

Abstract
The mitochondrial death pathway is triggered in cultured sympathetic neurons by deprivation of nerve growth factor (NGF), but the death mechanisms activated by deprivation of other neurotrophic factors are poorly studied. We compared sympathetic neurons deprived of NGF to those deprived of glial cell line–derived neurotrophic factor (GDNF). In contrast to NGF-deprived neurons, GDNF-deprived neurons did not die via the mitochondrial pathway. Indeed, cytochrome c was not released to the cytosol; Bax and caspase-9 and -3 were not involved; overexpressed Bcl-xL did not block the death; and the mitochondrial ultrastructure was not changed. Similarly to NGF-deprived neurons, the death induced by GDNF removal is associated with increased autophagy and requires multiple lineage kinases, c-Jun and caspase-2 and -7. Serine 73 of c-Jun was phosphorylated in both NGF- and GDNF-deprived neurons, whereas serine 63 was phosphorylated only in NGF-deprived neurons. In many NGF-deprived neurons, the ultrastructure of the mitochondria was changed. Thus, a novel nonmitochondrial caspase-dependent death pathway is activated in GDNF-deprived sympathetic neurons.
Keywords: glial cell line–derived neurotrophic factor; NGF; Bax; cytochrome c; caspases
Introduction
Programmed cell death is a process by which unwanted cells are intentionally removed due to either physiological or pathological reasons. Morphological appearance of the dying cells and the death program (molecular and cellular death pathways) can differ remarkably between cell types and death stimuli (Clarke, 1990; Leist and Jäättelä, 2001; Zimmermann et al., 2001). Currently, two death pathways have been described in detail, the death receptor (extrinsic) and mitochondrial (intrinsic) pathways. The extrinsic pathway is activated by tumor necrosis factor receptor superfamily death receptor ligation (Vincenz, 2001). The death-inducing signaling complex, assembled directly at the death receptors, activates the initiator caspase-8 that in turn activates caspase-3, -6, and -7. Activation of the intrinsic death pathway leads to release of cytochrome c (but also other apoptotic molecules) from the mitochondrial intermembrane space to the cytosol. Cytosolic cytochrome c triggers formation of the apoptosome that activates the initiator caspase-9 followed by activation of caspase-3, -6, and -7. It was shown recently that caspase-2 is activated upstream of mitochondria and may participate in the activation of mitochondria-related death events (Guo et al., 2002; Lassus et al., 2002; Read et al., 2002).
The mitochondrial death pathway is triggered by different modes of cellular stress and, in some cells, by removal (deprivation) of survival (trophic) factors. The well-characterized examples of such cells are the neonatal mouse or rat sympathetic neurons that critically depend on NGF for survival. Withdrawal of NGF from the cultured sympathetic neurons leads to the following events: the protein levels and phosphorylation of transcription factor c-Jun are increased (Estus et al., 1994; Ham et al., 1995; Virdee et al., 1997; Eilers et al., 1998), proapoptotic protein Bax is translocated from the cytosol to the mitochondria (Deckwerth et al., 1996; Putcha et al., 1999), and cytochrome c is released from the mitochondria to the cytosol (Deshmukh and Johnson, 1998; Neame et al., 1998; Martinou et al., 1999) with Smac/DIABLO, a protein that releases caspases from the inhibitor of apoptosis proteins (Deshmukh et al., 2002). As a result, caspase-9, -3 (Deshmukh et al., 2000, 2002), and -2 (Troy et al., 2001) are activated. All these events are critically required for the NGF deprivation-induced death. The neurons then exhibit classical features of apoptosis, including condensation of chromatin, cleavage of DNA, and increased autophagy (Martin et al., 1988; Pittman et al., 1993; Edwards and Tolkovsky, 1994; Xue et al., 1999), and they finally die in the culture by secondary necrosis.
In addition to these two pathways, several other death pathways exist (Clarke, 1990; Leist and Jäättelä, 2001), but those remain largely unknown. Cells in which the intrinsic apoptotic pathway is blocked can still be induced to die both in vitro and in vivo, often with nonapoptotic ultrastructure (Yaginuma et al., 2001; Oppenheim et al., 2001; Zaidi et al., 2001; Marsden et al., 2002). Recently, novel death pathways have been proposed for the dependence receptors that trigger death by a novel mechanism without their ligands, whereas this death was blocked in the presence of the ligands (Rabizadeh et al., 1993; Ellerby et al., 1999; Bordeaux et al., 2000; Llambi et al., 2001; Thibert et al., 2003). In the case of the deleted in colorectal cancer receptor, this mechanism includes direct interaction of caspases with the receptor and does not require the death receptors or mitochondrial pathways. Certainly, further death pathways exist.
NGF is currently the best-characterized neurotrophic factor. Although many more neurotrophic factors are known that promote survival of different types of neurons (Huang and Reichardt, 2001), the death pathways activated by their withdrawal are virtually unstudied. Glial cell line–derived neurotrophic factor (GDNF; Airaksinen and Saarma, 2002) is a neurotrophic factor that promotes survival of several neuronal populations, including neonatal rat sympathetic neurons (Kotzbauer et al., 1996). NGF and GDNF signal via different receptor systems, TrkA/p75 and Ret/GFRα1 complex, respectively. We compared the death programs triggered in the same cell type (sympathetic neurons) by removal of two different neurotrophic factors (NGF or GDNF). Surprisingly, we found that the death pathways activated in these two cases differ considerably.
Results
One third of neonatal rat superior cervical ganglion (SCG) neurons is GDNF responsive
When sympathetic neurons from 1- or 2-d-old rat SCG were cultured with GDNF, the number of neurons gradually and slowly decreased. By 6 days in vitro (DIV), ∼34% of the initially plated neurons had survived, and this number did not decrease further (Fig. 1 A). In similar conditions, ∼98% of the NGF-maintained neurons had survived (Fig. 1 A). Dose dependence study revealed that 100 ng/ml was a saturating concentration of GDNF for SCG neurons (Fig. 1 B). Removal of GDNF from the 6 DIV cultures leads to death of the neurons that was kinetically similar to the death of NGF-deprived neurons (Fig. 1 C). In all studies described throughout this paper, the neurons from newborn rat SCG were maintained in sister dishes with 100 ng/ml GDNF or 30 ng/ml NGF for 6 DIV, deprived of these factors for 48 or 72 h, and analyzed. NGF- and GDNF-responsive neurons were always analyzed in parallel and received identical treatment. The GDNF-responsive neurons appeared morphologically indistinguishable from NGF-responsive neurons. However, the data obtained from GDNF-responsive neurons were generally more variable and more cultures failed when compared with NGF-responsive neurons. Also, a small number of neurons in the GDNF-deprived dishes seemed to die nonspecifically due to the washing procedure. Thus, GDNF-responsive neurons seem to be more sensitive to small variations in the culture conditions than NGF-responsive neurons.
Figure 1.
