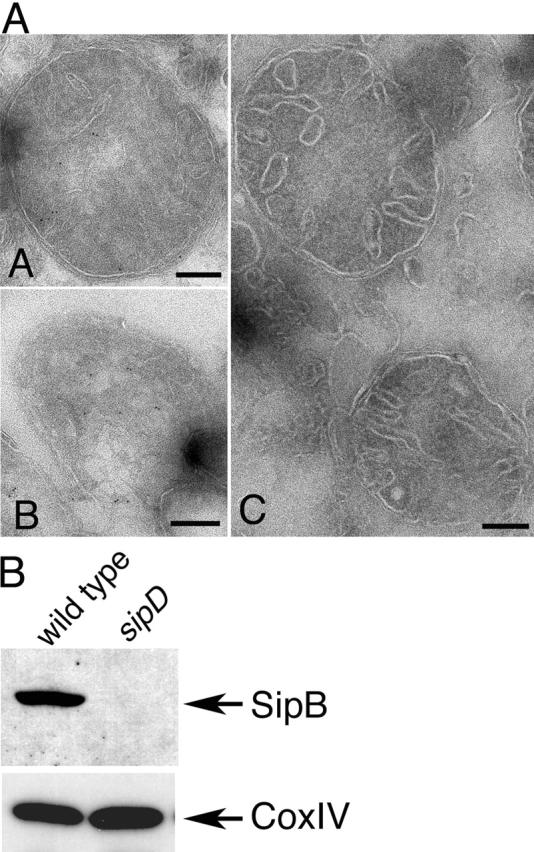
The S. typhimurium SPI-1 TTSS–secreted protein SipB localizes to the mitochondria of infected macrophages. (A) Bone marrow–derived macrophages were infected with either wild-type S. typhimurium or the TTSS translocation-defective sipD mutant strain with a multiplicity of infection of 50. 3 h after infection, macrophages were collected and processed for immuno-EM using gold-labeled antibodies. SipB could be readily detected in macrophages infected with wild-type (A and B), but not in macrophages infected with the sipD mutant strain (C). Bars, 100 nm. (B) Alternatively, mitochondria were isolated from the infected macrophages and the presence of SipB or the resident mitochondrial protein CoxIV, subunit IV, and were examined by Western immunoblot analysis using specific antibodies.
S. typhimurium infection of macrophages induces the formation of autophagosomes
In their multilamellar nature and in their association with ER markers and damaged mitochondria, the SipB-induced structures in transfected cells resemble autophagosomes, membranous structures normally induced to sequester and remove damaged organelles (Kim and Klionsky, 2000; Klionsky and Emr, 2000). Therefore, we investigated the presence of autophagosomes in macrophages during bacterial infection. BMDPM from caspase-1−/− mice were infected with wild-type S. typhimurium or the translocation-defective sipD mutant strain, and we examined them for the presence of autophagosomes using the autophagic vacuole marker monodansylcadaverine (MDC; Biederbick et al., 1995; Inbal et al., 2002). Macrophages infected with wild-type S. typhimurium, but not those infected with the translocation-defective isogenic sipD mutant strain, exhibited abundant MDC-stained structures (Fig. 7, panel I). In addition, electron microscopic analysis of macrophages infected with wild-type S. typhimurium revealed the presence of a large number of multivesicular structures resembling autophagosomes (Fig. 7, A–D), which were absent from macrophages infected with the translocation-defective sipD mutant (Fig. 7, E and F). The autophagosome-like structures exhibited a somewhat different appearance from that of the structures induced by SipB expression in resistant cells such as COS, most likely because macrophages rapidly die before structures like the ones observed in resistant cells could be visualized. Together, these results indicate that S. typhimurium infection of macrophages induces the formation of autophagosomes.
Figure 7.
