

Abstract
Over the past three decades the protein folding field has undergone monumental changes. Originally a purely academic question, how a protein folds has now become vital in understanding diseases and our abilities to rationally manipulate cellular life by engineering protein folding pathways. We review and contrast past and recent developments in the protein folding field. Specifically, we discuss the progress in our understanding of protein folding thermodynamics and kinetics, the properties of evasive intermediates, and unfolded states. We also discuss how some abnormalities in protein folding lead to protein aggregation and human diseases.
I. Introduction
Protein folding refers to the process by which a protein assumes its characteristic structure, known as the native state. The most fundamental question of how an amino acid sequence specifies both a native structure and the pathway to attain that state has defined the protein folding field. Over more than four decades the protein folding field has evolved (Fig. 1), as have the questions pertaining to it. This evolution can be divided into two predominant phases. During the first phase, research was focused on understanding the mechanisms of protein folding and uncovering the fundamental principles that govern the folding transition. While the first phase provided general answers to the protein folding question, new and no less ambitious questions arose: what are the mechanisms of protein folding in a context, such as under the influence of other biological molecules in the cellular environment? This next set of questions defined the second phase in protein folding field evolution.
Figure 1. Growth of the Protein Folding Field.
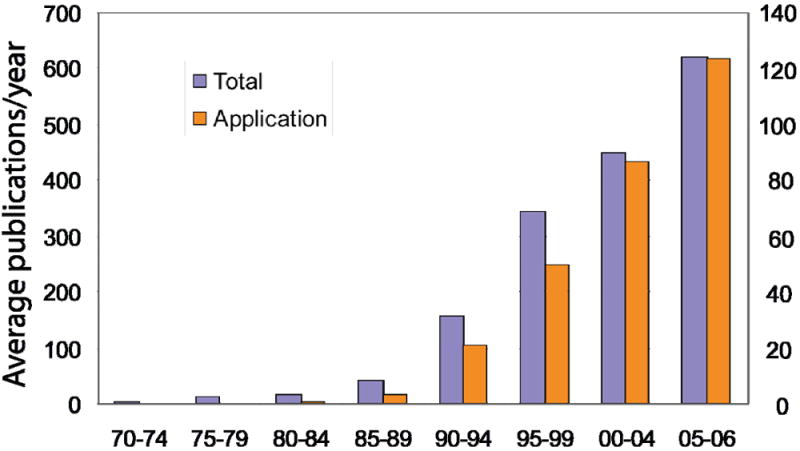
The average number of publications per year in protein folding field (left y axis) and the average number of publications per year that are dedicated to application (right y axis) were plotted every five years between 1970 and 2004, and 2005-2006. The first dataset was generated by searching articles in PubMed that contain the keyword ‘protein folding’ or ‘protein unfolding’ in either title or abstract. The second dataset was extracted from the previous dataset by searching with the following additional keywords: ‘engineering’, ‘design’, ‘misfolding’, ‘aggregation’, ’amyloid’ and ‘amyloid disease’.
The first phase is akin to a romantic stage of research, where the final goal of studies may not be directly applicable to a broader understanding, or exploitable in a relevant science. The final goal is to determine the basic principles that relate protein sequence and structure. The second phase is a more pragmatic stage of research, where the applications drive research in the field and the rational manipulation of derived knowledge allows engineering of tools for advancement of a relevant science. For example, understanding the functional intermediates that accompany the transition of a protein en route to its native state may allow rational manipulation of protein structure via protein design. This example not only relates protein sequence, structure and function, but also demonstrates the engineering aspect of the modern protein folding field.
Next, we survey the questions of the modern protein folding field. We attempt to describe a number of directions where understanding protein folding offers insights into more complex questions in molecular and cellular biology as well as medicine. We also describe new approaches and tools to address complexities associated with these new areas of research. We review studies of protein stability, folding kinetics, intermediate and unfolded states, and protein self-association and aggregation.
II. Studying protein folding
The protein folding field has witnessed significant changes and progress since the original work of Anfinsen showing that proteins can fold spontaneously [1, 2]. Early in vitro studies showed that the folding process typically occurs on a milliseconds-to-seconds time scale, much faster than the rate estimated assuming that folding proceeds by a random search of all possible conformations. Based upon this observation, Levinthal then proposed that a random conformation search does not occur in folding and that proteins fold by specific ‘folding pathways’ [3]. On these pathways, the protein molecule passes through well-defined partially-structured intermediate states. Based on this view, numerous experiments and simulations were conducted to test the existence of transient folding intermediates [4, 5]. It was expected that the determination of the structures and population of folding intermediates could help elucidate protein folding mechanisms. Earlier experimental studies on protein folding kinetics monitored the structural changes through relaxation of the protein’s spectroscopic properties after exposing the protein to folding or unfolding conditions. The data obtained from such experiments exhibit single- or multiple-exponential time-decay: a single-exponential decay is interpreted as a signature of two-state kinetics between the native state and the denatured state, whereas models involving more than two states are required to explain multiple-exponential decay data. These experiments generally probe only the average behavior of proteins, and they are not able to provide information about the folding/unfolding process in atomic details.
The discovery of a class of simple, single-domain proteins which fold via two-state kinetics without any detectable intermediates in the early 1990s [6, 7], the development of experimental techniques with improved spatial/temporal resolution[8-13], and the application of computer simulations using simplified lattice and off-lattice models[14, 15] greatly enhanced our understanding of various aspects of the protein folding problem. Based on the nucleation theory [16-18], one of the early proposed mechanisms for protein folding, the nucleation-condensation model was formulated[19-21]. In this scenario, a small number of residues (folding nucleus) need to form their native contacts in order for the folding reaction to proceed fast into the native state. The cooperativity of the protein folding process is analogous to that exhibited in first-order phase transitions, which proceed via a nucleation and growth mechanism [22]. Because of these similarities, terminology used in studies of phase transitions, such as energy landscapes and nucleation, was introduced into the discussion of protein folding. The concepts of the nucleation and the free energy landscape have promoted much of the recent progress in understanding the process of protein folding. Proteins are generally thought to have evolved to exhibit globally funneled energy landscapes [23-25] which allow proteins to fold to their native states through a stochastic process in which the free energy decreases spontaneously. The unfolded state, transition state, native state and possible intermediates correspond to local minima or saddle points in the free-energy landscape.
Advances in experimental techniques such as protein engineering, nuclear magnetic resonance (NMR), mass spectrometry, hydrogen exchange, fluorescence resonance energy transfer (FRET), atomic force microscopy (AFM), have made it possible to obtain detailed information about the different conformations occurring in the folding process[26, 27]. At the same time, computational methods have been developed to better interpret experimental data by using simulations to obtain structural information about the states which are populated during the folding process. In Table 1, we list several advances in experimental and computational methodologies used for investigating the folding of model proteins.
Table 1. Protein folding in select model systems.
A sampling of experimental and theoretical approaches for probing protein folding for three model protein systems: Chymotrypsin Inhibitor 2 (CI2), Lysozyme, and src/Fyn SH3 Domain.
| Theme | Relevant experiments and theoretical models | Contribution to Protein Folding Research |
|---|---|---|
| Mechanism of folding/unfolding | Equillibrium denaturation by guanidinium chloride[6] | CI2 unfolding and refolding follows two-state transition.
Lysozyme folding intermediates obstruct formation of transition state, but does not change the folding rate [248]. |
| Protein engineering, Φ-value analysis [249]. | CI2 folding supports nucleation-condensation model [19, 20].
CI2 transition state has secondary, tertiary structure elements [121]. Native topology and hydrogen bonds mediates SH3 folding [250, 251]. |
|
| Quantitative Φ–value analysis using MD simulations [122]. | Postulated structure of CI2 unfolding transition state. | |
| Free-energy landscape: protein folding funnel [23]. | Statistical description of protein folding process.
Role of water in facilitating protein folding [155]. |
|
| Transition State Structure | MD simulations of CI2 transition state [252]. | Folding of CI2 is cooperative. |
| Multiscalar modeling and DMD simulations. | Identification of src SH3 residues critical to folding nucleus [47]. | |
| Monte Carlo simulations on lattice models [253] | pfold as a reaction coordinate for protein folding. | |
| Folding Kinetics | Equillibrium and stopped-flow fluorescence [254]. | Src SH3 unfolding is cooperative; its denatured state may be compact under native conditions [254]. |
| Relaxation dispersion NMR of Fyn SH3 [174]. | Identified and characterized low-population folding intermediates | |
| Graph representation of CI2 and src-SH3 conformation [255]. | Protein network contact topology determines proteins’ ability to fold. | |
| Φ-value analysis of SH3 [256]. | Hydrophobic core composition is another determinant of protein folding rate | |
| Multiple MD simulations of CI2 unfolding [257]. | Preferred pathway for protein folding on a funnel-like average energy surface. | |
| Unfolded proteins structure | Unfolded proteins’ NMR [195]. Size of measurements of various unfolded proteins [192]. | Denatured proteins have a strong local conformational bias towards native state. On the other hand, the scaling of protein sizes in the unfolded state suggests a random-coil like conformations. |
| Computational models of denatured proteins [192, 203]. | Unfolded states features local native-like structures (short-rage correlations), but the correlations decays quickly. Protein behaves as a “renormalized” random coil after grouping local structures together. |
All-atom protein models with explicit or implicit solvents were developed to study the folding thermodynamics and the unfolding dynamics of specific proteins. Technological advances in computation allowed folding simulations of small proteins and peptides at atomic detail [28-30]. However, due to the complexity and vast dimensionality of protein conformational space, all-atom MD simulations have severe limitations on the time and length scales that can be studied. Novel simulation protocols have been proposed to improve conformational sampling efficiency, including biased sampling of the free-energy surface and non-equilibrium unfolding simulations [24]. In addition, world-wide parallel computing (e.g. Folding@Home [31]) and generalized ensemble sampling techniques that involve parallel simulations of molecular systems coupled with a Monte Carlo (MC) protocol [32, 33] have been successfully applied to protein folding [25, 34-36].
Multi-scale modeling approaches have also been used to combine efficient conformational sampling of coarse-grained models and accuracy of all-atom models to study protein folding pathways. In this approach, iterative simulations and inter-conversion between high and low-resolution protein models are performed. Feig et al. developed a multi-scale modeling tool set, MMTSB [37], which integrates a simplified protein model with the MC simulation engine, MONSSTER [38], and the all-atom MD packages AMBER [39] or CHARMM[40]. Using a combination of CHARMM and discrete molecular dynamics (DMD) [41-46], Ding et al. reconstructed the transition state ensemble of the src-SH3 protein domain through multi-scale simulations [47]. The protein folding studies can also be facilitated by sampling protein conformations near the native state. Several native-state sampling algorithms [48, 49] have been successfully utilized to study plasticity [50], cooperative interactions [51], and allostery [52] in proteins. Considering native-state ensemble naturally takes into account protein flexibility, which is shown to be crucial in structure based drug designs.
During the last five years, several tools for performing web-based analyses of protein folding dynamics have been developed. The Fold-Rate server (http://psfs.cbrc.jp/fold-rate/) [53] predicts rates of protein folding using the amino-acid sequence. The Parasol folding server (http://parasol.tamu.edu/groups/amatogroup/foldingserver) [54] predicts protein folding pathways using “probabilistic roadmaps”-based motion planning techniques. The iFold server (http://ifold.dokhlab.org) [55] allows discrete molecular dynamics (DMD) simulations of protein dynamics using simplified two-bead per residue protein models. These tools facilitate the second phase of protein folding research, whereby targeted simulations may be performed for probing the dynamics of protein folding and unfolding under controlled conditions.
