

Abstract
The NHPX protein is a nucleolar factor that binds directly to a conserved RNA target sequence found in nucleolar box C/D snoRNAs and in U4 snRNA. Using enhanced yellow fluorescent protein (EYFP)– and enhanced cyan fluorescent protein–NHPX fusions, we show here that NHPX is specifically accumulated in both nucleoli and Cajal bodies (CBs) in vivo. The fusion proteins display identical localization patterns and RNA binding specificities to the endogenous NHPX. Analysis of a HeLa cell line stably expressing EYFP–NHPX showed that the nucleolar accumulation of NHPX was preceded by its transient accumulation in splicing speckles. Only newly expressed NHPX accumulated in speckles, and the nucleolar pool of NHPX did not interchange with the pool in speckles, consistent with a unidirectional pathway. The transient accumulation of NHPX in speckles prior to nucleoli was observed in multiple cell lines, including primary cells that lack CBs. Inhibitor studies indicated that progression of newly expressed NHPX from speckles to nucleoli was dependent on RNA polymerase II transcription, but not on RNA polymerase I activity. The data show a specific temporal pathway involving the sequential and directed accumulation of NHPX in distinct subnuclear compartments, and define a novel mechanism for nucleolar localization.
Keywords: nucleolus; Cajal bodies; speckles; localization; nucleus
Introduction
The cell nucleus is the site at which chromosomes are located, and at which DNA replication and gene expression are coordinated and regulated. Many nuclear factors are organized into specific structures termed nuclear bodies (for review see Lamond and Earnshaw, 1998; Schul et al., 1998; Matera, 1999; Spector, 2001). Most nuclear bodies are dynamic, disassembling on entry to M phase and reassembling after mitosis (Dundr et al., 2000). Unlike cytoplasmic organelles, nuclear bodies are not enclosed by membranes. Factors can move in and out of nuclear bodies (for review see Misteli, 2001), and the bodies themselves can also move within the nucleoplasm (Boudonck et al., 1999; Platani et al., 2000; Snaar et al., 2000; Muratani et al., 2001).
The best-studied subnuclear body is the nucleolus (for reviews see Pederson, 1998; Scheer and Hock, 1999; Carmo-Fonseca et al., 2000; Olson et al., 2000; Visintin and Amon, 2000). Whereas ∼10% of protein-coding genes are active at any time, ∼80% of the total cellular transcription activity is devoted to expression of the conserved ribosomal DNA (rDNA)* repeats in nucleoli by RNA polymerase I (Blobel and Potter, 1967). Multiple rDNA transcription units are joined together by nontranscribed intergenic spaces to form the so-called nucleolar organizing regions (NORs) located in human chromosomes 13, 14, 15, 21, and 22. Nucleoli are assembled on these chromosomal NOR regions after every round of mitosis. The rDNA-containing region is transcribed to yield a precursor, the 45S pre-rRNA, which is processed in a series of posttranscriptional cleavage and modification reactions to generate mature rRNA species that form the catalytic core of the ribosome (for review see Warner, 2001). Various small nucleolar RNAs (snoRNAs), for example U3 and U14, are needed for these separate rRNA cleavage steps, each snoRNA being required at specific steps. The snoRNAs can be categorized into two main classes, box C/D (i.e., U3, U8, U14) and box H/ACA (i.e., U17, U19, U64), according to conserved sequence elements and the way in which they are assumed to fold into defined secondary structures (for review see Weinstein and Steitz, 1999). These snoRNAs assemble together with protein factors to form snoRNPs that have roles in guiding and catalyzing posttranscriptional RNA modifications. For example, box C/D snoRNPs direct 2′-O-ribose methylation and box H/ACA snoRNPs direct pseudouridylations of specific rRNA nucleotides (for review see Lafontaine and Tollervey, 1998; Lewis and Tollervey, 2000).
Several nucleolar proteins, including the snoRNP factor fibrillarin, have been found in Cajal bodies (CBs) (for review see Gall, 2000). These subnuclear structures, originally called nucleolar accessory bodies, or coiled bodies, are often associated with the nucleolar periphery, or even located inside the nucleolus (Malatesta et al., 1994; Ochs et al., 1994; Lyon et al., 1997; Sleeman et al., 1998). In addition to snoRNPs, they also contain splicing snRNPs and some transcription factors; however, they do not contain non-snRNP protein splicing factors or nascent pre-mRNA. Their snRNP components are specifically newly assembled particles (Carvalho et al., 1999; Gall et al., 1999; Sleeman and Lamond, 1999a; Sleeman et al., 2001). Fluorescently labeled U3, U8, and U14 snoRNAs, when microinjected into Xenopus oocytes, accumulate transiently in CBs prior to nucleoli, suggesting that newly imported snoRNAs flow from the CBs to nucleoli (Narayanan et al., 1999b). Therefore, CBs might be involved in transport and maturation of both snRNPs and snoRNPs.
In higher eukaryotes, most pre-mRNAs must be spliced to generate functional mRNAs. Splicing is a nuclear process carried out by a complex RNA protein machine termed the spliceosome (for review see Staley and Guthrie, 1998). Although splicing often occurs cotranscriptionally, the majority of pre-mRNA splicing factors are not localized at active transcription sites; instead, they are enriched in domains called speckles (for reviews see Sleeman and Lamond, 1999b; Misteli, 2000). Recruitment of splicing factors to sites of transcription is believed to involve a cycle of phosphorylation and dephosphorylation (for review see Misteli, 1999).
The advent of green fluorescent protein (GFP) technology allows the visualization of tagged proteins and their movement in living cells (for review see Swedlow and Lamond, 2001). The use of photobleaching techniques with the GFP fusion proteins also allows analysis of protein dynamics (Phair and Misteli, 2001; Reits and Neefjes, 2001). Here we describe a novel nuclear pathway shown by the nucleolar factor NHPX. The NHPX protein was recently identified as a putative human homologue of yeast NHP2p (Saito et al., 1996), and later, independently, as a 15.5-kD RNA-binding protein (Nottrott et al., 1999). Subsequent analyses indicated that NHPX is the human orthologue of the Saccharomyces cerevisiae Snu13p (Chang et al., 1999; Nottrott et al., 1999). The human NHPX protein was also independently identified in a directed proteomic analysis of nucleoli isolated from cultured HeLa cells (Andersen et al., 2002); it shares a common RNP structure that binds to both box C/D snoRNAs and spliceosomal U4 snRNA (Nottrott et al., 1999; Vidovic et al., 2000; Watkins et al., 2000). However, NHPX localizes in nucleoli by immunofluorescence (Chang et al., 1999). Here we analyze the in vivo pathway by which NHPX accumulates in nucleoli, and show that it involves an unexpected, transient interaction with splicing speckles. The movement of NHPX from speckles to nucleoli is dependent on pre-mRNA transcription. These data suggest that NHPX may be involved in, and possibly link, several parallel RNA metabolic pathways that occur in distinct nuclear domains.
Results
NHPX is localized to nucleoli and CBs
NHPX was selected for characterization as part of the analysis of the human nucleolar proteome (Andersen et al., 2002). To address its in vivo localization, the NHPX cDNA was isolated from a HeLa cDNA library and tagged at its NH2 terminus with the enhanced yellow fluorescent protein (EYFP) gene to generate plasmid pAL107EYFP–NHPX (see Materials and methods). The localization of the EYFP–NHPX fusion protein was analyzed by fluorescence microscopy after transient transfection of pAL107EYFP–NHPX in HeLa cells (Fig. 1), and compared with the nucleolar protein fibrillarin (FIB). Transiently expressed EYFP–NHPX was colocalized with FIB in the dense fibrillar component of nucleoli and also, unexpectedly, in CBs. (Fig. 1 A, arrow indicates nucleolus, arrowhead indicates CB). The CB localization was confirmed by counterstaining with anti-coilin antibodies (Fig. 2 A; unpublished data). The localization of NHPX to CBs and nucleoli was also confirmed by immunofluorescence (Figs. 1 B and S1, available at http://www.jcb.org/cgi/content/full/200201120/DC1). The tagged NHPX showed an identical localization to its endogenous counterpart, suggesting that NHPX is a component of both nuclear structures (Fig. 1 B).
Figure 1.