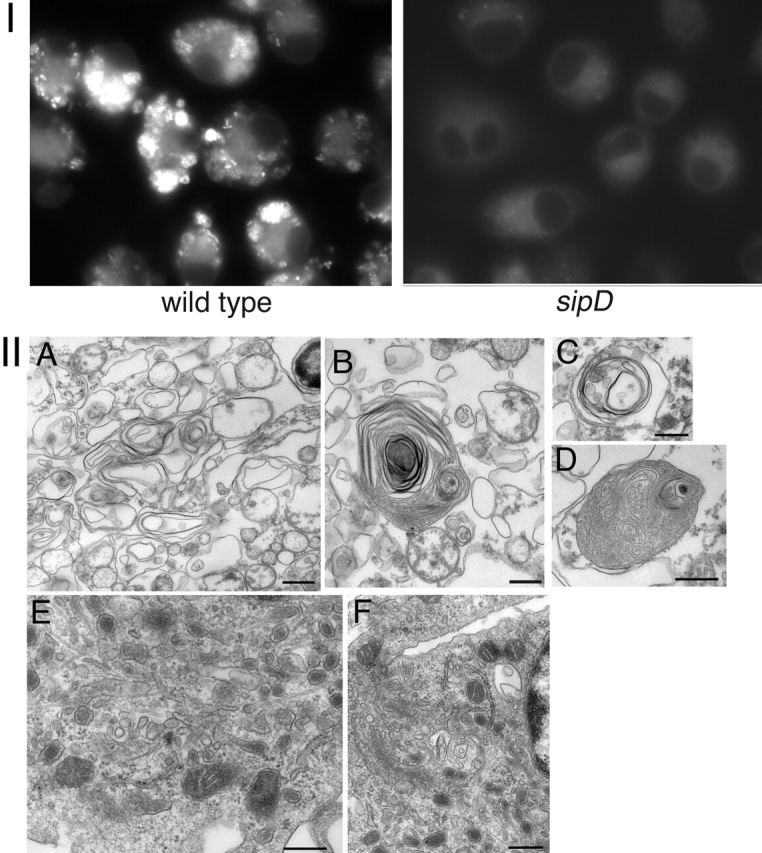
S. typhimurium-infected macrophages exhibited signs of autophagocytosis. Caspase-1−/− BMDPM were infected with wild-type S. typhimurium or it isogenic translocation-defective sipD mutant. 6 h after infection, macrophages were either stained with MDC to label autophagosomes (I) or processed for EM (II) as described in the legend to Fig. 3. Macrophages infected with wild-type S. typhimurium (but not those infected with the sipD mutant) exhibited a large number of vesicular structures that stained with MDC (I). Under the electron microscope (II), macrophages infected with wild-type Salmonella exhibited multivesicular structures resembling autophagosomes (A–D) Often, electron-dense material, presumably damaged organelles, were readily observed within the multivesicular structures (B–D). In contrast, macrophages infected with type III secretion-defective sipD mutant did not exhibit the large number of multivesicular structures observed in wild-type–infected cells. Bars (A and E) 500 nm; (B, C, D and F) 250 nm.
S. typhimurium cytotoxicity for macrophages does not require caspase activity
The observation that transient expression of SipB in cultured cells as well as infection of caspase-1–defective macrophages with S. typhimurium resulted in the formation of autophagosomes prompted us to consider the possibility that induction of autophagy may be linked to the induction of macrophage cell death. Autophagy is a highly ordered phenomenon that has been linked to the induction of programmed cell death, and is considered an important component of programmed tissue remodeling during development (Bursch et al., 2000; Inbal et al., 2002). Although the molecular mechanisms of the induction and execution of autophagy-mediated programmed cell death (also known as “type II” programmed cell death) are poorly understood, it has been shown that these mechanisms involve elements common to classical apoptosis or “type I” programmed cell death, as well as processes apparently unique to type II programmed cell death (Schwartz et al., 1993; Lemasters, 1999; Xue et al., 1999; Cohen et al., 2002). A distinctive feature of type II programmed cell death is its independence from the activity of caspases (Xue et al., 1999). Therefore, we examined the caspase dependence of Salmonella-induced macrophage cell death. BMDPM from caspase-1−/− mice were infected with wild-type S. typhimurium or the translocation-deficient sipD mutant in the presence or absence of the pan-caspase inhibitor Z-VAD. Consistent with the involvement of type II programmed cell death, the Salmonella-induced macrophage cell death was not significantly affected by the addition of a pan-caspase inhibitor (Fig. 8).