DMD approaches [43-46] with simplified structural models of proteins have been extensively used for investigating general principles of protein folding and unfolding [56-60]. Dokholyan et al.[61] have highlighted the differences between molecular dynamics and DMD approaches. As opposed to the traditional MD approach of iteratively solving Newtonian equations of motion for evolving protein folding trajectory, DMD simulations solve ballistic equations of motion with square-well approximation to inter-particle interaction potentials. DMD algorithm gains efficiency over traditional MD simulations in multiple ways. First, due to ballistic modeling of particle dynamics, a larger time step can be used in DMD simulations on average, which corresponds to the time interval between fastest ballistic interactions; secondly a faster inter-particle collision detection and velocity updating algorithm is used, since only the coordinates of colliding atoms need to be updated at each collision. Additionally, faster simulation speeds are attainable with the DMD approach through simplification of protein models. Overall, an increase in simulation speed of 5–10 orders of magnitude is attainable using DMD [62]. Jang et al. [56] used DMD and simplified protein models with Gō interactions [63] to probe protein folding kinetics. Protein folding kinetics studies using DMD simulations are reviewed in [42]. Recently, DMD simulations we used in uncovering the structural mechanisms of protein aggregation [64-66]. Among the fundamental challenges in studying protein folding using computer simulations are the time-scales and length-scales that can be investigated. DMD simulations have been shown to be useful for investigating long-timescale folding dynamics of complex biological systems such as poly-alanine aggregation [66, 67] and the nucleosome core particle [68].
In addition to the extensive in silico and in vitro studies of protein folding, significant progress has been made in understanding protein folding in vivo. There are two major differences between protein folding in vivo and in vitro. First, protein folding in vivo is usually assisted by molecular machinery, such as chaperones (in an ATP-dependent manner), and often involves small molecule cofactors. Molecular chaperones such as the heat shock protein Hsp70 and chaperonin proteins facilitate protein folding, in part, by isolating the proteins from bulk cytosol[69, 70]. Hartl and Horwich pioneered the research of chaperone-mediated protein folding[71], highlighting the differences between in vivo and in vitro folding mechanisms[69]. The mechanism of chaperonin GroEL mediated folding, including in vivo folding intermediates, has been extensively studied by Horwich and Gierasch [72, 73]. Work by Landry et al. [74] showed that chaperon binding promotes α-helix formation in partially folded polypeptide chains. Horowitz et al. have investigated the role of chaperonin Cpn60-mediated hydrophobic exposure in protein folding[75, 76]. Nearly one third of all proteins in living cells are coordinated to small molecule cofactors. The pioneering work of Wittung-Stafshede and coworkers on the role of cofactors in in vivo protein folding[77, 78] demonstrated that bound metals stabilize the native fold, suggesting cofactor binding to unfolded polypeptides dramatically accelerates folding timescales[77].
A second notable difference between in vivo and in vitro protein folding is the fact that the concentrations of macromolecular solutes in cells can reach hundreds of grams per liter in cells[79], but most in vitro studies are performed in buffered solution with <1% of the cellular macromolecule concentration. The crowding environment in vivo can have a significant impact on protein stability and the native structure by changing the energy landscape of protein folding[80, 81]. Dedmon et al.[82] showed that FlgM, a 97-residue protein from Salmonella typhimurium is unstructured in dilute solution, but in E.coli cells its C-terminal half is structured. McPhie et al.[83] found that a molten globular state of apomyoglobin at low pH is stabilized by high concentration of the inert polymer, dextran, compared to the unfolded state. Moreover, it was found that aggregate formation from human apolipoprotein C-II is significantly accelerated by the addition of dextran[84], suggesting a direct effect of molecular crowding on protein aggregation.
Over the past three decades, novel experimental techniques and simulations have yielded many significant insights in protein folding research. Important advances have been made, especially toward the understanding of folding and unfolding mechanisms, the structure of folding transition states, folding kinetics, the nature of folding pathways, and the structure of unfolded proteins and protein folding in vivo. Theoretical approaches to study protein folding have largely complemented experiments by providing experimentally testable hypotheses. In recent years, the rational manipulation of folding pathways and the association between protein folding and disease have marked a more applied phase of protein folding research.
III. Protein stability
The thermodynamic stability of a protein is measured by the free energy difference between the folded state and the unfolded state (ΔG = Gunfold - Gfold). It determines the fraction of folded proteins, thereby having a profound effect on protein function. Natural proteins are only marginally stable[85]. The energetic contributions from the favorable folding forces such as hydrophobic packing, hydrogen bonding and electrostatic interactions are nearly offset by the entropic penalization of folding. As a result, the measured the ΔG values of most proteins fall in the range of 3-15 kcal/mol [86]. Due to this subtle balance between various physical interactions, a single mutation may shift the balance and significantly affect the stability of the whole protein. The accurate estimation of protein stability changes induced by mutations, measured as ΔΔG = ΔGWT − ΔGMut, still remains a significant challenge for computational biologists.
Experimentally, ΔG values can be obtained from denaturing experiments [6, 87-90] where the protein unfolds by increasing temperature or by adding denaturing agents such as urea and guanidinium HCl (GdHCl). Theoretically, given the interactions, free energy can be obtained from statistical mechanics using the partition function, Z, as G = -RT lnZ. Analytical calculations of partition functions require integration over all degrees of freedom in the protein’s conformational space, which is impossible in practice, except for simple models. Advances in computational biology have made possible the direct calculation of ΔΔG by MD or Monte Carlo simulations[28, 91-94], a comprehensive review of which can be found in Ref. [95]. Using MD simulations, a ΔΔG value has been calculated for T157V mutant of T4 lysozyme which is in close agreement with experimental measurements [92].
The computational cost of direct ΔΔG estimation is still too high, however, preventing it from being applied to a large number of mutations for protein engineering. More heuristic approaches have been adapted which try to describe the free energy using empirical or effective functions taking advantage of the vast amount of known protein structures and stability measurements. Such simplifications significantly decrease the computational overhead and allow ΔΔG calculations of large numbers of mutants that can be compared with experimental results. In recent years, various methods have been proposed for large-scale ΔΔG predictions with reasonable prediction accuracies (which are commonly assessed by the linear correlation coefficient between the predicted and measured ΔΔG values). A comparison between these methods is listed in Table 2.
Table 2. Comparison of different ΔΔG calculation approaches.
The approaches are evaluated base on the calculation speed, dependence on parameter training using existing stability data, side-chain and backbone flexibility modeling capability, and transferability to study other protein properties.
| Methods | Speed | Parameter Training | Side-chain Flexibility | Backbone Flexibility | Transferability |
|---|---|---|---|---|---|
| Ab initio Simulation | Slow | No | Yes | Yes | Yes |
| Statistical Potential | Fast | No | No | No | Yes |
| Empirical Function | Fast | Yes | No | No | Yes |
| Machine Learning | Fast | Yes | No | No | No |
One approach utilizes statistical potentials that are developed using information from known protein structures, where the frequency distribution of amino acids conformations (such as pairwise distances and torsion angles) is used to extract effective potentials for free energy evaluations[96]. Gilis and Rooman first applied database-derived backbone dihedral potentials to study the change of thermodynamic stability upon point mutations[97-99]. They found that torsion-angle potentials predict ΔΔG accurately for mutations of solvent-exposed residues and that distance-dependent statistical potentials are more accurate for predicting the ΔΔG of buried residues. They obtained correlation coefficients of 0.55 to 0.87 for a dataset of 238 mutations. Zhou et al. [100] developed a knowledge-based potential using the distance-scaled finite ideal-gas reference state (DFIRE) approach and calculated ΔΔG for 895 mutants, which have a correlation of 0.67 with experimental measurements. Similarly, statistical potentials utilizing side-chain rotamer libraries[101], direction- and distance-dependent distributions[102], and four-body interactions[103] were also adapted for ΔΔG predictions and significant agreement with experimental measurements was achieved.
Another approach for large-scale ΔΔG predictions uses empirical functions to describe free energy changes induced by mutations and trains the parameters to recapitulate the experimental results. Guerois et al. [104] developed the FOLD-X energy function to study the stabilities of 1088 mutants. They used a comprehensive set of parameters to describe the van der Waals, solvation, hydrogen-bonding, electrostatic and entropic contribution to the protein stability, and obtained a correlation of 0.64 for the blind test set after training their parameters on 339 mutants[104]. Khatun et al. [105] utilized contact potentials to predict ΔΔG of three sets of 303, 658 and 1356 mutants and their prediction correlations varied between 0.45 to 0.78. Bordner and Abagyan [106] used a combination of physical energy terms, statistical energy terms and a structural descriptor with weight factors scaled to experimental data for ΔΔG predictions, and found a correlation of 0.59 on 908 test mutants. Saraboji et al. classified the available thermal denaturing data on mutations according to substitution types, secondary structures and the area of solvent accessibilities, and used the average value from each category for the prediction and obtained a correlation of 0.64 [107].
Taking advantage of the vast amount of experimental ΔΔG data now available, machine-learning techniques have been introduced for ΔΔG estimation. Capriotti et al. [108, 109] trained a support vector machine using temperature, pH, mutations, nearby residues and relative solvent accessible area as input vectors. The support vector machine, when applied to a test set, gives a prediction correlation of 0.71. Cheng et al. [110] improved the support vector machine model to directly include the sequence information and obtained higher correlation prediction accuracy.
There are two significant drawbacks with training-based studies. First, improvement of the prediction accuracy relies on the available experimental stability data for parameter trainings. It is questionable whether parameters obtained from these trainings are transferable to other studies [105] since the experimentally-available mutation data may be biased (e.g., towards substitutions of large residues for small ones). Second, some mutations introduce strains in the protein backbone. To properly estimate the ΔΔG values, it is necessary to estimate the structural rearrangement that a protein undergoes to release the strain. To our knowledge, protein dynamics and flexibility have not been explicitly modeled in previous methods. Ignoring protein flexibility prohibits the application of current prediction methods to a wide range of mutations [100, 104].
To address both these caveats, Yin et al. [111] developed a novel method, Eris (http://eris.dokhlab.org), for accurate and rapid evaluation of the ΔΔG values using the recently-developed Medusa redesign suite[112]. Eris features an all-atom force field developed from x-ray crystal structures, a fast side-chain packing algorithm, and a backbone relaxation method. The ΔΔG values of 595 mutants from five proteins were calculated and compared with the experimental data from the Protherm database and other sources [104, 105, 113, 114]. Significant correlations of ≈0.75 were found between predicted and experimental ΔΔG values. Eris identifies and efficiently relaxes strains in the backbone, especially when clashes and backbone strains are introduced by a small-to-large amino acid substitution. Interestingly, when high-resolution structures are not available, Eris allows refinement of the backbone structure, which yields better prediction accuracy. Compared with other ΔΔG prediction methods, the Eris method is a unique approach that combines physical energies with efficient atomic modeling, resulting in fast and unbiased ΔΔG predictions.
Despite remarkable progress in the last several decades, protein stability estimation methods are still imperfect. Several obstacles must be cleared in order to achieve a more reliable method for stability estimation. Due to the complexity of sampling multi-dimensional space, the entropic free energy of a protein is difficult to evaluate and is thereby often ignored or only roughly counted in current stability estimation methods. Furthermore, since protein stability is determined by the free energy difference between the folded and unfolded states, it is crucial to model the unfolded state and its effect on protein stability (cf. section on unfolded protein states). Most importantly, the prediction of large conformational changes upon mutations remains a major challenge in protein-stability estimation.
IV. Protein folding kinetics
To understand protein folding, important details must be taken into consideration as the protein proceeds from unfolded to native state. How fast does a protein fold? Are there multiple pathways accessed en route to its folded state? What are the structural characteristics that determine the path and rate of protein folding? In addition, as we realized in recent years, there is a broad class of human diseases that arises from failure of some proteins to adopt and remain in their native states, partly due to abnormal folding kinetics [115]. For example, a major cystic fibrosis-related deletion mutation of a single amino acid in the cystic fibrosis transmembrane conductance regulator (CFTR) affects its folding kinetics, but has minimal effect on its stability and structure [116]. Also, recent evidence [117] suggests that altered dynamics of superoxide dismutase (SOD1) mutants, which are non-destabilizing or even stabilizing, possibly cause the aggregation of mutants in familial amyloid sclerosis (FALS). Elucidation of protein folding kinetics has never been more important, particularly in the context of finding molecular mechanisms of protein misfolding diseases.
Probing the structural properties of intermediate states and determining folding pathways have been major experimental challenges. Nonetheless, there have been significant advances in experimental techniques. Experimental methods with different temporal resolutions allow a more detailed dissection of the folding process (Table 3). Other methods allow for investigation of protein structure (e.g. NMR, ultraviolet/visual light CD, site-directed mutagenesis, Φ-value analysis, isotope labeling) and global properties (e.g., mass spectroscopy, quasi elastic light scattering, ultracentrifugation) [118].
Table 3.
