

Abstract
The Notch and transforming growth factor-β (TGF-β) signaling pathways play critical roles in the control of cell fate during metazoan development. However, mechanisms of cross-talk and signal integration between the two systems are unknown. Here, we demonstrate a functional synergism between Notch and TGF-β signaling in the regulation of Hes-1, a direct target of the Notch pathway. Activation of TGF-β signaling up-regulated Hes-1 expression in vitro and in vivo. This effect was abrogated in myogenic cells by a dominant-negative form of CSL, an essential DNA-binding component of the Notch pathway. TGF-β regulated transcription from the Hes-1 promoter in a Notch-dependent manner, and the intracellular domain of Notch1 (NICD) cooperated synergistically with Smad3, an intracellular transducer of TGF-β signals, to induce the activation of synthetic promoters containing multimerized CSL- or Smad3-binding sites. NICD and Smad3 were shown to interact directly, both in vitro and in cells, in a ligand-dependent manner, and Smad3 could be recruited to CSL-binding sites on DNA in the presence of CSL and NICD. These findings indicate that Notch and TGF-β signals are integrated by direct protein–protein interactions between the signal-transducing intracellular elements from both pathways.
Keywords: Hes-1; C2C12; CSL; Smad4; neural stem cell
Introduction
The Notch and TGF-β signaling pathways are important for the control of cellular differentiation and display principal similarities in their mode of signaling (for review see Artavanis-Tsakonas et al., 1999; Massagué et al., 2000). As summarized in Fig. 1 A, the ligand-induced signal is in both cases transmitted via membrane-proximal components, i.e., the Notch intracellular domain (NICD) and Smads, respectively, which relocate from the cytoplasm to the nucleus to control gene activation. Binding of TGF-β to type I and II serine-threonine kinase receptors results in phosphorylation and dissociation of receptor-regulated Smads. This group of Smad proteins is ligand-specific, i.e., Smads 1, 5, and 8 mediate bone morphogenetic protein signaling, whereas Smad2 and 3 participate in TGF-β and activin signaling. Smad4 is an essential signaling component common to all TGF-β superfamily ligands that associates with phosphorylated receptor-regulated Smads. In the nucleus, the complex of receptor-regulated Smads and Smad4 cooperates with additional coactivators, corepressors, and tissue-specific factors to regulate transcription of target genes (Attisano and Wrana, 2002). Activation of Notch by cell-bound ligands (Delta or Serrate) results in proteolytic cleavage of the NICD and its translocation to the cell nucleus, where it is recruited to target genes via interaction with CSL (RBP-Jk/CBF1), an essential DNA-binding component of the Notch pathway (for review see Artavanis-Tsakonas et al., 1999).
Figure 1.

Induction of Hes-1 expression by TGF-β signaling in vivo and in cell culture. (A) Scheme summarizing principal similarities in membrane-to-nucleus signaling between the Notch and TGF-β signaling pathways. (B) Induction of c-hairy mRNA expression in embryonic chick heart (arrows) by TGF-β signaling. Images show whole-mount in situ hybridization c-hairy mRNA of E4 chick embryos electroporated with GFP (control) or GFP plus CA-ALK5 expression plasmids. Induction of c-hairy mRNA expression could be observed in six out of six embryos electroporated with CA-ALK5. In no case (four out of four embryos) was c-hairy mRNA expression observed in control embryos. (C) Real-time PCR analysis of c-hairy mRNA expression in electroporated embryonic chick brain. The histogram shows means of three independent experiments each performed in triplicate ± SEM. (D) Real-time PCR analysis of Hes-1 mRNA expression in adult mouse neural stem cells treated with 10 ng/ml TGF-β for 90 min before RNA extraction. Results are presented as the mean ± SD. (E) Real-time PCR analysis of Hes-1 mRNA expression in C2C12 mouse myoblasts treated with 10 ng/ml TGF-β for 60 min before RNA extraction. Cycloheximide treatment (+ Chx) was begun 10 min before TGF-β stimulation. Results are presented as the mean ± SD of triplicate determinations.