Figure 8.
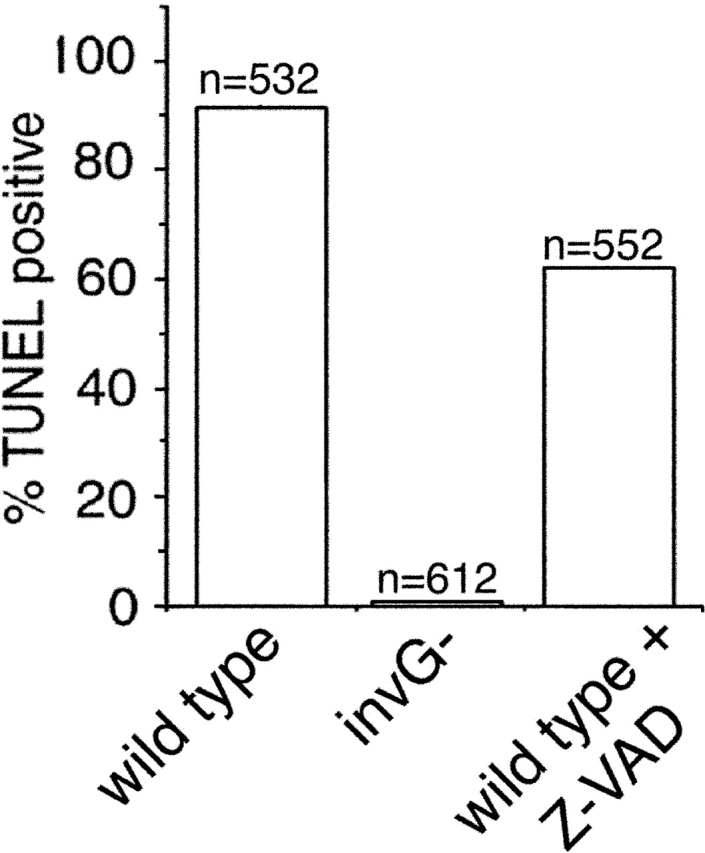
The S. typhimurium-induced macrophage death does not require caspase activity. BMDPM were infected with wild-type S. typhimurium or the TTSS-defective invG mutant in the presence or absence of 50 μM of the pan-caspase inhibitor Z-VAD, and cell death was measured as indicated in the legend to Fig. 1.
Discussion
Salmonella induces programmed cell death in macrophages by complex mechanisms that involve distinct pathways and require the bacterial TTSS encoded in the pathogenicity island 1 (SPI-1 TTSS). One death pathway, which results in rapid macrophage death with features of necrosis, is dependent on caspase-1 and the SPI-1 TTSS–secreted protein SipB (Hersh et al., 1999; Brennan and Cookson, 2000; Jesenberger et al., 2000). However, macrophages from caspase-1−/− mice also undergo programmed cell death, although with delayed kinetics and different morphological features. Here, we have described another death pathway induced by Salmonella that is independent of caspase-1. A systematic bacterial genetic analysis led us to conclude that this cell death pathway is also dependent on SipB, a protein with membrane fusion activity that is delivered into host cells by the SPI-1 TTSS. This analysis was complicated by the known dual function of SipB, which acts both as an effector of virulence within cells and as a protein translocase required for the transfer of other effector proteins into host cells. Nevertheless, we found that a strain of Salmonella defective in the production of all known effector proteins delivered by the SPI-1 TTSS retained its ability to kill caspase-1−/− macrophages. More importantly, expression of SipB in caspase-1−/− macrophages resulted in their death. The involvement of SipB is also supported by the observation that a saturation mutagenesis screen for S. typhimurium genes involved in macrophage cytotoxicity exclusively yielded mutations in genes that encode components or regulators of the SPI-1 TTSS or the protein translocases themselves (unpublished data). The latter indicates that it is unlikely that there is an unknown SPI-1 effector responsible for the induction of macrophage cell death.