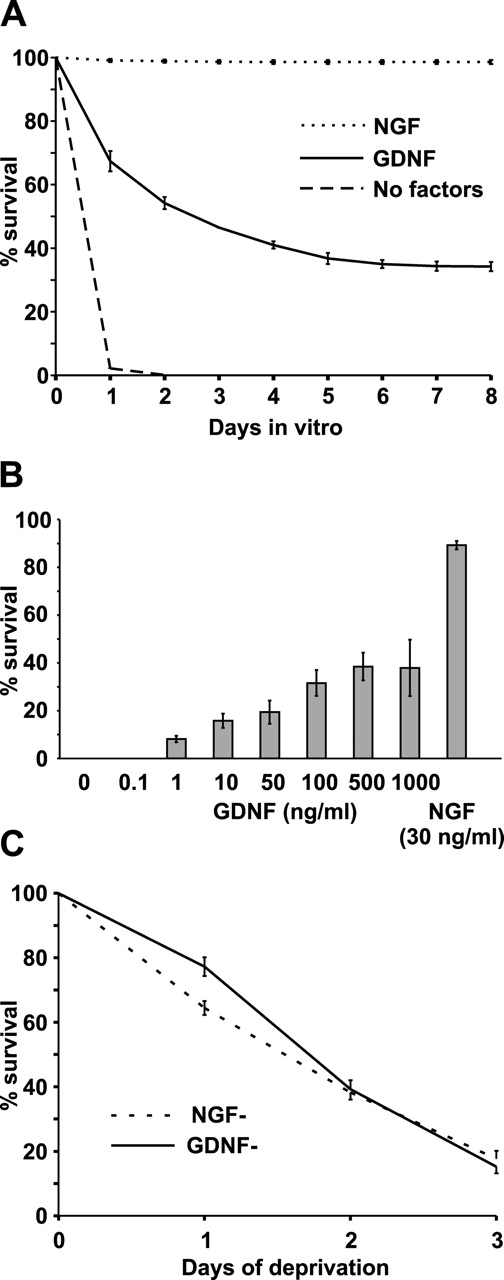
Culture conditions for GDNF-responsive newborn rat sympathetic neurons from the superior cervical ganglion. (A) Newly isolated neurons were grown with 100 ng/ml GDNF, 30 ng/ml NGF, or without neurotrophic factors for 8 d. The living neurons were counted daily and expressed as a percentage of initial neurons counted 2 h after plating. The mean ± SEM of three independent cultures is shown. (B) Newly isolated neurons were maintained with different doses of GDNF for 6 d. The living neurons were counted daily and expressed as a percentage of initial neurons counted 2 h after plating. For comparison, survival with 30 ng/ml NGF is also shown. The mean ± SEM of three independent cultures is shown. (C) Neurons were first maintained with 100 ng/ml GDNF or 30 ng/ml NGF for 6 d. Neurotrophic factors were removed (0 d of deprivation), and the neurons were grown further without them. Living neurons were counted daily and expressed as a percentage of initial neurons counted immediately after factor deprivation. The mean ± SEM of six independent cultures is shown.
Mitochondrial death pathway is not activated in GDNF-deprived sympathetic neurons
To study the localization of cytochrome c, we removed GDNF or NGF from the respective neurons for 48 h in the presence of broad-range caspase inhibitor boc-aspartyl(OMe)-fluoromethylketone (BAF) and stained the neurons with anti–cytochrome c antibodies. Only a small number of GDNF-deprived neurons showed faint diffuse cytochrome c staining characteristic of its cytosolic localization (Fig. 2). In contrast, removal of NGF dramatically reduced the number of neurons with punctate mitochondrial cytochrome c localization (Fig. 2), as shown previously (Deshmukh and Johnson, 1998; Neame et al., 1998; Martinou et al., 1999). Similar results were obtained at 72 h after neurotrophic factor deprivation (unpublished data).
Figure 2.
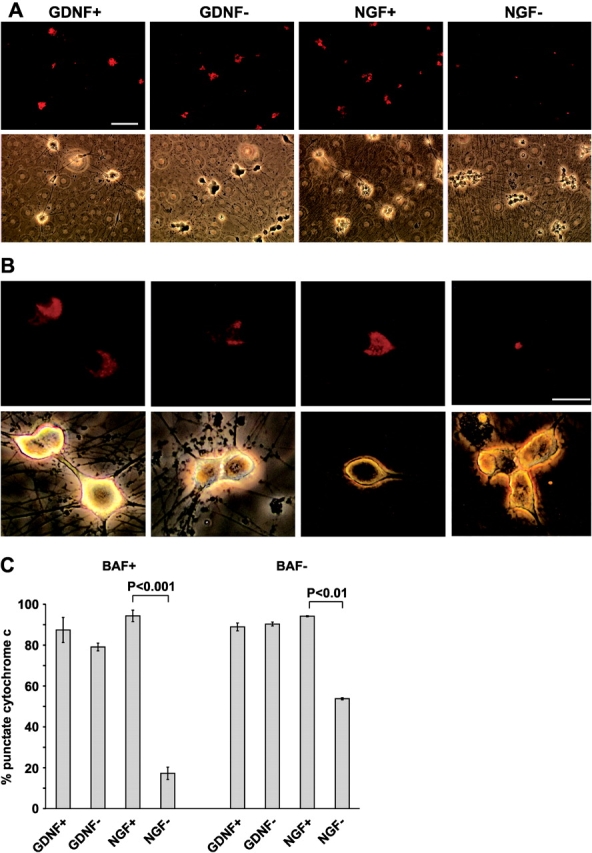
Cytochrome c is not released from the mitochondria of GDNF-deprived sympathetic neurons. (A) Micrographs of the neurons deprived of GDNF or NGF for 48 h in the presence of caspase inhibitor BAF, or maintained with these factors, and immunostained with cytochrome c antibodies are shown in the top row. Corresponding phase-contrast images are shown on the bottom row. Note that weak and diffuse immunostaining is barely visible on the NGF-deprived neurons, whereas almost all GDNF-deprived neurons stain strongly like the neurons maintained with the factors. (B) Typical cytochrome c immunostaining patterns of GDNF- or NGF- deprived or -maintained neurons are shown in the top row. Corresponding phase-contrast images are shown on the bottom row. Note that in spite of pyknotic appearance, the GDNF-deprived neurons show strong punctate immunostaining (mitochondrial localization), whereas the NGF-deprived neurons stain weakly and diffusely (cytosolic localization). Levels of the images were equally enhanced with Adobe Photoshop software. (C) Quantitation of the neurons deprived of neurotrophic factors for 48 h with or without BAF and having punctate pattern of cytochrome c immunostaining, calculated as a percentage of all neurons. Experiments with or without BAF were performed separately (four independent experiments for both) and combined in the same figure. The mean ± SEM is shown. Statistical significance of the differences between factor-maintained and -deprived groups was estimated by t test. Bars: (A) 100 μm; (B) 10 μm.
The role of Bax in the GDNF-deprived neurons was studied by overexpressing Ku70, a protein that was recently shown to bind the NH2 terminus of Bax, thereby inhibiting its translocation to the mitochondria (Sawada et al., 2003a,b). Ku70 had no effect on the death of GDNF-deprived neurons, although it significantly blocked the death of NGF-deprived neurons at 72 h (Fig. 3 A). A similar effect was achieved with the Ku70-derived cell-permeable peptide V5 shown to block the activity of Bax (Sawada et al., 2003a), whereas the control peptide I5 had no effect (Fig. 3 B).
Figure 3.
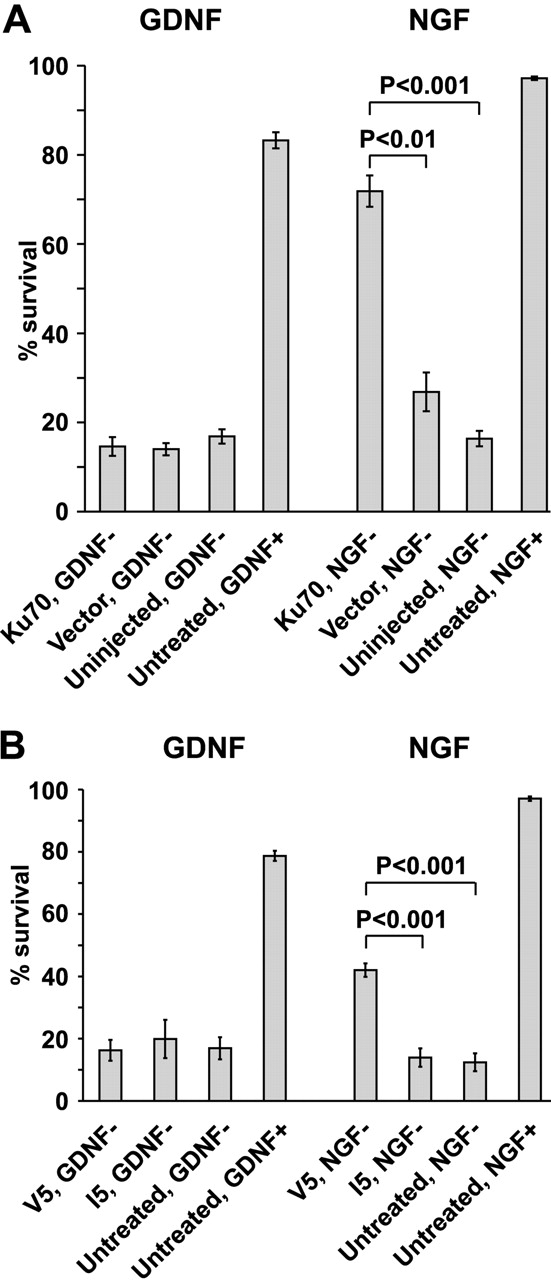
Bax is not required for death of GDNF-deprived sympathetic neurons. (A) GDNF- or NGF-deprived neurons were microinjected with expression plasmids for Ku70 or empty vector. (B) Neurons were deprived of GDNF or NGF in the presence of Ku70-derived Bax-blocking peptide V5 or control peptide I5. In A and B, living neurons were counted 72 h later and expressed as a percentage of initial neurons. The mean ± SEM of three (A) or four (B) independent experiments is shown. Statistical significance of the differences was estimated by one-way ANOVA and post hoc Tukey's honestly significant difference test.
