

Abstract
Escherichia coli reporter strains modeling the high-level type A and B vancomycin resistances of Enterococcus faecium BM4147 and Ent. faecalis have been developed to study the respective VanR-VanS two-component regulatory systems. PvanH-, PvanRa-, PvanY-, and PvanRb-lacZ fusions report on expression from the vancomycin-resistant enterococci promoters of the type A vanRSHAXYZ and type B vanRSYWHBX gene clusters. These strains also express from single-copy chromosomal genes vanRa, vanRb, or vanRSb behind their respective promoter (PvanRa or PvanRb) or vanSa or vanSb behind the rhamnose-inducible PrhaB. Results show that activation (phosphorylation) of the response regulator VanRa by its sensor kinase VanSa leads to transcriptional activation of both PvanH and PvanRa. Additionally, VanRb activates its cognate promoters PvanY and PvanRb, although this occurs only in the absence of VanSb and presumably is caused by VanRb phosphorylation by an unidentified endogenous E. coli kinase. Thus, VanSb interferes with activation of VanRb, probably by acting as a phospho-VanRb phosphatase. Although both VanRa and VanRb activate their cognate promoters, neither activates the heterologous PvanR, PvanH, or PvanY, arguing against the interchangeability of type A and B two-component regulatory switches in vancomycin-resistant enterococci. VanRa also is activated by the nonpartner kinase PhoR. Because this occurs in the absence of its inducing signal (Pi limitation), PhoR autophosphorylation apparently is regulated in vivo. Furthermore, the activation of VanRa caused by cross talk from PhoR in the absence of a signal allows distinction of cross talk from crossregulation as the latter, but not the former, responds to environmental cues.
Keywords: crossregulation/cross talk/PhoB/PhoR/sensor histidine kinase
Vancomycin-resistant enterococci (VRE) have become clinically problematic human pathogens with high mortality and incidence increasing alarmingly over 35-fold from 0.4% to 14% in hospital intensive-care units from 1989 to 1993 (1). Clinical isolates of VRE have been divided into three types based on the level of resistance (type A and B, high-level resistance; VanC, low- or moderate-level resistance), inducible versus constitutive resistance (type A and B, inducible; VanC, constitutive), and inducibility by both vancomycin and teicoplanin (type A) or inducibility by vancomycin only (type B; refs. 2–4). At the molecular level, types A and B resistance result from the same substitution of an ester linkage (d-Ala-d-lactate) for an amide linkage (d-Ala-d-Ala) at the termini of peptidoglycan chains involved in cell wall synthesis; VanC resistance instead involves a switch to d-Ala-d-Ser termini (5, 6).
Type A and B strains have six genes in common, ones encoding a two-component sensor kinase-response regulator system (VanSa, VanRa and VanSb, VanRb), three enzymes (VanH, VanA/VanB, and VanX) that bring about the switch to d-Ala-d-lactate peptidoglycan chains with a 1,000-fold lower affinity for vancomycin, and the d,d-carboxypeptidase (VanY). In type B VRE, vanY is upstream of vanH, whereas in type A the corresponding vanY comes after vanX. The consequence of this reordering is that while the promoters controlling the two-component regulatory system genes are PvanRa and PvanRb, respectively, the promoters for the structural genes are instead PvanH and PvanY (Fig. 1). Most previous genetic and enzymatic studies trying to define the molecular logic of how these proteins function in VRE have focused on the type A system. Courvalin and colleagues (7) also have demonstrated that mutations of type B VanSb can result in teicoplanin-inducible resistance.
Figure 1.
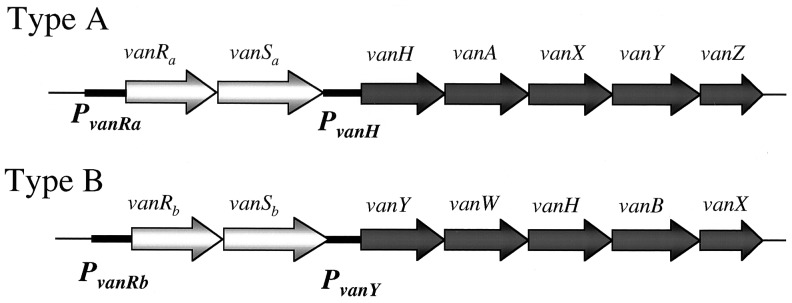
Organization of type A and type B VRE gene clusters.
To characterize the two-component VRE signal transducing proteins, we previously have studied VanSa and VanRa in vitro and in vivo by using Escherichia coli (8, 9). We now report studies on the in vivo function and selectivity of VanSa and VanRa along with similar ones of VanSb and VanRb on expression from the type A and B promoters (Fig. 1). We examined VanRa and VanRb for transcriptional activation of its cognate and noncognate promoters. We tested the requirements of VanSa and VanSb for activation (phosphorylation) of VanRa and VanRb. Additionally, we tested for cross talk between these heterologous VRE signaling proteins, comparing interactions between them to those with the response regulator PhoB and the sensor kinase PhoR of the E. coli Pho regulon (10) as a basis for distinguishing specific and nonspecific (cross talk) interactions. Our results indicate that, unlike the VanH, VanA/VanB and VanX enzymes required for vancomycin resistance, the type A and type B two-component regulatory systems evolved independently.
MATERIALS AND METHODS
Media, Chemicals, and Other Reagents.