Notch and TGF-β signaling converge in the regulation of a number of developmental processes, including myogenic, endothelial, pancreatic, and neuronal differentiation. However, it is at present assumed that the two systems act in parallel, largely independent pathways to regulate expression of target genes. A recent microarray survey of transcriptional changes in human keratinocytes exposed to TGF-β identified several components of the Notch pathway, including the basic helix-loop-helix transcription factor Hes-1, a direct target of Notch signaling (Zavadil et al., 2001). The rapid (≤60 min) induction of Hes-1 expression in human keratinocytes by TGF-β raised the possibility of an interaction between the two pathways.
Results and discussion
Electroporation of a constitutively active form of the type I TGF-β receptor (CA-ALK5) in the precardial mesoderm of embryonic day 2 (E2) chicken embryos resulted in ectopic expression of the chick Hes-1 homologue c-hairy in the heart at E4 (Fig. 1 B). In no case was ectopic expression of c-hairy detected after electroporation of a control construct (Fig. 1 B). Electroporation of CA-ALK5 in the mesencephalic vesicle resulted in a 2.3-fold increase in c-hairy expression in the midbrain, as evaluated by real-time PCR analysis of electroporated tissue (Fig. 1 C). stimulation of adult neural stem cells and C2C12 myoblasts with TGF-β induced a rapid (<60 min) increase in Hes-1 expression in both cell types (Fig. 1, D and E). Blockade of protein translation by prior treatment of C2C12 cells with cycloheximide did not affect induction of Hes-1 expression by TGF-β (Fig. 1 E), indicating that Hes-1 is a direct target of TGF-β signaling.
The requirement of Notch signaling was tested by transfecting C2C12 cells with a GFP expression plasmid and a dominant-negative CSL construct carrying a point mutation (R218H) that renders it unable to interact with DNA, and that has been shown to effectively block Notch signaling in other systems (Wettstein et al., 1997). CSL R281H abolished the effects of TGF-β on Hes-1 expression in GFP-positive cells isolated by cell sorting (Fig. 2 A), indicating the involvement of endogenous Notch signaling. Induction of PAI-1, a classical target of the TGF-β pathway, was unaffected by CSL R281H (Fig. 2 A). In addition, TGF-β stimulated transcription from a reporter construct containing 500 bp of upstream sequence from the Hes-1 promoter in C2C12 cells overexpressing NICD (Fig. 2 B), in agreement with a synergistic interaction between the two pathways.
Figure 2.
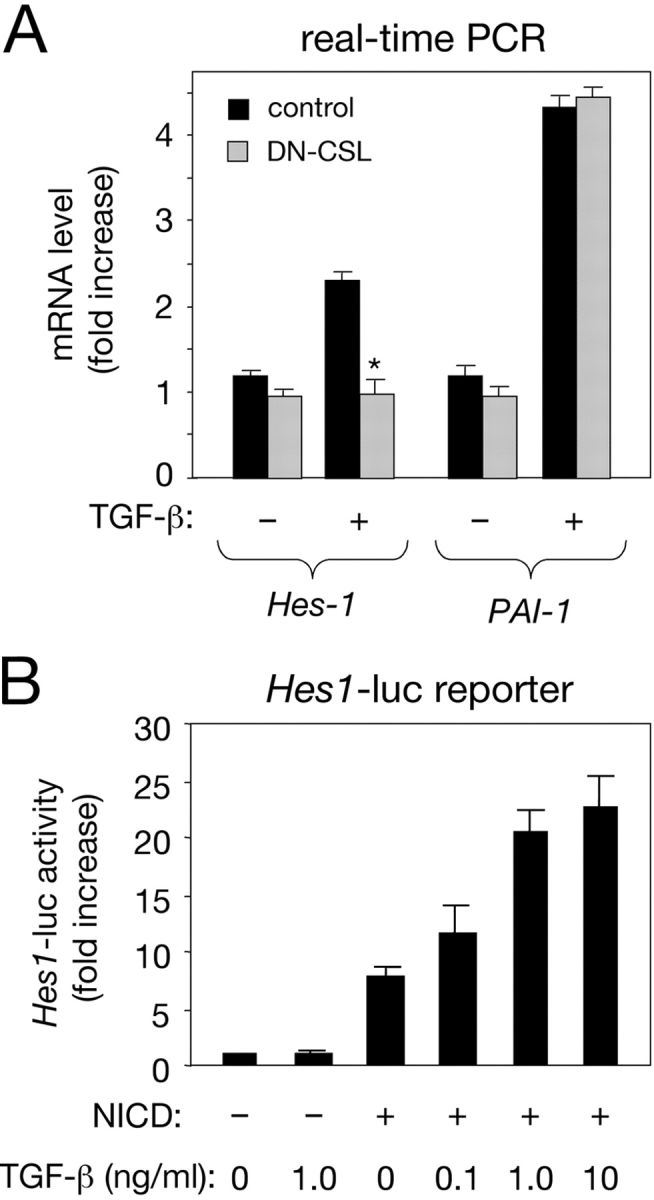