Involvement of caspase-9 and -3 in the death of neurotrophic factor–deprived sympathetic neurons was investigated. However, direct demonstration of activation of the caspases by Western blotting appeared impossible due to scarcity of the material. Therefore, we overexpressed dominant-negative mutants of these caspases in GDNF- or NGF-deprived neurons. Inhibition of either caspase-9 or -3 failed to inhibit the death of GDNF-deprived neurons, although they efficiently blocked the death of NGF-deprived neurons by 72 h after microinjection and NGF deprivation (Fig. 4).
Figure 4.

Inhibition of individual caspases in GDNF- or NGF-deprived sympathetic neurons. GDNF- or NGF-selected sympathetic neurons were microinjected with expression plasmids for dominant-negative (DN) mutants of indicated caspases, and the neurotrophic factors were then deprived. Living neurons were counted 72 h later and expressed as a percentage of initial neurons. The mean ± SEM of three (four for DN caspase-9) independent cultures is shown. Individual caspases were studied in different experiments and are combined in the same figure. Data of each DN caspase were compared with averaged vector controls and uninjected controls by one-way ANOVA and post hoc Tukey's honestly significant difference test.
The absence of cytochrome c release, together with a failure to observe the role of Bax and caspase-9 and -3, suggest that GDNF-deprived neurons die via a nonmitochondrial pathway. To confirm this, we overexpressed the antiapoptotic Bcl-2 family member Bcl-xL, which is shown to block the mitochondrial death pathway, in the GDNF- and NGF-deprived neurons. Overexpressed Bcl-xL did not rescue GDNF-deprived neurons (Fig. 5 A), further confirming that the mitochondrial death pathway is not activated. However, Bcl-xL efficiently blocked the death of NGF-deprived neurons (Fig. 5 A) as shown previously (Gonzalez-Garcia et al., 1995). GDNF-maintained neurons were killed by overexpressed proapoptotic protein Bax (Fig. 5 A), and Bax accelerated the death of GDNF-deprived neurons (not depicted).
Figure 5.

Involvement of proteins of the apoptotic machinery in the death of GDNF- or NGF-deprived sympathetic neurons. (A) Overexpressed Bcl-xL rescues NGF-deprived but not GDNF-deprived neurons, whereas overexpressed Bax kills both types of neurons. (B) Broad-range caspase inhibitor BAF (50 μg/ml) protects both GDNF- and NGF-deprived neurons. (C) Overexpressed XIAP protects NGF-deprived, but not GDNF-deprived, neurons. (D) Overexpressed dominant-negative FADD (DN FADD) does not protect GDNF- or NGF-deprived neurons. In A–D, the living neurons were counted 72 h after treatment and neurotrophic factor deprivation, and expressed as a percentage of initial neurons. The mean ± SEM of three independent cultures is shown. Data of Bcl-xL- (A), XIAP- (C), or DN-FADD–injected (D) neurons were compared with respective vector-injected or untreated neurotrophic factor-deprived controls by one-way ANOVA and post hoc Tukey's honestly significant difference test. Data of BAF-treated neurons (B) were compared with untreated controls by t test.
Caspase-2 and -7 are activated in GDNF-deprived sympathetic neurons
The broad-range caspase inhibitor BAF almost completely blocked death of both GDNF-deprived (Fig. 5 B) and NGF-deprived (Fig. 5 B; Deshmukh et al., 1996, 2000; Martinou et al., 1999) neurons, showing that some caspases are absolutely required for the death of GDNF-deprived neurons. To identify the relevant caspases, we overexpressed by microinjection the dominant-negative mutants of caspase-2, -3, -6, -7, and -8 in the GDNF-deprived, but also NGF-deprived, neurons. Blocking of caspase-2 and, to lesser extent, caspase-6 and -7 significantly inhibited death of NGF-deprived neurons, whereas dominant-negative caspase-8 had no effect (Fig. 4). A dominant negative mutant of caspase-2, and to lesser extent, caspase-7, also inhibited death of GDNF-deprived neurons, whereas blocking of caspase-6 and -8 had no effect (Fig. 4). Thus, GDNF deprivation-induced death requires caspase-2 and -7 in sympathetic neurons.
Caspase-2 was recently shown to be activated upstream of the mitochondria, and this event was required for the permeabilization of the mitochondria (Lassus et al., 2002). The presence of BAF in our culture medium may thus, via inhibition of caspase-2, block cytochrome c release in GDNF-deprived neurons and force them to chose another pathway. Therefore, we deprived the neurons of neurotrophic factors without BAF and applied cytochrome c immunocytochemistry. Although many neurons have disappeared, cytochrome c immunostaining was still mostly punctate in the remaining GDNF-deprived neurons (and in about half of the remaining NGF-deprived neurons; Fig. 2 C).
We also overexpressed the X chromosome–linked inhibitor of apoptosis protein (XIAP), a natural inhibitor of caspases, in GDNF- and NGF-deprived neurons. XIAP did not affect the death of GDNF-deprived neurons (Fig. 5 C), although, as expected (Yu et al., 2003), it rescued a significant portion of NGF-deprived neurons (Fig. 5 C). Overexpression of XIAP did not significantly affect the viability of NGF- or GDNF-maintained neurons (unpublished data). Thus, the caspases that execute death of GDNF-deprived neurons could not be blocked by overexpressed XIAP.
c-Jun is required for the death of, and is differently activated in, GDNF- and NGF-deprived sympathetic neurons
To study the activation of the transcription factor c-Jun, we deprived GDNF- or NGF-responsive BAF-saved neurons from their respective factors for 48 h, stained the neurons with antibodies to phosphorylated serines 63 or 73, and counted the neurons with strong nuclear immunoreactivity. Deprivation of GDNF significantly increased the number of neurons immunopositive for phosphorylated serine 73 (Fig. 6 A). However, the number of nuclei positive for phosphorylated serine 63 was unchanged in the GDNF-deprived neurons (Fig. 6, A and B). In control cultures, deprivation of NGF dramatically induced phosphorylation of serine 63 of c-Jun, as shown previously (Ham et al., 1995; Virdee et al., 1997; Eilers et al., 1998; Harris et al., 2002), and also of serine 73 (Fig. 6, A and B; Besirli and Johnson, 2003). As expected, only faint staining was obtained for the neurons maintained in the presence of GDNF or NGF (Fig. 6 A). Similar results were obtained at 72 h after neurotrophic factor deprivation (unpublished data).
Figure 6.

Activation of MLK and c-Jun is required for the death of GDNF-deprived sympathetic neurons. (A) Quantitation of neurons with strong nuclear immunostaining for phosphorylated c-Jun expressed as a percentage of all neurons. Neurons were deprived of neurotrophic factors in the presence of caspase inhibitor BAF for 48 h and immunostained with antibodies to phosphorylated serines 63 or 73 of c-Jun. Control neurons maintained with GDNF or NGF were stained as well. The mean ± SEM of four (for P-Ser-63) or three (for P-Ser-73) independent cultures is shown. Neurotrophic factor–maintained and –deprived groups were compared by t test. (B) Typical examples of weak (GDNF-deprived neurons) or strong (NGF-deprived neurons) nuclear immunostaining. Corresponding phase-contrast images are shown on the right column. Levels of the fluorescent images were equally enhanced with Adobe Photoshop software. Bar, 10 μm. (C) GDNF- or NGF- deprived sympathetic neurons were microinjected with expression plasmid encoding for dominant-negative form of c-Jun (DN-c-Jun). Living neurons were counted 72 h later and expressed as a percentage of initial neurons. The mean ± SEM of three independent cultures is shown. Statistical comparison of the DN-c-Jun–expressing group with vector-injected and uninjected groups was done by one-way ANOVA and post hoc Tukey's honestly significant difference test. (D) Survival of GDNF- or NGF-deprived sympathetic neurons in the presence or absence of 500 ng/ml CEP-1347. Living neurons were counted 72 h after neurotrophic factor deprivation and expressed as a percentage of initial neurons. The mean ± SEM of three independent cultures is shown. Statistical comparison of the means was performed by t test.