To gain insight into the mechanisms by which SipB kills macrophages, we expressed this protein in a cell line that is resistant to Salmonella-induced killing. We observed that in these cells, SipB induced the formation of multivesicular structures that resembled autophagosomes. These structures contained mitochondrial and ER markers, suggesting that mitochondria and ER-derived membranes are components of the SipB-induced vesicular structure. In fact, mitochondria were seen either in close association to or within the SipB-induced structures. These observations, in conjunction with the similarity between the SipB-induced membranous structures with those formed as a consequence of mitochondrial fusion such as Nebenkern (Hales and Fuller, 1997), suggest the intriguing possibility that SipB may have the capacity to induce mitochondrial fusion. In fact, a mutant form of SipB that is devoid of membrane fusion activity in vitro did not induce the formation of the multivesicular structures. In addition, expression of this mutant form of SipB, which in the in vitro liposome fusion assay has a dominant-negative effect over wild type (Hayward et al., 2000), prevented the formation of the multilamellar structures. These results strongly implicate the fusogenic activity of SipB in the formation of the multilamellar structures. The formation of Nebenkern is mediated by the product of the fuzzy onion gene (Hales and Fuller, 1997), which has mammalian counterparts known as mitofusins (Santel and Fuller, 2001). Interestingly, the modular organization of SipB (i.e., the arrangement of hydrophobic domains and coiled-coil regions), as well as its predicted topology when inserted into membranes (McGhie et al., 2002), are reminiscent of the mitofusins (Rojo et al., 2002). Therefore, it is possible that SipB exerts its function at least in part by mimicking some activities of these host cell proteins. More experiments will be required to investigate this hypothesis.
How is SipB targeted to the mitochondria? Examination of the primary amino acid sequence of SipB did not reveal canonical mitochondrial targeting sequences. Previous work has shown that SipB integration into liposomes, as well as its membrane fusion activity, is significantly influenced by the membrane lipid composition (Hayward et al., 2000). Importantly, the fusion activity of SipB was markedly enhanced when vesicles contained cardiolipin, a lipid that is abundant in mitochondrial membranes. Recent work has shown that the mitochondrial targeting of the proapoptotic Bcl-2 family members Bax (Kuwana et al., 2002) and BID (Lutter et al., 2000) is mediated by cardiolipin. It is intriguing to hypothesize that SipB may be targeted to mitochondria in a similar fashion.
What is the relationship between SipB's ability to localize to the mitochondria and form autophagosome-like structures in transient transfection experiments, and its ability to induce cell death in macrophages during bacterial infection? We found that during Salmonella infection, SipB localizes to mitochondria of infected macrophages, which appeared damaged and devoid of cristae. In addition, we found that macrophages undergoing cell death as a consequence of Salmonella infection exhibited the presence of numerous autophagosomes. A less well-understood form of programmed cell death, known as type II, has been linked to the induction of autophagocytosis (Schwartz et al., 1993; Lemasters, 1999; Xue et al., 1999; Cohen et al., 2002). Although many of the features of this type of programmed cell death are similar to those of conventional apoptosis (i.e., chromatin condensation, DNA breakage, etc.), there are features that appear to be unique to type II programmed cell death. For example, type II programmed cell death does not require caspase activation because addition of pan-caspase inhibitors does not prevent this type of cell death (Xue et al., 1999). We found that addition of a pan-caspase inhibitor did not prevent Salmonella-induced killing of caspase-1−/− macrophages. We propose from these analyses that Salmonella-induced macrophage cell killing is the result of autophagy-mediated or type II programmed cell death triggered by the activity of SipB in the mitochondria, and perhaps in other organelles such as the ER. We postulate that SipB may trigger this response by damaging mitochondria and/or by altering the balance between mitochondria fusion and fission, which are known stimulators of autophagy (Frank et al., 2001; Tolkovsky et al., 2002).