Luria–Bertani, 3-(N-morpholino)propanesulfonic acid (Mops), and tryptone-yeast extract media are described elsewhere (11). Ampicillin was added at 100 μg per ml; kanamycin, at 12.5 or 50 μg per ml, gentamicin, at 5 or 15 μg per ml, and chloramphenicol, at 5 and 20 μg per ml to select antibiotic-resistant integrants or maintain plasmids, respectively. l-rhamnose was used at 1.1 mM for induction. After integration, the integrants were grown without an antibiotic. Pfu DNA polymerase (Stratagene) was used to generate DNAs for cloning. All PCR-amplified fragments were cloned and sequenced as double-stranded plasmid DNAs in a core facility.
Bacteria.
All bacteria are derivatives of E. coli K-12 BW13711 (11). Conditional replication (oriRR6Kγ) plasmids were propagated at low or high copy number in the pir+ host BW23473, its isogenic pir-116 host BW23474, or similar hosts (12, 13). BW24381 (lacIqrrnBT14 ΔlacZWJ16 ΔphoBR580 ΔcreABCD154 rpoS (Am) ΔaraBADAH33 ΔrhaBADLD78 Δ(ackA pta)160) is a derivative of BW23660 (9) into which the ΔrhaBADLD78 (14) and Δ(ackA pta)160 (9) mutations were introduced by P1kc transduction essentially as described elsewhere (15). Its derivative BW25124 was made by recombining the PvanH-lacZ fusion in pJS110 onto the chromosome via allele replacement (13) and then introducing the ΔphoR574 mutation (9) by P1kc transduction. pJS110 was made by cloning the PstI–BsiWI, PvanH-lacZ DNA fragment from pJS79 into the allele-replacement lacZ transcriptional fusion vector pWJ18 (W. Jiang, A.H., and B.L.W., unpublished results).
Construction of lacZ Transcriptional Fusions.
The PvanH and PvanRa promoters were PCR-amplified by using pAT87 (16) as template and the primer JS-P27 (GCAGTCGAC-CGGAAAGCAATGATAACTATACGACG) with JS-P29 (GCAGGATCC-TCTGAAGAACGAAAACGGCTCGTTC) and JS-P31 (GCAGTCGAC-ATGTATCTAGGGCTTCATTATACAGG) with JS-P32 (GCAGGATCC-CTTAATAATTTATCAGATTATAGGGCCG), respectively (extensions containing a restriction site precede hyphens). The 240-bp PvanH and 255-bp PvanRa PCR products were cloned as BamHI–SalI fragments into pAH125 to make pJS79 and pJS81, respectively. The PvanY and PvanRb regions were amplified by using genomic DNA of Ent. faecalis V583 (from Daniel Sahm, Jewish Hospital, St Louis, MO; ref. 17) as template and the primer JS-P21 (GCAGTCGAC-TCACAGATATTCCAGCCGGACAAATTGTCC) with JS-P26 (GCAGGATCC-TTTGCAATAAAACTACGATTTGTGGC) and JS-P15 (GCAGTCGAC-TTAAACGGTATATTTCGGAAGAAC) with JS-P19 (GCAGGATCC-ATTTAAGAAGATAACATAACAGTCTG), respectively. The 554-bp PvanY and 182-bp PvanRb PCR products were similarly cloned into pAH125 to make pJS52 and pJS51, respectively. To integrate these plasmids at attP21, a fragment encoding chloramphenicol resistance of pCAH54 (18) and attPP21 of pAH95 (A.H. and B.L.W., unpublished results) was introduced by exchanging the NheI–SphI fragments with the corresponding one in pIADL46 (from Ivan Lessard, Harvard Medical School, Boston, MA) to make pJS94, pJS96, pJS97, and pJS98, respectively. pAH125 is a derivative of pSK49 (12) for making lacZ transcriptional fusions (S.-K. Kim, A.H., and B.L.W., unpublished results).
Plasmids.
pAH85, pAH66, and pAH67 (12) are derivatives of pSK49 that synthesize PhoBwt, PhoBM17V, E87D, and PhoBT97A, respectively, under PphoB control. pAH151 synthesizes PhoR under PrhaB control (14) in a pSK49 derivative with attPHK022 and encoding gentamicin resistance of pAH70 and pAH143 (A.H. and B.L.W., unpublished results). pMP1 is a derivative of pSK49 carrying the same PrhaB-phoR+ fusion. pJS87 synthesizes full-length VanSa with a C-terminal His6 tag (vanSH6) under PrhaB control. pMP3 is a derivative of pSK49 carrying the same PrhaB-vanSH6 fusion. The vanSH6 fragment was constructed by PCR using pSLF40 (9) and the primers JS-P3 (GAATTC-TGTACGGAGTCAAGCCATATGTTG) and JS-P7 (A ATAGTCGAC-TTATTAgtggtggtggtggtggtgGGACCTCCTTTTATCAACCAAGTC) according to conditions above (the His6 coding region is in lowercase). The 1,216-bp vanSH6 product was cloned into pUC19 as an EcoRI–SalI fragment to make pJS60 for sequencing and then into pAH151 as an NdeI–SalI fragment to make pJS87. pJS126 is a derivative of pSLF55 with attPP21 and encoding chloramphenicol resistance pSLF55 synthesizes VanRa from its native promoter (9). pMP2 is a derivative of pSK49 with the PrhaB-vanSb fusion. pJS107 and pJS109 are similar plasmids that express vanRb and vanRSb, respectively, behind PvanRb. The vanRb and vanSb fragments were generated by using Ent. faecalis V583 genomic DNA and the primer JS-P15 (GCAGTCGAC-TTAAACGGTATATTTCGGAAGAAC) with JS-P16 (CGCTCTAGA-ATAAGACACAAATTGCTGTGC) and JS-P17 (GCACTGCAG-CATATGACCATGGCGGGTGTAGGTTACCGATTGG) with JS-P18 (TTCTCTAGA-TTGTTTCATATGCCGTTTGTG), respectively. The 964-bp vanRb and 1,641-bp vanSb PCR products were cloned with SalI and XbaI or PstI and XbaI into pAH143, to make pJS45 and pJS46, respectively. The PvanRb-vanRb fragment was subcloned as an SphI–BamHI fragment from pJS45 into pAH85 to make pJS107. The SacII–XbaI vanSb fragment was subcloned from pJS46 into pJS45 to make the PvanRb-vanRSb plasmid pJS47. The PvanRb-vanRSb fragment then was subcloned as an SphI–BamHI fragment from pJS47 into pAH85 to make pJS109.
attP Plasmid Integration and PCR Testing of Copy Number.