To test whether c-Jun is necessary for the death of the neurons, we overexpressed a dominant-negative c-Jun mutant FLAGΔ169 in the NGF- or GDNF-deprived sympathetic neurons. As previously described (Ham et al., 1995; Eilers et al., 1998), ∼60% of the NGF-deprived neurons were rescued by 72 h (Fig. 6 C). A similar fraction of GDNF-deprived neurons were rescued by overexpressed FLAGΔ169 (Fig. 6 C), whereas the survival of GDNF- or NGF-maintained neurons was not affected (not depicted).
Phosphorylation of c-Jun is catalyzed by c-Jun NH2-terminal kinases whose activity is in turn regulated by several upstream kinases, including the mixed lineage kinases (MLK). We treated GDNF- or NGF-deprived neurons with 500 ng/ml CEP-1347, a semisynthetic derivative of the indolocarbazole K252a that is a selective inhibitor of MLK (Maroney et al., 2001), and counted the living neurons 72 h later. As previously described (Maroney et al., 1999; Harris et al., 2002), almost all NGF-deprived neurons were rescued by CEP-1347 (Fig. 6 D). CEP-1347 rescued GDNF-deprived neurons with equal efficiency (Fig. 6 D). When inspected during the third day of treatment, both GDNF- and NGF-deprived (Harris et al., 2002) neurons that were rescued by CEP-1347 had large phase-bright cell bodies, similar to those of factor-maintained neurons, whereas the nontreated factor-deprived control neurons had a pyknotic appearance (unpublished data). In summary, our data show that activation of c-Jun is necessary for the death of GDNF-deprived neurons, although it is activated differently from NGF-deprived neurons.
Death receptor pathway is not activated in GDNF-deprived sympathetic neurons
The aforementioned data show that GDNF-deprived sympathetic neurons die via a nonmitochondrial death pathway where c-Jun, as well as caspase-2 and -7, is involved. Another well-characterized pathway, the death receptor–mediated pathway, can be efficiently blocked by dominant-negative mutant of Fas-associated protein with death domain (FADD)/MORT1, an adapter that links pro–caspase-8 to most death receptors (Strasser and Newton, 1999; Vincenz, 2001). Overexpression of this mutant FADD in GDNF- and NGF-deprived sympathetic neurons did not change the death rate (Fig. 5 D), nor did it affect the viability of neurons maintained with NGF or GDNF (not depicted). Also, overexpression of dominant-negative caspase-8 did not affect death of neurons deprived of either factor (Fig. 4). Thus, the death receptor pathway is probably not activated in the NGF- or GDNF-deprived sympathetic neurons. In accordance with that finding, activation of death receptor tumor necrosis factor-α or Fas on the surface of NGF-maintained sympathetic neurons did not induce their death (Putcha et al., 2002).
Ultrastructure of GDNF- and NGF-deprived sympathetic neurons
To investigate the ultrastructural changes caused by removal of GDNF or NGF in sympathetic neurons, we deprived the cultures of neurotrophic factors for 48 h and analyzed the neurons by transmission electron microscopy. Neurons maintained with GDNF or NGF were analyzed as well. As a general observation, the cytoplasm of GDNF-deprived neurons was much more electron dense than that of NGF-deprived neurons in the sister culture or the neurons maintained with either neurotrophic factor. Removal of GDNF led to a marked increase in the number of different autophagic profiles, including double-membraned autophagosomes and single-membraned autolysosomes that often contained swirled packs of undigested membranes (Fig. 7). Thus, on average, nine autolysosomes per neuron, but no autophagosomes, were found in GDNF-maintained neurons (n = 25); whereas on average, 3 autophagosomes and 14 autolysosomes were found per GDNF-deprived neuron (n = 39). NGF-deprived neurons also exhibited an increased number of autolysosomes: average of 18 per NGF-deprived neuron (n = 63) versus 9 per NGF-maintained neuron (n = 39), but the autophagosomes were only rarely found (0.3 per average NGF-deprived neuron, and none in NGF-maintained neurons). Normal endoplasmic reticulum and Golgi complex were still found in the GDNF- and NGF-deprived neurons with enhanced autophagy.
Figure 7.
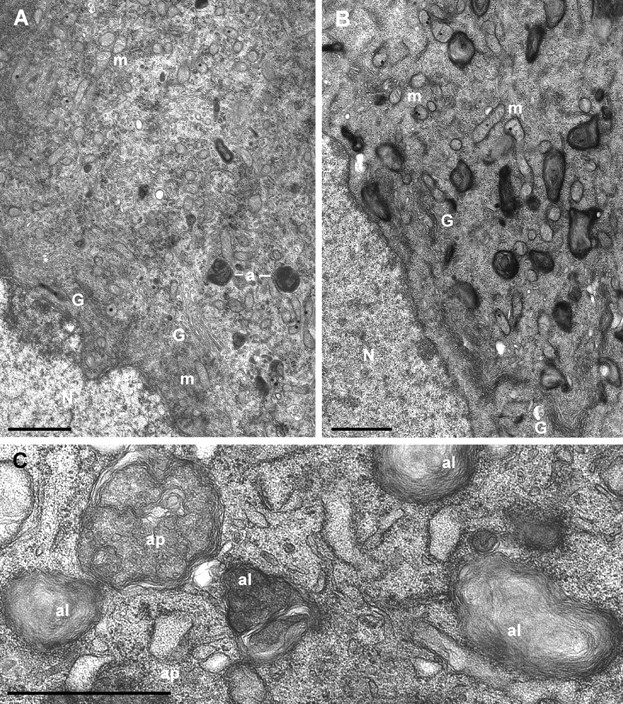
Autophagy is greatly enhanced in GDNF-deprived sympathetic neurons. (A) Ultrastructure of a typical GDNF-maintained neuron with normal mitochondria (m), Golgi complex (G), and two dark autolysosomes (a) that are sparse in these neurons. (B) Typical GDNF-deprived neuron from sister dish showing largely increased number of autolysosomes, but normal mitochondria and Golgi complex. N, nucleus. (C). A detail from another GDNF-deprived neuron with double-membraned autophagosomes (ap) and single-membraned autolysosomes (al) containing swirled packages of undigested membranes. Bars, 1 μm.
The mitochondria of GDNF-deprived neurons (Fig. 8 A) were similar to those of GDNF-maintained (and also NGF-maintained) neurons (not depicted), having mostly the orthodox configuration, including elongated shape and clear cristae, and were not clustered. Such normal mitochondria were found in all GDNF-deprived neurons, including those with massive autophagy (Fig. 8 A). However, the mitochondria in NGF-deprived neurons were often round-shaped and clustered (Fig. 8 B). The cristae of such mitochondria were markedly reorganized, often observed to be round, vesicular, and rare in the number, and sometimes only one mitochondrial membrane was discernible (Fig. 8 C). Some mitochondria in these clusters contained different numbers of normal rod-like cristae along with the mitochondria with changed vesicular cristae (Fig. 8 D). In many neurons with such mitochondrial clusters, few elongated nonclustered mitochondria with the orthodox configuration were found, whereas some neurons contained only normal mitochondria (unpublished data). Of all NGF-deprived neurons analyzed in two experiments (n = 63), 43% had round-shaped, clustered mitochondria with changed cristae, 19% had the mitochondria in orthodox configuration, and in 38%, both types of mitochondria were found. Appearance of the round clustered mitochondria seems to not be a feature of final death phase, as these were often found in neurons with normal endoplasmic reticulum, Golgi complex, and nucleus without condensed chromatin. We did not observe swelling and disruption of the mitochondria in NGF-deprived neurons.
Figure 8.