Methods for observing fast folding events [Adapted from [118], p. 77]
| Technique | Approximate timescale probed |
|---|---|
| LASER flash photolysis | 100 fs -1 ms |
| Electron-transfer-induced refolding | 1 μs – 1 ms |
| Acoustic relaxation | 1 ns – 1 ms |
| Dielectric relaxation | 1 ns – 1 s |
| LASER T-jump | 1 ns – 100 ms |
| Electrical discharge T-jump | 100 ns – 10 s |
| Mixing | 10 μs - ∞ |
| Pressure-jump | 60 μs – 1 s |
| NMR line broadening | 100 μs – 100 ms |
As experimental data on folding rates of various proteins accumulated over the years, people sought the determinants of folding rates. Plaxco and co-workers [119] showed that there is a high correlation between the folding rate and the structural properties of proteins, as defined by contact order CO, , where L is the sequence length, N is the total number of inter-residue atomic contacts within a cutoff distance, ΔLij is the sequence separation of contacting residues i and j. Interestingly, assessment of other protein folding rate determinants such as local and long-range contacts were found to perform equally well as the contact order[53]. Also, contact order is a geometric property which does not take into account the distribution and strength of the interactions on the rate [120]. Fersht and coworkers found that specific interactions in the folding nucleus are equally important determinants of folding rates. For example, mutations in the folding nucleus of CI2 did not change its contact order, but result in a three order magnitude increase in the folding rate [90, 121, 122]. Sequence-based prediction of folding rates has also been proposed and was found to be of comparable performance to that of contact order [123, 124]. Thus, there is still a debate as to whether structure-based or sequence-based prediction is a more reliable predictor of folding rates [123, 124].
Extensive studies have likewise been made on the prediction of folding rates using molecular dynamics simulations. The primary bottleneck in this approach was sampling the time scales where the folding transitions are observable. Thus, early studies pioneered by Caflisch et al. [125-127] employed continuum solvent with low viscosity to observe multiple folding transitions. However, there is a nonlinear relationship between the folding time and viscosity [128], hence, the precise effect of very low viscosity on the protein folding kinetics of various systems remains unclear. To circumvent this problem, Pande et al. used “coupled ensemble dynamics” to simulate the folding of a β-hairpin from protein G using continuum solvent model [129] and united atom force-field [130] with water-like viscosity. In this and other subsequent simulations of other β-hairpins, the calculated folding rate was in close agreement with experimental measurements. A remarkable simulation in this class is a 1 μs folding simulation on the villin headpiece by Duan and Kollman [28]. Folding rate predictions of this type have been limited to small two-state proteins. As protein size increases, it is difficult to computationally study folding kinetics. Rate predictions have been likewise performed using molecular dynamics simulations using explicit water models such as the TIP3P [131] and SPC [132] to gain additional insight into folding kinetics. Examples of MD simulations using explicit solvent which yielded experimentally consistent rates were performed by Pande et al., who observed helix-coil transitions [133] and protein folding [34]. However, a potential drawback in the use of water models is that they are parameterized to a single temperature (~298 K), and thus may bias the dynamics in non-native temperature simulations. Overall, theoretical rate determinations and their subsequent comparisons with experiments provide a test of our understanding of protein folding kinetics.
Structural investigation of the transition state ensemble (TSE) is extremely challenging experimentally, since the TSE is an unstable state whose experimental detection is very difficult. Computationally, transition state conformations may be identified using unfolding simulations (cf. section on unfolded protein states), projection into one or two reaction coordinates, validation of putative transition states through calculation of the probability to fold (Pfold), and path sampling. The intuitive appeal of low-dimensional energy landscapes in explaining simple chemical reactions inspired people to develop a similar formulation for protein folding. However, unlike simple molecules, the high degrees of freedom of a protein make the analysis formidable. Thus, several groups proposed dimensional reduction by projecting the multi-dimensional energy landscape into few relevant coordinates. The proposed reaction coordinates could be the volume of the molecule [134], the fraction of amino acids in their native conformation [135], the number of contacts between amino acids [136, 137], and the fraction of native contacts in a conformation [136, 137]. Some others directly tackled the transition state by developing rigorous path sampling techniques [138]. They constructed a large ensemble of transition paths, and through statistical analysis, they determined conformations whose Pfold=0.5. Although this method is computationally expensive, it is advantageous since there is no presupposed reaction coordinate.
Characteristics of the transition state ensemble have mainly been investigated by Φ-analysis[139], which involves measuring the folding kinetics and equilibrium thermodynamics of mutants containing amino acid substitutions throughout a protein. This method provides means to identify interactions mediated by specific amino acid side chains that stabilize the folding transition state[140]. Φ-analysis has been applied to a large number of proteins (such as BPTI, myoglobin, protein A, ubiquitin, SH3 domain, and the WW domain) and, recently, even to amyloidogenesis[141, 142]. However, despite the prevalence of the methodology, there is debate regarding the validity and conventional interpretation of Φ-analysis, especially when the ΔΔG between wild type and mutant is less than 1.7 kcal/mol [2, 42, 143, 144]. However, Fersht and coworkers argued that reliable ϕ-values can be derived from mutations in suitable proteins with 0.6 < ΔΔG < 1.7 kcal/mol[145]. Plaxco and coworkers disproved the assumed independence of the changes in free energy of transition and folded states when calculating error estimates in Φ-values [143]. They proposed a new method of error estimation that accounts for the interdependence of changes in free energy of transition and folded states.
Using simplified protein models and rapid sampling DMD, Dokholyan et al. directly observed and characterized the transition state ensemble of Src homology 3 (SH3) (Fig. 2) [47, 59, 146, 147]. To probe the contribution of each amino acid residue to the transition state ensemble, they calculated the Φ-values, and found high correlation between simulation and experimental Φ-values. Moreover, they also predicted that the two most kinetically important residues in folding are L24 and G64. Both L24 and G64 are experimentally-verified to be important kinetically [148].
Figure 2. Fast and slow folding pathways.
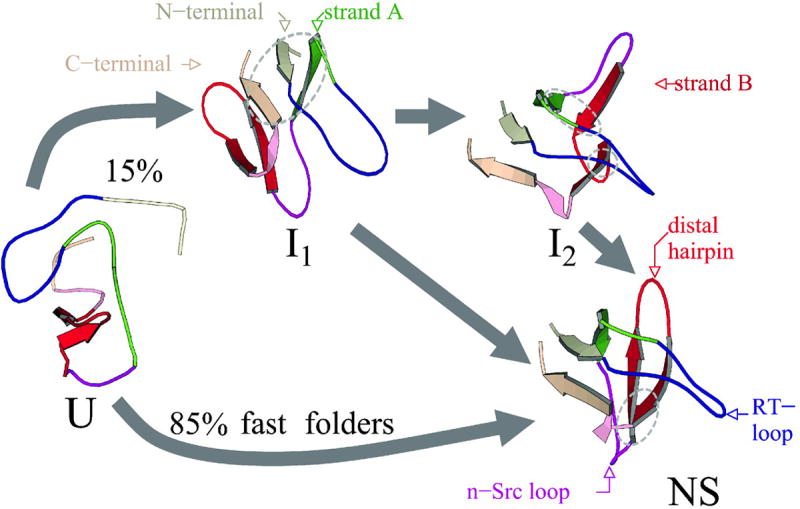
Molecular dynamics simulations of the c-Crk SH3 folding show multiple folding pathways via only one or two intermediates. [Reprinted with permission from ref. [258]. Copyright Biophysical Society.]
Experiments suggest that proteins may be kinetically trapped en route to the native state[149-151], but how do proteins avoid kinetic traps? By using a Go-model scaled to include sequence-specific interactions, Khare et al. [152] found that the residues which contribute most to Cu, Zn SOD1 stability also function as “gatekeepers” that avoid kinetic traps and protein misfolding. “Gatekeeper” residues were also identified in later computational studies of the ribosomal protein S6. Stoycheva et al. [153] and Matysiak et al.[154] show that the mutations of gatekeeper residues can alter the folding landscape of S6 and shift the balance between its folding and aggregation. All these computational studies are fully consistent with experimental observations and the existence of gatekeeper residues suggests a selective pressure on avoiding misfolding in natural proteins.
The close interplay of computational and experimental efforts has advanced our knowledge of protein folding kinetics, including predicting the protein folding rate, identifying the kinetically-important residues, and characterizing the multiple pathways. For example, recent studies have demonstrated an agreement between theoretical and experimental folding free energy landscapes [154-160]. The characterization of molecular interactions responsible for different pathways opens the possibility to manipulate folding pathways. Current strategies of manipulating the folding pathway includes addition of denaturants, point mutations, and circular permutations [161]. Specifically, Kuhlman and Baker rationally engineered multiple mutations in protein L to alter its folding pathways [162]. Lowe and Itzhaki likewise recently redesigned the folding pathways of the repeat protein myotrophin [163]. Hence, one strategy to tackle kinetics-related folding abnormalities is to rationally engineer the folding pathway after the full characterization of a protein’s folding kinetics.
V. Protein intermediate states
Proteins sample ensembles of heterogeneous conformations in solution. This emerging view of the one-to-many correspondence between protein primary sequence and its possible three-dimensional conformations also challenges the traditional paradigm that protein function is dictated by the native state. In recent years, it has been found that even for the conventionally observed small two-state proteins (~100 amino acids or less) there exist partially unfolded intermediates on the folding pathways. These intermediates are generally undetectable in kinetic folding experiments [8, 116, 164-168] and, therefore, are called “hidden intermediates”. In addition, mounting evidence has indicated that the intermediate states formed during protein folding and unfolding may have significant roles in protein functions (Fig. 3). The folding and unfolding intermediates impact the physiological functions of proteins by exposing cryptic post-translational modifications or ligand binding sites. The intermediates are usually weakly-populated (thermodynamic intermediates) or short-lived (kinetic intermediates), and their characterization presents a significant challenge with current experimental methods. Recent synergies between computational and experimental studies have greatly facilitated the unprecedented structural characterization of rare intermediates and suggested a functional role for these evasive conformations.
Figure 3. Protein intermediate states.

Intermediate states are meta-stable in protein free energy landscapes (N: the native state; I*, I**: intermediates). They may play a significant role in protein function, including exposing cryptic posttranslational modifications or ligand binding sites.
Among the traditional experimental methods, hydrogen exchange (HX)[169] is a unique tool that allows the detection and characterization of not only kinetic, but also weakly populated thermodynamic intermediates. In a typical equilibrium HX experiment, the rate at which an individual main-chain amide hydrogen exchanges with solvent deuterons is measured by NMR or mass spectrometry [170]. The exchange rates of different amide hydrogens are sensitive to local and global structural changes of proteins, and thus contain useful structural information about different protein conformations. From the amide hydrogen exchange rates measured by HX it is possible to detect and characterize weakly-populated intermediates which are inaccessible by bulk methods. Over the years, HX methods have provided considerable insight into the coarse features of intermediate state conformations for a wide variety of proteins[169]. However, HX is limited in its ability to describe the detailed structures of intermediates, which severely restricts its applications. In contrast, computational methods can reveal structural information at much higher resolutions that cannot be accessed by experiments. Also, unlike bulk experimental methods like HX, computational methods are able to study protein motions at the single-molecule level, which enables the detection of heterogeneous conformations in an ensemble of molecules.
In recent studies, Gsponer et al.[171] and Dixon et al.[172] have developed new computational approaches to incorporate HX protection factors from NMR experiments as constraints into MD simulations to detect and/or characterize the conformations of intermediate states. Using their new approach, Gsponer et al.[171] were able to define the thermodynamic folding intermediates of the bacterial immunity protein Ig7. A structural comparison between this thermodynamic intermediate and kinetic intermediate determined from other methods indicates that the kinetic and thermodynamic intermediates of Ig7 are similar. In a parallel study, Dixon et al.[172] computationally predicted and characterized a folding intermediate of the focal adhesion targeting (FAT) domain of focal adhesion kinase (FAK), which plays critical roles in cell proliferation and migration. The detected intermediate was hypothesized to expose a cryptic phosphorylation site in the FAT domain and regulate the localization of FAK. The existence of this intermediate and the predicted structural features were later experimentally confirmed [165]. Besides HX methods, the site-specific structural information obtained from other NMR-based techniques such as relaxation dispersion spectroscopy [173], has also been incorporated into MD simulations to study weakly-populated folding intermediates [174].
