

Abstract
Transfection of a Kaposi's sarcoma (KS) herpesvirus (KSHV) Bacterial Artificial Chromosome (KSHVBac36) into mouse bone marrow endothelial lineage cells generates a cell (mECK36) that forms KS-like tumors in mice. mECK36 expressed most KSHV genes and were angiogenic, but didn't form colonies in soft agar. In nude mice, mECK36 formed KSHV-harboring vascularized spindle-cell sarcomas that were LANA+/podoplanin+, overexpressed VEGF and Angiopoietin ligands and receptors, and displayed KSHV and host transcriptomes reminiscent of KS. mECK36 that lost the KSHV episome reverted to non-tumorigenicity. siRNA suppression of KSHV vGPCR, an angiogenic gene up-regulated in mECK36 tumors, inhibited angiogenicity and tumorigenicity. These results show that KSHV malignancy is in vivo growth-restricted and reversible, defining mECK36 as a biologically sensitive animal model of KSHV-dependent KS.
SIGNIFICANCE
Kaposi's sarcoma (KS) herpesvirus (KSHV) is the etiologic agent of KS, an angiogenic spindle-cell sarcoma associated with AIDS. The mechanisms driving KSHV carcinogenesis could be uncovered through reproduction of KS by KSHV-infection of normal cells. Our results defining this cell and animal model of KS show that KSHV-induced neoplasia is: 1) Reversible and KSHV-dependent. 2) In vivo-restricted: Upregulation of KSHV genes, such as the pro-angiogenic vGPCR, under in vivo growth conditions provides a selective advantage to KSHV-harboring cells leading to episome maintenance and tumorigenesis. Thus, mECK36 tumors are phenotypic, molecular, and viral surrogates of KS suitable for analyzing the role of viral and host genes in KS pathogenesis, and for preclinical testing of anti-KS drugs.
INTRODUCTION
Human herpesvirus-8 or Kaposi’s sarcoma associated herpesvirus (KSHV) is associated with three AIDS-related malignancies: Kaposi’s sarcoma (KS) (Boshoff and Weiss, 1998; Chang et al., 1994; Ganem, 2006); Multicentric Castlemans disease (Dupin et al., 1999), and Primary effusion lymphoma (Cesarman et al., 1995). In HIV-infected individuals, predominantly in the male homosexual population, KS incidence dramatically increases and can manifest as an advanced disseminated cancer with increased morbidity and mortality (Gallo, 1998). The development of pathogenesis-based therapies is important to improve current KS treatments (Pantanowitz and Dezube, 2004).
KS presents itself as multifocal lesions in the skin, lungs and gastrointestinal tract (Safai et al., 1985). It is characterized by intense VEGF-mediated angiogenesis, spindle cell proliferation, and erythrocyte extravasation (Gallo, 1998; Safai et al., 1985). There are many key unanswered issues in Kaposi’s sarcoma pathogenesis that are matters of controversy and intense research: 1-The neoplastic nature of KS, 2-The identity of the normal cell type that upon infection with KSHV becomes a malignant KS spindle cell, 3-The connection between KSHV gene expression and the KS phenotype, 4- The relationship between KSHV biology and other KS co-factors, such as immunosuppression, HIV infection and sexual transmission. Primary to answering these questions is the development of an experimental model of KSHV infection that reproduces the main pathogenic phenotypes of KS in vitro and in animals.
While spindle cells are generally considered the tumor cell in KS lesions, their origin and true malignant nature is controversial, because they lack many features of neoplastic cells such as aneuploidy, tumorigenicity (Gallo, 1998) or clonality (Gill et al., 1998). KS spindle cells express phenotypic markers that belong to many cell lineages including endothelium, smooth muscle cells, macrophages, and hematopoietic cells (Boshoff and Weiss, 2002; Gallo, 1998). Since KS could present itself as a multifocal disease and KS spindle cells express the hematopoietic stem cell marker CD34, it has been proposed that the KS progenitor cell is a circulating cell of the hematopoietic-endothelial lineage (Barozzi et al., 2003; Boshoff and Weiss, 2002; Browning et al., 1994). Spindle cells in the lesions express markers characteristic of lymphatic endothelial cell lineage, such as LYVE-1, podoplanin and VEGF-R3. It has been recently suggested that they originate from lymphatic endothelium (Dupin et al., 1999; Skobe et al., 1999), trans-differentiated vascular endothelium or a common endothelial cell precursor (Hong et al., 2004; Wang et al., 2004).
It is now well established that KSHV is an etiologic co-factor strictly necessary for KS (Boshoff and Weiss, 1998; Chang et al., 1994; Ganem, 2006), indicating that KSHV genetic expression is responsible for the KS angiogenic phenotype. Although the KSHV genome encodes for genes that can induce cell transformation (Gao et al., 1997; Lee et al., 1998; Wang et al., 2006) immune-deregulation (Moore et al., 1996; Nicholas et al., 1997), and angiogenesis activation (Aoki et al., 1999; Bais et al., 1998; Bais et al., 2003; Boshoff et al., 1997; Montaner et al., 2003; Yang et al., 2000), KSHV infection leads to KS in limited circumstances.
Since KSHV is an endothelial-tropic virus several KSHV-infection models using endothelial cells (ECs) have been described (Ciufo et al., 2001; Flore et al., 1998; Hong et al., 2004; Lagunoff et al., 2002; Moses et al., 1999; Naranatt et al., 2004; Wang et al., 2004). In contrast to the oncogenicity and tumorigenicity of its viral genome, KSHV infection of ECs leads to the induction of KS markers, spindle cell phenotype and signs of transformation but does not result in the acute angiogenicity and tumorigenicity characteristic of KSHV-infected spindle cells in lesions. This suggests that key oncogenic events are not recapitulated in the KSHV-infected EC cultures. Plausible explanations for this are the tendency for either latency or productivity of the in vitro infections, the possibility that terminally differentiated ECs may not be suitable KS progenitors; or that in vivo growth conditions not mimicked in culture are essential for KSHV oncogenicity.
Here we report that bacterial artificial chromosome (KSHVBac36) transfection of normal mouse bone marrow pro-angiogenic endothelial-lineage cells induces an angiogenic phenotype and KS-like KSHV-dependent tumorigenicity. This identifies a cell population containing putative KS progenitors and establishes a KS model with the following characteristics: 1) The pathological phenotype is a consequence of KSHV gene expression in normal progenitor cells subjected to in vivo growth conditions 2) The histopathologic phenotype of the tumors resembles KS lesions 3) The model is suitable for genetic analysis of viral pathogenesis.
RESULTS
Bac36-transfection of mouse bone marrow endothelial-hematopoietic cells leads to KSHV latent and lytic gene expression
To create a mouse model of KS, we used mouse bone marrow adherent-cell preparations enriched in endothelial cells (mEC), their progenitors and pro-angiogenic hematopoietic cells, as targets of KSHV infection (Boshoff and Weiss, 2002; Rafii and Lyden, 2003). To be able to select and track infected cells, we transfected mEC with KSHVBac36, which encodes the full infectious KSHV genome in a hygromycin-resistant and EGFP-encoding bacterial artificial chromosome (KSHVBac36) (Zhou et al., 2002). mEC transfected with the Bac backbone vector (mEC-V) were used as control. After three weeks of hygromycin selection both KSHVBac36-transfected mEC (mECK36) and mEC-V formed contact-inhibited stromal cell-like monolayers. We found that mECK36 cultures were over 95% EGFP+ (Figure 1A) indicating the presence of KSHVBac36. To determine if the EGFP+ cell population was infected by KSHV, we analyzed the expression of the latent genes LANA and Kaposin by immunofluorescence. As shown in Figure 1A-B, all EGFP+ mECK36 were positive for LANA, displaying the classic nuclear punctuated pattern produced by the tethering of KSHV episomes to the host chromosome (Ballestas et al., 1999). As shown in Figure 1C, mECK36 also stained for the latent oncogene Kaposin (Muralidhar et al., 1998), displaying its characteristic perinuclear localization.
Figure 1. KSHVBac36-transfection of mouse bone marrow endothelial-hematopoietic cells leads to KSHV latent and lytic gene expression.