Plasmids were integrated into the chromosome of cells expressing the respective integrase and verified by PCR to have a single integrated plasmid as described elsewhere (9, 12, 18).
Cell Growth and Enzyme Assay.
Cells were grown and assayed for bacterial alkaline phosphatase and β-galactosidase (BG) as described (11). Units are nanomoles of product made per min per cell culture at OD420 at 37°C and 28°C, respectively.
Purification of VanSb and Protein Phosphorylation Experiments.
A maltose binding protein (MBP) fusion to the cytosolic domain of VanSb (′VanSb, residues V146M to L447) was constructed by using pJS46 as template and the primer MKP5 (GCAGGTGTGGGATTGCTTCATATG-GGGCTGACAATTCGG) with MKP6 (GTACCCGGGGATCC-TCTAGATTGTTTCATATGC). The 1,224-bp ′vanSb fragment was cloned with NdeI into a derivative of pMAL-c (New England Biolabs) called pIADL16 (from I. Lessard) to make pJS119. Overproduction, purification, and autophosphorylation of MBP-′VanSb was accomplished as described for MBP-′VanSa (19).
RESULTS
An Improved E. coli Reporter System for Studying VRE Two-Component Systems.
lacZ transcriptional fusions to PvanH and PvanRa were made to improve on the signal-to-noise ratio of previously engineered strains. The basal levels of earlier PvanH- and PvanRa-lacZ fusions ranged from 28.5 ± 9.8 to 69.3 ± 6.4 BG units (BGU; ref. 9). These elevated levels interfered with their use in experiments directed toward probing structural determinants for VanRS interactions. Their high background probably resulted from an RNase III processing site upstream of lacZ in pWJ19 that stabilizes the mRNA and is derived from pTL61 (20). The new fusions lack this site as the lacZ junction in pAH125 is from pRS415 (21). Basal levels of the new type A fusions in the absence of VanRa were 0.7 ± 0.1 BGU for PvanH and 12.8 ± 0.9 BGU for PvanRa (Table 1). The basal levels of the type B PvanY-lacZ and PvanRb-lacZ fusions were 7.6 ± 1.2 and 8.5 ± 0.9 BGU, respectively (Table 2).
Table 1.
Activation of PvanH and PvanRa by VanRa
| Regulator, kinase | lacZ fusion* | β-Galactosidase Sp
Act
|
|
|---|---|---|---|
| −Rha | +Rha | ||
| None | PvanH | 0.7 ± 0.1 | N.D. |
| VanRa | PvanH | 0.9 ± 0.3 | N.D. |
| VanRa, PrhaB-vanSa | PvanH | 0.8 ± 0.1 | 31.2 ± 4.1 |
| None | PvanRa | 12.8 ± 0.9 | N.D. |
| VanRa | PvanRa | 14.7 ± 1.7 | N.D. |
| VanRa, PrhaB-vanSa | PvanRa | 10.4 ± 1.7 | 46.7 ± 4.2 |
Cells were assayed after 16–20 hr growth in 0.10% glycerol-Mops-2 mM Pi medium without (−Rha) or with (+Rha) rhamnose. Specific activity (Sp Act) units are nanomoles of product formed per cell optical density at 420 nm. Means of three or more determinations with SD are given. N.D., not determined.
Strains are integrants of BW24381 with single copies of the plasmids in parenthesis: JCS182 (pJS94), JCS184 (pJS96), JCS196 (pJS94, pSLF55), JCS207 (pJS96, pSLF55), JCS230 (pJS94, pSLF55, pJS87), and JCS345 (pJS96, pSLF55, pJS87).
Table 2.
Activation of PvanRb and PvanY by VanRb
| Regulator, kinase | lacZ fusion* | Carbon source | β-Galactosidase Sp Act
|
|
|---|---|---|---|---|
| −Rha | +Rha | |||
| None | PvanY | Fru | 7.6 ± 1.2 | N.D. |
| VanRb | PvanY | Fru | 42.5 ± 3.8 | N.D. |
| VanRSb | PvanY | Fru | 2.6 ± 0.5 | N.D. |
| None | PvanRb | Fru | 8.5 ± 0.9 | N.D. |
| VanRb | PvanRb | Fru | 40.8 ± 0.5 | N.D. |
| VanRSb | PvanRb | Fru | 2.3 ± 0.4 | N.D. |
| VanRb, PrhaB-vanSb | PvanY | Gly | 59.2 ± 3.0 | 6.7 ± 0.8 |
| VanRb, PrhaB-vanSb | PvanY | Fru | 44.7 ± 2.6 | 12.5 ± 1.3 |
| VanRb, PrhaB-vanSb | PvanY | Glu | 47.9 ± 0.4 | 40.5 ± 2.2 |
| VanRa | PvanY | Fru | 8.7 ± 1.3 | N.D. |
| VanRa, PrhaB-vanSa | PvanY | Fru | 7.4 ± 0.8 | 7.6 ± 1.0 |
| VanRa | PvanRb | Fru | 11.0 ± 1.4 | N.D. |
| VanRa, PrhaB-vanSa | PvanRb | Fru | 8.4 ± 0.9 | 8.7 ± 0.7 |
| VanRb | PvanH | Fru | 0.9 ± 0.5 | N.D. |
| VanRb | PvanRa | Fru | 13.6 ± 3.5 | N.D. |
Cells were assayed after growth in 0.06% glucose (Glu)-, 0.10% glycerol (Gly)-, or 0.06% fructose (Fru)-Mops-2 mM Pi medium as in Table 1. N.D., not determined.