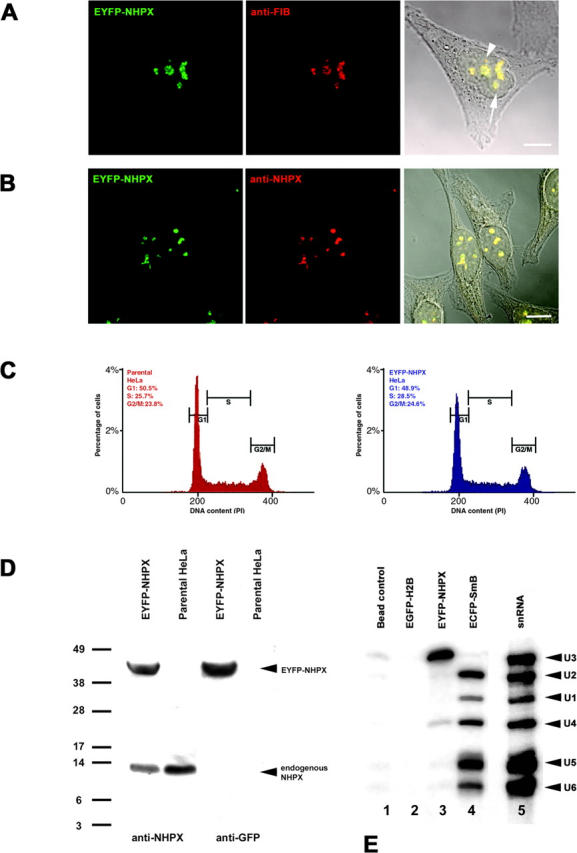
Transient and stable expression of EYFP–NHPX in HeLa cell line. HeLa cells transiently transfected with pAL107EYFP–NHPX for 16 h were fixed and counterstained with (A) anti-FIB antibody 72B9 and (B) affinity-purified anti-NHPX serum R86. Arrowhead indicates CB and arrow indicates nucleolus. (C) DNA content of HeLaEYFP–NHPX cells were analyzed by FACS analysis and (D) its expression level by immunoblot using antiserum R86 and anti-GFP. (E) The in vivo RNA binding activity of EYFP-NHPX was assayed by immunoprecipitation and the binding of snRNAs U1, U2, U4, U5, and U6 as well as snoRNA U3 were tested by Northern hybridization. Bars, 5 μm.
Figure 2.
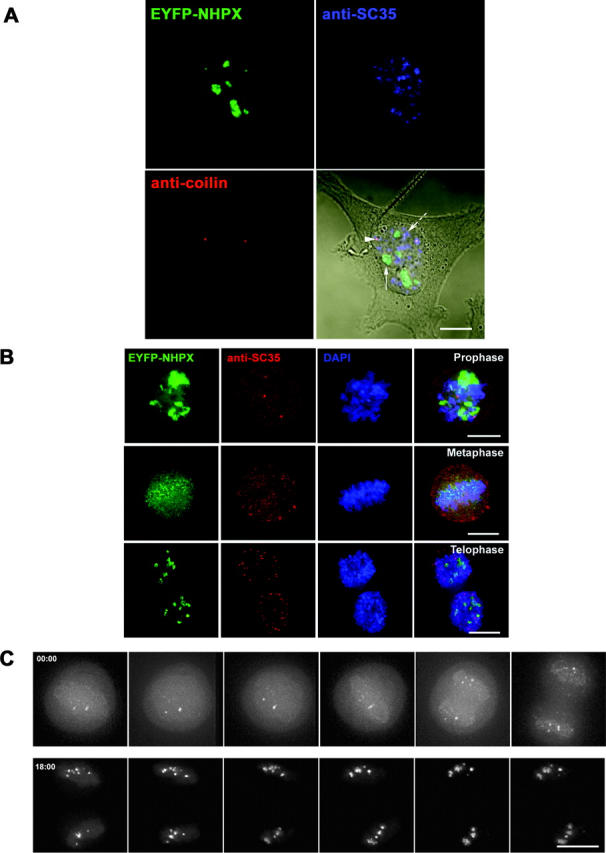
Localization of NHPX during interphase and mitosis. The patterns of EYFP–NHPX in HeLaEYFP–NHPX were analyzed (A) during interphase and (B) in different stages of mitosis. Anti-coilin 204/10 was used to denote CBs, anti-SC35 to denote speckles and DAPI to show the condensed chromosome in mitotic stages. Arrowhead indicates CB, arrow indicates nucleolus, and broken arrow indicates speckles. (C) The pattern of EYFP–NHPX in HeLaEYFP–NHPX during mitosis was followed after metaphase in a live cell imaged every 3 min. Bars, 5 μm.
To facilitate in vivo analysis of NHPX, we established a HeLa cell line stably expressing EYFP–NHPX (see Materials and methods). As shown by fluorescence microscopy, the EYFP–NHPX protein localized specifically in nucleoli and CBs in the HeLaEYFP–NHPX stable cell line (Fig. 2). The HeLaEYFP–NHPX cell line grows and divides at the same rate as the parental HeLa cell line without any apparent cell cycle defect, as shown by FACS analysis (Fig. 1 C; unpublished data). The expression level of EYFP–NHPX is comparable to that of the endogenous NHPX protein in the HeLaEYFP–NHPX cell line (Fig. 1 D).
To confirm that the EYFP–NHPX fusion protein expressed in the HeLaEYFP–NHPX cell line behaves biochemically as the endogenous protein, its in vivo RNA-binding specificity was investigated. An extract from HeLaEYFP–NHPX cells was immunoprecipitated using an anti-GFP antibody. RNAs in the immunoprecipitate were separated by Urea-polyacrylamide gel electrophoresis, transferred to nylon membrane, and hybridized with probes for U1, U2, U4, U5, and U6 snRNAs and U3 snoRNA (Fig. 1 E). This showed that U3 snoRNA and U4 snRNA, but not U1, U2, U5, or U6 snRNAs, were preferentially isolated with the anti-GFP antibody (Fig. 1 E, lane 3). Control experiments, i.e., bead control and an HeLaEGFP–H2B cell extract, showed that although the anti-GFP antibody still pulled down the fluorescent protein fusion protein (unpublished data), it did not pull down any of the RNAs tested from these extracts (Fig. 1 E, lane 1 and 2), whereas the same anti-GFP antibody pulled down each of the U1, U2, U4, U5, and U6 snRNAs, but not U3 snoRNA, from a HeLaECFP–SmB cell extract (Fig. 1 E, lane 4). These data show that the EYFP–NHPX fusion protein in vivo is specifically complexed with the same RNA targets that NHPX was shown to bind directly in vitro (Nottrott et al., 1999; Watkins et al., 2000). Given that U4 and U6 snRNAs usually exist as a duplex inside the nucleus, the immunoprecipitation of U4 snRNA alone is surprising. The disruption of the U4/U6 interaction in this assay is unlikely, because a non-Sm RNA, i.e., U6 snRNA, can be coimmunoprecipitated with U4 from the ECFP–SmB cell extract under the same experimental conditions and using the same anti-GFP antibody (Fig. 1 E, lane 4). Another interesting observation is that only a small amount of the total U4 snRNA was pulled down in these experiments (Fig. 1 E, lane 3), even though U4 snRNA is present in similar abundance to U3 snoRNA inside HeLa cells (Reddy and Bush, 1988). This suggests that only a subset of U4, which is likely not bound to U6, may be interacting with NHPX in vivo. Alternatively, the fraction of NHPX bound to the U4/U6 duplex could be inaccessible to the anti-GFP antibodies.
NHPX is colocalized with U3-containing snoRNPs, rather than U4-enriched splicing speckles, during interphase
The ability of NHPX to bind both U3 snoRNA and U4 snRNA, which normally are located in different subnuclear structures, prompted us to investigate further the localization of NHPX under different conditions (Fig. 2). The binding of NHPX to the spliceosomal U4 snRNA suggests that it should colocalize, at least in part, with other splicing factors. However, EYFP–NHPX in the HeLaEYFP–NHPX cell line does not show a speckled nuclear pattern similar to other human splicing factors such as SC35 and U1A (Fig. 2 A, arrowhead indicates CB, arrow indicates nucleolus, broken arrow indicates speckles). The anti-NHPX antiserum also does not label speckles (Fig. 1 B, Chang et al., 1999). Instead, EYFP–NHPX is accumulated in nucleoli and CBs and colocalizes with the snoRNP protein FIB (Figs. 1 A and 2 A ; unpublished data). Also, nucleolar segregation, caused by treating the cells with the transcription inhibitor actinomycin D, results in the relocation of NHPX to the nucleolar periphery along with FIB, but does not cause it to colocalize with either SC35, or other splicing factors (unpublished data).
Analysis of the HeLaEYFP–NHPX cells at different stages of mitosis also showed no evidence for the association of NHPX with splicing factors (Fig. 2 B). Time-lapse analysis of live HeLaEYFP–NHPX cells showed that NHPX associated with snoRNP-containing nucleoli immediately after the nuclear envelope reforms (Fig. 2 C; unpublished data). Combined with other immunofluorescence data of fixed cells counterstained with anti-FIB (unpublished data), we conclude that EYFP–NHPX does not accumulate with snRNPs in speckles, but instead colocalizes with the snoRNP protein FIB at all cell cycle stages and metabolic conditions tested. The only colocalization of NHPX with splicing snRNPs detected in vivo was specifically in CBs, structures known to accumulate newly imported snRNPs and snoRNPs when they first enter the nucleus.