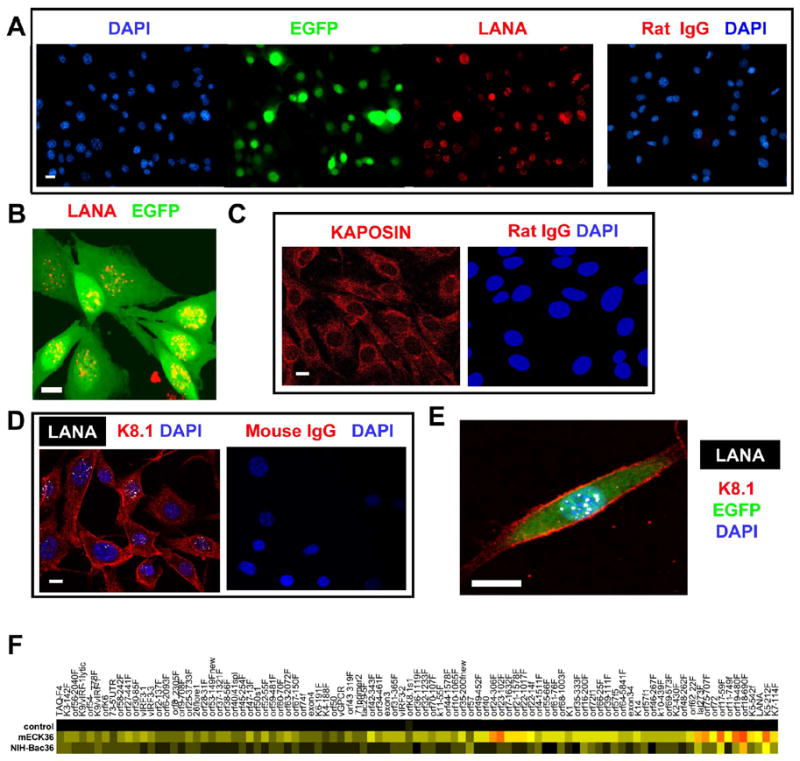
(A) Immunofluorescence staining of LANA. (B) Confocal image showing punctuated LANA staining. (C) Immunofluorescence staining of Kaposin. (D) Immunofluorescence staining of K8.1 and LANA. (E) Detailed confocal image of LANA and K8.1. (F) Heatmap representation of the Real Time RT-PCR analysis for the whole KSHV transcriptome in Bac36KSHV transfected cells. Ctrl: mock-transfected NIH3T3 cells. Red indicates highest, yellow intermediate, and black lowest relative mRNA levels. Scale bar = 10 μM
To estimate the integrity of the KSHV genome in mECK36, we performed Real Time RT-PCR analysis for the whole KSHV transcriptome of mECK36 (Dittmer, 2003; Fakhari and Dittmer, 2002). We compared the expression pattern in the endothelial lineage (mECK36) cells with non-KSHV targets (Bac36-transfected NIH3T3) (CC, AM and EAM unpublished). mRNA levels were 218 fold higher (CV: 45…391, n=100, p 10−14) in mECK36 than in Bac36-transfected NIH3T3 cells. In contrast, KSHV latent mRNAs (LANA, lat273f, orf72, Taq-F4, 73-5'UTR, orf72f1) were present at about equal levels, with a mean difference of 2.24 fold (CV: −1.04..5.51, n=6, p 0.29). This shows that mECK36 have increased lytic gene expression. All KSHV genes are expressed over background levels (Figure 1F), indicating that the KSHV transcriptome is complete in mECK36 cells, further suggesting that the Bac has not undergone major deletions or translocations that would affect KSHV gene expression.
Although lytic gene transcription in mECK36 is abundant, they did not produce KSHV virions as indicated by transmission electron microscopy analysis and the absence of cytopathic effect (CPE) (data not shown). Double immunofluorescence staining for the latent antigen LANA and the late lytic antigen K8.1 shows a co-expression pattern (Figure 1D,E) in most of the cells; which, together with mECK36 KSHV transcription profile, is indicative of an abortive-lytic transcription status.
KSHVBac36-transfected bone marrow endothelial lineage cells are angiogenic
The abortive lytic status of mECK36 is expected to be highly oncogenic, since it combines the expression of latent and lytic KSHV genes with transforming and angiogenesis-inducing potential. Both mEC-V and mECK36 became immortalized in culture and could be passaged infinitely (current passages > 50). Since murine cells express telomerase, they generally become immortalized as a consequence of cell culture shock (Sherr and DePinho, 2000). KSHV genes can up-regulate telomerase expression (Knight et al., 2001); therefore, we tested telomerase activity in mEC-V and mECK36 and found increased levels in mECK36 (Figure 2A). Transformed cells have increased telomerase expression; yet, the finding that mECK36 were contact-inhibited suggested that they were not transformed. To identify transformed clones among the mECK36 population, we carried out an anchorage-independent growth assay in soft agar using NIH3T3 and RasV12-NIH3T3 transformed cells as negative and positive controls, respectively. While 92% RasV12-transformed NIH3T3 formed colonies in soft agar, neither mECK36, mEC-V, nor control NIH3T3 formed colonies suggesting that mECK36 are not transformed. To investigate KSHVBac36-induced angiogenicity, we determined VEGF secretion levels, mRNA expression of angiogenic markers and skin angiogenicity in mEC-V and mECK36. As shown in Figure 2B-E, KSHVBac36 transfection of mEC increased VEGF secretion levels, upregulated ligands and receptors of the VEGF and Angiopoietin family, and displayed a significantly increased ability (P<0.05) to induce microvessel formation in mice skin. Taken together these results indicate that KSHV expression in mEC induces an angiogenic phenotype.
Figure 2. KSHVBac36-transfection of mECs induces an angiogenic phenotype.

(A) ELISA-based Telomerase Repeat Amplification Protocol (TRAP) assay of mEC-V and mECK36. Bars indicate mean of duplicates +/− SEM. (B) VEGF secretion levels of mEC-V and mECK36. Bars indicate mean of duplicates +/− SEM. (C) Intradermal skin angiogenesis assay (see methods) of mEC-V and mECK36 cells. The bar graphs show the mean microvessel density (vessels/mm2) +/− SEM. Total N (both flanks) = 10. (*** indicates p<0.05). (D) KSHVBac36-induced upregulation of angiogenic gene expression determined by Real Time qRT-PCR. Bars represent mean fold increase (triplicates +/−SEM) in mRNA levels between mECK36 and mECs. (E) Appearance at a dissected inoculation site in the skin of a mouse injected with mEC cells or mECK36 cells.
KSHVBac36-transfected bone marrow endothelial hematopoietic cells form KSHV-infected tumors that resemble Kaposi’s sarcoma
Cell angiogenicity is key for tumor growth. To determine if mECK36 were tumorigenic in mice, they were injected subcutaneously in nude mice using mEC-V as controls. We found that mECK36 formed solid tumors three weeks after injection in contrast to mEC-V which did not (Figure 3 A-C). On dissection, we found that the tumors were green-yellow indicating a significant percentage of EGFP+ cells (Figure 3B). Tumors were analyzed in a blind fashion and found to be “vascularized spindle cell sarcomas”, which is the histological presentation of human KS tumors (Figure 3D). Advanced invasive KS often appears as multi-focal lesions on the lungs and the gastrointestinal tract (Safai et al., 1985). To determine whether mECK36 could also induce invasive KS, we injected passage 30 cells intravenously in irradiated SCID/NOD mice. Three months after injection, mECK36-injected animals were sick while mEC-V-injected animals did not show any signs of ailment. Necropsy of mECK36-injected animals revealed multi-focal invasive spindle cell sarcoma lesions in lungs (9-20 foci per lung) (Figure 3E-F), a presentation reminiscent of advanced visceral KS.
Figure 3. mECK36 induce vascularized spindle cell sarcomas in immunocompromised mice.