Strains are integrants of BW24381: JCS186 (pJS97), JCS188 (pJS98), JCS197 (pJS97, pSLF55), JCS198 (pJS98, pSLF55), JCS199 (pJS94, pJS107), JCS200 (pJS96, pJS107), JCS201 (pJS97, pJS107), JCS202 (pJS98, pJS107), JCS237 (pJS97, pJS107, pJS102), JCS240 (pJS97, pJS109), JCS241 (pJS98, pJS109), JCS347 (pJS97, pSLF55, pJS87), and JCS349 (pJS98, pSLF55, pJS87).
It is generally more meaningful to study regulatory effects when the relevant genes are in single copy. This is especially true in studies of the VRE and Pho two-component systems. Abnormal regulatory effects have been repeatedly observed when using multicopy phoB, phoR, or vanS plasmids, which presumably result from effects of plasmid copy number on synthesis of their gene products (15, 22, 23). It therefore was critical to construct reporter strains in which both the promoter fusions and regulatory genes are in single copy. The use of strains with the vanR and vanS genes stably recombined on the chromosome also afforded tighter control of these regulatory genes and circumvented problems that can arise from inconsistent gene dosages.
All reporter strains express vanR alone or together with vanS from its native promoter (PvanRa or PvanRb). Many express vanR from its promoter and vanS or phoR under tight control from the rhamnose-inducible promoter PrhaB (14). These genes and fusions were recombined onto the chromosome by use of conditional replication, integration, and modular plasmids that have different phage attachment (attP) sites and antibiotic resistances (refs. 9, 12, 14, and 18; Materials and Methods; A.H. and B.L.W., unpublished results). The promoter-lacZ fusions were recombined onto the chromosome either by use of an analogous lacZ vector or one that provides homologous sequences for allele replacement at the lac locus.
Activation of PvanH and PvanRa by VanRa and VanSa.
Previous studies had shown that VanRa and VanSa both were required for ca. 6- to 45-fold activation of a PvanH-lacZ fusion in E. coli (9). Although no activation of a PvanRa-lacZ fusion was apparent in that study, the basal level was high. As shown in Table 1, the new PvanH-lacZ fusion was activated ca. 40-fold only in the presence of both VanRa and VanSa and the new PvanRa-lacZ fusion was similarly activated ca. 5-fold. Apparently, the high basal activities of earlier fusion strains interfered with detecting activation of PvanRa by VanRa. Activation of PvanRa is consistent with phospho-VanRa (P-VanRa) binding PvanRa with higher affinity than free VanRa (19).
Activation of PvanY and PvanRb by VanRb.
Both the PvanY- and PvanRb-lacZ fusions show ca. 5-fold activation by VanRb. Unexpectedly, this activation occurs in the absence of VanSb (Table 2). Many response regulators are phosphorylated by nonpartner histidine kinases (24). It therefore is likely that VanRb activation results from its being phosphorylated by an unidentified E. coli kinase. Although VanRb also might have been activated by acetyl phosphate (25), this was ruled out here because our reporter strains are genetically blocked in acetyl phosphate synthesis.
Evidence of an in Vivo VanSb Phosphatase.
Many two-component sensor proteins act both as a kinase and as phosphatase toward their partner response regulators. Using analogous reporter strains, we previously showed that VanSa acts as a P-VanRa phosphatase by demonstrating that VanSa blocked activation of VanRa by PhoR or acetyl phosphate (9). Accordingly, VanSb was tested for interference with the activation of VanRb. As shown in Table 2, no activation of the PvanY- or PvanRb-lacZ fusion was apparent when vanRb and vanSb were expressed together from PvanRb. Similar ca. 17-fold decreases were seen for both type B VRE promoters. Curiously, the expression levels of both fusions in presence of PvanRb-vanRSb were reduced ca. 3-fold below the basal levels in the absence of VanRb, suggesting that free VanRb might act as a repressor. Further evidence that VanSb is responsible for inhibiting activation comes from experiments in which vanRb and vanSb were expressed independently. The PrhaB-vanSb strains also showed reduced activation of PvanY with the levels varying inversely with catabolite repression conditions. Glycerol results in the lowest, fructose results in an intermediate, and glucose results in the highest catabolite repression of PrhaB (14). In agreement, greatest inhibition occurred upon induction in glycerol medium and the lowest, in glucose medium (Table 2). Together, these data suggest that VanSa and VanSb are poised at different kinase-phosphatase set points; VanSa acts as a kinase under conditions where VanSb is a phosphatase. Finally, VanSa and PhoR were tested for phosphatase function toward P-VanRb. As expected, no interference by them was seen.
Promoter Specificity of the VRE Response Regulators.
Both type A and type B VRE promoters were tested for activation by their cognate and noncognate response regulators. Conditions leading to activation of PvanH and PvanRa by VanRa (Table 1) were without effect on expression of PvanY or PvanRb (Table 2). Likewise, conditions leading to activation of PvanY and PvanRb by VanRb were without effect on PvanH and PvanRa (Table 2).