A major limitation of traditional experimental methods such as HX is that they can only measure the average behavior of an ensemble of molecules and often cannot distinguish individual folding and unfolding routes or intermediates in the ensemble. By contrast, single-molecule methods enable the observation of the folding and unfolding of individual molecules and play increasingly important roles in studying intermediates[175]. Two single-molecule techniques: fluorescence resonance energy transfer [176] and force spectroscopy including optical tweezers and AFM[177], have been utilized to probe intermediate states of proteins [178-180]. Direct comparisons of results from single-molecule stretching experiments by AFM and computational simulations shed light on the mechanical unfolding of proteins and how intermediates contribute to protein function.
Using AFM, Marszalek et al. [179] uncovered a force-induced unfolding intermediate of Ig domain I27 from titin, a modular protein which is responsible for muscle elasticity. This experimentally-discovered intermediate was also predicted by steered-molecular dynamics (SMD) simulations [179]. Based on the results of SMD, the authors further predicted that the hydrogen-bonding network between strand A’ and G in I27 mainly determines its mechanical stability. This prediction was verified in a mutagenesis study where Li et al. [181] showed that the point mutations on the residues participating in this hydrogen-bonding network dramatically altered the mechanical stability of I27. In a similar study of another protein, domain 10 of type III fibronectin module, which plays a pivotal role in mechanical coupling between cell surface and extracellular matrix (ECM), Gao et al. [182] predicted force-induced unfolding intermediates using SMD simulations. The discovered intermediates were considered to expose binding sites which are necessary for the assembly of ECM fibronectin fibrils. One of the computationally-predicted unfolding intermediates is in excellent agreement with the one observed in a more recent AFM stretching experiment [178].
The finding that folding and unfolding intermediates can have significant contributions to protein function is challenging the conventional understanding of protein structure and function that is centered on the native state. It is expected that a close synergy between computational and experimental approaches will continue to play essential roles in characterizing these evasive protein states.
VI. Unfolded protein states
Unveiling the structural and dynamic properties of denatured proteins is crucial for understanding the protein folding [183, 184] and misfolding [185] problems. For example, the computational determination of a protein’s thermodynamic stability requires an accurate approximation of the denatured state as the reference state. NMR hydrogen exchange experiments also rely on models of protein unfolded states to tabulate the intrinsic hydrogen exchange rate [186]. Furthermore, understanding the structural properties of unfolded proteins may shed light on the early events of protein folding and protein aggregation [187].
It has long been postulated that the denatured state of proteins is composed of an ensemble of featureless random coil-like conformations. According to Flory’s random coil theory[188], the size of a random coil polymer, characterized by the radius of gyration Rg, follows a power law dependence on the length of the polymer chain, n, Rg = R0nν. Here, R0 is the scaling constant, which is a function of persistence length, and ν represents the power law scaling exponent. Flory predicted the exponent to be 0.6 and later a more accurate renormalization calculation obtained ν=0.588 [189]. Tanford et al. first confirmed this random coil scaling behavior for denatured proteins [190]. Using intrinsic viscosity measurements for 12 proteins denatured by 5-6 M GuHCl, the authors obtained a scaling exponent ν=0.67±0.09. Wilkins et al. [191] showed that the hydrodynamic radii of sets of 8 highly denatured, disulfide-free proteins follow a power law scaling with ν=0.58±0.11. Recently, Kohn et al. [192] reassessed the scaling behavior of denatured proteins using small angle x-ray scattering for 17 proteins of lengths varying from 8 to 549 residues. They found the scaling exponent to be ν=0.598±0.029. All these experimental results confirm the random coil scaling of denatured proteins. In a random coil model of the denatured state, proteins are believed to lack persistent structure both locally and globally. The distribution of end-to-end distances or radii of gyration can be fit by a Gaussian distribution. A recent computational study [193] confirmed this behavior by generating an ensemble of protein conformations whereby only steric interactions between amino acids were considered for four different proteins. A scaling exponent of 0.58±0.02 was obtained.
The random coil scaling behavior [188, 194] was originally derived for homopolymers. However, proteins are heteropolymers for which specific interactions between amino acids play an important role and determine a unique native structure. Hence, while the scaling of the sizes of denatured proteins follows the random coil scaling as shown in experiments, it does not necessarily exclude the possibility that the denatured proteins can have residual native-like structures. Under denaturing conditions, the protein can still exhibit native conformational bias and retain a certain amount of residual native structures. Mounting experimental evidence [168, 195-198] supports residual native-like structural elements in the denatured state for a variety of proteins. Using residual dipolar coupling from NMR measurements, Shortle and Ackerman [195] showed that native-like topology persists under strong denaturing conditions as high as 8 M Urea for a truncated staphylococcal nuclease. By applying quasi-elastic neutron-scattering on α-lactalbumin, Bu et al. [196] demonstrated residual helical structure and tertiary-like interactions even in the absence of disulfide bonds and under highly denaturing conditions. Similarly, by using triple-resonance NMR, native-like topology has also been observed in protein L [197].
Several theoretical and computational studies [199-202] have addressed the role of specific interactions in conformational biasing toward the native state in the denatured states. Using a simple force field with only steric and hydrogen bond interactions, Pappu et al. [200] demonstrated that denatured protein states have a strong preference for the native structure. It was suggested [199-202] that the conformational bias of native structures in the denatured state is a possible explanation of the Levinthal’s paradox [3].
To reconcile the seemingly controversial properties of denatured proteins — the random coil scaling of their sizes and the presence of residual native structures — several computational works have recently been reported [203-207]. Fitzkee and Rose [206] reproduced the random coil scaling exponent using a denatured protein model with fixed secondary structure elements for a set of proteins. Tran et al. [204, 207] constructed the protein denatured state ensemble at atomic resolution in the excluded volume limit. Jha et al. [205] built the denatured state using a statistical coil library. Both the Pappu and Sosnick groups found that the putative denatured state ensemble features transient local structures such as turns, strand, and helices. In the mean time, the dimension of the denatured state follows the experimentally-observed random-coil scaling exponent. Ding et al. [203] developed a computational method to model denatured proteins using a structure-based potential [63]. This interaction model is commonly used in thermodynamic and kinetics studies of protein folding [58, 208-210] to model amino acid interactions. This study [203] suggested that denatured proteins follow the random coil scaling sizes and retain residual secondary structures akin to those observed in native protein states. Hence, these computational works provide a conceptual reconciliation between two seemingly mutually-exclusive views of protein unfolded states.
What is the physical origin of the random coil scaling of protein size along with the seemingly contradictory persistence of local structures in denatured proteins? Previous computational studies [203-205, 207] suggest that the residual structures in the denatured state are limited to short-range elements, which only extend approximately seven to ten residues. The correlated fluctuation of residual structures diminishes quickly along the sequence and long-range contact formation is purely governed by the random diffusion of peptide chains. Hence, a coarse-graining process, which groups locally-interacting amino acids along the polypeptide chain into renormalized structural units (Fig. 4) reduces a denatured protein to a renormalized homopolymer. The renormalization process will result in an effective homopolymer which forms contacts due to chain diffusion. Protein sizes follow the renormalized power law scaling as proposed by Flory [188]: Rg=R0(N/L)ν=(R0L-ν)Nν. Thus, we expect that the scaling exponent of denatured proteins ν is the same as for homopolymers, whose structural units are locally interacting amino acids.
Figure 4. Unfolded protein states.

The unfolded state of a protein features residual local structures, which span a short segment of approximately 10 residues (L~10). This structural correlation quickly decays as the segment length increases. A renormalization process, which groups the local amino acid residues into a coarse-grained bead, reduces the unfolded protein into an effective non-interacting polymer.
The observation of residual native secondary structures in thermally-denatured protein states is consistent with a “guided-folding” scenario [211], where the rate-limiting process is the packing of the preformed secondary structures into the correct fold. In contrast, a random coil model of the denatured state without residual native-like structures implies that a protein has to overcome an excessive entropic barrier to form both the secondary and tertiary structures upon folding. The existence of persistent native-like secondary structures in the denatured state may also be responsible for the recent success of protein structure prediction using small secondary structure segments derived from the protein data bank [212]. The existence of residual native-like structures in the denatured state also provides a novel way to manipulate a protein’s stability by stabilizing or destabilizing the residual structures [213].
VII. Protein self-association and aggregation
While all the information needed for proteins to fold is encoded in their amino acid sequence [1], there are many more elements that play a part in vivo. In a crowded cellular environment, surrounded by interacting proteins, nascent polypeptides face a formidable challenge in finding the correct interactions that result in a folded and functional protein. Many become “trapped” in meta-stable intermediate structures which are usually recognized by proteasomal machinery and degraded or refolded by chaperones. Alternatively, they can associate with similar misfolded proteins to form aggregates.
Extant protein sequences are the result of a long history of evolutionary refinement establishing a set of interactions defining the native state. However, the same inherent recognition that occurs between sequences within a protein is the basis for a type of self-association termed 3-dimensional domain swapping ([214], extensively catalogued in 2002 by Liu and Eisenberg [215]). Domain swapping is an important phenomenon, taking part in both normal and disease-related processes, and is intimately tied to protein folding. Domain swapping may be viewed as a natural mechanism for dealing with instability due to evolutionary changes in the amino acid sequence [216]. For example, a mutation that rigidifies a loop connecting two parts of a protein induces strain, which can be relieved without the loss of function by “swapping” the portion of the protein on one side of the loop with the corresponding part of a similar protein. Dimerization by this mechanism has advantages including a high local concentration of enzymatic activity, since two functional proteins are joined together.
Many diseases are now associated with protein aggregation and particularly with a form of ordered aggregate called the amyloid fibrils, which, regardless of the native sequence and structure of the precursor proteins, share distinct structural characteristics. From a protein folding standpoint, the inherent properties of the polypeptide chain that allow proteins with little or no sequence or structural similarity to misfold and assemble into similar high-order structures are of vital interest. Studies of aggregate structure reveal defined characteristics such as extensive hydrogen bond networks perpendicular to the fiber axis, called a cross-β conformation[217], and an α to β transition known to occur during the oligomerization of amyloid-forming proteins with significant helical content. Evidence for domain swapping as an early step in the aggregation process has been reported for several proteins [218-220]. In several aggregation-associated diseases including familial Amyotrophic Lateral Sclerosis and the transthyretin amyloidoses, the dissociation of a protein from its multimeric native state is known to be the rate limiting step for aggregation, suggesting a method for preventing aggregation by stabilizing the native interfaces of these assemblies [221-223].
Amyloid fibrils have alternately been deemed responsible for the pathologies of their various associated diseases and, more recently, credited with delaying or counteracting the observed pathologies by acting as a sink for highly cytotoxic soluble oligomers [224]. The viewpoint that soluble oligomers act as cytotoxic species has garnered widespread attention since at least 1999 when it was noted that the abundance of soluble Aβ 1-42 oligomers is inversely correlated with neuronal degeneration in Alzheimer’s disease whereas amyloid levels do not correlate [225, 226]. In 2003, Glabe and co-workers discovered that soluble oligomeric species from several disease-related proteins shared a common structural epitope to which an antibody was developed [227]. Later studies showed that soluble oligomers are able to disrupt the polarity of cellular membranes [144, 228, 229], one possible basis for disease-associated toxicity.
Various cellular protective mechanisms have evolved to ensure the proper folding of proteins. Molecular chaperones, for example, recognize misfolded proteins and provide an environment conducive to the formation of the appropriate native contacts [230]. Vast proteosomal machinery clears proteinaceous debris from the cell using ubiquitin ligases to tag misfolded proteins for degradation and removal [231, 232]. One hypothesis formulated to explain the prevalence of protein aggregation in neurodegenerative diseases is that the ubiquitin proteasome loses efficiency over time causing a buildup of protein aggregates and debris in post-mitotic cells such as neurons [233]. Also, parkin, an E3 ubiquitin ligase was found to be mutated in at least half of autosomal recessive juvenile parkinsonism patients, suggesting that a deficit in the clearance of its target protein leads to an early onset of symptoms [234].