Newly imported NHPX transiently colocalizes with splicing factors
Next, we investigated the localization of newly synthesized NHPX because recent data have shown a temporal pathway for snRNP and snoRNP localization in nuclei (Fig. 3; Carvalho et al., 1999; Narayanan et al., 1999a, 1999b; Sleeman and Lamond, 1999a; Sleeman et al., 2001). To our surprise, microinjection of the expression vector pAL107EYFP–NHPX into the cultured cell lines, followed by examination in the fluorescence microscope, revealed that 1 h after microinjection, EYFP–NHPX is accumulated in splicing speckles and CBs (Fig. 3). 2–6 h postmicroinjection, EYFP–NHPX is also detected in nucleoli, whereas the level of EYFP–NHPX in speckles shows a concomitant decrease. At later time points, the signal in speckles can no longer be detected and EYFP–NHPX accumulates specifically in nucleoli and CBs, resembling the pattern observed in the HeLaEYFP–NHPX cell line during interphase.
Figure 3.
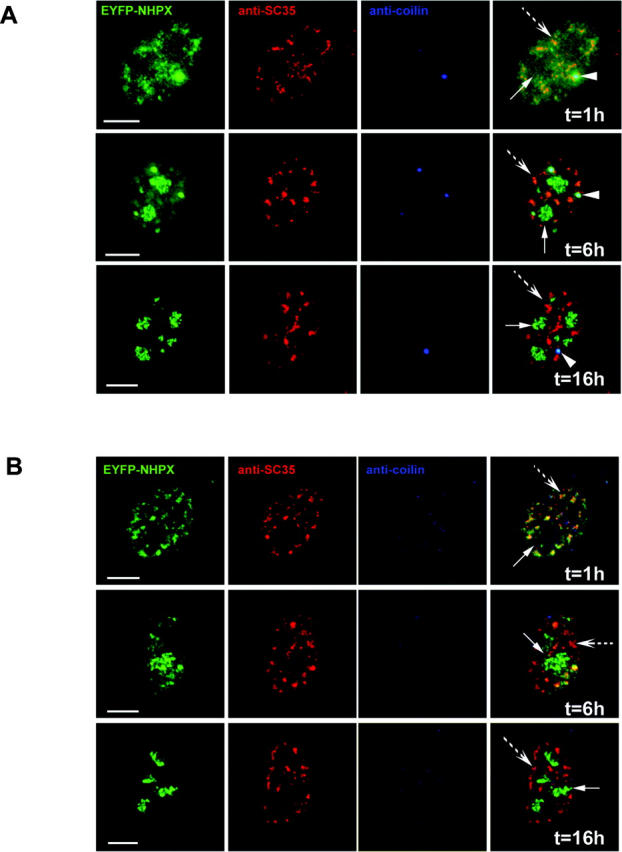
NHPX transiently accumulates in splicing speckles. Microinjection of pAL107EYFP–NHPX into (A) transformed cell line MCF7 and (B) primary fibroblast htert1787. The microinjected cells were fixed at different time points (1 h, 6 h and 16 h) and counterstained with anti-SC35 to denote speckles and anti-coilin 204/10 to denote CBs. Arrowheads indicate CBs, arrows indicate nucleoli and broken arrows indicate speckles. Bars, 5 μm.
This novel nuclear pathway for NHPX was observed not only in parental HeLa cells, but also when newly synthesized EYFP–NHPX was expressed after either microinjection or transient transfection in other transformed cell lines, including MCF7 (Fig. 3 A, arrowhead indicates CB, arrow indicates nucleolus, broken arrow indicates speckles) and HEK293 and in primary cell lines, i.e., human foreskin fibroblasts and primary human fibroblasts expressing telomerase htert1787 (Fig. 3 B; unpublished data). Some cell lines, i.e., htert1787, do not contain prominent CBs, and therefore provide an opportunity to test whether the presence of CBs is required for the transport and/or localization of NHPX in nucleoli. Microinjection of pAL107EYFP–NHPX into htert1787 cells did not induce the formation of CBs, and EYFP–NHPX still relocated in the same temporal sequence from speckles to nucleoli (Fig. 3 B, arrow indicates nucleolus, broken arrow indicates speckles). Therefore, the presence of CBs is apparently not required for the directional movement of NHPX from speckles to nucleoli.
Newly synthesized NHPX does not colocalize with U3 snoRNP in speckles
Next, we examined whether the speckle localization of newly synthesized NHPX is a consequence of a previously unknown behavior of U3 snoRNP. To test this, an expression vector, pAL118ECFP–FIB, encoding ECFP-tagged FIB, a known U3 snoRNP component, was cotransfected with pAL107EYFP–NHPX into the parental HeLa cell line and analyzed at various time points posttransfection (Fig. 4). At 1 h posttransfection, ECFP–FIB had already accumulated in nucleoli and CBs, whereas in the same cells, EYFP–NHPX accumulated in speckles and CBs, but not in nucleoli (Fig. 4 A, arrow indicates nucleolus, broken arrow indicates speckles). Several hours later, EYFP–NHPX began to accumulate in nucleoli and the signal in speckles decreased, whereas the ECFP–FIB remained specifically in nucleoli, and both proteins were detected in CBs (unpublished data). After one cell cycle, EYFP–NHPX and ECFP–FIB both quantitatively colocalized in the dense fibrillar component inside nucleoli and in CBs (Figs. 1 A and 4 B, arrowhead indicates CB, arrow indicates nucleolus, broken arrow indicates speckles; unpublished data). Therefore, the transient presence of newly expressed NHPX in nuclear speckles is not explained by its association with U3 snoRNP.
Figure 4.

Separate targeting pathways for nuclear factors. pAL107EYFP–NHPX and pAL118ECFP–FIB were cotransfected into HeLa cells and fixed after (A) 1 and (B) 24 h. (C) HeLaEYFP–NHPX and HeLaECFP–FIB were fused to form heterokaryon using PEG and were fixed at 15 min after fusion. To show the relative distribution of EYFP and ECFP components in each nucleus, the central panel shows the same two nuclei within a heterokaryon as the side panels, but in the opposite fluorescence channels. Arrowheads indicate CBs, arrows indicate nucleoli, and broken arrows indicate speckles; dotted ovals outline nuclei in the heterokaryon in the central panel. Bars, 5 μm.
The differential timing in the nucleolar entry of NHPX and FIB is confirmed by analysis of heterokaryons formed between HeLaECFP–FIB and HeLaEYFP–NHPX stable cell lines (Fig. 4 C). Formation of heterokaryons between two cell lines expressing FP-tagged proteins allows the gradual introduction of EYFP–NHPX into the HeLaECFP–FIB cells and vice versa (Sleeman et al., 2001). The advantages of using this heterokaryon approach are that it minimizes the possible effect of overexpression that can occur in both microinjection and transient transfection assays, and allows the dynamic exchange of two fluorophores to be observed under the same conditions. ECFP–FIB accumulated directly in nucleoli as soon as 15 min after fusion, whereas EYFP–NHPX accumulated instead in speckles at the same time (Fig. 4 C, arrowheads indicate CBs, arrows indicate nucleoli, broken arrows indicate speckles; unpublished data). Therefore, the NHPX protein detected in speckles is unlikely to be associated with U3 snoRNP.
The expression of exogenous NHPX raises the possibility that the target RNAs it binds to may be upregulated, and thereby results in the observed temporal sequence of nuclear localization. Therefore, we investigated the in vivo localizations of U3 and U4 RNAs in the HeLa cells that were microinjected with pAL107EYFP–NHPX using complementary 2′-O-methyl antisense RNAs (Fig. 5; for review see Lamond, 1993). The U3 in microinjected cells remained localized in the dense fibrillar component of nucleoli and CBs at both early and late time points, similar to control, nonmicroinjected cells (Fig. 5 A and B, arrowhead indicates CB, arrow indicates nucleolus, broken arrow indicates speckles; unpublished data). The U4 snRNA was detected in speckles and CBs in both the microinjected cells at different time points and in control, nonmicroinjected cells (Fig. 5, C and D). Because the localization of U3 and U4 remained unaltered, the differential localization of NHPX at different time points is likely not due to the movement or relocalization of either of these NHPX target RNAs. These data are consistent with NHPX binding U4 snRNA in speckles and U3 snoRNA in the nucleolus. However, we cannot exclude that NHPX also binds to different and/or unknown target RNAs at the different nuclear structures.
Figure 5.

Localization of endogenous snRNPs and snoRNPs. pAL107EYFP–NHPX was microinjected into HeLa cells and fixed after (A and C) 2 and (B and D) 24 h. The cells were then counterstained with antisense 2′-O-methyl RNA (A and B) U3 and (C and D) U4 and anti-SC35 to show the location of speckles. Arrowheads indicate CBs, arrows indicate nucleoli, and broken arrows indicate speckles. Bars, 5 μm.