Requirement of Notch signaling for the effects of TGF-β on Hes-1 expression. (A) Real-time PCR analysis of Hes-1 mRNA expression in C2C12 mouse myoblasts expressing a dominant-negative CSL construct (DN-CSL) after treatment with TGF-β. Cells expressing DN-CSL (cotransfected with a GFP construct) were selected by FACS® analysis and treated with TGF-β for 60 min before RNA extraction. Results are presented as the mean ± SD of triplicate determinations. (B) Activation of the Hes-1 promoter in C2C12 myoblasts by NICD and TGF-β. A 500-bp construct of the Hes-1 promoter coupled to a luciferase reporter was introduced into C2C12 cells along with NICD (from mouse Notch1) as indicated. Treatment with TGF-β resulted in potentiation of NICD activity in a dose-dependent manner. Results are presented as the mean ± SD of triplicate determinations.
TGF-β augmented transcriptional activity from a reporter construct carrying multimerized CSL-binding sites (12xCSL-luc; Wallberg et al., 2002) in C2C12 myoblasts and C17.2 neural stem cells overexpressing NICD (Fig. 3, A and B). In the absence of NICD, overexpression of Smad proteins had no effect on the activity of the 12xCSL-luc reporter. However, in the presence of NICD, Smad3 increased the relative responsiveness of the 12xCSL-luc reporter to TGF-β (Fig. 3, A and B), indicating a synergistic cooperation of the two factors to regulate ligand-dependent gene transcription. Despite the inability of overexpressed Smad4 to potentiate NICD activity in C2C12 cells (Fig. 3, A and B), TGF-β failed to enhance NICD-mediated 12xCSL-luc activity in a human breast cancer cell line (MDA468) that lacks a functional Smad4 protein (Schutte et al., 1996; Fig. 3 C). Ligand-dependent activation of 12xCSL-luc could be restored upon transfection of Smad3 together with Smad4, but not Smad3 alone (Fig. 3 C), suggesting that Smad4 is nevertheless required for the functional interaction between the Notch and TGF-β signaling pathways. A point mutant of Smad3 that is unable to bind DNA (K81R Smad3; Morén et al., 2000) was also capable of potentiating the response of the 12xCSL-luc reporter to Notch and TGF-β signaling (Fig. 3, A and B), suggesting that Smad3 may not need to bind DNA directly in order to cooperate with NICD to regulate gene transcription.
Figure 3.

Cooperation of NICD and Smad3 in the activation of Notch- and Smad-specific synthetic promoters. (A) Role of individual Smad proteins in the regulation of the 12xCSL-luc reporter by NICD and TGF-β ligands in C2C12 myoblasts. Smad expression plasmids as indicated were transfected along with the 12xCSL-luc reporter construct in the presence or absence of NICD and TGF-β ligands at 10 ng/ml. T, TGF-β1; B, BMP-4. Normalized results are expressed relative to control as the mean ± SD of triplicate determinations. (B) Role of individual Smad proteins in the regulation of the 12xCSL-luc reporter by NICD and TGF-β ligands in C17.2 neural stem cells. (C) Activation of the 12xCSL-luc reporter in MDA468 breast cancer cells (lacking Smad4) by NICD and Smad3. (D) Activation of the Gal4-luc reporter in C2C12 myoblasts by Gal4–NICD and Smad3. (E) Activation of the Smad3-specific CAGA-luc reporter in C17.2 neural stem cells by TGF-β and potentiation by increasing amounts of NICD.