(A) Mice showing subcutaneous mECK36 tumor four weeks after inoculation (B) Dissection site showing green-yellow tumor (EGFP expression). (C) Subcutaneous tumor growth in nude mice injected with mECK36 cells (closed circles) or mEC-V (open squares). Data indicate mean tumor size (N=7) +/− SEM. (D) Microscopic image of an histological section of a mECK36 tumor stained with hematoxylin & eosin (H&E). (V, microvessel). (E) Image of a spindle cell sarcoma foci (T) growing in the lung (L) stained with H&E. (F) Image of an spindle cell tumor in the lungs stained with H&E. E, blood extravasation. V, microvessel. Scale bar = 50 μM
The fact that subcutaneous tumors showed strong EGFP fluorescence (Figure 4A) indicated the presence of mECK36 in the tumors. Although mECK36 did not display typical signs of cell transformation in vitro, they were malignant in vivo, forming vascularized tumors that led to significant morbidity and mortality. To determine the presence of KSHV in the EGFP+ spindle cells of mECK36 tumors, we carried out immunohistochemical determination of KSHV LANA expression. Figure 4B-C shows that LANA+ cells in mECK36 tumors were in the order of 30% to 50%, which is also reminiscent of KS lesions (Dupin et al., 1999). To further characterize the phenotype of the KSHV-harboring cells in tumors and the status of their KSHV infection, we co-stained for KSHV LANA, podoplanin and K8.1. All EGFP+ cells co-expressed LANA and podoplanin (Figure 4B). Confocal images at different Z planes revealed that up to 90% of nuclei were LANA+ and displayed the punctuated pattern indicative of episomal infection (Figure 4D). More than 20% of the tumor cells were also K8.1+ (Figure 4E). To determine changes in the status of KSHV infection in mECK36 due to in vivo growth conditions, we compared the KSHV transcription profiles from three different tumors and from mECK36 in culture (Figure 4F) normalized for LANA in order to include only KSHV-harboring cells (Dittmer, 2003). Compared to cultured mECK36, the tumors showed a 15.6 fold increased expression of lytic genes versus a 1.1 fold increase of latent genes. Since the average variation among the three tumors was only 2.13 fold (CV: 1.93..2.33, n=91), the increase in lytic mRNA levels must represent a biological response of the viral genome to in vivo growth conditions, rather than a variation introduced by inter-tumor variation. Despite increased lytic transcription, mECK36 did not show increased viral genome replication, evidenced by the lack of variations in KSHV viral load and absence of KSHV virions in the tumors (data not shown). This data indicates that similar to mECK36 cells in culture, mECK36 tumors express latent and lytic genes consistent with an abortive lytic infection, but display increased expression of KSHV lytic genes.
Figure 4. mECK36 induce KSHV-bearing spindle cell sarcomas resembling KS tumors.
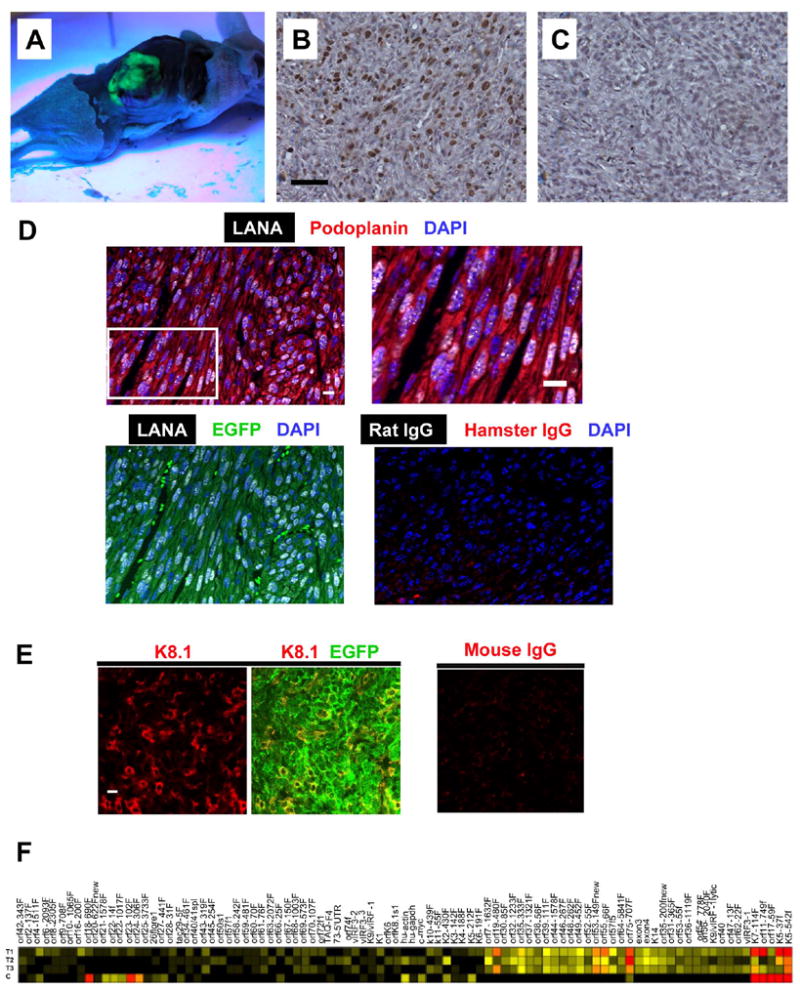
(A) Dissected mouse showing subcutaneous fluorescent tumor observed under long wave UV light. (B) LANA immunohistochemistry of paraffin embedded mECK36 tumors (C) Rat IgG control for LANA immuohistochemistry. (D) Top left: Confocal images showing podoplanin staining in the cytoplasm and LANA staining in the nucleus. Top right: higher magnification image showing the specked pattern of LANA immunofluorescence in podoplanin+ cells. Bottom left: colocalization of LANA with EGFP. Bottom right: an isotype-matched control. (E) Confocal images showing K8.1 staining in the membrane (left) and colocalizing with EGFP (middle). (Right) Mouse IgG control. (F) Heatmap representation of KSHV transcriptome in mECK36 tumors and mECK36 cells grown in culture quantified using Real Time qRT-PCR. C: mECK36 cells grown in culture. T1, T2, and T3: primary mECK36 tumors. Red indicates highest, yellow intermediate, and black lowest relative mRNA levels. Scale bars: 50 μM (B-C), 10 μM (D-E).
mECK36 Tumors express KS and angiogenic markers
To further determine if the phenotypic markers of the mECK36 sarcomas corresponded to those typically found in human KS lesions, we immunostained for CD31/PECAM (pan-endothelial cell marker), VEGF-R2 (EPCs, angiogenically active vessels, KS), CD34 (EPC, microvascular endothelium and KS) (Boshoff and Weiss, 2002; Brown et al., 1996; Rafii and Lyden, 2003). The presentation of these markers (Figure 5A) corresponded to a highly vascularized tumor (CD31+ vessels), bearing some VEGF-R2+ vessels as well as VEGF-R2+ and CD34+ cells. To assess the expression of VEGF-receptors in EGFP+ cells, we used immunofluorescence and cytoflurometry analysis for the VEGF-R1 (endothelial cells, pro-angiogenic hematopoietic cells), VEGF-R2 and VEGF-R3 (lymphatic endothelium and KS) (Hong et al., 2004; Rafii and Lyden, 2003; Wang et al., 2004). A significant percentage of EGFP+ cells displayed the KS and angiogenic markers VEGF-R1, VEGF-R2 and VEGF-R3 (Figure 5A). Cytofluorometric determinations of a representative tumor also showed EGFP+ cells, which comprised 56% of the tumor, were 60, 45 and 40% VEGF-R1, VEGF-R2 and VEGF-R3 positive, respectively (Figure 5D-E) as compared to 1.5, 2.0 and 4.0% of EGFP- cells, comprised by endothelial and stromal cells from the tumor (Figure 5E). To assess the possible upregulation of this angiogenic phenotype in vivo, mRNA levels of VEGF and Angiopoietin family ligands and receptors in mECK36 cells grown culture and from nude mice tumors were quantified by Real Time RT-PCR. Most of the angiogenic mediators that were up-regulated by KSHVBac36 transfection, were further up-regulated in mECK36 tumors (Figures 2D and 5F). These results indicate that KSHV infection synergizes with in vivo growth conditions to induce the expression of an angiogenic phenotype reminiscent of KS.