Little is known about the biological significance of the different forms of programmed cell death triggered by Salmonella and their relative contribution to pathogenesis and/or host defense. It is unclear whether they constitute a host defense response to the incoming pathogen to halt its replication or a process triggered by the pathogen to avoid host defense mechanisms. The specific impact of the caspase-1–dependent and –independent forms of cell death in the innate and acquired immune response to Salmonella is also not known. Undoubtedly, the understanding of the mechanisms by which the different forms of cell death triggered by Salmonella come about will help to understand the biological and immunological consequences of these different forms of cell death.
Previous works have shown that effector proteins delivered via TTSSs modulate cellular functions by virtue of precisely mimicking host cell proteins (Stebbins and Galán, 2001). In some cases, the bacterial effectors share extensive amino acid sequence similarity with host cell proteins (e.g., tyrosine phosphatases and inositol polyphosphatases), whereas in others the similarity is not apparent at the primary amino acid sequence level, but is revealed by their crystal structures. SipB may be another example of a bacterial protein that mimics the function of a host cell protein to secure the pathogen's replication. The ability of this bacterial protein to remodel mitochondria and trigger programmed cell death is a remarkable example of the ability of microbial pathogens to modulate cellular functions.
Materials and methods
Bacterial strains and plasmids
The wild-type, invG-, and sipD S. typhimurium strains have been described previously (Kaniga et al., 1994, 1995). The effectorless strain containing null mutations in the sopE, sopE2, sopB, avrA, sptP, sipA, sopA, sopD, and sopD2 genes was constructed by conjugation and P22-mediated allele replacement strategies according to standard procedures (Kaniga et al., 1994). Sequences encoding SipB, SipB428–593, SipC, or SipD were amplified by PCR and cloned into the BamHI-XbaI sites of pEYFP-C1 or pECFP-C1 (CLONTECH Laboratories, Inc.) by standard techniques. The plasmid pECFP-Sec61 was a gift from Walther Mothes (Yale University, New Haven, CT).
Isolation of mitochondria from infected macrophages
Mitochondria were isolated as indicated previously (Scaffidi et al., 1998) with some modifications. In brief, BMDPM from caspase-1−/− macrophages were seeded overnight on 15-cm plates at a density of ∼2 × 106 cells per plate. Cells were infected with a multiplicity of infection of ∼15 with wild-type S. typhimurium or mutant derivatives defective in type III secreted protein translocation (sipD) or lacking the TTSS effector proteins (effectorless). Infections were performed for 50 min at 37°C. Plates were washed three times with HBSS, and were then incubated with media containing 100 μg/ml gentamicin for an additional 50 min. Plates were washed three times with HBSS, and were then incubated at 37°C in 10 ml HBSS containing 200 μg proteinase K. After 20 min, 10 ml ice-cold HBSS containing 2 mM PMSF was added to each plate. Cells were harvested by scraping, and mitochondria were isolated as described previously (Scaffidi et al., 1998).
Fluorescence and electron microscopy
Plasmids expressing SipB, SipB428–593, or Sec61 were transfected into Cos-2 cells using FuGENE™ 6 (Roche) following the manufacturer's instructions. For immunofluorescence analyses, cells were fixed 18 h after transfection with 3.7% PFA, mounted, and visualized under a confocal microscope (LSM510; Carl Zeiss MicroImaging, Inc.) equipped with a META detector. For immuno-EM, cells were fixed 18 h after infection and processed as described previously (Seemann et al., 2002) using an antibody specific for either GFP (provided by L. Pelletier, Yale University, New Haven, CT) or SipB, and a gold-labeled anti–rabbit secondary antibody.