Next, we used a Gal4–NICD fusion construct, which bypasses the requirement of CSL for the recruitment of NICD to DNA (Beatus et al., 2001), together with a reporter construct carrying a multimerized Gal4-binding site (MH100, herein referred to as Gal4-luc). Transfection of Gal4–NICD activated this reporter, and this effect could be potentiated by cotransfection of Smad3 (Fig. 3 D), which on its own had no effect on the activity of the Gal4-luc reporter (unpublished data). These data suggested that a synergistic interaction between the Notch and TGF-β signaling pathways may take place even in the absence of CSL, as long as NICD and Smad3 (and presumably also Smad4) are present. We also used a reporter construct carrying nine tandem copies of the Smad-binding element from the PAI-1 promoter (CAGA-luc) that is specific for Smad3 and highly responsive to TGF-β (Dennler et al., 1998). Although largely insensitive to NICD in the absence of TGF-β, the transcriptional activity of this reporter in response to TGF-β could be further enhanced by NICD in a dose-dependent manner (Fig. 3 E).
The possibility that NICD and Smad3 may be able to interact directly was first investigated in vitro using GST–Smad fusion proteins produced in bacteria and 35S-labeled NICD produced by in vitro translation. A GST fusion of full-length Smad3, but not of Smad1, Smad4, or GST alone, was able to pull down 35S-labeled NICD (Fig. 4 A), indicating that Smad3 and NICD have the capacity of interacting with each other in the absence of additional components. In mammalian cells expressing myc-tagged NICD and Flag-tagged Smad3, immunoprecipitation with anti-Flag antibodies allowed the recovery of myc-tagged NICD only from extracts of cells that had also received the Flag–Smad3 construct (Fig. 4 B), demonstrating the formation of a complex between NICD and Smad3 in intact cells. Using deletion constructs of Smad3, the domain mediating its interaction with NICD was mapped to the COOH-terminal portion of the molecule, containing the MH2 domain and the linker (Fig. 4 C). A deletion analysis of NICD indicated that neither the most COOH-terminal transactivation domain (Kurooka et al., 1998) nor the region mediating p300/CBP binding (RE/AC; Oswald et al., 2001) are involved in interactions with Smad3 (Fig. 4 D). In parental C2C12 myoblasts, low but detectable levels of Smad3 could be recovered by coimmunoprecipitation with endogenous Notch1 at basal conditions. This interaction could be augmented by treatment with TGF-β (Fig. 5 A). In a stable line of C2C12 cells expressing higher levels of full-length Notch1—and displaying elevated basal 12xCSL-luc activity (Chapman, G., personal communication)—higher levels of endogenous Smad3 could be recovered in Notch1 immunoprecipitates (Fig. 5 A). Importantly, this interaction could be further enhanced by treatment with TGF-β (Fig. 5 A), demonstrating a ligand-dependent association between Smad3 and Notch in myoblast cells.
Figure 4.

Physical interaction between NICD and Smad3. (A) Full-length 35S-labeled NICD produced by in vitro translation was used in precipitation assays together with equal amounts of the indicated GST–Smad fusion proteins. A GST fusion of the zinc-finger transcription factor GATA-3 was used as a negative control. (B) Coimmunoprecipitation of myc-NICD and Flag-Smad3 in total lysates of transfected COS cells. The anti-Flag antibody directed against Flag-tagged Smad3 brings down myc-tagged NICD only in cells that also received the Flag-tagged Smad3 construct (top). The panels below show immunoblots of 20% of the lysates. (C) In vitro pull-down of myc-tagged NICD from COS cell lysates with GST fusions of the MH1 and MH2 domains of Smad3, including the intervening linker region. (D) In vitro pull-down of Flag-tagged Smad3 from COS cell lysates using GST fusions of full-length NICD or a COOH-terminally truncated NICD construct lacking the transactivation and p300/CBP-binding domains (ΔC NICD).
Figure 5.