Computational studies of protein aggregation have traditionally been inadequate due to the massive complexity of the system in both time and length scale. Several approaches have been used to overcome this complexity (Fig. 5). Traditional all-atom molecular dynamics simulations have been carried out to model the aggregation of disease-related peptides such as Aβ and polyglutamine [235-237]. Other techniques seek to identify which parts within larger proteins are responsible for their aggregation behavior, resulting in the identification of sequence stretches in various proteins that are “amyloidogenic,” or “hot spots” for aggregation [238-240]. Underlying this work is the idea that evolution acts to prevent aggregation by burying aggregation-prone protein sequences or otherwise prohibiting their apposition in protein structures and during folding1. To study the nature of subunit assembly and extension during aggregation, which are out of reach for all-atom molecular dynamics in both the size scale and time scale, simplified protein models are being utilized [65]. By accessing aggregation events that are out of the reach of experimentalists, computational studies of aggregation are an essential compliment to the experimental findings regarding aggregate structure and formation mechanisms.
Figure 5. Computational studies of protein self-association and aggregation.
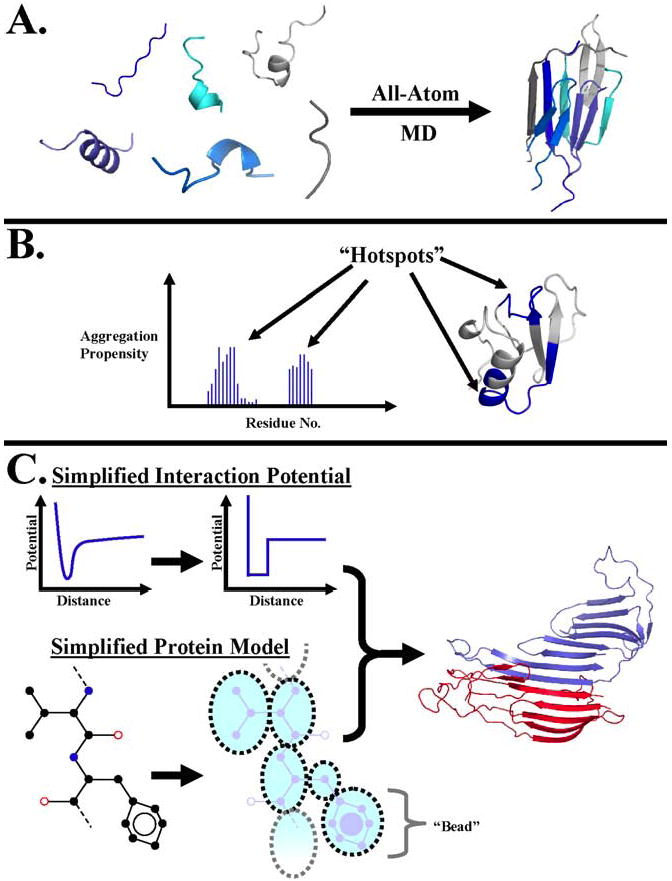
Different computational techniques have been utilized to study various aspects of protein self-association and aggregation. (A) All-atom molecular dynamics is used to model the aggregation of short peptides. (B) Simulations of peptides from within larger proteins are used to suggest aggregation “hotspots.” (C) By combining simplified interaction models and protein models, aggregating systems that are inaccessible by traditional molecular dynamics due to size and time limitations can be studied. The curves show the approximation of a continuous interaction potential by a square well as used in DMD [58]. The dotted circles represent “beads” in the model which take the place of several atoms in the original protein. The structure to the right shows the self-association of two identical proteins forming an extended β-sheet.
Protein aggregation is now widely viewed as a fundamental property of the polypeptide chain, meaning that all of the considerations discussed in the earlier sections of this review must, to some degree, apply to the study of aggregating proteins. Therefore, the study of protein self-association and aggregation really is the study of protein folding in the context of external influences like protein concentration, localization, or evolution. As this field develops and knowledge of protein aggregation as a general phenomenon accumulates, we stand to gain not only vital tools for treating specific diseases, but also insight into the behavior of all proteins with respect to their environment.
VIII. Conclusions
Perhaps the most significant four measures of success in the natural sciences are our abilities (i) to observe natural phenomena, (ii) to explain natural phenomena, (iii) to predict the effect of relevant variables on a specific phenomenon, and (iv) to rationally manipulate these phenomena. The protein folding field has seen significant breakthroughs according to all of these measures. Research in the protein folding field uncovered folding pathways and states that accompany the transition from unfolded to folded proteins, revealed the origin of the cooperative folding transition, allowed prediction of folding rates and changes in thermodynamic stability upon mutation, and permitted rational alteration of protein structure and folding pathways.
Given the recognition of many human maladies as “diseases of protein folding” over the past two decades, the wealth of new knowledge about the folding process is driving the study of protein folding back into its native environment, i.e. inside living organisms. The effect of the cellular environment on protein folding, e.g., how proteins fold in vivo and especially the behavior of “intrinsically-disordered” proteins is a highly active area of inquiry [82, 242, 243],. More applied research is ongoing in the design of animal models of diseases associated with protein folding such as cystic fibrosis, ALS, Alzheimer’s disease, and the prion diseases [244-246], and the design of small molecules for use in clinical trials for treating diseases like the transthyretin amyloidoses [247]. The future of the protein folding field lies in its direct application to such medical problems, and for a growing number of protein systems, the future is now.
Acknowledgments
We thank Oana Lungu for reading the manuscript. This work was supported by the American Heart Association grant No. 0665361U, the North Carolina Biotechnology Center Grant No. 2006-MRG-1107, Cystic Fibrosis Foundation grant DOKHOL07I0, and the National Institutes of Health grants RO1GM080742-01 and R01CA084480-07.
Footnotes
It is interesting to note here that in the case of Pmel17, which aggregates to form a “functional amyloid” involved in melanin biosynthesis, a protein seems to have evolved to aggregate at an incredible rate, perhaps to minimize the population time in a soluble oligomer form [241].
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimersthat apply to the journal pertain.
References
- 1.Anfinsen CB, Harber E, Sela M, White FH., Jr Proc Natl Acad Sci U S A. 1961;47:1309–1314. doi: 10.1073/pnas.47.9.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anfinsen CB. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 3.Levinthal C. J Chim Phys. 1968;65:44–45. [Google Scholar]
- 4.Ikai A, Tanford C. Nature. 1971;230:100–102. doi: 10.1038/230100a0. [DOI] [PubMed] [Google Scholar]
- 5.Tsong TY, Baldwin RL, Elson EL. Proc Natl Acad Sci U S A. 1971;68:2712–2715. doi: 10.1073/pnas.68.11.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackson SE, Fersht AR. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 7.Jackson SE, Fersht AR. Biochemistry. 1991;30:10436–10443. doi: 10.1021/bi00107a011. [DOI] [PubMed] [Google Scholar]
- 8.Bai Y, Sosnick TR, Mayne L, Englander SW. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bai Y, Englander SW. Proteins. 1996;24:145–151. doi: 10.1002/(SICI)1097-0134(199602)24:2<145::AID-PROT1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 10.Roder H, Elove GA, Englander SW. Nature %20. 1988;335:700–704. doi: 10.1038/335700a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miranker A, Robinson CV, Radford SE, Aplin RT, Dobson CM. Science. 1993;262:896–900. doi: 10.1126/science.8235611. [DOI] [PubMed] [Google Scholar]
- 12.Jones CM, Henry ER, Hu Y, Chan CK, Luck SD, Bhuyan A, Roder H, Hofrichter J, Eaton WA. Proc Natl Acad Sci U S A. 1993;90:11860–11864. doi: 10.1073/pnas.90.24.11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matouschek A, Kellis JT, Serrano L, Bycroft M, Fersht AR. Nature. 1990;346:440–445. doi: 10.1038/346440a0. [DOI] [PubMed] [Google Scholar]
- 14.Mirny L, Shakhnovich E. Annu Rev Biophys Biomol Struct. 2001;30:361–396. doi: 10.1146/annurev.biophys.30.1.361. [DOI] [PubMed] [Google Scholar]
- 15.Shakhnovich E. Chemical Reviews. 2006;106:1559–1588. doi: 10.1021/cr040425u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wetlaufer DB. Proc Natl Acad Sci U S A. 1973;70:697–701. doi: 10.1073/pnas.70.3.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wetlaufer DB. Trends Biochem Sci. 1990;15:414–415. doi: 10.1016/0968-0004(90)90275-g. [DOI] [PubMed] [Google Scholar]
- 18.Go N. Annu Rev Biophys Bioeng. 1983;12:183–210. doi: 10.1146/annurev.bb.12.060183.001151. [DOI] [PubMed] [Google Scholar]
- 19.Abkevich VI, Gutin AM, Shakhnovich EI. Biochemistry. 1994;33:10026–10036. doi: 10.1021/bi00199a029. [DOI] [PubMed] [Google Scholar]
- 20.Fersht AR. Curr Opin Struct Biol. 1997;7:3–9. doi: 10.1016/s0959-440x(97)80002-4. [DOI] [PubMed] [Google Scholar]
- 21.Shakhnovich EI. Curr Opin Struct Biol. 1997;7:29–40. doi: 10.1016/s0959-440x(97)80005-x. [DOI] [PubMed] [Google Scholar]
- 22.Lifshits EM, Pitaevskii LP. Physical Kinetics. Pergamon Press; Oxford: 1981. [Google Scholar]
- 23.Onuchic JN, Luthey-Schulten Z, Wolynes PG. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 24.Shea JE, Brooks CL., III Annu Rev Phys Chem. 2001;52:499–535. doi: 10.1146/annurev.physchem.52.1.499. [DOI] [PubMed] [Google Scholar]
- 25.Zhou RH, Berne BJ, Germain R. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:14931–14936. doi: 10.1073/pnas.201543998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fersht AR, Daggett V. Cell. 2002;108:573–582. doi: 10.1016/s0092-8674(02)00620-7. [DOI] [PubMed] [Google Scholar]
- 27.Dinner AR, Sali A, Smith LJ, Dobson CM, Karplus M. Trends Biochem Sci. 2000;25:331–339. doi: 10.1016/s0968-0004(00)01610-8. [DOI] [PubMed] [Google Scholar]
- 28.Duan Y, Kollman PA. Science. 1998;282:740–744. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 29.Ghosh A, Elber R, Scheraga HA. Proc Natl Acad Sci U S A. 2002;99:10394–10398. doi: 10.1073/pnas.142288099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cavalli A, Ferrara P, Caflisch A. Proteins. 2002;47:305–314. doi: 10.1002/prot.10041. [DOI] [PubMed] [Google Scholar]
- 31.Pande VS, Baker I, Chapman J, Elmer SP, Khaliq S, Larson SM, Rhee YM, Shirts MR, Snow CD, Sorin EJ, Zagrovic B. Biopolymers. 2003;68:91–109. doi: 10.1002/bip.10219. [DOI] [PubMed] [Google Scholar]
- 32.Hansmann UHE. International Journal of Quantum Chemistry. 2002;90:1515–1521. [Google Scholar]
- 33.Hansmann UHE. Computer Physics Communications. 2002;147:604–607. [Google Scholar]
- 34.Snow CD, Nguyen N, Pande VS, Gruebele M. Nature. 2002;420:102–106. doi: 10.1038/nature01160. [DOI] [PubMed] [Google Scholar]
- 35.Gront D, Kolinski A, Skolnick J. Journal of Chemical Physics. 2001;115:1569–1574. [Google Scholar]
- 36.Rhee YM, Pande VS. Biophysical Journal. 2003;84:775–786. doi: 10.1016/S0006-3495(03)74897-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feig M, Karanicolas J, Brooks CL. Journal of Molecular Graphics & Modelling. 2004;22:377–395. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 38.Skolnick J, Kolinski A, Ortiz AR. Journal of Molecular Biology. 1997;265:217–241. doi: 10.1006/jmbi.1996.0720. [DOI] [PubMed] [Google Scholar]