Bacterial infections and macrophage cell death assay
Wild-type or caspase-1−/− mouse BMDPM were seeded overnight on glass coverslips. The next morning, cells were infected with the different strains of S. typhimurium for 25 min at a multiplicity of infection of 50:1 at 37°C. Cells were washed, 100 μg/ml gentamicin was added to the media to kill any extracellular bacteria, and 6 h after this, cells were fixed with 3.7% PFA. Nuclei were stained with propidium iodide (red) and fragmented chromatin was labeled by TUNEL staining as described previously (Chen et al., 1996). To visualize autophagosomes, caspase-1−/− BMDPM were infected with wild-type or sipD (effector-protein-translocation defective) Salmonella for 25 min with a multiplicity of infection of 50:1 at 37°C. 6 h after infection, MDC was added to the media to a final concentration of 100 μM, and cells were incubated at 37°C for an additional 60 min. Cells were washed three times with HBSS, fixed with 3.7% PFA, and visualized under a fluorescence microscope. To evaluate the toxicity of SipB, SipC, or SipD, caspase-1−/− BMDPM were transfected with plasmids encoding these proteins using the Human CD34 Cell Nucleofector™ Kit according to the manufacturer's instructions (Amaxa Biosystems). In brief, 8 × 106 cells were resuspended in 100 μl Nucleofector™ Solution. 2 μg plasmid DNA (pEYFP-C1, PEYFP-SipB, pEYFP-SipC, or pEYFP-SipD) was added, and the cells were transferred to a cuvette (Amaxa Biosystems). Cells were electroporated using the U-08 program on the Nuclefector™ device, resuspended in media (RPMI 1640 containing 20% FBS and 30% L cell supernatant), and incubated overnight on glass coverslips. Cells were then fixed with 3% PFA, stained with an anti-GFP antibody (CLONTECH Laboratories, Inc.) and DAPI, and examined for signs of toxicity by fluorescence microscopy.
Acknowledgments
We thank members of the Galán laboratory for careful review of this manuscript, Walther Mothes for various reagents, and L. Pelletier for the anti-GFP antibody.
This work was supported by a National Institutes of Health grant GM52543 (to J.E. Galán) and a fellowship from the Damon Runyon-Walter Winchell Foundation (to L.D. Hernandez).
Abbreviations used in this paper: BMDPM, bone marrow–derived primary macrophages; MDC, monodansylcadaverine; SPI-1, Salmonella pathogenicity island 1; TTSS, type III secretion system.
References
- Biederbick, A., H. Kern, and H. Elsasser. 1995. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur. J. Cell Biol. 66:3–14. [PubMed] [Google Scholar]
- Boise, L., and C. Collins. 2001. Salmonella-induced cell death: apoptosis, necrosis or programmed cell death? Trends Microbiol. 9:64–67. [DOI] [PubMed] [Google Scholar]
- Brennan, M.A., and B.T. Cookson. 2000. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol. Microbiol. 38:31–40. [DOI] [PubMed] [Google Scholar]
- Bursch, W., A. Ellinger, C. Gerner, U. Frohwein, and R. Schulte-Hermann. 2000. Programmed cell death (PCD). Apoptosis, autophagic PCD, or others? Ann. NY Acad. Sci. 926:1–12. [DOI] [PubMed] [Google Scholar]
- Chen, L.M., K. Kaniga, and J.E. Galán. 1996. Salmonella spp. are cytotoxic for cultured macrophages. Mol. Microbiol. 21:1101–1115. [DOI] [PubMed] [Google Scholar]
- Cohen, I., M. Castedo, and G. Kroemer. 2002. Tantalizing Thanatos: unexpected links in death pathways. Trends Cell Biol. 12:293–295. [DOI] [PubMed] [Google Scholar]
- Collazo, C., and J.E. Galán. 1997. The invasion-associated type III system of Salmonella typhimurium directs the translocation of Sip proteins into the host cell. Mol. Microbiol. 24:747–756. [DOI] [PubMed] [Google Scholar]