Ligand-dependent interaction between endogenous Smad3 and Notch, and recruitment of Smad3 to specific DNA sites by CSL and NICD. (A) Parental C2C12 cells or a stable C2C12 transfectant overexpressing full-length Notch1 (C2C12-N1) were either left untreated or stimulated with 10 ng/ml TGF-β1 for 50 min before lysis and immunoprecipitation with anti-Notch1 antibodies. Immunoblots were probed with anti-Smad3 antibodies and reprobed with anti-Notch1, detected as its transmembrane and intracellular (TMIC) domain. It should be noted that to date, nuclear NICD has been very difficult to detect by biochemical or in situ methods in normal cells, possibly because it is present in very low amounts and/or has a very short half-life (Rand et al., 2000). (B) A biotinylated oligonucleotide containing two tandem CSL-binding sites (2xCSL) was used to pull down GST–Smad3, 35S-labeled NICD, and 35S-labeled CSL in different combinations as indicated. A mutant oligonucleotide (mut) was used as a control. Note that GST–Smad3 (detected by immunoblot with anti-GST antibodies) could only be recovered using the wild-type (wt) oligonucleotide in the presence of both NICD and CSL. Weak levels of 35S-labeled NICD could also be detected in the same lane. (C) A mechanism for the integration of TGF-β and Notch signaling by direct interaction between Smad3 and NICD. Although the Smad3–Smad4 complex and the NICD can translocate to the nucleus independently, our results do not rule out the possibility that their interaction could already take place in the cytoplasm.
Finally, we used a biotinylated oligonucleotide containing two tandem CSL-binding sites to probe combinations of GST–Smad3 and in vitro–translated NICD and CSL in a cell-free system. GST–Smad3 could only be precipitated by the biotinylated oligonucleotide in the presence of both NICD and CSL, but not NICD or CSL alone (Fig. 5 B), indicating that Smad3 depends on its ability to interact with NICD for its recruitment to CSL DNA-binding sites. Importantly, a mutated oligonucleotide carrying three point mutations in the consensus site of CSL binding was unable to recover GST–Smad3 under any condition (Fig. 5 B), underlying the specificity of these protein–DNA interactions.
Together, these results suggest that Smad3 can be recruited to the promoter regions of Notch target genes via direct interaction with NICD (Fig. 5 C). In a reciprocal situation, our observations suggest a mechanism by which Notch signaling could influence the expression of TGF-β target genes. Although previous work has revealed several examples of cross-regulatory interactions between Notch and other pathways, including Wnt (Espinosa et al., 2003) and Ras (Shaye and Greenwald, 2002), Notch signaling had until now not been shown to be involved in a direct cross-talk mediated by defined protein–protein interactions with signaling components from other major ligand systems.
Notch signaling profoundly influences the differentiation of many cell types, and there are several situations in which Notch and TGF-β signaling are known to converge. During myogenesis, induction of Hes-1 by Notch inhibits the expression of myogenic regulatory factors, such as the basic helix-loop-helix transcription factor MyoD (Kuroda et al., 1999). TGF-β signaling also inhibits myogenesis, induction of muscle-specific gene expression, and myotube formation in cultured myoblasts without affecting cell proliferation (Massagué et al., 1986). Although Smad3 has been shown to repress the activity of MyoD through direct protein–protein interactions (Liu et al., 2001), the results presented here highlight a more upstream point of convergence between the two pathways. Notch and TGF-β signaling are also known to converge in the regulation of several other differentiation events, for example during endothelial (Goumans et al., 2002), pancreas (Kim and Hebrok, 2001), and neural (Shah et al., 1996) development. Our findings indicate an unexpected level of cross-talk between two major signaling pathways, and warrant further investigations on the roles of Smad3–NICD interactions in the coordination of metazoan development.