- 39.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, Debolt S, Ferguson D, Seibel G, Kollman P. Computer Physics Communications. 1995;91:1–41. [Google Scholar]
- 40.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. Journal of Computational Chemistry. 1983;4:187–217. [Google Scholar]
- 41.Zhou Y, Karplus M. Proc Natl Acad Sci U S A. 1997;94:14429–14432. doi: 10.1073/pnas.94.26.14429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dokholyan NV, Borreguero JM, Buldyrev SV, Ding F, Stanley HE, Shakhnovich EI. Methods Enzymol. 2003;374:616–638. doi: 10.1016/S0076-6879(03)74025-7. [DOI] [PubMed] [Google Scholar]
- 43.Alder BJ, Wainwright TE. Journal of Chemical Physics. 1959;31:459–466. [Google Scholar]
- 44.Rapaport DC. The art of molecular dynamics simulations. Cambridge University Press; Cambridge: 1997. [Google Scholar]
- 45.Rapaport DC. Journal of Chemical Physics. 1979;71:3299–3303. [Google Scholar]
- 46.Bellemans A, Orban J, Vanbelle D. Molecular Physics. 1980;39:781–782. [Google Scholar]
- 47.Ding F, Guo WH, Dokholyan NV, Shakhnovich EI, Shea JE. Journal of Molecular Biology. 2005;350:1035–1050. doi: 10.1016/j.jmb.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 48.Hilser VJ, Garcia-Moreno EB, Oas TG, Kapp G, Whitten ST. Chem Rev. 2006;106:1545–1558. doi: 10.1021/cr040423+. [DOI] [PubMed] [Google Scholar]
- 49.Ma J. Structure. 2005;13:373–380. doi: 10.1016/j.str.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 50.Cui Q, Li G, Ma J, Karplus M. J Mol Biol. 2004;340:345–372. doi: 10.1016/j.jmb.2004.04.044. [DOI] [PubMed] [Google Scholar]
- 51.Liu T, Whitten ST, Hilser VJ. Proc Natl Acad Sci U S A. 2007;104:4347–4352. doi: 10.1073/pnas.0607132104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Freire E. National Acad Sciences. 2000 [Google Scholar]
- 53.Gromiha MM, Thangakani AM, Selvaraj S. Nucleic Acids Res. 2006;34:W70–W74. doi: 10.1093/nar/gkl043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thomas S, Song G, Amato NM. Phys Biol. 2005;2:S148–S155. doi: 10.1088/1478-3975/2/4/S09. [DOI] [PubMed] [Google Scholar]
- 55.Sharma S, Ding F, Nie H, Watson D, Unnithan A, Lopp J, Pozefsky D, Dokholyan NV. Bioinformatics. 2006 doi: 10.1093/bioinformatics/btl460. [DOI] [PubMed] [Google Scholar]
- 56.Jang H, Hall CK, Zhou Y. Biophys J. 2004;86:31–49. doi: 10.1016/S0006-3495(04)74081-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith AV, Hall CK. Proteins. 2001;44:376–391. doi: 10.1002/prot.1103. [DOI] [PubMed] [Google Scholar]
- 58.Dokholyan NV, Buldyrev SV, Stanley HE, Shakhnovich EI. Folding & Design. 1998;3:577–587. doi: 10.1016/S1359-0278(98)00072-8. [DOI] [PubMed] [Google Scholar]
- 59.Borreguero JM, Dokholyan NV, Buldyrev SV, Shakhnovich EI, Stanley HE. Journal of Molecular Biology. 2002;318:863–876. doi: 10.1016/S0022-2836(02)00136-5. [DOI] [PubMed] [Google Scholar]
- 60.Jang H, Hall CK, Zhou Y. Biophys J. 2002;83:819–835. doi: 10.1016/S0006-3495(02)75211-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dokholyan NV. Curr Opin Struct Biol. 2006 [Google Scholar]
- 62.Ding F, Dokholyan NV. Trends Biotechnol. 2005;23:450–455. doi: 10.1016/j.tibtech.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 63.Go N, Abe H. Biopolymers. 1981;20:991–1011. doi: 10.1002/bip.1981.360200511. [DOI] [PubMed] [Google Scholar]
- 64.Urbanc B, Borreguero JM, Cruz L, Stanley HE. Methods Enzymol. 2006;412:314–338. doi: 10.1016/S0076-6879(06)12019-4. [DOI] [PubMed] [Google Scholar]
- 65.Peng S, Ding F, Urbanc B, Buldyrev SV, Cruz L, Stanley HE, Dokholyan NV. Phys Rev E Stat Nonlin Soft Matter Phys. 2004;69:041908. doi: 10.1103/PhysRevE.69.041908. [DOI] [PubMed] [Google Scholar]
- 66.Nguyen HD, Hall CK. Proc Natl Acad Sci U S A. 2004;101:16180–16185. doi: 10.1073/pnas.0407273101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nguyen HD, Hall CK. J Biol Chem. 2005;280:9074–9082. doi: 10.1074/jbc.M407338200. [DOI] [PubMed] [Google Scholar]
- 68.Sharma S, Ding F, Dokholyan NV. Biophys J. 2007;92 doi: 10.1529/biophysj.106.094805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frydman J, Hartl FU. Science. 1996;272:1497–1502. doi: 10.1126/science.272.5267.1497. [DOI] [PubMed] [Google Scholar]
- 70.Martin J, Hartl FU. Curr Opin Struct Biol. 1997;7:41–52. doi: 10.1016/s0959-440x(97)80006-1. [DOI] [PubMed] [Google Scholar]
- 71.Bukau B, Horwich AL. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 72.Weissman JS, Rye HS, Fenton WA, Beechem JM, Horwich AL. Cell. 1996;84:481–490. doi: 10.1016/s0092-8674(00)81293-3. [DOI] [PubMed] [Google Scholar]
- 73.Feltham JL, Gierasch LM. Cell. 2000;100:193–196. doi: 10.1016/s0092-8674(00)81557-3. [DOI] [PubMed] [Google Scholar]
- 74.Landry SJ, Gierasch LM. Biochemistry. 1991;30:7359–7362. doi: 10.1021/bi00244a001. [DOI] [PubMed] [Google Scholar]
- 75.Horowitz PM, Hua S, Gibbons DL. J Biol Chem. 1995;270:1535–1542. [PubMed] [Google Scholar]
- 76.Gorovits BM, Horowitz PM. J Biol Chem. 1995;270:13057–13062. doi: 10.1074/jbc.270.22.13057. [DOI] [PubMed] [Google Scholar]
- 77.Wittung-Stafshede P. Acc Chem Res. 2002;35:201–208. doi: 10.1021/ar010106e. [DOI] [PubMed] [Google Scholar]
- 78.Leckner J, Bonander N, Wittung-Stafshede P, Malmstrom BG, Karlsson BG. Biochim Biophys Acta. 1997;1342:19–27. doi: 10.1016/s0167-4838(97)00074-5. [DOI] [PubMed] [Google Scholar]
- 79.Luby-Phelps K. Int Rev Cytol. 2000;192:189–221. doi: 10.1016/s0074-7696(08)60527-6. [DOI] [PubMed] [Google Scholar]
- 80.Minton AP. J Biol Chem. 2001;276:10577–10580. doi: 10.1074/jbc.R100005200. [DOI] [PubMed] [Google Scholar]
- 81.Minton AP. J Pharm Sci. 2005;94:1668–1675. doi: 10.1002/jps.20417. [DOI] [PubMed] [Google Scholar]
- 82.Dedmon MM, Patel CN, Young GB, Pielak GJ. Proc Natl Acad Sci U S A. 2002;99:12681–12684. doi: 10.1073/pnas.202331299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McPhie P, Ni YS, Minton AP. J Mol Biol. 2006;361:7–10. doi: 10.1016/j.jmb.2006.05.075. [DOI] [PubMed] [Google Scholar]
- 84.Hatters DM, Minton AP, Howlett GJ. J Biol Chem. 2002;277:7824–7830. doi: 10.1074/jbc.M110429200. [DOI] [PubMed] [Google Scholar]
- 85.Taverna DM, Goldstein RA. Proteins. 2002;46:105–109. doi: 10.1002/prot.10016. [DOI] [PubMed] [Google Scholar]
- 86.Pakula AA, Sauer RT. Annual Review of Genetics. 1989;23:289–310. doi: 10.1146/annurev.ge.23.120189.001445. [DOI] [PubMed] [Google Scholar]
- 87.Privalov PL. Adv Protein Chem. 1979;33:167–241. doi: 10.1016/s0065-3233(08)60460-x. [DOI] [PubMed] [Google Scholar]
- 88.Becktel WJ, Schellman JA. Biopolymers. 1987;26:1859–1877. doi: 10.1002/bip.360261104. [DOI] [PubMed] [Google Scholar]
- 89.Makhatadze GI, Privalov PL. J Mol Biol. 1992;226:491–505. doi: 10.1016/0022-2836(92)90963-k. [DOI] [PubMed] [Google Scholar]
- 90.Jackson SE, Moracci M, elMasry N, Johnson CM, Fersht AR. Biochemistry. 1993;32:11259–11269. doi: 10.1021/bi00093a001. [DOI] [PubMed] [Google Scholar]
- 91.Bash PA, Singh UC, Langridge R, Kollman PA. Science. 1987;236:564–568. doi: 10.1126/science.3576184. [DOI] [PubMed] [Google Scholar]
- 92.Dang LX, Merz KM, Kollman PA. Journal of the American Chemical Society. 1989;111:8505–8508. [Google Scholar]
- 93.Kollman PA, Massova I, Reyes C, Kuhn B, Huo SH, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE. Accounts of Chemical Research. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 94.Vorobjev YN, Hermans J. Biophysical Chemistry. 1999;78:195–205. doi: 10.1016/s0301-4622(98)00230-0. [DOI] [PubMed] [Google Scholar]
- 95.Kollman P. Chemical Reviews. 1993;93:2395–2417. [Google Scholar]
- 96.Lazaridis T, Karplus M. Curr Opin Struct Biol. 2000;10:139–145. doi: 10.1016/s0959-440x(00)00063-4. [DOI] [PubMed] [Google Scholar]
- 97.Gilis D, Rooman M. Journal of Molecular Biology. 1996;257:1112–1126. doi: 10.1006/jmbi.1996.0226. [DOI] [PubMed] [Google Scholar]
- 98.Gilis D, Rooman M. Journal of Molecular Biology. 1997;272:276–290. doi: 10.1006/jmbi.1997.1237. [DOI] [PubMed] [Google Scholar]
- 99.Gilis D, Rooman M. Protein Engineering. 2000;13:849–856. doi: 10.1093/protein/13.12.849. [DOI] [PubMed] [Google Scholar]
- 100.Zhou HY, Zhou YQ. Protein Sci. 2003;12:2121. [Google Scholar]
- 101.Ota M, Isogai Y, Nishikawa K. Protein Engineering. 2001;14:557–564. doi: 10.1093/protein/14.8.557. [DOI] [PubMed] [Google Scholar]
- 102.Hoppe C, Schomburg D. Protein Sci. 2005;14:2682–2692. doi: 10.1110/ps.04940705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Carter CW, LeFebvre BC, Cammer SA, Tropsha A, Edgell MH. J Mol Biol. 2001;311:625–638. doi: 10.1006/jmbi.2001.4906. [DOI] [PubMed] [Google Scholar]
- 104.Guerois R, Nielsen JE, Serrano L. Journal of Molecular Biology. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 105.Khatun J, Khare SD, Dokholyan NV. J Mol Biol. 2004;336:1223–1238. doi: 10.1016/j.jmb.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 106.Bordner AJ, Abagyan RA. Proteins-Structure Function and Bioinformatics. 2004;57:400–413. doi: 10.1002/prot.20185. [DOI] [PubMed] [Google Scholar]
- 107.Saraboji K, Gromiha MM, Ponnuswamy MN. Biopolymers. 2006;82:80–92. doi: 10.1002/bip.20462. [DOI] [PubMed] [Google Scholar]
- 108.Capriotti E, Fariselli P, Calabrese R, Casadio R. Bioinformatics. 2005;21:54–58. doi: 10.1093/bioinformatics/bti1109. [DOI] [PubMed] [Google Scholar]
- 109.Kuhlman B, Baker D. Current Opinion in Structural Biology. 2004;14:89–95. doi: 10.1016/j.sbi.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 110.Cheng JL, Randall A, Baldi P. Proteins-Structure Function and Bioinformatics. 2006;62:1125–1132. doi: 10.1002/prot.20810. [DOI] [PubMed] [Google Scholar]
- 111.Yin SY, Ding F, Dokholyan NV. Nature Methods. 2007;4:2. doi: 10.1038/nmeth0607-466. [DOI] [PubMed] [Google Scholar]
- 112.Ding F, Dokholyan NV. Public Library of Science Computational Biology. 2006;2:e85. doi: 10.1371/journal.pcbi.0020085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Edgell MH, Sims DA, Pielak GJ, Yi F. Biochemistry. 2003;42:7587–7593. doi: 10.1021/bi034063g. [DOI] [PubMed] [Google Scholar]