Figure 5. mECK36 spindle cell sarcomas express KS and angiogenic markers.
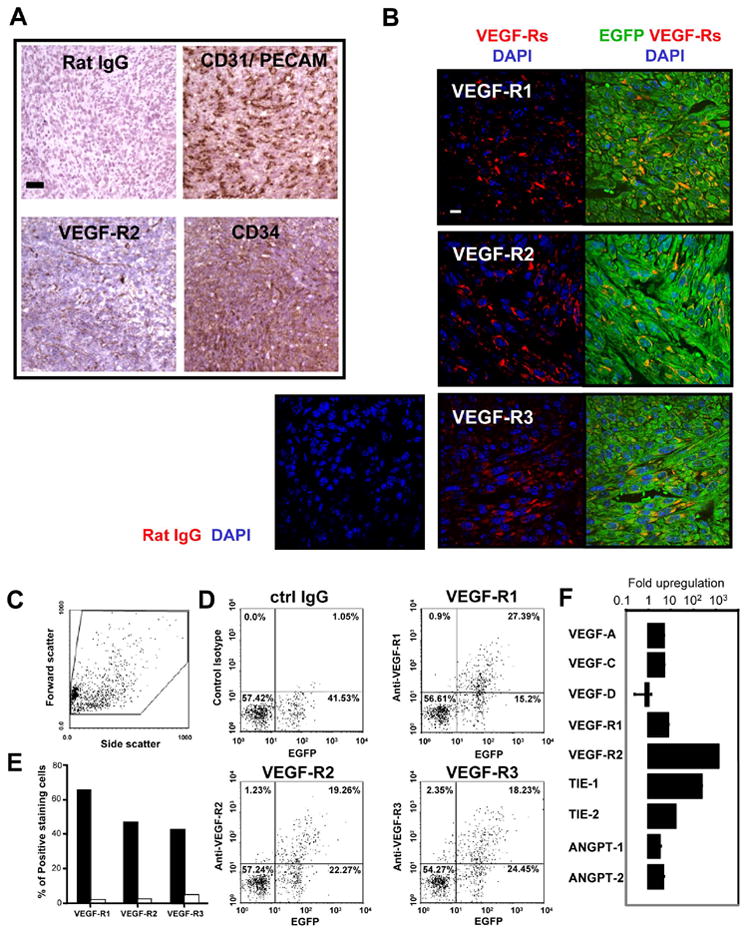
(A) Frozen sections of tumors stained with indicated antibodies. (B) Paraffin-embedded tissue sections stained with indicated antibody and developed with Cy3-conjugated anti-IgG. (C) Side and forward scatter of VEGF receptors on single cell suspensions from mECK36 tumors by Flow Cytometry. (D) Cells double positive for EGFP and indicated VEGFR from tumors in (C). (E) Percentage of cells expressing VEGFR among EGFP+ (black bars) and EGFR- (white bars) population. (F) Up-regulation of angiogenic gene expression in mECK36 tumors compared to cultured mECK36. Bars represent mean fold increase (triplicates +/- SEM) in mRNA levels quantified by Real time qRT-PCR. Scale bars: 50 μM (A), 10 μM (B).
mECK36 mouse spindle cell sarcomas display KSHV and host transcription profiles reminiscent of Kaposi's sarcoma
To gain a perspective of how closely mECK36 KS-like sarcomas resemble primary KS lesions from the standpoint of KSHV expression, we compared the pattern of KSHV transcription in mECK36 sarcomas and in biopsies from KS lesions. Merging data reported here with data from our previous study (Dittmer, 2003), we found that the three mECK36 tumors grouped together, indicating the high reproducibility of the mouse sarcomas (Figure 6A). Six of the primary KS lesions clustered closer to the mECK36 mouse tumors than the five other KS lesions, which in turn clustered with cultured mECK36. These results indicate that the pattern of KSHV gene expression obtained in mECK36 cultures and tumors can be considered representative of sub-populations defined by different degrees of lytic gene expression that are found among KS human tumors.
Figure 6. Viral and host global gene expression of mECK36 tumors resembles KS.
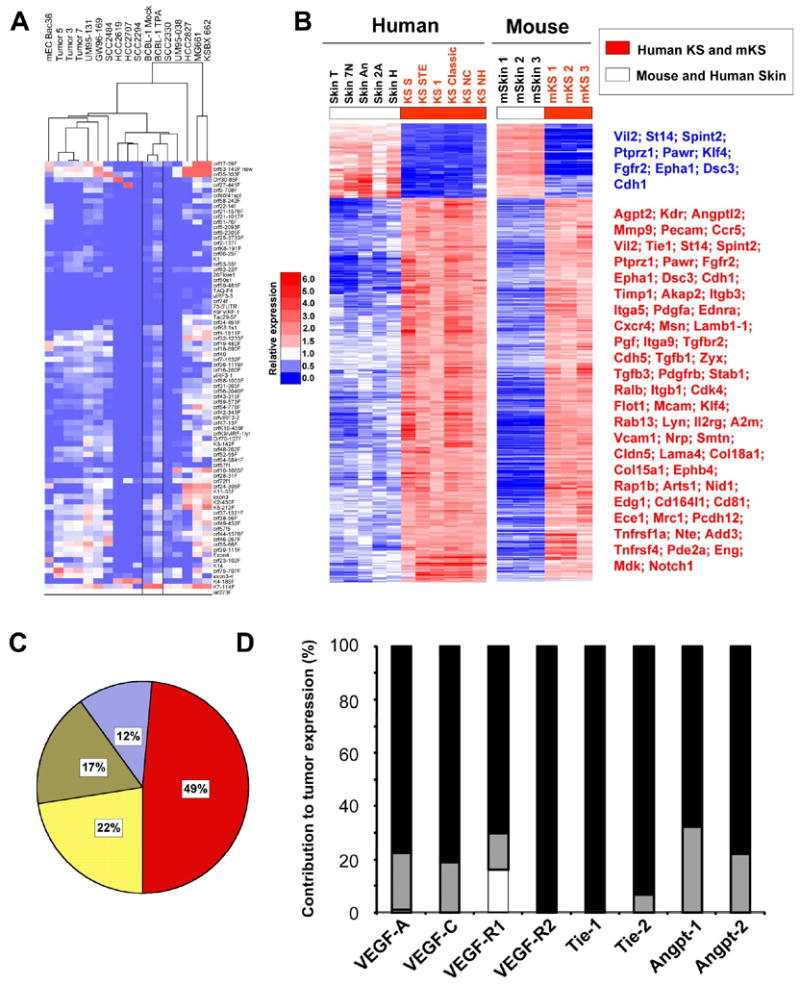
(A) Clustering of primary KS with mEC-Bac36 tumors. Heatmap represents Real Time qRT-PCR results. Tumor 5, 3 and 7 are three mECK36 spindle cell sarcomas. BCBL Mock and TPA are BCBL-1 uninduced and induced with TPA, respectively. (B) Heatmap representation of 562 genes (q<0.05) of the human KS Signature that are also mECK36 tumor signature (equally up or down regulated). 723 genes from the human KS signature were ortholog to mouse genes present in the mouse Affymetrix Array. Among these, 691 genes were able to differentiate mECK36 spindle cell sarcomas and mouse skin (q<0.05) for at least a two fold difference. 471 genes (68%) and 91 genes (13%) were upregulated (red) and downregulated (blue), respectively, in both mECK36 tumors and human KS. Selected downregulated genes are shown in blue and upregulated genes in red. The full list of genes is available in Supplementary Table 1. (C) Distribution of mouse KS signature. Blue: genes shared between mECK36 tumors and mEC-V. Yellow: genes shared between mECK36 tumors and mECK36 in culture. Olive: genes shared by mEC-V, mECK36 in culture and tumors. Red: mECK36 tumors only. Gene intensity from the mouse KS signature was set to 100%, and the percentage of genes that contribute to the mouse KS signature was calculated using 10% difference in intensity between the different samples and tumors (GeneSpring 7 package). (D) Angiogenic gene expression in mEC-V and in mECK36 relative to that in mECK36 tumors. mRNA levels were determined by Real Time qRT-PCR. mRNA levels in tumors were set at 100% (closed bar). Overlapped are bars representing relative mRNA levels in mECK36 cells in culture (grey bars) and in mEC-V cells (open bars) (mean of triplicates +/− SEM). Relative levels were calculated as 1/ fold-increase in tumors X 100.