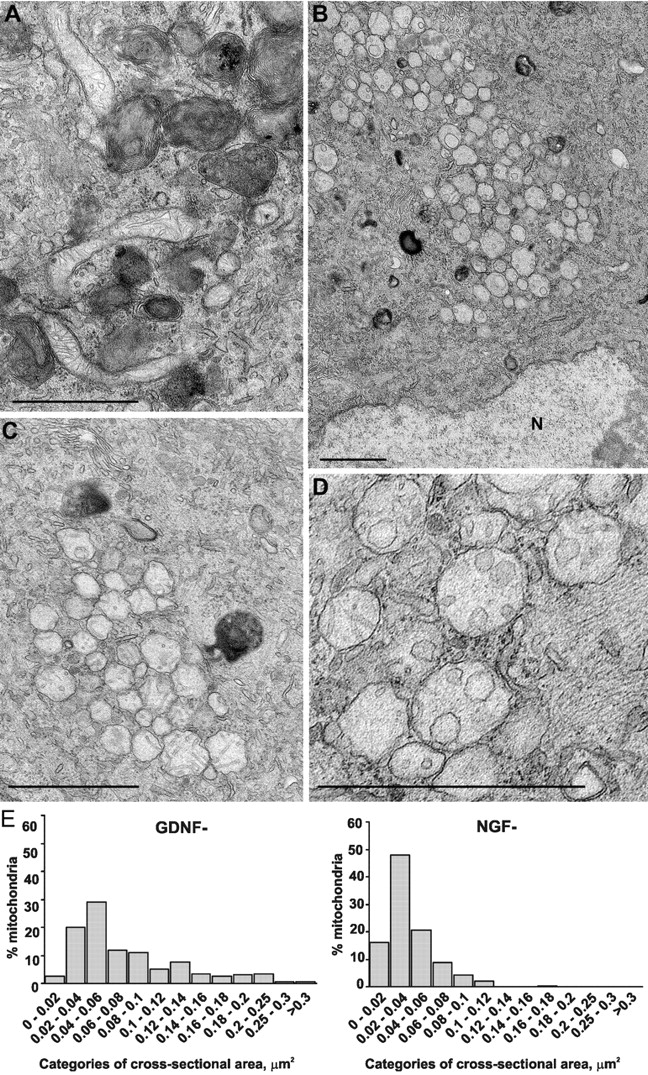
Mitochondria of NGF-deprived, but not GDNF-deprived, sympathetic neurons are structurally changed. (A) Typical view of a GDNF-deprived neuron with numerous dark autophagic profiles and several nonclustered elongated mitochondria with normal cristae. (B) An NGF-deprived neuron with several dark autolysosomes and large number of round clustered mitochondria with changed cristae. N, nucleus. (C) Higher magnification of the mitochondrial cluster with vesicular cristae and one membrane in an NGF-deprived neuron. (D) Mitochondria whose cristae and inner membrane are altered to a different extent in an NGF-deprived neuron. (E) Distribution of mitochondrial profiles from the sections of GDNF-deprived (n = 201) and NGF-deprived (n = 317) neurons according to their cross-sectional areas. Size categories are shown as a percentage of all mitochondrial profiles. Bars, 1 μm.
The average cross-sectional area of the mitochondria was 0.0781 ± 0.0039 μm2 (mean ± SEM, n = 201) in the GDNF-deprived neurons and 0.038 ± 0.0013 μm2 (n = 317) in the NGF-deprived neurons. These values, as determined from thin sections, do not directly indicate the length or size of the mitochondria. In each section, there is a collection of profiles from smallest perpendicular round profiles to longer oval-shape or even branched profiles, depending on the orientation of the mitochondria in relation to sectioning angle. To illustrate the difference in the size of the mitochondria, we plotted the distribution of profiles according to their cross-sectional area (Fig. 8 E). About 50% of all profiles in sections from the NGF-deprived neurons fit into the category of 0.02–0.04 μm2, and there were few profiles larger than 0.12 μm2, whereas a broad range of mitochondrial profiles up to 0.3 μm2 and larger were found from the GDNF-deprived neurons.
We did not find nuclei with condensed chromatin in the GDNF-deprived neurons analyzed by electron microscopy, although 24% of the NGF-deprived neurons had the nuclei with DNA condensed to a different extent (unpublished data). We also stained the neurons maintained with or deprived of GDNF or NGF with Hoechst 33258 and counted the neurons having typical fragmented nuclei with condensed chromatin. As shown in Fig. 9, a progressively increasing number of NGF-deprived neurons with apoptotic nuclei was observed during a 3-d period, whereas only a small fraction (5–7%) of the GDNF-deprived or –maintained neurons had fragmented nuclei. This number did not increase with time and most probably shows nonspecific death. Virtually all NGF-maintained neurons had normal nuclei (unpublished data). Thus, we did not observe nuclear changes in the GDNF-deprived neurons.
Figure 9.

GDNF deprivation does not induce chromatin condensation and nuclear fragmentation in the sympathetic neurons. 6 DIV neurons were deprived of or maintained with GDNF or NGF. The cultures were fixed daily and stained with Hoechst 33258. The neurons with fragmented nuclei and condensed chromatin were counted and expressed as a percentage of all neurons. The mean ± SEM of four independent experiments is shown.
Discussion
We found that removal of GDNF from the cultured sympathetic neurons triggers a novel nonmitochondrial MLK-, c-Jun–, and caspase-dependent death pathway, although removal of NGF from the sympathetic neurons activates the mitochondrial pathway. To our knowledge, this is the first description of a nonmitochondrial pathway activated by withdrawal of a survival factor.
GDNF deprivation-induced death requires MLK–c-Jun pathway, caspase-2 and -7, and involves increased autophagy
The mitochondrial pathway is not activated in GDNF-deprived sympathetic neurons. Indeed, cytochrome c is not released from the mitochondria to cytosol; Bax and caspase-9 and -3 are not involved in death execution; overexpression of Bcl-xL does not protect the neurons; and the ultrastructure of the mitochondria in GDNF-deprived neurons is not changed. We have currently found several proteins involved in the death of GDNF-deprived neurons. MLK and c-Jun are activated and seem to be similarly required for death of both NGF- and GDNF-deprived neurons. Surprisingly, we did not detect increase in phosphoserine 63 immunoreactivity of c-Jun in GDNF-deprived neurons, although both serines 63 and 73 were phoshorylated in NGF-deprived neurons. The kinases that phosphorylate c-Jun in GDNF-deprived neurons remain to be studied.
Caspase-2 and -7 are involved in the death of GDNF-deprived sympathetic neurons. Very little is known about the mechanism of caspase-2 activation. However, it is tempting to speculate that caspase-2 functions as an initiator and caspase-7 as an executioner in these neurons. Recent reports that caspase-2 is activated upstream of mitochondrial events in some apoptotic cell types (Guo et al., 2002; Lassus et al., 2002; Read et al., 2002) are in accordance with our data. Overexpressed XIAP did not rescue GDNF-deprived neurons, although XIAP can inactivate caspase-7 in cell-free systems (Deveraux et al., 1997), suggesting that caspase-7 is not available for XIAP in the GDNF-responsive sympathetic neurons. It should also be stressed that, although the dominant-negative caspase isoforms used here should be specific for given caspases, some nonspecific effects cannot be completely excluded.
Many cells in which the main mitochondrial death pathway is genetically or pharmacologically disabled can still die via an alternative, autophagic pathway that is often caspase independent and with increased autophagy leading to largely vacuolized cytoplasm (Sperandio et al., 2000; Oppenheim et al., 2001; Yaginuma et al., 2001; Zaidi et al., 2001; Marsden et al., 2002). However, dying GDNF-deprived neurons seem to differ from those “classical” autophagic death patterns because the caspases are clearly involved. Indeed, we observed markedly increased autophagy in the GDNF-deprived neurons, but no remarkable vacuolization of the cytoplasm was found, at least before the short final death execution phase. Few neurons in the terminal state of death that were retained in our electron microscopic preparations showed typical features of secondary necrosis, similar for both GDNF- and NGF-deprived neurons (unpublished data). We found increased autophagy also in the NGF-deprived neurons, as described previously (Martinou et al., 1999; Xue et al., 1999; Kirkland et al., 2002). Thus, both NGF- and GDNF-deprived neurons die in a caspase-dependent manner with enhanced autophagy.