- 114.Bava KA, Gromiha MM, Uedaira H, Kitajima K, Sarai A. Nucleic Acids Research. 2004;32:D120–D121. doi: 10.1093/nar/gkh082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chiti F, Dobson CM. Annual Review of Biochemistry. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 116.Thibodeau PH, Brautigam CA, Machius M, Thomas PJ. Nature Structural & Molecular Biology. 2005;12:10–16. doi: 10.1038/nsmb881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Khare SD, Dokholyan NV. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:3147–3152. doi: 10.1073/pnas.0511266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nolting B. Protein Folding Kinetics. Springer-Verlag; Berlin, Germany: 2006. [Google Scholar]
- 119.Plaxco KW, Simons KT, Baker D. Journal of Molecular Biology. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 120.Munoz V, Eaton WA. PNAS. 1999;96:11311–11316. doi: 10.1073/pnas.96.20.11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Otzen DE, Itzhaki LS, elMasry NF, Jackson SE, Fersht AR. Proc Natl Acad Sci U S A. 1994;91:10422–10425. doi: 10.1073/pnas.91.22.10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li A, Daggett V. Proc Natl Acad Sci U S A. 1994;91:10430–10434. doi: 10.1073/pnas.91.22.10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ivankov DN, Finkelstein AV. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8942–8944. doi: 10.1073/pnas.0402659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kuznetsov IB, Rackovsky S. Proteins-Structure Function and Genetics. 2004;54:333–341. doi: 10.1002/prot.10518. [DOI] [PubMed] [Google Scholar]
- 125.Ferrara P, Apostolakis J, Caflisch A. J Phys Chem B. 2000;104:5000–5010. [Google Scholar]
- 126.Ferrara P, Caflisch A. Proc Natl Acad Sci U S A. 2000;97:10780–10785. doi: 10.1073/pnas.190324897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hiltpold A, Ferrara P, Gsponer J, Caflisch A. J Phys Chem B. 2000;104:10080–10086. [Google Scholar]
- 128.Zagrovic B, Pande V. Journal of Computational Chemistry. 2003;24:1432–1436. doi: 10.1002/jcc.10297. [DOI] [PubMed] [Google Scholar]
- 129.Qiu D, Shenkin PS, Hollinger FP, Still WC. J Phys Chem A. 1997;101:3005–3014. [Google Scholar]
- 130.Jorgensen WL, Tiradorives J. Journal of the American Chemical Society. 1988;110:1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 131.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. The Journal of Chemical Physics. 1983;79:926–935. [Google Scholar]
- 132.Berendsen H, Potsma J, van Gunsteren W, Hermans J. Intermolecular Forces. Dordrecht: Reidel; 1981. pp. 331–334. [Google Scholar]
- 133.Sorin EJ, Pande VS. Biophysical Journal. 2005;88:2472–2493. doi: 10.1529/biophysj.104.051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shakhnovich EI, Finkelstein AV. Biopolymers. 1989;28:1667–1680. doi: 10.1002/bip.360281003. [DOI] [PubMed] [Google Scholar]
- 135.Bryngelson JD, Wolynes PG. Proc Natl Acad Sci U S A. 1987;84:7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shakhnovich E, Farztdinov G, Gutin AM, Karplus M. Phys Rev Lett. 1991;67:1665. doi: 10.1103/PhysRevLett.67.1665. [DOI] [PubMed] [Google Scholar]
- 137.Sali A, Shakhnovich E, Karplus M. Nature. 1994;369:248–251. doi: 10.1038/369248a0. [DOI] [PubMed] [Google Scholar]
- 138.Bolhuis PG, Chandler D, Dellago C, Geissler PL. Annual Review of Physical Chemistry. 2002;53:291–318. doi: 10.1146/annurev.physchem.53.082301.113146. [DOI] [PubMed] [Google Scholar]
- 139.Matouschek A, Kellis JT, Serrano L, Fersht AR. Nature. 1989;340:122–126. doi: 10.1038/340122a0. [DOI] [PubMed] [Google Scholar]
- 140.Fersht AR, Matouschek A, Serrano L. Journal of Molecular Biology. 1992;224:771–782. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- 141.Day R, Daggett V. Protein Simulations. 2003;66:373–403. doi: 10.1016/s0065-3233(03)66009-2. [DOI] [PubMed] [Google Scholar]
- 142.Scheraga HA. Febs Journal. 2005;272:359. [Google Scholar]
- 143.Ruczinski I, Sosnick TR, Plaxco KW. Protein Sci. 2006;15:2257–2264. doi: 10.1110/ps.062230106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Baglioni S, Casamenti F, Bucciantini M, Luheshi LM, Taddei N, Chiti F, Dobson CM, Stefani M. J Neurosci. 2006;26:8160–8167. doi: 10.1523/JNEUROSCI.4809-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fersht AR, Sato S. PNAS. 2004;101:7976–7981. doi: 10.1073/pnas.0402684101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ding F, Dokholyan NV, Buldyrev SV, Stanley HE, Shakhnovich EI. Biophysical Journal. 2002;83:3525–3532. doi: 10.1016/S0006-3495(02)75352-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ding F, Dokholyan NV, Buldyrev SV, Stanley HE, Shakhnovich EI. Journal of Molecular Biology. 2002;324:851–857. doi: 10.1016/s0022-2836(02)01112-9. [DOI] [PubMed] [Google Scholar]
- 148.Di Nardo AA, Korzhnev DM, Stogios PJ, Zarrine-Afsar A, Kay LE, Davidson AR. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:7954–7959. doi: 10.1073/pnas.0400550101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chen YR, Clark AC. Protein Science. 2004;13:2196–2206. doi: 10.1110/ps.03521504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chen YR, Clark AC. Biochemistry. 2003;42:6310–6320. doi: 10.1021/bi0340752. [DOI] [PubMed] [Google Scholar]
- 151.Reader JS, Van Nuland NAJ, Thompson GS, Ferguson SJ, Dobson CM, Radford SE. Protein Sci. 2001;10:1216–1224. doi: 10.1110/ps.52801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Khare SD, Ding F, Dokholyan NV. J Mol Biol. 2003;334:515–525. doi: 10.1016/j.jmb.2003.09.069. [DOI] [PubMed] [Google Scholar]
- 153.Stoycheva AD, Brooks CL, III, Onuchic JN. J Mol Biol. 2004;340:571–585. doi: 10.1016/j.jmb.2004.04.073. [DOI] [PubMed] [Google Scholar]
- 154.Matysiak S, Clementi C. J Mol Biol. 2006;363:297–308. doi: 10.1016/j.jmb.2006.07.088. [DOI] [PubMed] [Google Scholar]
- 155.Shea JE, Onuchic JN, Brooks CL., III Proc Natl Acad Sci U S A. 2002;99:16064–16068. doi: 10.1073/pnas.242293099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nguyen H, Jager M, Moretto A, Gruebele M, Kelly JW. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:3948–3953. doi: 10.1073/pnas.0538054100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Karanicolas J, Brooks CL. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:3954–3959. doi: 10.1073/pnas.0731771100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fernandez-Escamilla AM, Cheung MS, Vega MC, Wilmanns M, Onuchic JN, Serrano L. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2834–2839. doi: 10.1073/pnas.0304180101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chavez LL, Onuchic JN, Clementi C. Journal of the American Chemical Society. 2004;126:8426–8432. doi: 10.1021/ja049510+. [DOI] [PubMed] [Google Scholar]
- 160.Onuchic JN, Wolynes PG. Current Opinion in Structural Biology. 2004;14:70–75. doi: 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 161.Lindberg M, Oliveberg M. Current Opinion in Structural Biology. 2007;17:21–29. doi: 10.1016/j.sbi.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 162.Nauli S, Kuhlman B, Baker D. Nature Structural Biology. 2001;8:602–605. doi: 10.1038/89638. [DOI] [PubMed] [Google Scholar]
- 163.Lowe AR, Itzhaki LS. PNAS. 2007;104:2679–2684. doi: 10.1073/pnas.0604653104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Feng HQ, Vu ND, Bai YW. Journal of Molecular Biology. 2005;346:345–353. doi: 10.1016/j.jmb.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 165.Zhou Z, Feng H, Bai Y. Proteins. 2006;65:259–265. doi: 10.1002/prot.21107. [DOI] [PubMed] [Google Scholar]
- 166.Fersht AR. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:1525–1529. doi: 10.1073/pnas.97.4.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kato H, Feng HQ, Bai YW. Journal of Molecular Biology. 2007;365:870–880. doi: 10.1016/j.jmb.2006.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Religa TL, Markson JS, Mayor U, Freund SM, Fersht AR. Nature. 2005;437:1053–1056. doi: 10.1038/nature04054. [DOI] [PubMed] [Google Scholar]
- 169.Englander SW. Annu Rev Biophys Biomol Struct. 2000;29:213–238. doi: 10.1146/annurev.biophys.29.1.213. [DOI] [PubMed] [Google Scholar]
- 170.Englander SW. J Am Soc Mass Spectrom. 2006;17:1481–1489. doi: 10.1016/j.jasms.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gsponer J, Hopearuoho H, Whittaker SB, Spence GR, Moore GR, Paci E, Radford SE, Vendruscolo M. Proc Natl Acad Sci U S A. 2006;103:99–104. doi: 10.1073/pnas.0508667102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Dixon RD, Chen Y, Ding F, Khare SD, Prutzman KC, Schaller MD, Campbell SL, Dokholyan NV. Structure. 2004;12:2161–2171. doi: 10.1016/j.str.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 173.Mittermaier A, Kay LE. Science. 2006;312:224–228. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- 174.Korzhnev DM, Salvatella X, Vendruscolo M, Di Nardo AA, Davidson AR, Dobson CM, Kay LE. Nature. 2004;430:586–590. doi: 10.1038/nature02655. [DOI] [PubMed] [Google Scholar]
- 175.Zhuang X, Rief M. Curr Opin Struct Biol. 2003;13:88–97. doi: 10.1016/s0959-440x(03)00011-3. [DOI] [PubMed] [Google Scholar]
- 176.Selvin PR. Nat Struct Biol. 2000;7:730–734. doi: 10.1038/78948. [DOI] [PubMed] [Google Scholar]
- 177.Clausen-Schaumann H, Seitz M, Krautbauer R, Gaub HE. Curr Opin Chem Biol. 2000;4:524–530. doi: 10.1016/s1367-5931(00)00126-5. [DOI] [PubMed] [Google Scholar]
- 178.Li L, Huang HH, Badilla CL, Fernandez JM. J Mol Biol. 2005;345:817–826. doi: 10.1016/j.jmb.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 179.Marszalek PE, Lu H, Li H, Carrion-Vazquez M, Oberhauser AF, Schulten K, Fernandez JM. Nature. 1999;402:100–103. doi: 10.1038/47083. [DOI] [PubMed] [Google Scholar]
- 180.Cecconi C, Shank EA, Bustamante C, Marqusee S. Science. 2005;309:2057–2060. doi: 10.1126/science.1116702. [DOI] [PubMed] [Google Scholar]
- 181.Li H, Carrion-Vazquez M, Oberhauser AF, Marszalek PE, Fernandez JM. Nat Struct Biol. 2000;7:1117–1120. doi: 10.1038/81964. [DOI] [PubMed] [Google Scholar]
- 182.Gao M, Craig D, Lequin O, Campbell ID, Vogel V, Schulten K. Proc Natl Acad Sci U S A. 2003;100:14784–14789. doi: 10.1073/pnas.2334390100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Proteins. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 184.Baldwin RL. Adv Protein Chem. 2002;62:361–367. doi: 10.1016/s0065-3233(02)62014-5. [DOI] [PubMed] [Google Scholar]