In Vivo Competition Between VanRa and PhoB.
Previous kinetic studies of MBP-′VanSa with VanRa and PhoB provided a quantitative estimate of the overall efficiency and specificity of phosphotransfer from P-VanSa (8). By acting as a slow heterologous substrate for P-MBP-′VanSa with a kxfer of 0.2 min−1 and KM of 95 ± 30 μM, PhoB was a competitive inhibitor of P-VanR formation. In those studies, VanRa had a kxfer of ca. 60 min−1 and KM of 3 μM for phosphotransfer from P-MBP-′VanSa. Experiments therefore were done to see whether competition occurs between VanRa and PhoB in vivo. As shown in Fig. 2, VanSa activated phoA and PvanH-lacZ expression ca. 1,120- and 18-fold, respectively, when only PhoB or VanRa was present. When both were present, VanSa activated phoA and PvanH-lacZ expression ca. 760- and 36-fold, respectively. Although a modest reduction of PhoB activation by VanSa occurred in the presence of VanRa, it is unclear why VanRa was stimulated, as these experiments were carried out in the absence of deliberate stimulation of VanSa.
Figure 2.

Competition between VanR and PhoB. Expression of phoA (encoding bacterial alkaline phosphatase, Bap) and a PvanH-lacZ fusion report on PhoB and VanRa activation, respectively. VanSa or PhoR were synthesized under PrhaB control in cells expressing phoB, vanRa, or both behind its native promoter. Bap and BG were assayed as in Table 1 in medium with rhamnose and 2 mM (Excess) or 0.1 mM (Limited) Pi. Arrows on the right are in proportion to Sp Act values (nmol product made per min per OD420). With no rhamnose, Bap and BG Sp Act values of all strains were ca. 0.1 ± 0.1 and 1.0 ± 0.2, respectively. Strains are integrants of BW25053 (ΔphoBR) or BW25124 (ΔphoR): JCS289 (pJS126, pMP1), JCS297 (pMP3), JCS304 (pJS126, pMP3), JCS338 (pMP1), and JCS342 (pMP3).
Results from similar experiments with PhoR provided convincing evidence of in vivo competition. With excess Pi, PhoR activated phoA and PvanH-lacZ expression ca. 7- and 90-fold, respectively, when only PhoB or VanRa was present (Fig. 2). Activation of phoA under these conditions was an apparent consequence of PhoR overproduction from PrhaB (data not shown). When both PhoB and VanRa were present, activation of VanRa by PhoR was <2-fold, whereas activation of PhoB was unaffected. The ca. 40-fold reduced activation of PvanH caused by PhoB indicates that a PhoB-PhoR interaction interferes with PhoR autophosphorylation when Pi is in excess.
Under limited Pi conditions, PhoR activated phoA and PvanH-lacZ expression ca. 2,900- and 90-fold, respectively, when only PhoB or VanRa was present (Fig. 2). High activation of PhoB under these conditions was anticipated. These results also showed that activation of VanRa by PhoR is unresponsive to Pi limitation. In agreement, competition between PhoB and VanRa occurred with limited Pi as well. An ca. 2,600-fold activation of phoA expression was accompanied with only a ca. 7-fold activation of PvanH-lacZ expression when both response regulators were present. That is, ca. 13-fold reduction of PvanH-lacZ expression resulted because of the presence of PhoB.
Absence of Cross Talk from VanSb in Vivo.
VanSb was tested for activation of both VanRa and PhoB. No activation of VanRa by VanSb occurred under conditions leading to activation of VanRa by VanSa or PhoR (Table 3). VanSb also was tested for activation of PhoBwt, PhoBM17V, E87D, and PhoBT97A. The latter are altered recognition (AR) mutants showing 200- to 400-fold enhanced activation by ′VanSa (12). Yet, no activation by VanSb occurred (data not shown).
Table 3.
Activation of VanRa and VanRb by VanSa, VanSb, and PhoR
| Regulator, kinase | lacZ fusion* | Carbon source | β-Galactosidase Sp Act
|
|
|---|---|---|---|---|
| −Rha | +Rha | |||
| None | PvanH | Fru | 0.7 ± 0.1 | N.D. |
| VanRa | PvanH | Fru | 0.9 ± 0.3 | N.D. |
| VanRa, PrhaB-vanSa | PvanH | Fru | 0.7 ± 0.0 | 15.2 ± 1.9 |
| VanRa, PrhaB-vanSb | PvanH | Fru | 0.8 ± 0.1 | 0.8 ± 0.1 |
| VanRa, PrhaB-vanSb | PvanH | Gly | 0.7 ± 0.1 | 0.7 ± 0.1 |
| VanRa, PrhaB-phoR | PvanH | Fru | 0.7 ± 0.2 | 21.2 ± 1.3 |
| VanRa, PrhaB-phoR | PvanH | Gly | 0.8 ± 0.2 | 47.6 ± 8.4 |
| None | PvanY | Fru | 7.6 ± 1.2 | N.D. |
| VanRb | PvanY | Fru | 42.5 ± 3.8 | N.D. |
| VanRb, PrhaB-vanSa | PvanY | Fru | 38.7 ± 2.4 | 43.1 ± 1.8 |
| VanRb, PrhaB-vanSa | PvanY | Gly | 22.7 ± 2.7 | 37.4 ± 3.3 |
| VanRb, PrhaB-phoR | PvanY | Fru | 32.2 ± 2.3 | 34.2 ± 1.1 |
| VanRb, PrhaB-phoR | PvanY | Gly | 26.0 ± 5.2 | 37.0 ± 2.6 |
Cells were assayed as in Table 2. N.D., not determined.