Molecular signatures are a useful way to evaluate the phenotypic identity of tumors (Klein et al., 2001). The KS signature was recently established by identifying 1256 KS genes (1482 probes) which differentiate KS and skin sample groups (Wang et al., 2004). This group of differentially expressed genes is thought to determine key phenotypic differences defining KS, and thus they are primary to the biology of KS tumors. To evaluate the extent to which our mouse model mimics the KS signature, we first determined the transcriptional profile of mECK36 tumors using genomic arrays and we identified which genes of the human KS signature were able to differentiate mECK36 tumors and mouse skin (q<0.05) (Figure 6B) defining the mouse KS-like signature. Then we compared the human and mouse KS-like signature and we found that 81% of the transcripts, including many key genes for KS pathophysiology, were equally up-or down-regulated in human KS and mouse mECK36 tumors compared to skin (Figure 6B). Determination of the degree of contribution to the KS-like signature from mEC-V, mECK36 and mECK36 tumor microarray data, indicates that the major contribution to the signature—close to 50%-- was by mECK36 tumors (Figure 6C). In agreement with this microarray analysis, Real time qRT-PCR comparison of angiogenesis-mediating gene expression in mECK36 tumors, cultured mECK36 and mEC-V show that most of angiogenic gene upregulation takes place when mECK36 grow in vivo to form tumors. Taken together, these results show that KSHV is sufficient to confer to infected cells in vivo angiogenic, tumorigenic and phenotypic characteristics of KS.
mECK36 belong to the endothelial cell lineage
The KS-like phenotype mECK36 cells tumors suggests that Bac36 was originally transfected to a cell lineage closely related to the one that in human KSHV infection leads to KS. More accurate determination of the cell lineage that gave rise to mECK36 could point to a KS spindle cell progenitor. Genomic profiling provides a useful way to trace the cell of origin of transformed cells (Klein et al., 2001). To further identify the progenitor(s) cell type that were targeted by KSHVBac36 transfection among the heterogeneous mEC population, we used the genome wide transcription profile of mEC-V and mECK36 and clustered it to several mouse cell types that can be present in the mECs including endothelial cells, endothelial progenitors and hematopoietic progenitors. As shown in Figure S1 (Supplementary information) mEC-V and mECK36 cluster in between endothelial progenitors and mature vascular endothelial cells further supporting the endothelial lineage origin of mECK36.
mECK36 tumorigenicity is reversible and strictly KSHV dependent
mECK36 cultures and tumors are a valuable tool for defining the function of KSHV pathogenic genes and their relationship with host responses and KSHV biology. The phenotypic differences between mEC-V and mECK36 is thought to be a consequence of KSHVBac36 transfection with concomitant KSHV gene expression in normal cell progenitors, and it is consistent with the proposed KSHV-induced phenotype in KS. However, to demonstrate a link between KSHV and mECK36 tumorigenesis, it is necessary to show the dependency between KSHV gene expression and the KS-like phenotype. This is because mECK36 could have accumulated host gene oncogenic alterations that may contribute to the malignant phenotype independently of KSHV. This can be demonstrated by showing that mECK36 that have lost the KSHV genome lose the ability to form KS-like tumors. It was previously shown that episomal KSHV is lost from cells in culture unless it is maintained by antibiotic selection (Grundhoff and Ganem, 2004). Episomally-infected mECK36 are routinely selected in hygromycin (Hyg)-containing media for the maintenance of the Hygr KSHVBac36. To test whether mECK36 cultured in the absence of antibiotic selection lose the KSHVBac36 episome, cultured mECK36 and mECK36 explanted from tumors were cultured in the presence or the absence of Hyg. We found that both mECK36 and explanted mECK36 tumor cells lost the BacKSHV episome after four weeks of culture without Hyg, as evidenced by loss of EGFP fluorescence (Figure 7A-B). To demonstrate that this effect was actual episome loss and not outgrowth by EGFP negative cells, we carried out a colony formation assay in plastic, in which we monitored colonies originated from a single EGFP+ mECK36. EGFP+ mECK36 single cell clones grown without Hyg lost their episomes and became EGFP- as they divided (Figure 7A, peripheral cells). These results have several implications: 1) The rapid loss of the episome in vitro suggest that mECK36 cells that lost the KSHV episome had a selective advantage over KSHV positive cells. 2) The generation of non-fluorescent and hygromycin-sensitive cultured and tumor-explanted cells indicates that the KSHVBac36 is only present as an episome and it is not integrated in the host genome. 3) KSHV provides an in vivo selective advantage equivalent to Hyg in vitro since mECK36 explanted from the tumor lose the episome and tumors are grown in the absence of hygromycin. We compared KSHV+ mECK36 cells with mECK36 that have lost the KSHV episome (KSHV-null mECK36). While KSHV-null mECK36 or mEC-V have a higher growth rate in vitro than mECK36 cells (Figure 7C), KSHV-null mECK36 completely lost tumorigenicity in mice and a mixture of 1:1000 of mECK36:KSHV-null mECK36 have a delayed tumor growth in mice but leads to the formation of tumors similar in size to those induced by mECK36 alone (Figure 7D). Analysis of these tumors shows up to 30% EGFP+/KSHV LANA+ mECK36 cells (data not shown). These results demonstrate that KSHV provides a selective advantage in vivo but not in vitro, and that mECK36 tumorigenesis is strictly dependent on KSHV since it can be reversed by loss of the KSHV episome.
Figure 7. mECK36 tumorigenicity is reversible and strictly KSHV-dependent.
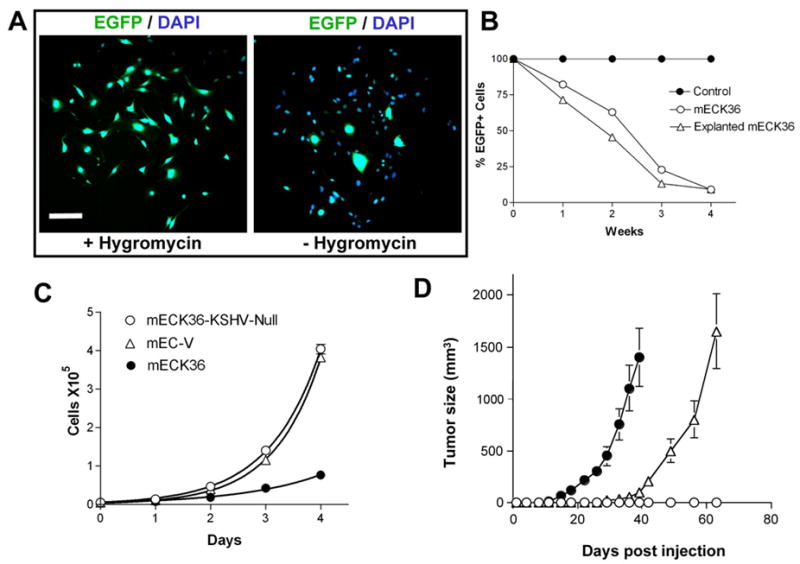
(A) Many cells in colonies each originated from a single EGFP+ mECK36 cell became EGFP- when grown in the absence of hygromycin. (B) EGFP levels of cultured and explanted mECK36 grown with or without hygromycin determined by flow cytometry. mECK36 grown in hygromycin media was used as a control and set at 100% for every passage. (C) In vitro growth rate of mECK36, mEC-V, and mECK36-KSHV-Null. Doubling times were 22, 14, and 15 hours, respectively. (D) Tumor formation of mECK36-KSHV-Null cells (open circles), mECK36 cells (closed circles), and 1:1000 ratio of mECK36:mECK36-KSHV-Null (open triangles). Data indicate mean tumor size +/− SEM (N=5). Scale bar: 100 μM.