Materials and methods
In ovo electroporation of chicken embryos
Embryos from fertilized white leghorn eggs were electroporated at Hamburger and Hamilton (HH) stages 8–10 (≈E2) with constructs at a final concentration of 0.4 μg/ml in the presence of 0.4 μg/ml EGFP (CLONTECH Laboratories, Inc.) in 1× PBS and 0.2% Fast Green using an Electro-Square Porator (model ECM830; Genetronics, Inc.) at 20 V with five pulses of 15 ms. Stage 8–9 embryos were used for targeting the precardiac mesoderm in the vicinity of the cephalic mesenchyme, whereas stage 10 embryos were injected in the mesencephalic vesicle for analysis of brain expression. 2 d after electroporation (HH stages 21–23), surviving embryos with the appropriate targeting of GFP expression (i.e., heart or brain) were subjected to either whole-mount in situ hybridization (heart) or real-time PCR (brain) for analysis of c-hairy mRNA expression. Chick embryos were photographed with a digital camera (Kodak) mounted to a dissection microscope (Carl Zeiss MicroImaging, Inc.) at 2×.
In situ hybridization and real-time PCR
Whole-mount in situ hybridization for c-hairy expression was performed according to previously published procedures (Palmeirim et al., 1997), but omitting proteinase K digestion. 1 d after transfection, C2C12 cells were mechanically detached from the plate, and GFP-positive cells were sorted by FACS® analysis in a FACSVantage™ SE System (Becton Dickinson). GFP-positive cells were cultured for another 24 h and then treated with TGF-β for 60 min before RNA isolation and cDNA synthesis. Real-time PCR was performed in a LightCycler system (PerkinElmer). PCR primer sequences are available on request. All results are expressed relative to GAPDH values obtained in parallel reactions.
Plasmid constructs, cell transfection, and reporter assays
The Hes-1 reporter construct contains 0.5 kb of upstream sequence of the Hes-1 gene followed by a luciferase reporter (Jarriault et al., 1995). The 12xCSL-luc reporter contains a hexameric 50-bp repeat of the EBNA2 response element of the TP-1 promoter (each containing two CSL-binding sites) in front of the minimal β-globin promoter driving the luciferase gene (Kato et al., 1997; Wallberg et al., 2002). The CAGA reporter contains nine tandem copies of the Smad-binding element from the PAI-1 promoter (Dennler et al., 1998). All Smad expression plasmids have been described elsewhere (Morén et al., 2000). The NICD constructs used for reporter and pull-down assays were derived from the intracellular domain of the mouse Notch1 receptor (Kopan et al., 1994; Beatus et al., 2001; Wallberg et al., 2002).
COS cells were transfected by the calcium phosphate method. C2C12, MDA468, and C17.2 cells were transfected in complete medium with FuGENE™ 6 (Roche). After 24 h incubation, cell monolayers were washed with serum-free medium and incubated for a further 16 h in 0.1% serum-containing medium supplemented with 10 ng/ml TGF-β1 or 40 ng/ml BMP-4 (R&D Systems) as indicated. Cycloheximide (Sigma-Aldrich) was used at 25 μg/ml. Reporter assays were performed and analyzed as described previously (Blokzijl et al., 2002). All treatments and transfection conditions were analyzed in triplicate.
Pull-down and coimmunoprecipitation assays
GST fusions were produced in Escherichia coli and purified by chromatography on glutathione-conjugated agarose beads (Amersham Biosciences). In vitro–translated products were produced using a kit from Promega. Anti-Flag mAb was from Kodak or Sigma-Aldrich, anti-myc 9E1 mAb was from Covance or BD Biosciences. Cell lysates and immunoprecipitations were done as described previously (Blokzijl et al., 2002). Parental and Notch1-transfected C2C12 cells were starved in serum-free medium for 4 h before treatment with 10 ng/ml TGF-β1 for 50 min. Triton X-100 cell lysates were immunoprecipitated with anti-Notch1 antibodies (M-20; Santa Cruz Biotechnology, Inc.) that had previously been covalently coupled to CNBr-activated Sepharose beads (Amersham Biosciences). Immunoblots were probed with an anti-Smad3 pAb (Zymed Laboratories). Oligonucleotide pull-down assays were performed with biotinylated 50-mer oligonucleotides containing the EBNA2 response element of the TP-1 promoter (equivalent to two CSL-binding sites) with sequence as follows: 5′-GATCCCGACTCGTGGGAAAATGGGCGGAAGGGCACCGTGGGAAAATAGTA-3′. As control, a mutant oligonucleotide carrying three point mutations in the CSL consensus site was used with sequence as follows: 5′-GATCCCGACTCTACGGAAAATGGGCGGAAGGGCACCTACGGAAAATAGTA-3′ (mutations in bold). Different combinations of GST–Smad3, 35S-labeled NICD, and 35S-labeled CSL produced as above were mixed 1:1 with H buffer (20 mM Hepes, 50 mM KCl, 20% glycerol, 0.1% NP-40, and 1 mM DTT) plus biotinylated oligonucleotide and polydIdC (50 μg/ml). After 2 h at 4°C, streptavidin beads (Pierce Chemical Co.) were added, followed by an additional 1 h of incubation, precipitation, and washing in H buffer with additional salt (up to 300 mM). Immunoblots were processed by autoradiography and subsequently probed with anti-GST antibodies to detect GST-tagged Smad3.