Reciprocal movement of nuclear proteins
The NHPX pathway appears complementary to that previously described for Sm proteins (Fig. 6; Sleeman and Lamond, 1999a; Sleeman et al., 2001). By making heterokaryons between HeLaECFP–SmB and HeLaEYFP–NHPX, we observed that EYFP–NHPX entered into speckles directly, whereas ECFP–SmB accumulated specifically in CBs shortly after fusion (Fig. 6, A and B; arrowheads indicate CBs, arrows indicate nucleoli, broken arrows indicate speckles). At 2 h after fusion, ECFP–SmB remained in CBs, whereas the EYFP–NHPX signal inside nucleoli increased (Fig. 6 C, arrows indicate nucleoli). In some cells, ECFP–SmB was also detected inside nucleoli, as previously reported (Fig. 6 D; Sleeman and Lamond, 1999a; Sleeman et al., 2001). At later time points (∼36 h), the ECFP–SmB signal in speckles increased whereas the EYFP–NHPX signal in speckles decreased to a very low/undetectable level (Fig. 6 E; unpublished data). Therefore, we conclude that both nuclear pathways, though operating in different directions, function simultaneously inside a single cell nucleus. The pathways can also be observed by live cell imaging over a period of 12 h (unpublished data). This demonstrates the directed movement of proteins between separate, membrane free nuclear compartments.
Figure 6.
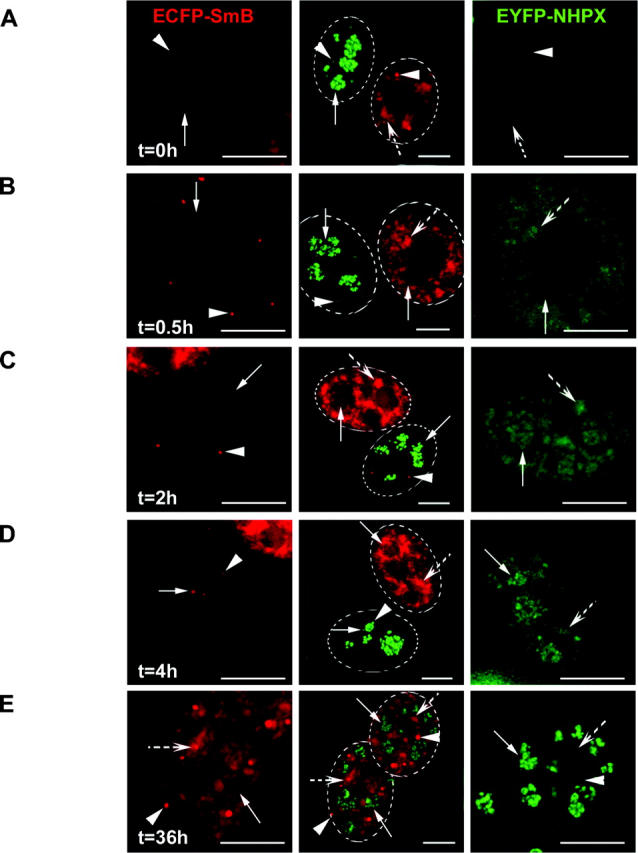
Reciprocal movement of nucleolar proteins SmB and NHPX. HeLaEYFP–NHPX and HeLaECFP–SmB were fused to form heterokaryon using PEG and were fixed at different time points: 0 h (A); 0.5 h (B); 2 h (C); 4 h (D); and 36 h (E). Panel representation as of Fig. 4 C. Arrowheads indicate CBs, arrows indicate nucleoli, and broken arrows indicate speckles; dotted ovals outline nuclei of the heterokaryon in the central panel. Bars, 5 μm.
Newly expressed NHPX localizes to speckles
We next investigated whether the directed movement of NHPX is either restricted to newly synthesized proteins, or whether it is a reversible relocation of existing proteins (Fig. 7). To test this, pAL214ECFP–NHPX was transfected into HeLa cells and left for 24 h, such that ECFP–NHPX was already accumulated in nucleoli and CBs before microinjection of pAL107EYFP–NHPX. Microinjection of pAL107EYFP–NHPX provided a pulse of newly synthesized EYFP–NHPX that accumulated in splicing speckles and CBs, whereas the previously expressed ECFP–NHPX accumulated instead in nucleoli and CBs (Fig. 7 A; arrowhead indicates CB, arrow indicates nucleolus and broken arrow indicates speckles). Gradually, EYFP–NHPX appeared in nucleoli, whereas the signal in speckles subsided. The nucleolar pattern of ECFP–NHPX remained unaltered after microinjection of pAL107EYFP–NHPX (Fig. 7 B, arrow indicates nucleolus). At 24 h postmicroinjection, EYFP–NHPX completely colocalized with the existing ECFP–NHPX (Fig. 7 C, arrowhead indicates CB, arrow indicates nucleolus). These data indicate that only newly synthesized NHPX accumulates in splicing speckles, and further argue that this association is transient. Thus, the presence of NHPX in speckles is likely not a result of protein relocation due to exogenous expression.
Figure 7.

Newly expressed NHPX localizes to speckles. HeLa cells were transfected with pAL214ECFP–NHPX for 24 h before microinjecting pAL107EYFP–NHPX and the microinjected cells were fixed after 1.5 h (A), 4 h (B), and 24 h (C), and counterstained with anti-SC35 to denote speckles. Arrowheads indicate CBs, arrows indicate nucleoli, and broken arrows indicate speckles. Bars, 5 μm.
To test further whether or not pools of NHPX in speckles and nucleoli interchange, we generated micronuclei by treating the HeLaEYFP–NHPX cells with the spindle-disrupting drug colcemid (Fig. 8). Colcemid inhibits the progress of mitosis and renders the segregation of chromosomes into many micronuclei without preventing DNA replication, mRNA transcription, splicing, and protein translation (Ferreira et al., 1997). Nucleoli are assembled only on the nucleolar organizer regions in 5 out of 23 chromosomes in human nuclei (Introduction), and therefore the colcemid treatment allows the generation of a subset of micronuclei without nucleoli. To locate those micronuclei, we screened with an antibody specific for nucleolar antigen B23. If the two pools of NHPX in splicing speckles and nucleoli freely exchange, NHPX originally from nucleoli would be expected by default to accumulate back in splicing speckles in the micronuclei lacking nucleoli. Interestingly, EYFP–NHPX does not accumulate in splicing speckles, even in those micronuclei lacking nucleoli (Fig. 8, arrows indicate speckles, arrowheads indicate NHPX localizations, inset shows a micronucleus that lacks nucleoli); instead, they are localized in spot-like structures that also contain the snoRNP protein FIB, but not the CB marker coilin (unpublished data). Microinjection of pAL107EYFP–NHPX into colcemid-treated parental HeLa cells showed the same temporal sequence of localization in speckles prior to nucleoli as seen for untreated cells (unpublished data). Therefore, this differential localization is apparently not a result of colcemid modifying the NHPX pathway and the pools of NHPX localized in splicing speckles and nucleoli appear not to interchange.
Figure 8.

NHPX does not accumulate in speckles in micronuclei lacking nucleoli. (A) HeLaEYFP–NHPX cells were fixed after treating with colcemid for 31 h and counterstained with anti-SC35 to denote speckles and anti-B23 to denote nucleoli. Arrows indicate speckles, whereas arrowheads indicate the locations of EYFP–NHPX; dotted ovals outline the micronuclei. Bar, 5 μm.
The NHPX pathway is unidirectional
The noncycling behavior of NHPX between speckles and nucleoli prompted us to further investigate the directionality of the localization pathway. We performed fluorescence loss in photobleaching (FLIP) analyses of different nuclear structures in the HeLaEYFP–NHPX cells in which one area of the cell is repeatedly bleached while collecting images of the entire cell (Fig. 9). If fluorescent molecules from other regions of the cell diffuse into the bleached area (Fig. 9, white circle indicates bleach zone), loss of fluorescence will occur from both places, indicating that the regions are connected (for review see Reits and Neefjes, 2001). First, we tested whether NHPX inside speckles is moving into the nucleolus (Fig. 9 A, a and c). The positions of speckles were defined by DsRED-U1A in live cells (Fig. 9 A, b and d). The fluorescence intensity of EYFP–NHPX in speckles outside the bleached region decreased, indicating that NHPX diffuses between these nuclear domains (Fig. 9 A, curve b). In comparison, the signals inside nucleoli only showed a minor decrease (Fig. 9 A, curve a). This is consistent with the expected movement of NHPX from speckles to nucleoli. The small change in nucleolar fluorescence may be because the directed movement of NHPX from speckles to nucleoli is slow (hours) compared with the experimental time (∼15 min), and/or because it accounts only for a small fraction of the total NHPX signal in nucleoli. However, repeated photobleaching of the nucleolus (Fig. 9 B, white circle indicates bleach zone) resulted in the immediate loss of signal in neighboring nucleoli, indicating that the nucleolar pool of NHPX can cycle between different nucleoli (Fig. 9 B, curve e). The constant level of fluorescence observed in the speckles in the same experiment further strengthens the argument in favor of a unidirectional movement of NHPX from speckles to nucleoli (Fig. 9 B, curve f).
Figure 9.