NGF-deprived sympathetic neurons die via the mitochondrial pathway
We confirmed the published data that NGF-deprived neurons die via the mitochondrial pathway, including cytosolic localization of cytochrome c (Deshmukh and Johnson, 1998; Neame et al., 1998; Martinou et al., 1999), involvement of Bax (Deckwerth et al., 1996; Putcha et al., 1999) and caspase-9 and -3 (Deshmukh et al., 2000, 2002), and inhibition of death by Bcl-xL (Gonzalez-Garcia et al., 1995). In addition, we showed for the first time that caspase-6 and -7 are also necessary for NGF deprivation–induced death, and we confirmed the role of caspase-2 (Troy et al., 2001).
Our overexpression studies (Yu et al., 2003; this paper) are in agreement with the current concept that in NGF-responsive sympathetic neurons, critical caspases are blocked with inhibitor of apoptosis proteins, e.g., XIAP. Withdrawal of NGF releases caspases from that block by proteasome-mediated degradation, but also by removal of XIAP from the caspases by Smac/DIABLO that is released from the mitochondria together with cytochrome c (Troy et al., 2001; Deshmukh et al., 2002; Yu et al., 2003). Most probably, a large amount of overexpressed XIAP replaces the degraded bulk and keeps the caspases inactivated in our experiment.
We found remarkable ultrastructural changes in the mitochondria of many NGF-deprived neurons; they gradually become round, clustered, and their cristae change considerably. Appearance of round mitochondria as a result of increased fission has been described previously in the NGF-deprived neurons (Martinou et al., 1999) and in other cells (Karbowski et al., 2002). Also, the clustering of mitochondria in NGF-deprived neurons has been described previously (Tolkovsky et al., 2002), although the mechanism remained obscure. However, our observation that the cristae in the small clustered mitochondria of NGF-deprived neurons were often round, vesicular, and reduced, whereas the inner membrane was sometimes found to be missing, have not been described previously (Martin et al., 1988; Martinou et al., 1999; Xue et al., 1999; Kirkland et al., 2002). We do not know whether this discrepancy results from differences in the culture conditions, genetic background of the animals, or from other conditions; but this ultrastructural pattern was repeatedly observed in our cultures. Mitochondria with orthodox and altered ultrastructure were found in the same sample, sometimes even in the same neuron, ruling out the possibility of a processing artifact. The mitochondrial cristae are dynamic structures that can considerably change their shape (Frey et al., 2002). In the apoptotic cells, these changes are proposed to facilitate the release of cristae-associated cytochrome c into the intermembrane space (Scorrano et al., 2002). It is tempting to speculate that the mitochondria with altered cristae have already released their cytochrome c. We also stress that our data do not support the release of apoptotic proteins from the mitochondria via their swelling and rupture.
In summary, we propose that GDNF-deprived sympathetic neurons die by caspase-dependent nonmitochondrial death pathway that has not been described previously. More studies are required to characterize the molecular and cellular components of this pathway. How an exposure of SCG neurons to different neurotrophic factors dictates the death program, is currently unknown. It was recently shown that an apoptotic fragment, generated from unligated Ret by caspase-3, can trigger apoptosis in some cell lines (Bordeaux et al., 2000). However, overexpression of Ret or apoptotic fragment of Ret in the sympathetic neurons did not induce their death in our model (unpublished data), suggesting that death-promoting activity of unligated Ret is not manifested in the sympathetic neurons. Ret, similarly to deleted in colorectal cancer (Forcet et al., 2001), may be able to recruit and activate caspases directly, so that mitochondrial pathway is not required. Alternatively, exposure of the neurons to GDNF for 6 d may differentiate the neurons so that the mitochondrial pathway is nonfunctional. Whether and how the nonmitochondrial death pathway is used in vivo is currently unknown, as virtually nothing is yet known about the biological role of GDNF for the SCG neurons.
Materials and methods
Culture of sympathetic neurons and the survival assays
Culture of the SCG neurons was performed as described previously (Hamner et al., 2001; Lindahl et al., 2001; Sun et al., 2001). In brief, the neurons of postnatal day 1–2 Han/Wi strain rats were grown 6 DIV on polyornithine-laminin–coated dishes or glass coverslips with 100 ng/ml of human GDNF (PeproTech) or 30 ng/ml of 2.5 S mouse NGF (Promega). Four to five times more neurons were initially plated for GDNF experiments compared with NGF controls, so that by the day of neurotrophic factor deprivation, the number of neurons in both groups was similar. To reduce the number of nonneuronal cells, 1 μM cytosine arabinoside (Sigma-Aldrich) was always included the next day after plating, but was not added to factor-deprived neurons. To deprive GDNF, the cultures were washed gently three times with GDNF-free culture medium. To remove NGF, the cultures were washed once with NGF-free medium and function-blocking anti-NGF antibodies (Roche) were added. The compounds of interest were added and initial neurons were counted immediately after neurotrophic factor deprivation. Living neurons were counted daily by a “blind” experimenter who was not aware of the identity of experimental groups. Compounds were assayed as follows: broad-range caspase inhibitor BAF (Enzyme Systems Products) at 50 μM; CEP-1347 (Cephalon, Inc.) at 500 ng/ml; and V5 and I5 peptides (a gift of S. Matsuyama, Medical College of Wisconsin, Milwaukee, WI) at 200 μM. When the compounds were dissolved in DMSO, the same amount of DMSO was always added to the control cultures.
Microinjections
The neurons were pressure-microinjected with 50 ng/μl of expression plasmids encoding the proteins of interest together with 10 ng/μl of enhanced GFP-encoding plasmid as an indicator of successful injection. The relevant empty vector (pcDNA3.1 or pCR3.1) without the insert, as well as uninjected controls, were always included. When neurotrophic factor–deprived neurons were analyzed, the factor-maintained uninjected neurons were always included to show that the neurons do not die due to poor culture conditions. Neurons tolerating the injection procedure were counted 4–6 h later according to the map drawn with the help of squares scratched to the bottom of the culture dish, and considered as initial neurons. The next morning, the few living injected neurons that did not show GFP fluorescence were subtracted from the initial neurons. On average, 25–80 neurons were successfully injected per experimental point. All experiments were repeated at least three times on the independent cultures. The results were expressed as the mean ± the SEM and were tested for the significance by either one-way ANOVA and post hoc Tukey's honestly significant difference tests or two-tailed t test with two-sample unequal variance. The null hypothesis was rejected at P < 0.05.
The following expression plasmids were injected: full-length human Bax and full-length human Bcl-xL expression plasmids were a gift from M. Simonen (Novartis Pharma AG, Basel, Switzerland). Dominant-negative FADD plasmid was a gift of C. Vincenz (University of Michigan, Ann Arbor, MI). Cys287Ala mutant of caspase-9 was a gift of Y. Lazebnik (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). Cys320Ser mutant of mouse caspase-2 was obtained from T. Örd (Estonian Biocentre, Tartu, Estonia). Dominant-negative c-Jun pCD FLAGΔ169 plasmid was obtained from J. Whitfield (Eisai London Research Laboratories Ltd., London, UK). Plasmid for Ku70 was a gift of S. Matsuyama. Also, the expression plasmids for human XIAP (Yu et al., 2003) and dominant-negative mutants of caspase-3, -6, -7, and -8 (active center cysteine mutated to alanine in all cases; Forcet et al., 2001) were used.
Immunocytochemistry
The neurons were grown on round glass coverslips, fixed with fresh 4% PFA in PBS, permeabilized with 1% Triton X-100, and blocked with 5% of donkey serum (Jackson ImmunoResearch Laboratories) in PBS. Antibodies to the following antigens were used: cytochrome c (BD Biosciences), phosphorylated serine 63 of c-Jun (Cell Signaling Technology), and phosphorylated serine 73 of c-Jun (Cell Signaling Technology). Cy3-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) were used for visualization and the specimens were mounted in Vectashield (Vector Laboratories). Images were captured at RT with an inverted microscope (model DM-IRB; Leica) using PL FLUOTAR objective (63×/0.70) or, for Fig. 2 A, HC PL FLUOTAR (10×/0.30), and a 3CCD color video camera (model DXC-950P; Sony) under the control of Image-Pro Plus software version 3.0 (Media Cybernetics). Individual images were processed and assembled with Photoshop 7.0 (Adobe Systems). In some experiments, the fixed cultures were treated with 1 μg/ml of Hoechst 33258 (Molecular Probes) for 15 min, mounted, and observed for the nuclear morphology.