- 185.Dobson CM. Semin Cell Dev Biol. 2004;15:3–16. doi: 10.1016/j.semcdb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 186.Bai Y, Milne JS, Mayne L, Englander SW. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Uversky VN, Fink AL. Biochim Biophys Acta. 2004;1698:131–153. doi: 10.1016/j.bbapap.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 188.Flory PJ. Principles of Polymer Chemistry. Cornell University Press; Ithaca, NY: 1965. [Google Scholar]
- 189.Le Guillou JC, Zinn-Justin J. Phys Rev Lett. 1977;39:95–98. [Google Scholar]
- 190.Tanford C, Kawahara K, Lapanje S, Hooker TM, Jr, Zarlengo MH, Salahuddin A, Aune KC, Takagi T. J Am Chem Soc. 1967;89:5023–5029. doi: 10.1021/ja00995a034. [DOI] [PubMed] [Google Scholar]
- 191.Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ. Biochemistry. 1999;38:16424–16431. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- 192.Kohn JE, Millett IS, Jacob J, Zagrovic B, Dillon TM, Cingel N, Dothager RS, Seifert S, Thiyagarajan P, Sosnick TR, Hasan MZ, Pande VS, Ruczinski I, Doniach S, Plaxco KW. Proc Natl Acad Sci U S A. 2004;101:12491–12496. doi: 10.1073/pnas.0403643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Goldenberg DP. J Mol Biol. 2003;326:1615–1633. doi: 10.1016/s0022-2836(03)00033-0. [DOI] [PubMed] [Google Scholar]
- 194.de Gennes PG. Scaling Concepts in Polymer Physics. Cornell University Press; Ithaca, NY: 1979. [Google Scholar]
- 195.Shortle D, Ackerman MS. Science. 2001;293:487–489. doi: 10.1126/science.1060438. [DOI] [PubMed] [Google Scholar]
- 196.Bu Z, Cook J, Callaway DJ. J Mol Biol. 2001;312:865–873. doi: 10.1006/jmbi.2001.5006. [DOI] [PubMed] [Google Scholar]
- 197.Yi Q, Scalley-Kim ML, Alm EJ, Baker D. Journal of Molecular Biology. 2000;299:1341–1351. doi: 10.1006/jmbi.2000.3816. [DOI] [PubMed] [Google Scholar]
- 198.Platt GW, McParland VJ, Kalverda AP, Homans SW, Radford SE. J Mol Biol. 2005;346:279–294. doi: 10.1016/j.jmb.2004.11.035. [DOI] [PubMed] [Google Scholar]
- 199.Zwanzig R, Szabo A, Bagchi B. Proc Natl Acad Sci U S A. 1992;89:20–22. doi: 10.1073/pnas.89.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Pappu RV, Srinivasan R, Rose GD. Proc Natl Acad Sci U S A. 2000;97:12565–12570. doi: 10.1073/pnas.97.23.12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Srinivasan R, Rose GD. Biophys Chem. 2002;101-102:167–171. doi: 10.1016/s0301-4622(02)00147-3. [DOI] [PubMed] [Google Scholar]
- 202.Fitzkee NC, Rose GD. Protein Sci. 2004;13:633–639. doi: 10.1110/ps.03503304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ding F, Jha RK, Dokholyan NV. Structure. 2005;13:1047–1054. doi: 10.1016/j.str.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 204.Tran HT, Wang X, Pappu RV. Biochemistry. 2005;44:11369–11380. doi: 10.1021/bi050196l. [DOI] [PubMed] [Google Scholar]
- 205.Jha AK, Colubri A, Freed KF, Sosnick TR. Proc Natl Acad Sci U S A. 2005;102:13099–13104. doi: 10.1073/pnas.0506078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Fitzkee NC, Rose GD. Proc Natl Acad Sci U S A. 2004;101:12497–12502. doi: 10.1073/pnas.0404236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Tran HT, Pappu RV. Biophys J. 2006;91:1868–1886. doi: 10.1529/biophysj.106.086264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Clementi C, Nymeyer H, Onuchic JN. Journal of Molecular Biology. 2000;298:937–953. doi: 10.1006/jmbi.2000.3693. [DOI] [PubMed] [Google Scholar]
- 209.Yang S, Cho SS, Levy Y, Cheung MS, Levine H, Wolynes PG, Onuchic JN. Proc Natl Acad Sci U S A. 2004;101:13786–13791. doi: 10.1073/pnas.0403724101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Dokholyan NV, Buldyrev SV, Stanley HE, Shakhnovich EI. Journal of Molecular Biology. 2000;296:1183–1188. doi: 10.1006/jmbi.1999.3534. [DOI] [PubMed] [Google Scholar]
- 211.Baldwin RL, Rose GD. Trends in Biochemical Sciences. 1999;24:77–83. doi: 10.1016/s0968-0004(98)01345-0. [DOI] [PubMed] [Google Scholar]
- 212.Bradley P, Chivian D, Meiler J, Misura KM, Rohl CA, Schief WR, Wedemeyer WJ, Schueler-Furman O, Murphy P, Schonbrun J, Strauss CE, Baker D. Proteins. 2003;53(Suppl 6):457–468. doi: 10.1002/prot.10552. [DOI] [PubMed] [Google Scholar]
- 213.Bowler BE. Mol Biosyst. 2007;3:88–99. doi: 10.1039/b611895j. [DOI] [PubMed] [Google Scholar]
- 214.Bennett MJ, Choe S, Eisenberg D. Proc Natl Acad Sci U S A. 1994;91:3127–3131. doi: 10.1073/pnas.91.8.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Liu Y, Eisenberg D. Protein Sci. 2002;11:1285–1299. doi: 10.1110/ps.0201402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Schymkowitz JW, Rousseau F, Wilkinson HR, Friedler A, Itzhaki LS. Nat Struct Biol. 2001;8:888–892. doi: 10.1038/nsb1001-888. [DOI] [PubMed] [Google Scholar]
- 217.Blake C, Serpell L. Structure. 1996;4:989–998. doi: 10.1016/s0969-2126(96)00104-9. [DOI] [PubMed] [Google Scholar]
- 218.Eakin CM, Attenello FJ, Morgan CJ, Miranker AD. Biochemistry. 2004;43:7808–7815. doi: 10.1021/bi049792q. [DOI] [PubMed] [Google Scholar]
- 219.Knaus KJ, Morillas M, Swietnicki W, Malone M, Surewicz WK, Yee VC. Nat Struct Biol. 2001;8:770–774. doi: 10.1038/nsb0901-770. [DOI] [PubMed] [Google Scholar]
- 220.Sanders A, Jeremy CC, Higgins LD, Giannini S, Conroy MJ, Hounslow AM, Waltho JP, Staniforth RA. J Mol Biol. 2004;336:165–178. doi: 10.1016/j.jmb.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 221.Johnson SM, Wiseman RL, Sekijima Y, Green NS, mski-Werner SL, Kelly JW. Acc Chem Res. 2005;38:911–921. doi: 10.1021/ar020073i. [DOI] [PubMed] [Google Scholar]
- 222.Khare SD, Caplow M, Dokholyan NV. Proc Natl Acad Sci U S A. 2004;101:15094–15099. doi: 10.1073/pnas.0406650101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Ray SS, Nowak RJ, Strokovich K, Brown RH, Walz T, Lansbury PT. Biochemistry. 2004;43:4899–4905. doi: 10.1021/bi030246r. [DOI] [PubMed] [Google Scholar]
- 224.Rao KS, Hegde ML, Anitha S, Musicco M, Zucca FA, Turro NJ, Zecca L. Prog Neurobiol. 2006;78:364–373. doi: 10.1016/j.pneurobio.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 225.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 227.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 228.Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. J Biol Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- 229.Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. J Biol Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 230.Bukau B, Weissman J, Horwich A. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 231.Berke SJ, Paulson HL. Curr Opin Genet Dev. 2003;13:253–261. doi: 10.1016/s0959-437x(03)00053-4. [DOI] [PubMed] [Google Scholar]
- 232.Rubinsztein DC. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 233.Keller JN, Gee J, Ding Q. Ageing Res Rev. 2002;1:279–293. doi: 10.1016/s1568-1637(01)00006-x. [DOI] [PubMed] [Google Scholar]
- 234.Lucking CB, Durr A, Bonifati V, Vaughan J, De MG, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A. N Engl J Med. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- 235.Han W, Wu YD. J Am Chem Soc. 2005;127:15408–15416. doi: 10.1021/ja051699h. [DOI] [PubMed] [Google Scholar]
- 236.Khare SD, Ding F, Gwanmesia KN, Dokholyan NV. PLoS Comput Biol. 2005;1:230–235. doi: 10.1371/journal.pcbi.0010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Marchut AJ, Hall CK. Comput Biol Chem. 2006;30:215–218. doi: 10.1016/j.compbiolchem.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 238.Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Nat Biotechnol. 2004;22:1302–1306. doi: 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- 239.Khare SD, Wilcox KC, Gong P, Dokholyan NV. Proteins. 2005;61:617–632. doi: 10.1002/prot.20629. [DOI] [PubMed] [Google Scholar]
- 240.Thompson MJ, Sievers SA, Karanicolas J, Ivanova MI, Baker D, Eisenberg D. Proc Natl Acad Sci U S A. 2006;103:4074–4078. doi: 10.1073/pnas.0511295103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Fowler DM, Koulov AV, ory-Jost C, Marks MS, Balch WE, Kelly JW. PLoS Biol. 2006;4:e6. doi: 10.1371/journal.pbio.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Bryant JE, Lecomte JT, Lee AL, Young GB, Pielak GJ. Biochemistry. 2005;44:9275–9279. doi: 10.1021/bi050786j. [DOI] [PubMed] [Google Scholar]
- 243.Gething MJ, Sambrook J. Nature. 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- 244.Hensley K, Mhatre M, Mou S, Pye QN, Stewart C, West M, Williamson KS. Antioxid Redox Signal. 2006;8:2075–2087. doi: 10.1089/ars.2006.8.2075. [DOI] [PubMed] [Google Scholar]
- 245.Games D, Buttini M, Kobayashi D, Schenk D, Seubert P. J Alzheimers Dis. 2006;9:133–149. doi: 10.3233/jad-2006-9s316. [DOI] [PubMed] [Google Scholar]
- 246.Harris DA, Chiesa R, Drisaldi B, Quaglio E, Migheli A, Piccardo P, Ghetti B. Clin Lab Med. 2003;23:175–186. doi: 10.1016/s0272-2712(02)00069-0. [DOI] [PubMed] [Google Scholar]
- 247.Sekijima Y, Dendle MA, Kelly JW. Amyloid. 2006;13:236–249. doi: 10.1080/13506120600960882. [DOI] [PubMed] [Google Scholar]
- 248.Wildegger G, Kiefhaber T. J Mol Biol. 1997;270:294–304. doi: 10.1006/jmbi.1997.1030. [DOI] [PubMed] [Google Scholar]
- 249.Itzhaki LS, Otzen DE, Fersht AR. J Mol Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 250.Riddle DS, Grantcharova VP, Santiago JV, Alm E, Ruczinski I, Baker D. Nat Struct Biol. 1999;6:1016–1024. doi: 10.1038/14901. [DOI] [PubMed] [Google Scholar]
- 251.Grantcharova VP, Riddle DS, Santiago JV, Baker D. Nat Struct Biol. 1998;5:714–720. doi: 10.1038/1412. [DOI] [PubMed] [Google Scholar]
- 252.Daggett V, Li A, Itzhaki LS, Otzen DE, Fersht AR. J Mol Biol. 1996;257:430–440. doi: 10.1006/jmbi.1996.0173. [DOI] [PubMed] [Google Scholar]
- 253.Du R, Pande VS, Grosberg AY, Tanaka T, Shakhnovich ES. Journal of Chemical Physics. 1998;108:334–350. [Google Scholar]
- 254.Grantcharova VP, Baker D. Biochemistry. 1997;36:15685–15692. doi: 10.1021/bi971786p. [DOI] [PubMed] [Google Scholar]
- 255.Dokholyan NV, Li L, Ding F, Shakhnovich EI. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8637–8641. doi: 10.1073/pnas.122076099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Northey JGB, Di Nardo AA, Davidson AR. Nature Structural Biology. 2002;9:126–130. doi: 10.1038/nsb748. [DOI] [PubMed] [Google Scholar]
- 257.Lazaridis T, Karplus M. Science. 1997;278:1928–1931. doi: 10.1126/science.278.5345.1928. [DOI] [PubMed] [Google Scholar]
- 258.Borreguero JM, Ding F, Buldyrev SV, Stanley HE, Dokholyan NV. Biophysical Journal. 2004;87:521–533. doi: 10.1529/biophysj.104.039529. [DOI] [PMC free article] [PubMed] [Google Scholar]