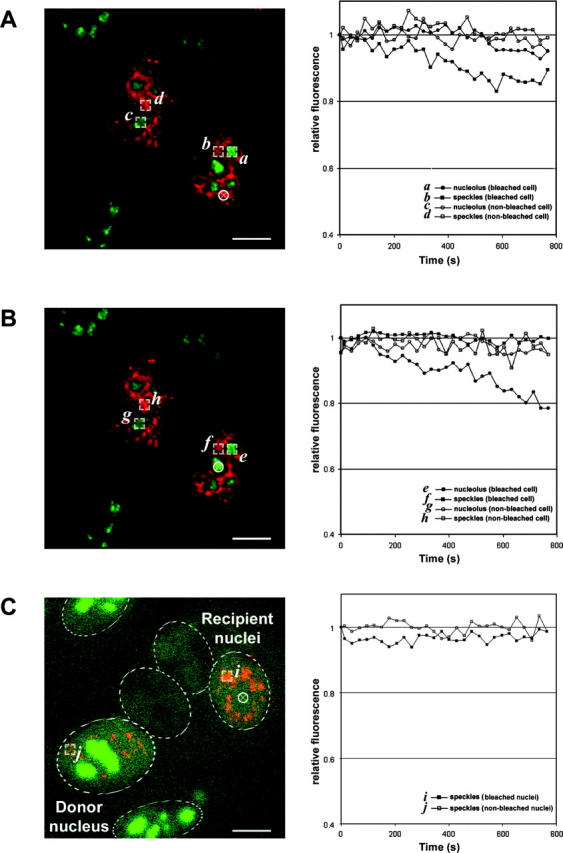
FLIP analysis of HeLaEYFP–NHPX. A region in the (A) speckles and (B) nucleolus was photobleached repetitively every 20 s and the fluorescence intensities of EYFP–NHPX were analyzed over 15 min. The positions of speckles were located in the live cells using DsRED-U1A that were transfected into the cell lines for 24 h before photobleaching and the selected region for photobleaching were highlighted by the white circle in left panel. The fluorescence intensities of EYFP–NHPX in different regions of the bleached and nonbleached cells were compared in right panel. (C) FLIP analysis of the newly imported EYFP–NHPX in speckles of the heterokaryon formed between HeLaEYFP–NHPX and HeLa cells that were both transfected with pDsRED-U1A for 24 h. The position of the nucleolus for photobleaching (left panel, white circle) in the recipient nuclei were located by both phase contrast microscopy and the absence of DsRED-U1A. The fluorescent intensities of EYFP-NHPX in speckles of bleached and nonbleached nuclei of the heterokaryon were analyzed and shown on the right panel. Dotted ovals outline nuclei in the heterokaryon. Bars, 5 μm.
The difference in fluorescence intensity between speckles and nucleoli raises the possibility that the flow from nucleoli to speckles was not observable in the HeLaEYFP–NHPX cells. To address this, we performed FLIP analysis on heterokaryons formed between HeLaEYFP–NHPX cells and the parental HeLa cells (Fig. 9 C). The locations of nucleoli in the recipient nuclei were positioned both by phase contrast microscopy and by the absence of splicing factor U1A. Shortly after the fusion, as shown previously (Figs. 4 C and 6, A–E), EYFP–NHPX first appeared in splicing speckles but was absent from nucleoli in the recipient nuclei. Repeated photobleaching inside the nucleoli did not change the fluorescence level of EYFP–NHPX inside the speckles of the recipient nuclei (Fig. 9 C, curve i, white circle indicates bleach zone). Therefore, NHPX either does not cycle from nucleoli to speckles, or else does so at a rate/level that cannot be detected in this assay. In summary, the photobleaching analyses, combined with the other data presented here, suggest a unidirectional movement of NHPX from splicing speckles to nucleoli. However, pools of EYFP–NHPX appear freely diffusible between separate components of the same nuclear structure, indicating the regulated entry of nuclear proteins into different domains inside the nucleus.
The progression of NHPX from speckles to nucleoli is dependent on Pol II, but not Pol I transcription
Next, we tested whether the progression of NHPX from speckles to nucleoli requires gene expression, including both Pol I and Pol II transcription (Fig. 10). We again employed the heterokaryon approach between HeLaECFP–FIB and HeLaEYFP–NHPX cells. At 4 h postfusion, only a small amount of EYFP–NHPX was in speckles, whereas most accumulated in nucleoli (Figs. 6 D and 10 A, arrowheads indicate CBs, arrows indicate nucleoli, broken arrows indicate speckles). The heterokaryons formed between HeLaEYFP–NHPX and HeLaECFP–FIB cells were subjected to transcription inhibitors targeted to specific polymerases. Low levels of actinomycin D cause the segregation of nucleoli and inhibit rRNA transcription, but not pre-mRNA transcription. Newly synthesized EYFP–NHPX moved to the speckles of recipient HeLaECFP–FIB nuclei, prior to accumulating in the segregated nucleoli (Fig. 10 B, arrows indicate segregated nucleoli, broken arrows indicate speckles), suggesting that pol I transcription and/or ribosome biogenesis is not a prerequisite for the NHPX pathway. Similarly, the immunosuppressant rapamycin, which inhibits transcription of a subset of ribosomal protein genes and hence ribosome assembly, gave the same results (unpublished data). However, when RNA pol II transcription was inhibited, either by α-amanitin or DRB, progression of the newly synthesized NHPX from speckles to the nucleolus was blocked (Fig. 10 C, arrows indicate nucleoli, broken arrows indicate speckles; unpublished data). This suggests that one or more factors must be continually synthesized to allow the newly imported NHPX to move from speckles to nucleoli.
Figure 10.

Transcription-dependent relocation of NHPX from speckles to nucleoli. Heterokaryon formed between HeLaEYFP–NHPX and HeLaECFP–FIB were treated with different transcription inhibitors: control (A); Pol I inhibitor Actinomycin D (0.04 μg/ml) (B); and Pol II inhibitor DRB (100 μM) (C). Panel representation as of Fig. 4 C. Arrowheads indicate CBs, arrows indicate nucleoli, and broken arrows indicate speckles; dotted ovals outline nuclei of the heterokaryon in the central panel. Bars, 5 μm.
Discussion
In this study we have identified a novel nuclear pathway that leads to the nucleolar accumulation of the NHPX protein. The pathway was detected in multiple mammalian cultured cell lines, including both primary and transformed cells. NHPX was analyzed in vivo, fused to either EYFP or ECFP fluorescent protein tags, and the resulting fusion proteins were shown to have similar localization patterns and RNA binding specificities to the endogenous NHPX. A stably transformed HeLa cell line that expressed EYFP–NHPX was established and used to demonstrate that, upon its initial entry into the nucleus, newly expressed NHPX transiently accumulates in splicing speckles prior to a later, steady-state accumulation in nucleoli. Further characterization ofHeLaEYFP–NHPX cells indicated that the NHPX protein in speckles was not associated with U3 snoRNP and required RNA pol II transcription for efficient relocation to nucleoli. Additional photobleaching experiments showed that the nucleolar pool of NHPX did not interchange with the pool in nuclear speckles, suggesting a unidirectional pathway.
Our recent proteomic analysis of nucleoli isolated from cultured HeLa cells shows that they contain >270 different proteins (Andersen et al., 2002). So far there has been no nucleolar targeting motif identified common to all of these factors and it seems likely that multiple, parallel nucleolar localization pathways can operate. Nonetheless, analyses of proteins that show a steady-state accumulation in nucleoli have shown that they usually move rapidly into the nucleolus when they enter the nucleus. This is illustrated here by the rapid nucleolar accumulation of EYFP–FIB when it is transiently expressed in vivo (Fig. 4). The finding that newly expressed NHPX accumulates in nuclear speckles transiently before accumulating specifically in the nucleolus defines a new localization pathway for nucleolar proteins. It is interesting to compare this with the recently reported pathway for nucleolar localization of snoRNAs, which showed that multiple snoRNAs accumulate in CBs prior to nucleoli upon initial entry into the nucleus (Narayanan et al., 1999a, 1999b; Verheggen et al., 2001). However, none of the snoRNAs showed a transient accumulation in speckles, consistent with our finding that the snoRNP protein FIB also does not transiently accumulate in speckles prior to nucleoli (Fig. 4). We also observed that NHPX localizes to CBs as well as speckles upon its initial entry into the nucleus, but unlike its transient association with speckles, NHPX is also detected in CBs at later stages of expression when the bulk of the protein is concentrated in nucleoli. At present, we cannot distinguish whether NHPX accumulates in CBs prior to speckles, or in both structures at the same time. However, the CB association does not appear to be obligatory for the NHPX localization pathway because a similar transient association with speckles prior to nucleolar accumulation is observed in cell lines lacking prominent CBs (Fig. 3 B). However, similar molecular events may occur either within the nucleoplasm or in CBs that are too small to detect.