Electron microscopy
The neurons were grown in the 4-well plates (Nunc) and deprived of neurotrophic factors. The medium of the control neurons was also changed and the factors were added back. The cultures were fixed 48 h after neurotrophic factor deprivation with 2% of glutaraldehyde. To avoid detachment of loosely attached dying neurons, fixative with twofold strength was carefully added to the culture medium. The cultures were processed for transmission electron microscopy as described previously (Yu et al., 2003). From each sample, we chose one section cut from the middle third of the depth of the neuron, and all neurons within that section were analyzed by transmission electron microscope (model FEI Tecnai F12; Philips Electron Optics) operated at 80 kV. In two independent experiments, altogether 39 GDNF-deprived, 63 NGF-deprived, 25 GDNF-maintained, and 39 NGF-maintained neurons were analyzed. Very similar results were obtained from two independent experiments. For estimation of the size of mitochondria from GDNF- or NGF-deprived neurons, mitochondria at a final magnification of 33,000 were manually traced onto transparencies that were scanned. Cross-sectional area of the mitochondria was measured using Image-Pro Plus version 3.0.
Acknowledgments
We are thankful to the following people for the expression plasmids: Marjo Simonen (Bcl-xL and Bax), Claudius Vincenz (dominant-negative FADD), Yuri Lazebnik (dominant-negative caspase-9), Tõnis Örd (dominant-negative caspase-2), and Shigemi Matsuyama (Ku70 and V5 and I5 peptides). Eisai London Research Laboratories Ltd. is appreciated for the plasmid for dominant-negative c-Jun, and Cephalon, Inc. is appreciated for CEP-1347. Mervi Lindman is appreciated for excellent technical assistance and Matthew Phillips for language correction.
This work was supported by the Institute of Biotechnology, Academy of Finland programme 44896 (Finnish Centre of Excellence Programme 2000–2005) and Sigrid Jusélius Foundation grant. M. Saarma is the Biocentrum Helsinki fellow.
Abbreviations used in this paper: BAF, boc-aspartyl(OMe)-fluoromethylketone; DIV, days in vitro; FADD, Fas-associated protein with death domain; GDNF, glial cell line–derived neurotrophic factor; MLK, mixed lineage kinases; SCG, superior cervical ganglion; XIAP, X chromosome–linked inhibitor of apoptosis protein.
References
- Airaksinen, M.S., and M. Saarma. 2002. The GDNF family: signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 3:383–394. [DOI] [PubMed] [Google Scholar]
- Besirli, C.G., and E.M. Johnson, Jr. 2003. JNK-independent activation of c-Jun during neuronal apoptosis Induced by multiple DNA-damaging agents. J. Biol. Chem. 278:22357–22366. [DOI] [PubMed] [Google Scholar]
- Bordeaux, M.C., C. Forcet, L. Granger, V. Corset, C. Bidaud, M. Billaud, D.E. Bredesen, P. Edery, and P. Mehlen. 2000. The RET proto-oncogene induces apoptosis: a novel mechanism for Hirschsprung disease. EMBO J. 19:4056–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, P.G. 1990. Developmental cell death: morphological diversity and multiple mechanisms. Anat. Embryol. (Berl.). 181:195–213. [DOI] [PubMed] [Google Scholar]
- Deckwerth, T.L., J.L. Elliott, C.M. Knudson, E.M. Johnson, Jr., W.D. Snider, and S.J. Korsmeyer. 1996. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 17:401–411. [DOI] [PubMed] [Google Scholar]
- Deshmukh, M., and E.M. Johnson, Jr. 1998. Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 21:695–705. [DOI] [PubMed] [Google Scholar]
- Deshmukh, M., J. Vasilakos, T.L. Deckwerth, P.A. Lampe, B.D. Shivers, and E.M. Johnson, Jr. 1996. Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE family proteases. J. Cell Biol. 135:1341–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh, M., K. Kuida, and E.M. Johnson. 2000. Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J. Cell Biol. 150:131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh, M., C. Du, X. Wang, and E.M. Johnson, Jr. 2002. Exogenous Smac induces competence and permits caspase activation in sympathetic neurons. J. Neurosci. 22:8018–8027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveraux, Q.L., R. Takahashi, G.S. Salvesen, and J.C. Reed. 1997. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 388:300–304. [DOI] [PubMed] [Google Scholar]
- Edwards, S.N., and A.M. Tolkovsky. 1994. Characterization of apoptosis in cultured rat sympathetic neurons after nerve growth factor withdrawal. J. Cell Biol. 124:537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers, A., J. Whitfield, C. Babij, L.L. Rubin, and J. Ham. 1998. Role of the Jun kinase pathway in the regulation of c-Jun expression and apoptosis in sympathetic neurons. J. Neurosci. 18:1713–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellerby, L.M., A.S. Hackam, S.S. Propp, H.M. Ellerby, S. Rabizadeh, N.R. Cashman, M.A. Trifiro, L. Pinsky, C.L. Wellington, G.S. Salvesen, et al. 1999. Kennedy's disease: caspase cleavage of the androgen receptor is a crucial event in cytotoxicity. J. Neurochem. 72:185–195. [DOI] [PubMed] [Google Scholar]
- Estus, S., W.J. Zaks, R.S. Freeman, M. Gruda, R. Bravo, and E.M. Johnson, Jr. 1994. Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J. Cell Biol. 127:1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcet, C., X. Ye, L. Granger, V. Corset, H. Shin, D.E. Bredesen, and P. Mehlen. 2001. The dependence receptor DCC (deleted in colorectal cancer) defines an alternative mechanism for caspase activation. Proc. Natl. Acad. Sci. USA. 98:3416–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey, T.G., C.W. Renken, and G.A. Perkins. 2002. Insight into mitochondrial structure and function from electron tomography. Biochim. Biophys. Acta. 1555:196–203. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Garcia, M., I. Garcia, L. Ding, S. O'Shea, L.H. Boise, C.B. Thompson, and G. Nunez. 1995. bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc. Natl. Acad. Sci. USA. 92:4304–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Y., S.M. Srinivasula, A. Druilhe, T. Fernandes-Alnemri, and E.S. Alnemri. 2002. Caspase-2 induces apoptosis by releasing proapoptotic proteins from mitochondria. J. Biol. Chem. 277:13430–13437. [DOI] [PubMed] [Google Scholar]
- Ham, J., C. Babij, J. Whitfield, C.M. Pfarr, D. Lallemand, M. Yaniv, and L.L. Rubin. 1995. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 14:927–939. [DOI] [PubMed] [Google Scholar]
- Hamner, S., U. Arumäe, Y. Li-Ying, Y.F. Sun, M. Saarma, and D. Lindholm. 2001. Functional characterization of two splice variants of rat bad and their interaction with Bcl-w in sympathetic neurons. Mol. Cell. Neurosci. 17:97–106. [DOI] [PubMed] [Google Scholar]
- Harris, C.A., M. Deshmukh, B. Tsui-Pierchala, A.C. Maroney, and E.M. Johnson, Jr. 2002. Inhibition of the c-Jun N-terminal kinase signaling pathway by the mixed lineage kinase inhibitor CEP-1347 (KT7515) preserves metabolism and growth of trophic factor-deprived neurons. J. Neurosci. 22:103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, E.J., and L.F. Reichardt. 2001. Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24:677–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski, M., Y.J. Lee, B. Gaume, S.Y. Jeong, S. Frank, A. Nechushtan, A. Santel, M. Fuller, C.L. Smith, and R.J. Youle. 2002. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J. Cell Biol. 159:931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland, R.A., R.M. Adibhatla, J.F. Hatcher, and J.L. Franklin. 2002. Loss of cardiolipin and mitochondria during programmed neuronal death: evidence of a role for lipid peroxidation and autophagy. Neuroscience. 115:587–602. [DOI] [PubMed] [Google Scholar]