Acknowledgments
We thank Olivier Pourquie (Stowers Institute for Medical Research, Kansas City, MO) for providing the c-hairy probe, Peter ten Dijke (Netherlands Cancer Institute, Amsterdam, Netherlands) for Smad constructs and for sharing unpublished results, Gavin Chapman for analysis of C2C12 cells expressing Notch1, and Xiaoli Li for secretarial help.
This work was supported by grants from the Swedish Foundation for Strategic Research (to C.F. Ibáñez and U. Lendahl), Swedish Cancer Society (3872-B02-09XAC to C.F. Ibáñez), Human Frontier Science Program (to U. Lendahl), and European Union project QLRT-1999-31471 (to U. Lendahl).
A. Blokzijl and C. Dahlqvist contributed equally to this paper.
Abbreviations used in this paper: E, embryonic day; NICD, Notch intracellular domain.
References
- Artavanis-Tsakonas, S., M.D. Rand, and R.J. Lake. 1999. Notch signaling: cell fate control and signal integration in development. Science. 284:770–776. [DOI] [PubMed] [Google Scholar]
- Attisano, L., and J.L. Wrana. 2002. Signal transduction by the TGF-β superfamily. Science. 296:1646–1647. [DOI] [PubMed] [Google Scholar]
- Beatus, P., J. Lundkvist, C. Oberg, K. Pedersen, and U. Lendahl. 2001. The origin of the ankyrin repeat region in Notch intracellular domains is critical for regulation of HES promoter activity. Mech. Dev. 104:3–20. [DOI] [PubMed] [Google Scholar]
- Blokzijl, A., P. ten Dijke, and C.F. Ibáñez. 2002. Physical and functional interaction between GATA-3 and Smad3 allows TGF-β regulation of GATA target genes. Curr. Biol. 12:35–45. [DOI] [PubMed] [Google Scholar]
- Dennler, S., S. Itoh, D. Vivien, P. ten Dijke, S. Huet, and J.M. Gauthier. 1998. Direct binding of smad3 and smad4 to critical TGFβ-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 17:3091–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa, L., J. Ingles-Esteve, C. Aguilera, and A. Bigas. 2003. Phosphorylation by glycogen synthase kinase-3β down-regulates Notch activity, a link for Notch and Wnt pathways. J. Biol. Chem. 278:32227–32235. [DOI] [PubMed] [Google Scholar]
- Goumans, M.J., G. Valdimarsdottir, S. Itoh, A. Rosendahl, P. Sideras, and P. ten Dijke. 2002. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J. 21:1743–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarriault, S., C. Brou, F. Logeat, E.H. Schroeter, R. Kopan, and A. Israel. 1995. Signalling downstream of activated mammalian Notch. Nature. 377:355–358. [DOI] [PubMed] [Google Scholar]
- Kato, H., Y. Taniguchi, H. Kurooka, S. Minoguchi, T. Sakai, S. Nomura-Okazaki, K. Tamura, and T. Honjo. 1997. Involvement of RBP-J in biological functions of mouse Notch1 and its derivatives. Development. 124:4133–4141. [DOI] [PubMed] [Google Scholar]
- Kim, S.K., and M. Hebrok. 2001. Intercellular signals regulating pancreas development and function. Genes Dev. 15:111–127. [DOI] [PubMed] [Google Scholar]
- Kopan, R., J.S. Nye, and H. Weintraub. 1994. The intracellular domain of mouse Notch: a constitutively activated repressor of myogenesis directed at the basic helix-loop-helix region of MyoD. Development. 120:2385–2396. [DOI] [PubMed] [Google Scholar]