Role of KSHV gene expression and the angiogenic oncogene vGPCR in sarcomagenesis
KSHV dependency suggests that mECK36 tumors are a biologically sensitive model in which we can study the role of KSHV genes in KS pathogenesis. A mechanistic explanation for mECK36 tumorigenesis is that KSHV genes up-regulated during in vivo growth induce mECK36’s malignancy by activating cell proliferation, cell survival or angiogenic gene expression (Figures 5F, 6D). We found that the KSHV genes most up-regulated when mECK36 form tumors encode for proteins that have been implicated in inhibition of p53-induced apoptosis (vIRF-1) (Nakamura et al., 2001), JNK signaling (ORF36) (Hamza et al., 2004), and angiogenesis activation (vGPCR) (Figure 8A). Since vGPCR has been shown to induce a KS-like angiogenic phenotype by activating VEGF expression (Bais et al., 1998; Bais et al., 2003; Grisotto et al., 2006; Guo et al., 2003; Montaner et al., 2003; Montaner et al., 2006; Yang et al., 2000), we speculated on the possibility that vGPCR upregulation leading to VEGF secretion was causally linked to mECK36 tumorigenesis. As a proof of principle for the use of mECK36 in genetic studies of KSHV pathogenesis, we silenced vGPCR by RNA interference (RNAi) using a vGPCR short hairpin RNA (shRNA) (Figure 8B) and assessed the impact on mECK36 angiogenesis and tumorigenicity. To determine whether the RNAi suppression affected other KSHV pathogenic genes, we performed Real-Time PCR array analysis on shRNA or control transfected mECK36 (Figure 8C). While the level for most of the KSHV mRNAs remained unchanged as evidenced by the clustering of datapoints on the 45° line (m=1.00, r2=0.95, n=93), the vGPCR message was reduced 117-fold. We also observed a reduction of the mRNAs for orfs 7, 25, 26 and 27. The significant decrease on the mRNA levels of these orfs strongly suggests that cellular signaling pathways activated by vGPCR might in turn feed back on the virus to control the expression of these KSHV genes. shRNA-mediated silencing of vGPCR in mECK36 blocked their ability to secrete VEGF and significantly reduced the ability of the mECK36 to induce microvessel formation in skin (Figure 8D) (P<0.05). Although both vGPCR-shRNA- and control-shRNA-transfected mECK36 grew at a very similar rate in culture (not shown), vGPCR-shRNA mECK36 tumors showed delay and inhibition in tumor formation (Figure 8E), which is consistent with a reduction in angiogenicity (Folkman, 1997). These results show that activation of angiogenesis by KSHV vGPCR is causally implicated in mECK36 tumorigenesis and demonstrate that mECK36 can be genetically manipulated to study KSHV pathobiology.
Figure 8. RNAi-mediated suppression of KSHV vGPCR in mECK36 blocks angiogenicity and tumorigenesis.
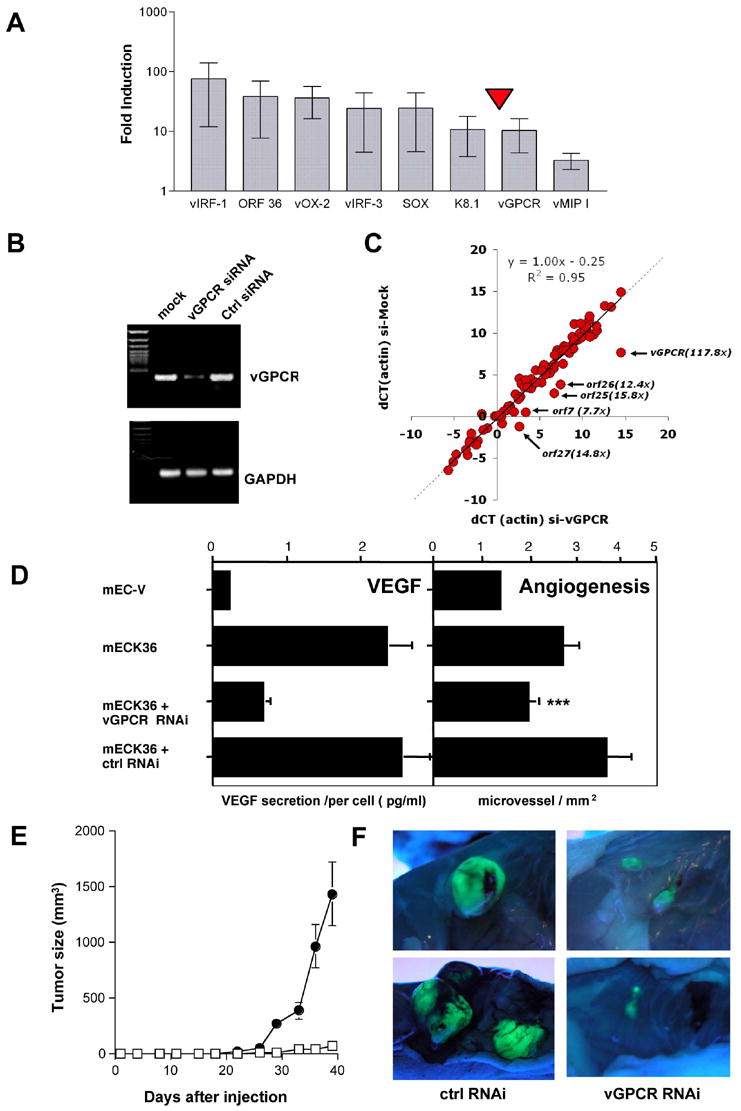
(A) mRNA levels of KSHV genes in mECK36 tumors compared to cultured mECK36 determined by Real time qRT-PCR. Mean values (+/− SD) from 3 tumor samples were plotted. (B) RT-PCR analysis of vGPCR and GAPDH for mECK36 cells transfected with control shRNA and vGPCR shRNA vector (C) Log2 scale plot (dCT) of the actin-normalized KSHV mRNA levels of mECK36 transfected with control shRNA (horizontal) or vGPCR shRNA (vertical). The arrows indicate transcripts that are specifically downregulated and the annotation shows the identity and fold downregulation. (D) Left panel: Levels of VEGF secretion for mECK36 transfected with either control shRNA or vGPCR shRNA vector measured by ELISA. Bars indicate mean of duplicate measures +/− range. Right panel: Angiogenesis in skin for mEC-V, mECK36, mECK36 cells transfected with either control shRNA or vGPCR shRNA vector. The bar graphs show the mean microvessel density (vessels/mm2) +/− SEM. Total N (both flanks) = 10. (***) indicates significant differences between vGPCR RNAi and control RNAi groups (P<0.05) (N=10). (E) Tumor formation of mECK36 cells transfected with control shRNA (closed circles) or with vGPCR shRNA vector (open squares). Data indicate mean tumor size +/− SEM (N=10). (F) Tumor appearance after dissection and lighting using a long-wave UV lamp for mECK36 cells transfected with control shRNA or with vGPCR shRNA vector.
DISCUSSION
A normal progenitor cell-derivative containing the whole KSHV genome, allowed for the reproduction of the tumorigenic and angiogenic characteristics of an infected KS spindle cell, and led to the generation of a cell and animal model of KSHV-induced KS. This new model not only illustrates several features that makes it useful for biological and preclinical studies, but it also challenges our current thinking on key issues of KSHV biology and KS pathogenesis.
We found that the KSHV genome is angiogenic and tumorigenic in endothelial lineage cells of adherent bone marrow cell preparations. These findings suggest that an endothelial lineage cell type is a natural target of KSHV infection and a progenitor of tumorigenic KS spindle cells. It should be added that although our approach allows us to “target” putative progenitors in the transfected population and may have been critical for inclusion of the right cell type(s) in the right microenvironment, it does not allow us to unequivocally identify the KSHV targets. Nevertheless, phenotypic marker expression and transcriptome profile clustering results are consistent with the KSHVBac36 being transfected to endothelial lineage cells that are among the bone marrow adherent cell population, including endothelial cells and endothelial cell progenitors (Rafii and Lyden, 2003).