It is also interesting to compare the NHPX pathway with the recently identified nuclear pathway for splicing snRNPs where FP-tagged snRNP Sm proteins accumulate in CBs, and nucleoli, prior to speckles, upon their initial nuclear entry (Sleeman and Lamond, 1999a; Sleeman et al., 2001). Therefore, this pathway appears to be complementary to that of NHPX. By analyzing heterokaryons formed between separate stable HeLa cell lines expressing EYFP–NHPX and ECFP–SmB, we could show that both these complementary pathways can operate simultaneously within the same nuclei (Fig. 6). These data confirm the specificity of the pathways and highlight the dynamic mechanisms operating to organize the distribution of proteins and RNPs in the nucleus. The results also point to the localization specificity of the separate subnuclear bodies, including nucleoli, CBs, and speckles, although they are not enclosed by membranes.
In order to answer why NHPX shows the observed transient accumulation in speckles prior to nucleoli, it may be important to consider that it is specifically newly expressed and imported NHPX protein that is detected in speckles. Several experiments showed that NHPX does not localize to speckles by default, and that the nucleolar pool of NHPX does not cycle continually to and from speckles. For example, in micronuclei that lack NOR-containing chromosomes (and hence do not have nucleoli), NHPX does not accumulate back in speckles or colocalize with splicing factors (Fig. 8). FLIP photobleaching experiments also showed that whereas nucleolar EYFP–NHPX can exchange rapidly between separate nucleoli within the same nucleus, little or no exchange occurs with the pool of NHPX in speckles (Fig. 9). This contrasts with the behavior of the nucleolar protein PSP1, which was recently shown to cycle continually between nucleoli and paraspeckles (Fox et al., 2002).
Our data strongly indicate that the association of NHPX with speckles is a temporal phenomenon linked to the entry of newly expressed NHPX into the nucleus. For example, transient expression of EYFP–NHPX in HeLa cells expressing ECFP–NHPX, which already accumulated in nucleoli, shows a transient accumulation of the EYFP–NHPX in speckles before it later colocalizes quantitatively with the existing nucleolar ECFP–NHPX (Fig. 7). Also, when the HeLaEYFP–NHPX cells undergo mitosis, EYFP–NHPX immediately relocalizes to the reforming nucleoli during telophase and does not accumulate in speckles in the postmitotic nuclei (Fig. 2). Therefore, the speckles association is not a result of nuclear import of NHPX per se, but rather relates to an effect specific for newly expressed protein. We propose that a likely explanation for this behavior of NHPX could be related to it having a function required for the assembly or maturation of some form of nuclear protein or RNP complex, prior to its subsequent stable association with U3 and/or other nucleolar snoRNPs. This could imply either that the affinity of NHPX for different target RNAs changes after it enters the nucleus for the first time or that its access to bind snoRNA targets is initially restricted.
Based upon the results of previous biochemical studies on the structure and binding specificity of NHPX, the U4 snRNA is a possible candidate target for NHPX in speckles. NHPX binds U4 snRNA in vitro via the 5′ stem loop sequence (Nottrott et al., 1999; Vidovic et al., 2000). Consistent with this idea, U4 snRNA has been localized to speckles in HeLa cells by hybridization experiments (Carmo-Fonseca et al., 1992; Fig. 5). The fact that we show here that EYFP-NHPX likely interacts in vivo with a form of U4 snRNA that is not stably associated with U6 snRNA suggests that NHPX may transiently interact in speckles with an immature form of U4 snRNP (Fig. 1 E). The observed requirement for gene expression in order for NHPX to move from speckles to nucleoli might reflect a requirement for other factors to be expressed to allow NHPX to complete its transient role in speckles (Fig. 10). Whether this is connected to U4 snRNP assembly and/or some other events remains to be established. Future studies will aim to analyze further the molecular mechanism involved in the novel nucleolar localization pathway detected for NHPX and to establish what biological role this may play.
Materials and methods
Plasmid constructs
NHPX cDNA (gi:26185777) was isolated for PCR amplification from Marathon-Ready HeLa library (CLONTECH Laboratories, Inc.) using specific primers with BglII and KpnI restriction site attached on the 5′ and 3′ primer, respectively. The amplified fragment was subsequently cloned to the BglII-KpnI fragment of EYFP–C1 and ECFP–C1 to form pAL107EYFP–NHPX and pALZ14ECFP–NHPX, respectively, and verified by DNA sequencing.
Cell culture, transfection, and establishment of stable cell line
HeLa, MCF7, HEK293, and primary fibroblast htert1787 were grown in DME supplemented with 10% FCS and 100 U/ml penicillin and streptomycin (Invitrogen). 2 μg plasmid construct per 100-mm dish was used for transfection using Effectene (QIAGEN) according to the manufacturer's instruction. EYFP–NHPX and EYFP–FIB stable cell lines were generated essentially as described in Sleeman et al. (2001). ECFP–SmB stable cell line (CFPSmBE8.8) was previously described (Sleeman et al., 2001). EGFP–H2B stable cell line was a gift from T. Kanda (The Salk Institute for Biological Studies, La Jolla, CA) (Kanda et al., 1998). Drug treatments were carried out as follows: Actinomycin D (0.04 μg/ml, 3 h; Sigma-Aldrich); DRB (100 μM, 4 h; Sigma-Aldrich); α-amanitin (40 μM, 4 h; Sigma-Aldrich); and colcemid (0.5 μg/ml, 31 h; Sigma-Aldrich).
FACS analysis
Parental HeLa and HeLaEYFP–NHPX cells were harvested by trypsinization, and fixed in 70% ethanol for 3 h at 4°C. Cells were stained with PI (25 μg/ml) containing RNase A (100 μg/ml). Fluorescence was measured using a FACScan (Becton Dickinson). Cell debris and fixation artifacts were gated out. Data analysis was done using Cell Quest software (Becton Dickinson).
Immunoprecipitation and immunoblotting
For immunoprecipitation, 100 μg protein G Sepharose (Amersham Pharmacia Biotech) was preincubated with 10 μg anti-GFP antibodies (Roche). Extracts were prepared using nuclear lysis buffer (Sleeman et al., 2001). Extracts were precleared with 100 μg protein G Sepharose and then incubated with antibody-bound protein G Sepharose for 16 h. Beads were then washed three times with the lysis buffer, and bound RNAs were released by Proteinase K (2.24 mg/ml) in extraction buffer (0.63% SDS, 26.25 mM EDTA, 26.25 mM Tris-HCl, pH 8.0, 0.28 mg/ml yeast tRNA) for 45 min at 65°C. RNAs were precipitated by adding 7 vol of EtOH/NH4OAc (86% EtOH, 0.57 M NH4OAc), and washed once with 70% EtOH. Northern hybridizations were done by standard procedures (Lamond et al., 1989).
For immunoblotting, protein samples were separated by 4–12% Bis-Tris gels (Novex), and subsequently transferred onto nitrocellulose membrane using a submarine system (Novex). After blocking with 5% milk powder in PBS+0.05% Tween-20, the membranes were incubated with either rabbit anti-NHPX R86 (1:100; Chang et al., 1999) or mouse monoclonal anti-GFP (1:1,000; Roche), and the bound antibody was then probed using anti–rabbit HRP (1:2,000; Pierce Chemical Co.) and anti–mouse HRP conjugate, respectively (1:5,000; Pierce Chemical Co.) in PBS containing 5% milk powder and 0.05% Tween-20, and detected via chemiluminescence with ECL Plus (Amersham Pharmacia Biotech).
Immunostaining and 2′-O-methyl RNA hybridization
Cells grown on coverslips were washed in PBS and fixed for 10 min in 3.7% (wt/vol) paraformaldehyde in CSK buffer (10 mM Pipes, pH 6.8, 10 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 2 mM EDTA) at RT, permeabilized with 1% Triton X-100 in PBS for 10 min at room temperature, mounted onto glass slides using VectorShield (Vector Lab), and imaged as described below. Antibodies used were anti-FIB monoclonal 72b9 (1:10; Turley et al., 1993); anti-NHPX peptide antibody R86 (1:100; Chang et al., 1999); anti-SC35 monoclonal (1:500; Sigma-Aldrich); anti-coilin 204/10 (1:300; Bohmann et al., 1995); anti-B23 (1:75; Santa Cruz Biotechnology); and TRITC-, Texas red–, and Cy5-conjugated secondary antibodies (Jackson Laboratories). Before being mounted on slides, coverslips were incubated with 1 μM DAPI (Sigma-Aldrich) for 30 s to stain DNA. Fluorescence microscopy of fixed cells was carried out using a 100× NA 1.4 Plan-Apochromat objective. Three-dimensional images and sections were recorded either on a LSM410 Confocal microscope (ZEISS) or on a Zeiss DeltaVision Restoration microscope (Applied Precision, Inc.). Images presented here are maximal intensity projections of the entire nuclear fluorescence.