Strains are integrants of BW24381; JCS182 (pJS94), JCS186 (pJS97), JCS196 (pJS94, pSLF55), JCS201 (pJS97, pJS107), JCS229 (pJS94, pSLF55, pAH151), JCS230 (pJS94, pSLF55, pJS87), JCS231 (pJS94, pSLF55, pJS102), JCS238 (pJS97, pJS107, pJS87), and JCS239 (pJS97, pJS107, pAH151).
In Vitro Analysis of VanSb.
Unable to demonstrate VanSb kinase in vivo, an MBP-′VanSb fusion protein was purified by using the same strategies used to study both VanSa (26) and PhoR (M.K.P., J.C.S., B.L.W., and C.T.W., unpublished results). Consistent with the in vivo results, MBP-′VanSb showed no autophosphorylation in the absence or presence of detergents intended to facilitate proper folding (0.002 to 1.0% SDS, Brij 58, or Nonidet P-40). To investigate whether the truncation or overproduction of MBP-′VanSb was deleterious to its folding, full-length VanSb also was overproduced as an N-terminal, His-tagged, or MBP-VanSb fusion protein. In no case would VanSb undergo autophosphorylation, even in the membrane fraction. Although it is possible that VanSb kinase is particularly labile, it is also reasonable to suppose that under these conditions VanSb is heavily biased as a P-VanRb phosphatase.
DISCUSSION
Although we previously have reported in vitro studies on purified native VanRa and an MBP-′VanSa fusion protein to the VanSa cytoplasmic domain (8, 19, 26) and in vivo studies on VanRa and VanSa in E. coli (9, 15), little information has been available describing in vitro and in vivo features of the type B regulatory proteins (17). When PvanRa- and PvanH-lacZ fusions were tested for activation by VanRa and VanSa in previous E. coli in vivo studies (9), activation of PvanH, but not PvanRa, was observed. Using reporter strains that provide improved signal-to-noise ratios, a 5-fold increase in PvanRa expression was seen when VanSa synthesis was driven from the tightly regulated PrhaB (14). This PvanRa response is about one-eighth as strong as the 40-fold activation of PvanH. These E. coli strains now provide a more faithful readout of the type A VRE promoters.
Initial results on VanRb in E. coli showed ca. 5-fold activation of both type B VRE promoters, PvanRb and PvanY, in the absence of VanSb, consistent with the phosphorylation of VanRb by an unidentified endogenous E. coli kinase. This cross talk to VanRb is in contrast to the behavior of VanRa in E. coli where no significant activation of PvanRa or PvanH has been observed in the absence of VanSa. That VanRa and VanRb are recognized by different E. coli kinases was unexpected; it is explicable by the fact that VanRa and VanRb share only 34% sequence similarity whereas the VanH, VanA/VanB and VanX resistance enzymes share more than 67% sequence similarity (17).
A second distinction between the type A and B VRE systems in E. coli was the behavior of the sensor kinases. In these studies, VanSb exhibited only a phosphatase and no discernable kinase activity toward its cognate response regulator. In contrast, VanSa exhibited both kinase and phosphatase activity under similar conditions (9). The issue of balance between phosphoaspartyl-response regulator-phosphatase and phosphohistidinyl-autophosphorylation-kinase has been noted elsewhere, but only for VanSa rather than VanSb (Fig. 3). While studying control of VanRa by VanSa in Ent. faecium BM4147, Arthur et al. (27) showed that VanRa was constitutively active in the absence of VanSa, presumably because of cross talk from an unidentified enterococcal kinase. In the presence of VanSa and absence of vancomycin or teicoplanin, VanSa acted predominantly as a phosphatase to reduce P-VanRa-dependent transcription. Only in the presence of a glycopeptide inducer did VanSa shift from a phosphatase to kinase.
Figure 3.

Signaling in the VanS-VanR phosphorelays and activation of the type A and B VRE promoters. Sensor kinase reaction scheme involves its autophosphorylation and phosphotransfer to the response regulator (thick solid arrows) leading to activation of the respective promoters (thick dashed arrow). Phosphatase reaction of the sensor kinase dephosphorylates the cognate response leading to down-regulation of the promoter in the absence of external stimulus (thin dashed arrows). While VanSa has both kinase and phosphatase activities, whether VanSb has a kinase activity has not been established (see text).
In this study, VanSb apparently was locked as a phosphatase. To what degree this reflects its true phosphatase/kinase balance in VRE remains to be determined. Along with the unrelieved VanSb phosphatase, there was no indication that VanSb phosphorylates VanRa or PhoB. In contrast, VanSa phosphorylates both. The inability of VanSb to activate PhoB was especially surprising. Under similar conditions, five other nonpartner kinases activate PhoBwt and PhoBAR, making VanSb the sole exception (A.H., S.-K. Kim, and B.L.W., unpublished results). Because of amplification of PhoB synthesis resulting from its autogenous positive control, PhoB activation should provide a very sensitive test for cross talk (10), providing further evidence that VanSb is in a kinase inactive state under these conditions. Although the absence of kinase activity for VanSb is an anomaly, more rigorous experiments are required to prove that VanSb is not a kinase. Nevertheless, these results support the hypothesis that the VanSb-VanRb system is regulated largely or exclusively through a phosphatase on/off balance rather than phosphatase/kinase balance. Accordingly, VanSb might act solely as a negative regulator of the type B system, whereby its phosphatase activity would be inhibited in the presence of vancomycin, thus permitting phosphorylation of VanRb by a different kinase in VRE and allowing P-VanRb to act as a positive regulator.