We showed that the KSHV genome can independently induce the KS phenotype when its genome is expressed within the context of an appropriate normal cell progenitor and when the KSHV-bearing cells are grown in vivo. mECK36 lead to the growth of subcutaneous tumors or multifocal KS pulmonary lesions consisting of vascular spindle cell sarcomas, that expressed angiogenic and KS phenotypic markers such as podoplanin and the VEGF and angiopoietin receptors and ligands. Moreover, genome wide transcriptome analysis of mECK36 tumors revealed that 81% of human KS signature genes behaved as mECK36 tumor signature genes, and were similarly up-or down-regulated in mouse and human tumors. These findings suggest that the KSHV genome encodes the ability to recreate the full KS pathophysiology including visceral invasiveness and disease localization.
The pattern of KSHV expression in tumors, as evidenced by RT-PCR array analysis and immunodetection of K8.1, was consistent with an increase in lytic transcripts. Although it was reported that most spindle cells in KS lesions are latently infected (Staskus et al., 1997), we found that a subpopulation of KS lesions (Dittmer, 2003) exhibits a pattern of KSHV expression similar to mECK36 spindle sarcomas. Thus, the KSHV expression profile of mECK36 in tumors can be biologically relevant. In future studies, it will be of importance to establish possible correlations between the mECK36-like KSHV expression profile and KS clinical presentation. Interestingly enough, the mECK36 pattern of KSHV gene expression was not accompanied by virus production indicating that mECK36 were in an abortive lytic replication state. Others have shown that in permissive human cells KSHVBac36 transfection leads to virion production (Zhou et al., 2002). In contrast, both in vitro and in vivo grown mECK36 showed an absence of viral replication. Since transcritptome analysis shows that the viral genome is intact, it is unlikely that the defects in viral replication were due to Bac36 alterations. Instead, viral replication and maturation events are most likely being blocked. A recent report (Parsons et al., 2006), points to productive KSHV infection in SCID/NOD mice upon injection of KSHV virions suggesting that the use of KSHVBac36 transfection may relate to the abortive lytic status of mECK36. Interestingly, KS lesions contain lytic replication-defective KSHV genomes that may play a role in KS tumorigenesis (Deng et al., 2004). Thus, the replication-defective infection achieved by Bac36 transfection may be relevant to actual occurrences of the disease and might have been critical to capture a KSHV-induced KS phenotype.
We found that KSHV episome-loss led to loss of tumorigenicity. This indicates that the mECK36 tumorigenic phenotype is strictly dependent on KSHV and reversible, since mECK36 reverted to a non-tumorigenic phenotype upon KSHV loss in vitro. This resembles explanted KS spindle cells that lose KSHV in culture becoming non-tumorigenic (Aluigi et al., 1996; Dictor et al., 1996; Ganem, 2006). It also suggests that KSHV-induced oncogenesis, at least in its initial stages, does not lead to the accumulation of further oncogenic hits which could contribute to a malignant phenotype independent of KSHV. This also resembles human KS lesions in which cellular oncogenic alterations such as p53 and ras mutations, or Bcl-2 overexpression together with increased clonality tend to be found only in advanced KS (Gill et al., 1998; Nicolaides et al., 1994; Pillay et al., 1999; Rabkin et al., 1997). It can also be concluded that the maintenance of the KSHV episome during tumor formation—which is carried out in the absence of hygromycin—indicates that KSHV does provide a selective advantage for in vivo growth that leads to tumorigenesis. This selective advantage of KSHV+ versus non-infected cells provides an alternative explanation to re-infection (Grundhoff and Ganem, 2004) for maintenance of the KSHV episome in infected spindle cells in KS lesions.
KSHVBac36-mediated tumorigenesis correlated with the upregulation of several KSHV lytic transcripts and the induction of a KS-like angiogenic phenotype upon implantation into nude mice. This further supports the notion, evidenced by recent publications, that in KS as well as in PEL (An et al., 2006; Staudt et al., 2004) the host microenvironment influences KSHV gene expression. However, the regulatory effects are indeed dialectic. We found that KSHV together with in vivo growth conditions trigger in the infected cell the expression of genes related to angiogenesis and malignancy which provides a selective advantage leading to tumor formation. We found that mECK36 grown in vitro did not display signs of cell transformation. When subjected to in vivo growth conditions, however, the same cells induced angiogenic spindle cell sarcomas with upregulation of key angiogenic markers (Figures 5F and 6D). In concordance, analysis of the mouse-KS-like signature of mECK36 tumors showed that almost 50% of it derives from genes that were only expressed in mECK36 tumors. The concomitant upregulation of KSHV lytic genes in vivo and the induction of an angiogenic phenotype in KS-like lesions pointed to a series of potentially pathogenic KSHV genes as critical determinants for KS-like tumorigenesis. We selectively inhibited one of these genes to prove its involvement in mECK36 tumorigenesis and to test the suitability of our model for genetic analysis of KSHV pathogenic function. siRNA-mediated suppression of the early lytic angiogenic oncogene vGPCR (Bais et al., 1998; Bais et al., 2003; Yang et al., 2000), blocked VEGF secretion and tumorigenesis, leading to significant retardation in tumor growth. It is unlikely that siRNA was also targeting the expression of orf K14, which is co-expressed with vGPCR in bi-cistronic messages (Kirshner et al., 1999), as orf K14 mRNA levels were not affected by the vGPCR shRNA (Figure 8C). These results with vGPCR silencing were verified by preliminary studies with vGPCR-null Bac36 mutants (LC, EM unpublished). Our results confirm single gene experiments with vGPCR (Bais et al., 1998; Bais et al., 2003; Grisotto et al., 2006; Guo et al., 2003; Montaner et al., 2003; Montaner et al., 2006; Yang et al., 2000), indicating that it plays a non-redundant role in angiogenesis and tumorigenesis within the context of full KSHV genome expression, and further supports the notion that this KSHV gene is a good therapeutic target. Our results with vGPCR suppression show that our model is sensitive to genetic manipulation and can be used to analyze the contribution of other KSHV genes in tumorigenesis. Furthermore, they point to mECK36 in vivo malignant phenotype as a consequence of upregulation of KSHV genes, such as vGPCR, that provide a selective advantage to the cell—in this case angiogenicity- leading to positive selection of the KSHV episome and KS-like tumor formation.
The suitability of mECK36 for genetic studies of KS pathogenesis could help to reveal the role of both KSHV and host genes implicated in the KS phenotype. A recent report showed that long-term latent KSHV infection of immortalized human endothelial cells leads to the outgrowth of transformed and tumorigenic clones (An et al., 2006). However, the use of hTERT-immortalized cells as substrate, and the sporadic occurrence of the in vitro-transformed outgrowths points to the accumulation of oncogenic alterations in the host genome; thus, making it difficult to strictly link the malignant phenotype to specific KSHV genes. In our case, the fact that Bac36-transfection of mEC leads to a non-productive infection also imposes limitations to the mECK36 model, in particular for studying viral entry and viral replication. However, several key characteristics make the mECK36 model appropriate for genetic studies of pathobiology and experimental therapeutics: 1) The progenitor target population(s) are normal mouse bone marrow cells, making it possible to use available knockout (K/O) or conditional K/O mice to generate different Bac36-transfected cells that could be used to identify genetic determinants of KS pathogenesis. 2) The KSHV dependence of the KS phenotype makes the mECK36 system highly sensitive for studies of KSHV genes critical for tumor growth, and makes it suitable for testing therapeutic strategies targeting KSHV pathogenic genes or host angiogenic machinery. 3) The mECK36 population expresses the full KSHV genetic complement, allowing for manipulation of latent and lytic KSHV genes using Bac or siRNA in functional studies. 4) mECK36 viral expression was stable through multiple passages (DD and EAM unpublished) and had reproducible in vivo pathogenic phenotypes.