For 2′-O-Methyl RNA hybridization (Carmo-Fonseca et al., 1992), cells were permeabilized with 0.5% Triton X-100 in CSK buffer containing Complete (Roche) on ice for 3 min, and were then fixed in freshly prepared 3.7% paraformaldehyde in CSK buffer for 10 min at room temperature. Cells were washed three times in PBS, one time with 6 × SSPE, and prehybridized with 6 × SSPE/5 × Denhardt's solution containing yeast tRNA (0.5 mg/ml) for 15 min. Cells were then hybridized with the same buffer with biotinylated 2′-O-methyl antisense oligonucleotide probe (2 μM) for 30 min, and then were washed three times with 6 × SSPE and rinsed with avidin wash buffer (0.03 M Hepes, pH 7.9, 0.15 M KCl, 0.05% Tween-20, 1% donkey serum) before incubating with Texas red–conjugated avidin DCS (Vector Labs) at 2 μg/ml for 30 min. They were then washed and mounted on slides for microscopic studies as above. (U3 probe 1011: 5′ – *C*CUUUCGGUGCUC*C*C – 3′; U4 probe 1012: 5′ – *C*CUGCCACUGCGCAAAGCU*C*C – 3′; * denotes biotinylated sites).
Mitotic studies of living cells
Cells were grown on 40-mm glass coverslips (Intracel) in medium containing 200 μg/ml G418. Cells were maintained at 37°C by use of a closed-system perfusion chamber (Bioptech) in DME media (20 mM Hepes, without Phenol red; Invitrogen). Images were collected using the 100× NA 1.4 Plan-Apochromat objective on the Zeiss-DeltaVision Restoration microscope. For each nucleus, 20–30 optical sections in the z-axis were recorded. The Hg lamp was attenuated with a 0.5-OD neutral density filter, and images were recorded every 3 min over a time period of 2 h (3 × 3 binning). Time-lapse images were viewed as maximal intensity projections of each time point (SoftWoRx; Applied Precision, Inc.).
Microinjection and heterokaryon formation
For microinjection, pAL107 was diluted to 20 μg/ml with injection buffer (100 mM glutamic acid, pH 7.2 [with citric acid], 140 mM KOH, 1 mM MgSO4, and 1mM DTT) prior to injection into living cells using an Eppendorf 5242 microinjector. For heterokaryon formation, two different cell lines expressing fluorescent proteins were mixed in a ratio of 1:1 in 100-mm diameter petri dishes containing coverslips and cultured until 80–90% confluent. The culture medium was drained, and 1.5 ml of 50% Polyethylene Glycol (PEG hybrimax; Sigma-Aldrich) was added. The dishes were rocked gently for 90 s and washed thoroughly by several changes of fresh culture medium (a modification of Sleeman et al. 2001).
Photobleaching analysis
Cells were grown on 42-mm glass coverslips (no. 1; Helmut Sauer) in medium containing 200 μg/ml G418. Cells were maintained at 37°C by use of a closed perfusion chamber (Bachofer) in DME media (20 mM Hepes, no Phenol red; Invitrogen). Photobleaching experiments were carried out on a Zeiss 510 confocal laser scanning microscope equipped with an argon-krypton laser (ZEISS). The 488-nm laser and a 63× plan Apolens with a 1.4 NA and a laser power of 2.5% was used for image acquisition, and 25% was used for photobleaching. An area of 16 × 16 pixels was bleached with an iteration of 250 (duration of bleach was 3 s). An image was collected after every bleaching event, with 20-s intervals between each bleaching event over a period of 15 min. To locate splicing speckles in vivo, plasmid pDsRED-U1A was transfected into the cells 24–36 h before imaging. Speckles were defined by red fluorescence, whereas nucleoli were defined by both phase contrast and the absence of U1A.
Online supplemental material
Fig. S1 (available at http://www.jcb.org/cgi/content/full/200201120/DC1) depicts immunofluorescence labeling of HeLa cells using anti-NHPX antibodies either with (A) or without (B) transient transfection with pAL107EYFP–NHPX. Panel A is identical to Fig. 1 B in the text.
Supplemental Material
Acknowledgments
We thank our colleges in the Lamond laboratory, Dr. Laura Trinkle-Mulcahy for plasmid DsRED-U1A, Dr. Judith Sleeman for the ECFP-SmB cell line, Ursula Ryder for help in RNA manipulation, and other colleagues in the laboratory and Prof. C Proud for helpful discussions. We would also like to thank Dr. T. Kanda for the EGFP-H2B cell line, Dr. K. Collins (University of California, Berkeley, Berkeley, CA) for the htert1787 primary fibroblast cell line, and Dr. B. Chen (Dana-Farber Cancer Institute, Boston, MA) for the affinity-purified anti-NHPX rabbit antiserum, Dr. B. Frenguelli for use of the Zeiss LSM 510 confocal microscope, and S. Shreeman for help in FACS analysis.
This work was supported by the Wellcome Trust. A.I. Lamond is a Wellcome Trust Principal Research Fellow. A.K.L. Leung is a Croucher Foundation Scholar (HK) and ORS awardee (UK).
Footnotes
Abbreviations used in this paper: CB, Cajal body; ECFP, enhanced cyan fluorescent protein; EYFP, enhanced yellow fluorescent protein; FIB, fibrillarin; FLIP, fluorescence loss in photobleaching; GFP, green fluorescent protein; NOR, nucleolar organizing regions; PEG, polyethene glycol; rDNA, ribosomal DNA; snoRNA, small nucleolar RNA.
References
- Andersen, J.S., C.E. Lyon, A.H. Fox, A.K. Leung, Y.W. Lam, H. Steen, M. Mann, and A.I. Lamond. 2002. Directed proteomic analysis of the human nucleolus. Curr. Biol. 12:1–11. [DOI] [PubMed] [Google Scholar]
- Blobel, G., and V.R. Potter. 1967. Ribosomes in rat liver: an estimate of the percentage of free and membrane-bound ribosomes interacting with messenger RNA in vivo. J. Mol. Biol. 28:539–542. [DOI] [PubMed] [Google Scholar]
- Bohmann, K., J.A. Ferreira, and A.I. Lamond. 1995. Mutational analysis of p80 coilin indicates a functional interaction between coiled bodies and the nucleolus. J. Cell Biol. 131:817–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudonck, K., L. Dolan, and P.J. Shaw. 1999. The movement of coiled bodies visualized in living plant cells by the green fluorescent protein. Mol. Biol. Cell. 10:2297–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmo-Fonseca, M., L. Mendes-Soares, and I. Campos. 2000. To be or not to be in the nucleolus. Nat. Cell Biol. 2:E107–E112. [DOI] [PubMed] [Google Scholar]
- Carmo-Fonseca, M., R. Pepperkok, M. Carvalho, and A. Lamond. 1992. Transcription-dependent colocalization of the U1, U2, U4/U6, and U5 snRNPs in coiled bodies. J. Cell Biol. 117:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho, T., F. Almeida, A. Calapez, M. Lafarga, M.T. Berciano, and M. Carmo-Fonseca. 1999. The spinal muscular atrophy disease gene product, SMN: A link between snRNP biogenesis and the Cajal (coiled) body. J. Cell Biol. 147:715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, M.S., H. Sasaki, M.S. Campbell, S.K. Kraeft, R. Sutherland, C.Y. Yang, Y. Liu, D. Auclair, L. Hao, and H. Sonoda, et al. 1999. HRad17 colocalizes with NHP2L1 in the nucleolus and redistributes after UV irradiation. J. Biol. Chem. 274:36544–36549. [DOI] [PubMed] [Google Scholar]
- Dundr, M., T. Misteli, and M.O. Olson. 2000. The dynamics of postmitotic reassembly of the nucleolus. J. Cell Biol. 150:433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira, J., G. Paolella, C. Ramos, and A.I. Lamond. 1997. Spatial organization of large-scale chromatin domains in the nucleus: a magnified view of single chromosome territories. J. Cell Biol. 139:1597–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox, A.H., Y.W. Lam, A.K. Leung, C.E. Lyon, J. Andersen, M. Mann, and A.I. Lamond. 2002. Paraspeckles. A novel nuclear domain. Curr. Biol. 12:13–25. [DOI] [PubMed] [Google Scholar]
- Gall, J.G. 2000. Cajal bodies: the first 100 years. Annu. Rev. Cell Dev. Biol. 16:273–300. [DOI] [PubMed] [Google Scholar]
- Gall, J.G., M. Bellini, Z. Wu, and C. Murphy. 1999. Assembly of the nuclear transcription and processing machinery: Cajal bodies (coiled bodies) and transcriptosomes. Mol. Biol. Cell. 10:4385–4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda, T., K.F. Sullivan, and G.M. Wahl. 1998. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 8:377–385. [DOI] [PubMed] [Google Scholar]