- Frank, S., B. Gaume, E. Bergmann-Leitner, W. Leitner, E. Robert, F. Catez, C. Smith, and R. Youle. 2001. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell. 1:515–525. [DOI] [PubMed] [Google Scholar]
- Fu, Y., and J.E. Galán. 1999. A salmonella protein antagonizes Rac-1 and Cdc42 to mediate host-cell recovery after bacterial invasion. Nature. 401:293–297. [DOI] [PubMed] [Google Scholar]
- Galán, J.E. 2001. Salmonella interaction with host cells: type III secretion at work. Annu. Rev. Cell Dev. Biol. 17:53–86. [DOI] [PubMed] [Google Scholar]
- Galán, J.E., and A. Collmer. 1999. Type III secretion machines: bacterial devices for protein delivery into host cells. Science. 284:1322–1328. [DOI] [PubMed] [Google Scholar]
- Hales, K., and M. Fuller. 1997. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. 90:121–129. [DOI] [PubMed] [Google Scholar]
- Hardt, W.-D., and J.E. Galán. 1997. A secreted Salmonella protein with homology to an avirulence determinant of plant pathogenic bacteria. Proc. Natl. Acad. Sci. USA. 94:9887–9892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardt, W.-D., L.-M. Chen, K.E. Schuebel, X.R. Bustelo, and J.E. Galán. 1998. a. Salmonella typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell. 93:815–826. [DOI] [PubMed] [Google Scholar]
- Hardt, W.-D., H. Urlaub, and J.E. Galán. 1998. b. A target of the centisome 63 type III protein secretion system of Salmonella typhimurium is encoded by a cryptic bacteriophage. Proc. Natl. Acad. Sci. USA. 95:2574–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward, R.D., E.J. McGhie, and V. Koronakis. 2000. Membrane fusion activity of purified SipB, a Salmonella surface protein essential for mammalian cell invasion. Mol. Microbiol. 37:727–739. [DOI] [PubMed] [Google Scholar]
- Hersh, D., D. Monack, M. Smith, N. Ghori, S. Falkow, and A. Zychlinsky. 1999. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc. Natl. Acad. Sci. USA. 96:2396–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inbal, B., S. Bialik, I. Sabanay, G. Shani, and A. Kimchi. 2002. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J. Cell Biol. 157:455–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesenberger, V., K.J. Procyk, J. Yuan, S. Reipert, and M. Baccarini. 2000. Salmonella-induced caspase-2 activation in macrophages: a novel mechanism in pathogen-mediated apoptosis. J. Exp. Med. 192:1035–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniga, K., J.C. Bossio, and J.E. Galán. 1994. The Salmonella typhimurium invasion genes invF and invG encode homologues to the PulD and AraC family of proteins. Mol. Microbiol. 13:555–568. [DOI] [PubMed] [Google Scholar]
- Kaniga, K., D. Trollinger, and J.E. Galán. 1995. Identification of two targets of the type III protein secretion system encoded by the inv and spa loci of Salmonella typhimurium that have homology to the Shigella IpaD and IpaA proteins. J. Bacteriol. 177:7078–7085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J., and D. Klionsky. 2000. Autophagy, cytoplasm-to-vacuole targeting pathway, and pexophagy in yeast and mammalian cells. Annu. Rev. Biochem. 69:303–342. [DOI] [PubMed] [Google Scholar]
- Klionsky, D., and S. Emr. 2000. Autophagy as a regulated pathway of cellular degradation. Science. 290:1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida, K., J. Lippke, G. Ku, M. Harding, D. Livingston, M. Su, and R. Flavell. 1995. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 267:2000–2003. [DOI] [PubMed] [Google Scholar]