- Kotzbauer, P.T., P.A. Lampe, R.O. Heuckeroth, J.P. Golden, D.J. Creedon, E.M. Johnson, Jr., and J. Milbrandt. 1996. Neurturin, a relative of glial-cell-line-derived neurotrophic factor. Nature. 384:467–470. [DOI] [PubMed] [Google Scholar]
- Lassus, P., X. Opitz-Araya, and Y. Lazebnik. 2002. Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science. 297:1352–1354. [DOI] [PubMed] [Google Scholar]
- Leist, M., and M. Jäättelä. 2001. Four deaths and a funeral: from caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol. 2:589–598. [DOI] [PubMed] [Google Scholar]
- Lindahl, M., D. Poteryaev, L. Yu, U. Arumäe, T. Timmusk, I. Bongarzone, A. Aiello, M.A. Pierotti, M.S. Airaksinen, and M. Saarma. 2001. Human glial cell line-derived neurotrophic factor receptor alpha 4 is the receptor for persephin and is predominantly expressed in normal and malignant thyroid medullary cells. J. Biol. Chem. 276:9344–9351. [DOI] [PubMed] [Google Scholar]
- Llambi, F., F. Causeret, E. Bloch-Gallego, and P. Mehlen. 2001. Netrin-1 acts as a survival factor via its receptors UNC5H and DCC. EMBO J. 20:2715–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroney, A.C., J.P. Finn, D. Bozyczko-Coyne, T.M. O'Kane, N.T. Neff, A.M. Tolkovsky, D.S. Park, C.Y. Yan, C.M. Troy, and L.A. Greene. 1999. CEP-1347 (KT7515), an inhibitor of JNK activation, rescues sympathetic neurons and neuronally differentiated PC12 cells from death evoked by three distinct insults. J. Neurochem. 73:1901–1912. [PubMed] [Google Scholar]
- Maroney, A.C., J.P. Finn, T.J. Connors, J.T. Durkin, T. Angeles, G. Gessner, Z. Xu, S.L. Meyer, M.J. Savage, L.A. Greene, et al. 2001. CEP-1347 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J. Biol. Chem. 276:25302–25308. [DOI] [PubMed] [Google Scholar]
- Marsden, V.S., L. O'Connor, L.A. O'Reilly, J. Silke, D. Metcalf, P.G. Ekert, D.C. Huang, F. Cecconi, K. Kuida, K.J. Tomaselli, et al. 2002. Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature. 419:634–637. [DOI] [PubMed] [Google Scholar]
- Martin, D.P., R.E. Schmidt, P.S. DiStefano, O.H. Lowry, J.G. Carter, and E.M. Johnson, Jr. 1988. Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J. Cell Biol. 106:829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinou, I., S. Desagher, R. Eskes, B. Antonsson, E. Andre, S. Fakan, and J.C. Martinou. 1999. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J. Cell Biol. 144:883–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neame, S.J., L.L. Rubin, and K.L. Philpott. 1998. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J. Cell Biol. 142:1583–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim, R.W., R.A. Flavell, S. Vinsant, D. Prevette, C.Y. Kuan, and P. Rakic. 2001. Programmed cell death of developing mammalian neurons after genetic deletion of caspases. J. Neurosci. 21:4752–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman, R.N., S. Wang, A.J. DiBenedetto, and J.C. Mills. 1993. A system for characterizing cellular and molecular events in programmed neuronal cell death. J. Neurosci. 13:3669–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha, G.V., M. Deshmukh, and E.M. Johnson, Jr. 1999. BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J. Neurosci. 19:7476–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha, G.V., C.A. Harris, K.L. Moulder, R.M. Easton, C.B. Thompson, and E.M. Johnson, Jr. 2002. Intrinsic and extrinsic pathway signaling during neuronal apoptosis: lessons from the analysis of mutant mice. J. Cell Biol. 157:441–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabizadeh, S., J. Oh, L.T. Zhong, J. Yang, C.M. Bitler, L.L. Butcher, and D.E. Bredesen. 1993. Induction of apoptosis by the low-affinity NGF receptor. Science. 261:345–348. [DOI] [PubMed] [Google Scholar]
- Read, S.H., B.C. Baliga, P.G. Ekert, D.L. Vaux, and S. Kumar. 2002. A novel Apaf-1–independent putative caspase-2 activation complex. J. Cell Biol. 159:739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada, M., P. Hayes, and S. Matsuyama. 2003. a. Cytoprotective membrane-permeable peptides designed from the Bax-binding domain of Ku70. Nat. Cell Biol. 5:352–357. [DOI] [PubMed] [Google Scholar]
- Sawada, M., W. Sun, P. Hayes, K. Leskov, D.A. Boothman, and S. Matsuyama. 2003. b. Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat. Cell Biol. 5:320–329. [DOI] [PubMed] [Google Scholar]
- Scorrano, L., M. Ashiya, K. Buttle, S. Weiler, S.A. Oakes, C.A. Mannella, and S.J. Korsmeyer. 2002. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell. 2:55–67. [DOI] [PubMed] [Google Scholar]
- Sperandio, S., I. de Belle, and D.E. Bredesen. 2000. An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. USA. 97:14376–14381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser, A., and K. Newton. 1999. FADD/MORT1, a signal transducer that can promote cell death or cell growth. Int. J. Biochem. Cell Biol. 31:533–537. [DOI] [PubMed] [Google Scholar]
- Sun, Y.F., L.Y. Yu, M. Saarma, T. Timmusk, and U. Arumäe. 2001. Neuron-specific Bcl-2 homology 3 domain-only splice variant of Bak is anti-apoptotic in neurons, but pro-apoptotic in non-neuronal cells. J. Biol. Chem. 276:16240–16247. [DOI] [PubMed] [Google Scholar]
- Thibert, C., M.A. Teillet, F. Lapointe, L. Mazelin, N.M. Le Douarin, and P. Mehlen. 2003. Inhibition of neuroepithelial patched-induced apoptosis by sonic hedgehog. Science. 301:843–846. [DOI] [PubMed] [Google Scholar]
- Tolkovsky, A.M., L. Xue, G.C. Fletcher, and V. Borutaite. 2002. Mitochondrial disappearance from cells: a clue to the role of autophagy in programmed cell death and disease? Biochimie. 84:233–240. [DOI] [PubMed] [Google Scholar]
- Troy, C.M., S.A. Rabacchi, J.B. Hohl, J.M. Angelastro, L.A. Greene, and M.L. Shelanski. 2001. Death in the balance: alternative participation of the caspase-2 and -9 pathways in neuronal death induced by nerve growth factor deprivation. J. Neurosci. 21:5007–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenz, C. 2001. Death receptors and apoptosis. Deadly signaling and evasive tactics. Cardiol. Clin. 19:31–43. [DOI] [PubMed] [Google Scholar]
- Virdee, K., A.J. Bannister, S.P. Hunt, and A.M. Tolkovsky. 1997. Comparison between the timing of JNK activation, c-Jun phosphorylation, and onset of death commitment in sympathetic neurones. J. Neurochem. 69:550–561. [DOI] [PubMed] [Google Scholar]
- Xue, L., G.C. Fletcher, and A.M. Tolkovsky. 1999. Autophagy is activated by apoptotic signalling in sympathetic neurons: an alternative mechanism of death execution. Mol. Cell. Neurosci. 14:180–198. [DOI] [PubMed] [Google Scholar]
- Yaginuma, H., N. Shiraiwa, T. Shimada, K. Nishiyama, J. Hong, S. Wang, T. Momoi, Y. Uchiyama, and R.W. Oppenheim. 2001. Caspase activity is involved in, but is dispensable for, early motoneuron death in the chick embryo cervical spinal cord. Mol. Cell. Neurosci. 18:168–182. [DOI] [PubMed] [Google Scholar]
- Yu, L., L. Korhonen, R. Martinez, E. Jokitalo, Y. Chen, U. Arumäe, and D. Lindholm. 2003. Regulation of sympathetic neuron and neuroblastoma cell death by XIAP and its association with proteasomes in neural cells. Mol. Cell. Neurosci. 22:308–318. [DOI] [PubMed] [Google Scholar]
- Zaidi, A.U., C. D'Sa-Eipper, J. Brenner, K. Kuida, T.S. Zheng, R.A. Flavell, P. Rakic, and K.A. Roth. 2001. Bcl-XL-caspase-9 interactions in the developing nervous system: evidence for multiple death pathways. J. Neurosci. 21:169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann, K.C., C. Bonzon, and D.R. Green. 2001. The machinery of programmed cell death. Pharmacol. Ther. 92:57–70. [DOI] [PubMed] [Google Scholar]