VanRa and VanRb are much less similar than the type A and B VanH, VanA/VanB and VanX enzyme pairs. Likewise, VanS similarities are low, with only 23% similarity between them (17). The disparity between the high similarities for the triad of peptidoglycan-modifying enzymes VanHAX and the low similarities for the regulatory proteins suggests that there was a single origin for VanHAX in VRE, e.g., from one of the vancomycin-producer organisms (28), but that distinct signaling systems were recruited independently for inducible resistance. This teleological explanation for the differences between type A and type B VRE is now supported by several complementary findings. Fig. 3 illustrates different inducers. The failure of type B VRE to acquire teicoplanin resistance results from an inability of teicoplanin to cause induction (7), presumably reflecting different ligands binding to VanSa and VanSb. Additionally, no apparent cross talk occurs (Fig. 4), so that mismatches of VanSa-VanRb or VanSb-VanRa would not produce peptidoglycan alterations. Finally, even though both pairs of VRE promoters function in E. coli, there was no cross activation.
Figure 4.

Type A and B VRE two-component regulatory circuits. A signal transduction event from one sensor (VanSa or VanSb) to its cognate response regulator (VanRa or VanRb) leads to activation of the type A (PvanH or PvanRa) and type B (PvanY or PvanRb) promoters without apparent cross talk between the two systems.
It remains uncertain whether VanSa or PhoR activates VanRb by cross talk. Because VanRb is activated by an unknown kinase in E. coli, only modest effects because of VanSa (1.6-fold) or PhoR (1.4-fold) were seen (Table 3). Greater effects might not have been evident if PvanY expression were already near maximally activated under these conditions.
Activation of VanRa by PhoR and PhoB by VanSa are clear examples of cross talk (Table 3, Fig. 2). It is noteworthy that high activation of VanRa by PhoR occurred only in the absence of PhoB. In an earlier study, PhoB was activated by the kinase EnvZ only in the absence of its cognate response regulator OmpR (23). Although activation of PhoB by EnvZ failed to respond to an environmental signal(s), the significance of that finding was unclear as the ligand(s) that stimulates EnvZ is unknown (29). It therefore is intriguing that activation of VanRa by PhoR was strongly inhibited by PhoB under both excess and limited Pi conditions. These results also show that PhoR autophosphorylation (or P-PhoR phosphotransfer) is somehow down-regulated under Pi excess conditions in a PhoB-dependent manner. Such an effect supports the model that PhoB and PhoR exist in an “inhibition complex” under these conditions (30). The absence of normal (ca. 2,000-fold) control by Pi over activation of VanRa by PhoR suggests that interactions between nonpartner proteins because of cross talk are unregulated. Yet, interactions between some nonpartner proteins are clearly physiologically regulated. Activation of PhoB by the kinase CreC responds to unknown catabolites (31). Likewise, nonpartner interactions between the NarX-NarL and NarQ-NarP two-component proteins are regulated (32). Hence, nonpartner interactions indicative of cross talk would appear to be uncontrolled whereas those indicative of “cross-regulation” would be physiologically regulated (25, 33).
There is substantial interest in developing inhibitors to one or more of the five necessary and sufficient proteins in the type A and type B VRE pathways to obviate vancomycin resistance (34). The sensor kinase-response regulator transcription factors are reasonable sites to target for interdiction of the signal that leads to glycopeptide antibiotic resistance. This work to elucidate the apparent differences between the VanRSa and VanRSb systems ultimately may help to define a compound that will inhibit both type A and B VRE. Thus, inhibitors of the Bacillus subtilis KinA-SpoOF two-component regulatory system recently have been shown to be antibacterial against Ent. faecalis ATCC 29212 and Ent. faecalis OC3041. It will be interesting to see whether compounds such as RWJ-49815 inhibit the VanRSa and VanRSb pairs equivalently, studies that will be enabled by using E. coli reporter systems similar to ones described here in in vivo assays.
Acknowledgments
We thank Daniel Sahm and Ivan Lessard for samples and Marisa Perez for assistance. This research was supported by National Institutes of Health Grants GM49338 to C.T.W., GM57695 and F33-AI10093 to B.L.W. and grants from Abbott Laboratories to C.T.W. and B.L.W.