- Kuwana, T., M. Mackey, G. Perkins, M. Ellisman, M. Latterich, R. Schneiter, D. Green, and D. Newmeyer. 2002. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 111:331–342. [DOI] [PubMed] [Google Scholar]
- Lemasters, J. 1999. V. Necrapoptosis and the mitochondrial permeability transition: shared pathways to necrosis and apoptosis. Am. J. Physiol. 276:G1–G6. [DOI] [PubMed] [Google Scholar]
- Lutter, M., M. Fang, X. Luo, M. Nishijima, X. Xie, and X. Wang. 2000. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat. Cell Biol. 2:754–761. [DOI] [PubMed] [Google Scholar]
- McGhie, E.J., P.J. Hume, R.D. Hayward, J. Torres, and V. Koronakis. 2002. Topology of the Salmonella invasion protein SipB in a model bilayer. Mol. Microbiol. 44:1309–1321. [DOI] [PubMed] [Google Scholar]
- Monack, D.M., B. Raupach, A.E. Hromockyj, and S. Falkow. 1996. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc. Natl. Acad. Sci. USA. 93:9833–9838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris, F.A., M.P. Wilson, T.S. Wallis, E.E. Galyov, and P.W. Majerus. 1998. SopB, a protein required for virulence of Salmonella dublin, is an inositol phosphate phosphatase. Proc. Natl. Acad. Sci. USA. 95:14057–14059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth, K., Z. Xu, M.B. Mudgett, Z.Q. Bao, L.E. Palmer, J.B. Bliska, W.F. Mangel, B. Staskawicz, and J.E. Dixon. 2000. Disruption of signaling by Yersinia effector YopJ, a ubiquitin-like protein protease. Science. 290:1594–1597. [DOI] [PubMed] [Google Scholar]
- Paumard, P., J. Vaillier, B. Coulary, J. Schaeffer, V. Soubannier, D. Mueller, D. Brethes, J. di Rago, and J. Velours. 2002. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 21:221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo, M., F. Legros, D. Chateau, and A. Lombes. 2002. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J. Cell Sci. 115:1663–1674. [DOI] [PubMed] [Google Scholar]
- Santel, A., and M. Fuller. 2001. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 114:867–874. [DOI] [PubMed] [Google Scholar]
- Scaffidi, C., S. Fulda, A. Srinivasan, C. Friesen, F. Li, K. Tomaselli, K. Debatin, P. Krammer, and M. Peter. 1998. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 17:1675–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz, L., S. Smith, M. Jones, and B. Osborne. 1993. Do all programmed cell deaths occur via apoptosis? Proc. Natl. Acad. Sci. USA. 90:980–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann, J., M. Pypaert, T. Taguchi, J. Malsam, and G. Warren. 2002. Partitioning of the matrix fraction of the Golgi apparatus during mitosis in animal cells. Science. 295:848–851. [DOI] [PubMed] [Google Scholar]
- Stebbins, C.E., and J.E. Galán. 2001. Structural mimicry in bacterial virulence. Nature. 412:701–705. [DOI] [PubMed] [Google Scholar]
- Stender, S., A. Friebel, S. Linder, M. Rohde, S. Mirold, and W.D. Hardt. 2000. Identification of SopE2 from Salmonella typhimurium, a conserved guanine nucleotide exchange factor for Cdc42 of the host cell. Mol. Microbiol. 36:1206–1221. [DOI] [PubMed] [Google Scholar]
- Tolkovsky, A., L. Xue, G. Fletcher, and V. Borutaite. 2002. Mitochondrial disappearance from cells: a clue to the role of autophagy in programmed cell death and disease? Biochimie. 84:233–240. [DOI] [PubMed] [Google Scholar]
- Xue, L., G. Fletcher, and A. Tolkovsky. 1999. Autophagy is activated by apoptotic signalling in sympathetic neurons: an alternative mechanism of death execution. Mol. Cell. Neurosci. 14:180–198. [DOI] [PubMed] [Google Scholar]
- Zhou, D., M. Mooseker, and J.E. Galán. 1999. Role of the S. typhimurium actin-binding protein SipA in bacterial internalization. Science. 283:2092–2095. [DOI] [PubMed] [Google Scholar]