- Lafontaine, D.L., and D. Tollervey. 1998. Birth of the snoRNPs: the evolution of the modification-guide snoRNAs. Trends Biochem. Sci. 23:383–388. [DOI] [PubMed] [Google Scholar]
- Lamond, A.I. 1993. 2′-O-alkyloligoribonucleotides: probes for studying the biochemistry and cell biology of RNA processing. Biochem. Soc. Trans. 21:1–8. [DOI] [PubMed] [Google Scholar]
- Lamond, A.I., and W.C. Earnshaw. 1998. Structure and function in the nucleus. Science. 280:547–553. [DOI] [PubMed] [Google Scholar]
- Lamond, A.I., B. Sproat, U. Ryder, and J. Hamm. 1989. Probing the structure and function of U2 snRNP with antisense oligonucleotides made of 2′-OMe RNA. Cell. 58:383–390. [DOI] [PubMed] [Google Scholar]
- Lewis, J.D., and D. Tollervey. 2000. Like attracts like: getting RNA processing together in the nucleus. Science. 288:1385–1389. [DOI] [PubMed] [Google Scholar]
- Lyon, C.E., K. Bohmann, J. Sleeman, and A.I. Lamond. 1997. Inhibition of protein dephosphorylation results in the accumulation of splicing snRNPs and coiled bodies within the nucleolus. Exp. Cell Res. 230:84–93. [DOI] [PubMed] [Google Scholar]
- Malatesta, M., C. Zancanaro, T.E. Martin, E.K. Chan, F. Amalric, R. Luhrmann, P. Vogel, and S. Fakan. 1994. Is the coiled body involved in nucleolar functions? Exp. Cell Res. 211:415–419. [DOI] [PubMed] [Google Scholar]
- Matera, A.G. 1999. Nuclear bodies: multifaceted subdomains of the interchromatin space. Trends Cell Biol. 9:302–309. [DOI] [PubMed] [Google Scholar]
- Misteli, T. 1999. RNA splicing: What has phosphorylation got to do with it? Curr. Biol. 9:R198–R200. [DOI] [PubMed] [Google Scholar]
- Misteli, T. 2000. Cell biology of transcription and pre-mRNA splicing: nuclear architecture meets nuclear function. J. Cell Sci. 113:1841–1849. [DOI] [PubMed] [Google Scholar]
- Misteli, T. 2001. Protein dynamics: implications for nuclear architecture and gene expression. Science. 291:843–847. [DOI] [PubMed] [Google Scholar]
- Muratani, M., D. Gerlich, S.M. Janicki, M. Gebhard, R. Eils, and D.L. Spector. 2001. Metabolic-energy-dependent movement of PML bodies within the mammalian cell nucleus. Nat. Cell Biol. 4:106–110. [DOI] [PubMed] [Google Scholar]
- Narayanan, A., A. Lukowiak, B.E. Jady, F. Dragon, T. Kiss, R.M. Terns, and M.P. Terns. 1999. a. Nucleolar localization signals of box H/ACA small nucleolar RNAs. EMBO J. 18:5120–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan, A., W. Speckmann, R. Terns, and M.P. Terns. 1999. b. Role of the box C/D motif in localization of small nucleolar RNAs to coiled bodies and nucleoli. Mol. Biol. Cell. 10:2131–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nottrott, S., K. Hartmuth, P. Fabrizio, H. Urlaub, I. Vidovic, R. Ficner, and R. Luhrmann. 1999. Functional interaction of a novel 15.5kD [U4/U6.U5] tri-snRNP protein with the 5′ stem-loop of U4 snRNA. EMBO J. 18:6119–6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs, R.L., T.W. Stein, Jr., and E.M. Tan. 1994. Coiled bodies in the nucleolus of breast cancer cells. J. Cell Sci. 107:385–399. [DOI] [PubMed] [Google Scholar]
- Olson, M.O., M. Dundr, and A. Szebeni. 2000. The nucleolus: an old factory with unexpected capabilities. Trends Cell Biol. 10:189–196. [DOI] [PubMed] [Google Scholar]
- Pederson, T. 1998. The plurifunctional nucleolus. Nucleic Acids Res. 26:3871–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phair, R.D., and T. Misteli. 2001. Kinetic modelling approaches to in vivo imaging. Nat. Rev. Mol. Cell Biol. 2:898–907. [DOI] [PubMed] [Google Scholar]
- Platani, M., I. Goldberg, J.R. Swedlow, and A.I. Lamond. 2000. In vivo analysis of Cajal body movement, separation, and joining in live human cells. J. Cell Biol. 151:1561–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy, R., and H. Bush. 1988. Small nuclear RNAs: RNA sequences, structure, and modifications. Small Nuclear Ribonucleoprotein Particles. M.L. Birnstiel, editor. Springer-Verlag, New York. 1–37.
- Reits, E.A., and J.J. Neefjes. 2001. From fixed to FRAP: measuring protein mobility and activity in living cells. Nat. Cell Biol. 3:E145–E147. [DOI] [PubMed] [Google Scholar]
- Saito, H., T. Fujiwara, S. Shin, K. Okui, and Y. Nakamura. 1996. Cloning and mapping of a human novel cDNA (NHP2L1) that encodes a protein highly homologous to yeast nuclear protein NHP2. Cytogenet. Cell Genet. 72:191–193. [DOI] [PubMed] [Google Scholar]
- Scheer, U., and R. Hock. 1999. Structure and function of the nucleolus. Curr. Opin. Cell Biol. 11:385–390. [DOI] [PubMed] [Google Scholar]
- Schul, W., L. de Jong, and R. van Driel. 1998. Nuclear neighbours: the spatial and functional organization of genes and nuclear domains. J. Cell. Biochem. 70:159–171. [DOI] [PubMed] [Google Scholar]
- Sleeman, J.E., and A.I. Lamond. 1999. a. Newly assembled snRNPs associate with coiled bodies before speckles, suggesting a nuclear snRNP maturation pathway. Curr. Biol. 9:1065–1074. [DOI] [PubMed] [Google Scholar]
- Sleeman, J.E., and A.I. Lamond. 1999. b. Nuclear organization of pre-mRNA splicing factors. Curr. Opin. Cell Biol. 11:372–377. [DOI] [PubMed] [Google Scholar]
- Sleeman, J.E, C.E. Lyon, M. Platani, J.P. Kreivi, and A.I. Lamond. 1998. Dynamic interactions between splicing snRNPs, coiled bodies and nucleoli revealed using snRNP protein fusions to the green fluorescent protein. Exp. Cell Res. 243:290–304. [DOI] [PubMed] [Google Scholar]
- Sleeman, J.E., P. Ajuh, and A.I. Lamond. 2001. snRNP protein expression enhances the formation of Cajal bodies containing p80-coilin and SMN. J. Cell Sci. 114:4407–4419. [DOI] [PubMed] [Google Scholar]
- Snaar, S., K. Wiesmeijer, A.G. Jochemsen, H.J. Tanke, and R.W. Dirks. 2000. Mutational analysis of fibrillarin and its mobility in living human cells. J. Cell Biol. 151:653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector, D.L. 2001. Nuclear domains. J. Cell Sci. 114:2891–2893. [DOI] [PubMed] [Google Scholar]
- Staley, J.P., and C. Guthrie. 1998. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell. 92:315–326. [DOI] [PubMed] [Google Scholar]
- Swedlow, J.R., and A.I. Lamond. 2001. Nuclear dynamics: where genes are and how they got there. Genome Biol. 2:0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turley, S.J., E.M. Tan, and K.M. Pollard. 1993. Molecular cloning and sequence analysis of U3 snoRNA-associated mouse fibrillarin. Biochim. Biophys. Acta. 1216:119–122. [DOI] [PubMed] [Google Scholar]
- Verheggen, C., J. Mouaikel, M. Thiry, J.M. Blanchard, D. Tollervey, R. Bordonne, D.L. Lafontaine, and E. Bertrand. 2001. Box C/D small nucleolar RNA trafficking involves small nucleolar RNP proteins, nucleolar factors and a novel nuclear domain. EMBO J. 20:5480–5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidovic, I., S. Nottrott, K. Hartmuth, R. Luhrmann, and R. Ficner. 2000. Crystal structure of the spliceosomal 15.5 kD protein bound to a U4 snRNA fragment. Mol. Cell. 6:1331–1342. [DOI] [PubMed] [Google Scholar]
- Visintin, R., and A. Amon. 2000. The nucleolus: the magician's hat for cell cycle tricks. Curr. Opin. Cell Biol. 12:372–377. [DOI] [PubMed] [Google Scholar]
- Warner, J.R. 2001. Nascent ribosomes. Cell. 107:133–136. [DOI] [PubMed] [Google Scholar]
- Watkins, N.J., V. Segault, B. Charpentier, S. Nottrott, P. Fabrizio, A. Bachi, M. Wilm, M. Rosbash, C. Branlant, and R. Luhrmann. 2000. A common core RNP structure shared between the small nucleoar box C/D RNPs and the spliceosomal U4 snRNP. Cell. 103:457–466. [DOI] [PubMed] [Google Scholar]
- Weinstein, L.B., and J.A. Steitz. 1999. Guided tours: from precursor snoRNA to functional snoRNP. Curr. Opin. Cell Biol. 11:378–384. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}