ABBREVIATIONS
- BG
β-galactosidase
- MBP
maltose binding protein
- VRE
vancomycin-resistant enterococci
References
- 1. Swartz M N. Proc Natl Acad Sci USA. 1994;91:2420–2427. doi: 10.1073/pnas.91.7.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leclercq R, Derlot E, Duval J, Courvalin P. N Engl J Med. 1988;319:157–161. doi: 10.1056/NEJM198807213190307. [DOI] [PubMed] [Google Scholar]
- 3.Leclercq R, Derlot E, Weber M, Duval J, Courvalin P. Antimicrob Agents Chemother. 1989;33:10–15. doi: 10.1128/aac.33.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reynolds P E, Snaith H A, Maguire A J, Dutka-Malen S, Courvalin P. Biochem J. 1994;301:5–8. doi: 10.1042/bj3010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arthur M, Courvalin P. Antimicrob Agents Chemother. 1993;37:1563–1571. doi: 10.1128/aac.37.8.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park I S, Lin C H, Walsh C T. Proc Natl Acad Sci USA. 1997;94:10040–10044. doi: 10.1073/pnas.94.19.10040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baptista M, Depardieu F, Reynolds P, Courvalin P, Arthur M. Mol Microbiol. 1997;25:93–105. doi: 10.1046/j.1365-2958.1997.4401812.x. [DOI] [PubMed] [Google Scholar]
- 8.Fisher S L, Kim S-K, Wanner B L, Walsh C T. Biochemistry. 1996;35:4732–4740. doi: 10.1021/bi9525435. [DOI] [PubMed] [Google Scholar]
- 9.Haldimann A, Fisher S L, Daniels L L, Walsh C T, Wanner B L. J Bacteriol. 1997;179:5903–5913. doi: 10.1128/jb.179.18.5903-5913.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wanner B L. In: Escherichia coli and Salmonella typhimurium Cellular and Molecular Biology. Neidhardt F C, Curtiss III R, Ingraham J L, Lin E C C, Low K B Jr, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Washington, DC: ASM Press; 1996. pp. 1357–1381. [Google Scholar]
- 11.Wanner B L. In: Methods in Molecular Genetics. Adolph K W, editor. Vol. 3. Orlando: Academic; 1994. pp. 291–310. [Google Scholar]
- 12.Haldimann A, Prahalad M K, Fisher S L, Kim S-K, Walsh C T, Wanner B L. Proc Natl Acad Sci USA. 1996;93:14361–14366. doi: 10.1073/pnas.93.25.14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Metcalf W W, Jiang W, Daniels L L, Kim S-K, Haldimann A, Wanner B L. Plasmid. 1996;35:1–13. doi: 10.1006/plas.1996.0001. [DOI] [PubMed] [Google Scholar]
- 14.Haldimann A, Daniels L L, Wanner B L. J Bacteriol. 1998;180:1277–1286. doi: 10.1128/jb.180.5.1277-1286.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fisher S L, Jiang W, Wanner B L, Walsh C T. J Biol Chem. 1995;270:23143–23149. doi: 10.1074/jbc.270.39.23143. [DOI] [PubMed] [Google Scholar]
- 16.Arthur M, Molinas C, Courvalin P. J Bacteriol. 1992;174:2582–2591. doi: 10.1128/jb.174.8.2582-2591.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evers S, Courvalin P. J Bacteriol. 1996;178:1302–1309. doi: 10.1128/jb.178.5.1302-1309.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu F, Schumacher M A, Arvidson D N, Haldimann A, Wanner B L, Zalkin H, Brennan R G. Biochemistry. 1997;37:971–982. doi: 10.1021/bi971942s. [DOI] [PubMed] [Google Scholar]
- 19.Holman T R, Wu Z, Wanner B L, Walsh C T. Biochemistry. 1994;33:4625–4631. doi: 10.1021/bi00181a024. [DOI] [PubMed] [Google Scholar]
- 20.Linn T, St. Pierre R. J Bacteriol. 1990;172:1077–1084. doi: 10.1128/jb.172.2.1077-1084.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simons R W, Houman F, Kleckner N. Gene. 1987;53:85–96. doi: 10.1016/0378-1119(87)90095-3. [DOI] [PubMed] [Google Scholar]
- 22.Wanner B L, Chang B-D. J Bacteriol. 1987;169:5569–5574. doi: 10.1128/jb.169.12.5569-5574.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim S-K, Wilmes-Riesenberg M R, Wanner B L. Mol Microbiol. 1996;22:135–147. doi: 10.1111/j.1365-2958.1996.tb02663.x. [DOI] [PubMed] [Google Scholar]
- 24.Ronson C W, Nixon B T, Ausubel F M. Cell. 1987;49:579–581. doi: 10.1016/0092-8674(87)90530-7. [DOI] [PubMed] [Google Scholar]
- 25.Wanner B L. J Bacteriol. 1992;174:2053–2058. doi: 10.1128/jb.174.7.2053-2058.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wright G D, Holman T R, Walsh C T. Biochemistry. 1993;32:5057–5063. doi: 10.1021/bi00070a013. [DOI] [PubMed] [Google Scholar]
- 27.Arthur M, Depardieu F, Gerbaud G, Galimand M, Leclercq R, Courvalin P. J Bacteriol. 1997;179:97–106. doi: 10.1128/jb.179.1.97-106.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marshall C G, Broadhead G, Leskiw B K, Wright G D. Proc Natl Acad Sci USA. 1997;94:6480–6483. doi: 10.1073/pnas.94.12.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pratt L A, Hsing W H, Gibson K E, Silhavy T J. Mol Microbiol. 1996;20:911–917. doi: 10.1111/j.1365-2958.1996.tb02532.x. [DOI] [PubMed] [Google Scholar]
- 30.Wanner B L. In: Metal Ions in Gene Regulation. Silver S, Walden W, editors. Sterling, VA: Chapman & Hall; 1997. pp. 104–128. [Google Scholar]
- 31.Wanner B L, Wilmes-Riesenberg M R. J Bacteriol. 1992;174:2124–2130. doi: 10.1128/jb.174.7.2124-2130.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stewart V, Rabin R S. In: Two-Component Signal Transduction. Hoch J A, Silhavy T J, editors. Washington, DC: ASM Press; 1995. pp. 233–252. [Google Scholar]
- 33.Wanner B L, Jiang W, Kim S-K, Yamagata S, Haldimann A, Daniels L L. In: Regulation of Gene Expression in Escherichia coli. Lin E C C, Lynch A S, editors. Austin, TX: Landes; 1996. pp. 297–315. [Google Scholar]
- 34.Barrett J F, Goldschmidt R M, Lawrence L E, Foleno B, Chen R, Demers J P, Johnson S, Kanojia R, Fernandez J, Bernstein J, et al. Proc Natl Acad Sci USA. 1998;95:5317–5322. doi: 10.1073/pnas.95.9.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]