- Kuroda, K., S. Tani, K. Tamura, S. Minoguchi, H. Kurooka, and T. Honjo. 1999. Delta-induced Notch signaling mediated by RBP-J inhibits MyoD expression and myogenesis. J. Biol. Chem. 274:7238–7244. [DOI] [PubMed] [Google Scholar]
- Kurooka, H., K. Kuroda, and T. Honjo. 1998. Roles of the ankyrin repeats and C-terminal region of the mouse notch1 intracellular region. Nucleic Acids Res. 26:5448–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, D., B.L. Black, and R. Derynck. 2001. TGF-β inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev. 15:2950–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué, J., S. Cheifetz, T. Endo, and B. Nadal-Ginard. 1986. Type beta transforming growth factor is an inhibitor of myogenic differentiation. Proc. Natl. Acad. Sci. USA. 83:8206–8210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué, J., S.W. Blain, and R.S. Lo. 2000. TGFβ signaling in growth control, cancer, and heritable disorders. Cell. 103:295–309. [DOI] [PubMed] [Google Scholar]
- Morén, A., S. Itoh, A. Moustakas, P. Dijke, and C.-H. Heldin. 2000. Functional consequences of tumorigenic missense mutations in the amino-terminal domain of Smad4. Oncogene. 19:4396–4404. [DOI] [PubMed] [Google Scholar]
- Oswald, F., B. Tauber, T. Dobner, S. Bourteele, U. Kostezka, G. Adler, S. Liptay, and R.M. Schmid. 2001. p300 acts as a transcriptional coactivator for mammalian Notch-1. Mol. Cell. Biol. 21:7761–7774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmeirim, I., D. Henrique, D. Ish-Horowicz, and O. Pourquie. 1997. Avian hairy gene expression identifies a molecular clock linked to vertebrate segmentation and somitogenesis. Cell. 91:639–648. [DOI] [PubMed] [Google Scholar]
- Rand, M.D., L.M. Grimm, S. Artavanis-Tsakonas, V. Patriub, S.C. Blacklow, J. Sklar, and J.C. Aster. 2000. Calcium depletion dissociates and activates heterodimeric notch receptors. Mol. Cell. Biol. 20:1825–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutte, M., R.H. Hruban, L. Hedrick, K.R. Cho, G.M. Nadasdy, C.L. Weinstein, G.S. Bova, W.B. Isaacs, P. Cairns, H. Nawroz, et al. 1996. DPC4 gene in various tumor types. Cancer Res. 56:2527–2530. [PubMed] [Google Scholar]
- Shah, N.M., A.K. Groves, and D.J. Anderson. 1996. Alternative neural crest cell fates are instructively promoted by TGFβ superfamily members. Cell. 85:331–343. [DOI] [PubMed] [Google Scholar]
- Shaye, D.D., and I. Greenwald. 2002. Endocytosis-mediated downregulation of LIN-12/Notch upon Ras activation in Caenorhabditis elegans. Nature. 420:686–690. [DOI] [PubMed] [Google Scholar]
- Wallberg, A.E., K. Pedersen, U. Lendahl, and R.G. Roeder. 2002. p300 and PCAF act cooperatively to mediate transcriptional activation from chromatin templates by Notch intracellular domains in vitro. Mol. Cell. Biol. 22:7812–7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettstein, D.A., D.L. Turner, and C. Kintner. 1997. The Xenopus homolog of Drosophila Suppressor of Hairless mediates Notch signaling during primary neurogenesis. Development. 124:693–702. [DOI] [PubMed] [Google Scholar]
- Zavadil, J., M. Bitzer, D. Liang, Y.C. Yang, A. Massimi, S. Kneitz, E. Piek, and E.P. Bottinger. 2001. Genetic programs of epithelial cell plasticity directed by transforming growth factor-β. Proc. Natl. Acad. Sci. USA. 98:6686–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]