In summary, our results show that the mEC Bac36 system and the mECK36 KSHV-harboring spindle sarcomas are good phenotypic, physiologic, molecular and viral surrogates of human KS that could unlock novel research avenues on KSHV pathobiology. They define mECK36 as a biologically sensitive cell and animal model of viral Kaposi’s sarcoma suitable for analyzing the role of KSHV and host cell genes in KS pathogenesis, as well as for preclinical testing of anti-KS drugs.
Materials and methods
Vectors and constructs
The empty vector pMHGP and the vector containing the insert with the genome of KSHV in Bacterial Artificial Chromosome (KSHV Bac36) were obtained as described previously (Zhou et al., 2002).: The empty vector pSilencer 2.1-U6 neo, shRNA control vector were obtained from Ambion (Austin, TX). To obtain the hairpin sequences that silence the expression of vGPCR, we selected candidate sequences for low homology to mouse transcripts by BLAST search. The selected sequence was: 5’-GATCCCATGTTCTTGGAAATGGATTTTCAAGAGAAATCCATTTCCAAGAACATTTTT TTGGAAA-3’ and 5’-AGCTTTTCCAAAAAATGTTCTTGGAAATGGATTTCTCTTGAAAATCCATTTCCAAGA ACATGG-3’, which created a vGPCR shRNA hairpin loops with BamHI and HindIII specific sticky ends to be cloned in the pSilencer 2.1-U6 neo. All the transfections were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Cells transfected with Bac36-KSHV or vector were selected with Hygromycin-B. For Silencer-2.1-U6 neo vGPCR or control vector, cells were double selected with Hygromycin-B and G418 (Sigma, Saint Louis, Missouri).
Cells
mEC were obtained from Balb/C An Ncr-nu mice (NCI, Bethesda, MD) bone marrow. Mice femurs were flushed twice with PBS, and the eluates were incubated in DMEM media plus 30% FBS (Gemini Bioproducts, Calabasas, CA), Endothelial Growth Factor (EGF) 0.2 mg/ml (Sigma, Saint Louis, MO), Endothelial Cell Growth Factor Supplement (ECGS) 0.2 mg/ml (Sigma, Saint Louis, Missouri), Heparin 1.2 mg/l (Sigma, Saint Louis, MO), Insulin transferrine selenium (Invitrogen, Carlsbad, CA), Penicilin-Streptomicin 1% (Invitrogen, Carlsbad, CA) and BME Vitamin (VWR Scientific, Rochester, NY). NIH3T3 cells were from ATCC. RasV12 transformed NIH3T3 cells were obtained as described (Bais et al., 1998).
Animals
Male Balb/C An Ncr-nu mice (NCI, Bethesda, MD), 8 weeks old, were used for angiogenesis assays and subcutaneous tumors. 12 weeks old male SCID/NOD ICRSC-M (Taconic, Germantown, NY), were used for intravenous injection of cells. Mice were kept under sterile conditions and used under protocols approved by the Institutional Animal Care and Use Committee
Angiogenesis assay
3x105 cells (two inoculation sites per mouse) were inoculated I.D. in Balb/C nude mice. Trypan blue (20%) was used to assess cell viability and mark the inoculation site. Mice (5 per group) were sacrificed 5 days later and the surface area of the injection site was exposed by dissection and photographed using a stereoscopic microscope (Nikon Corporation, Tokyo, Japan). To assess microvessel density the whole surface area of the injection site was examined for morphometric analysis following Auerbach’s criteria (Auerbach et al., 2000). To this extent each photograph slide was projected in a grid corresponding to 1 square mm and the total number of individual blood microvessels on all grids was counted in a blinded labeled fashion. The blood density was defined as the number of vessels per grid (D= total number of vessels / total number of grids counted).
Mouse tumor growth assays
Mice (N=7) were inoculated S.C. with cells (3x105 per mouse). Tumor growth was followed by caliper measurements of volume until date of sacrifice. mECK36 tumors were allowed to grow until they reached a volume of approximately 2 cm3. SCID/NOD mice (N=5) were irradiated (350 rad) and after 3 hours were inoculated intravenously (IV) in the tail vein (2x106 cells per mouse). Mice were followed for 3 months until symptoms of distress such as dyspnea and pain, at which point they were sacrificed and complete necropsies were performed.
Accession numbers
The microarray data can be accessed from NCBI's Gene Expression Omnibus (GEO) with accession number GSE6482.
Other experimental procedures
For details on immunofluorescence, immunohistochemistry, Real Time qRT-PCR, VEGF Elisa, flow cytometry of tumor, microarray procedures and analysis, and statistical methods, see Supplementary Data
Supplementary Material
Acknowledgments
This paper is dedicated to Philip Juan Browning (1953-2004) who made the initial observations on the existence of circulating hematopoietic KS progenitors. We would like to thank Rafael Tejada and David K Jin for their help and support. We are grateful to Dr. Don Ganem for his thoughtful critiques of earlier versions of this work and Dr. Ethel Cesarman for her helpful comments and her continuous support of our work. We are thankful to Dr. Beata Frydel and Brigitte Shaw from the Imaging and Molecular Core Facility (DRI, University of Miami). We thank Drs. Adam Asch, Dr. Roger Pierce, Anne Marie Valadere and Heidi Kiger for comments on the manuscript. We are indebted to Aileen Chen for her excellent edition job. We are thankful to Drs. David Salomon and Dr. Sunil Kurian for facilitating their array data sets. This work was supported by NIH grants CA 075918 to E.A.M, CA096512 to S.J.G. and CA110136 and CA109232 to D.P.D; by a grant from the Dorothy Rodbell Cohen Foundation for Sarcoma Research to E.A.M. and by the Viral Oncology Program of the Sylvester Comprehensive Cancer Center (E.A.M).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aluigi MG, Albini A, Carlone S, Repetto L, De Marchi R, Icardi A, Moro M, Noonan D, Benelli R. KSHV sequences in biopsies and cultured spindle cells of epidemic, iatrogenic and Mediterranean forms of Kaposi's sarcoma. Res Virol. 1996;147:267–275. doi: 10.1016/0923-2516(96)82285-0. [DOI] [PubMed] [Google Scholar]
- An FQ, Folarin HM, Compitello N, Roth J, Gerson SL, McCrae KR, Fakhari FD, Dittmer DP, Renne R. Long-Term-Infected Telomerase-Immortalized Endothelial Cells: a Model for Kaposi's Sarcoma-Associated Herpesvirus Latency In Vitro and In Vivo. J Virol. 2006;80:4833–4846. doi: 10.1128/JVI.80.10.4833-4846.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki Y, Jaffe ES, Chang Y, Jones K, Teruya-Feldstein J, Moore PS, Tosato G. Angiogenesis and hematopoiesis induced by Kaposi's sarcoma-associated herpesvirus-encoded interleukin-6 [see comments] Blood. 1999;93:4034–4043. [PubMed] [Google Scholar]
- Auerbach R, Akhtar N, Lewis RL, Shinners BL. Angiogenesis assays: problems and pitfalls. Cancer & Metastasis Reviews. 2000;19:167–172. doi: 10.1023/a:1026574416001. [DOI] [PubMed] [Google Scholar]
- Bais C, Santomasso B, Coso O, Arvanitakis L, Raaka EG, Gutkind JS, Asch AS, Cesarman E, Gershengorn MC, Mesri EA. G-protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature. 1998;391:86–89. doi: 10.1038/34193. [DOI] [PubMed] [Google Scholar]
- Bais C, Van Geelen A, Eroles P, Mutlu A, Chiozzini C, Dias S, Silverstein R, Rafii S, Mesri EA. Kaposi's sarcoma associated herpesvirus G protein coupled receptor immortalizes human endothelial cell by activation of the VEGF receptor-2/KDR. Cancer Cell. 2003;3:131–143. doi: 10.1016/s1535-6108(03)00024-2. [DOI] [PubMed] [Google Scholar]
- Ballestas ME, Chatis PA, Kaye KM. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science. 1999;284:641–644. doi: 10.1126/science.284.5414.641. [DOI] [PubMed] [Google Scholar]
- Barozzi P, Luppi M, Facchetti F, Mecucci C, Alu M, Sarid R, Rasini V, Ravazzini L, Rossi E, Festa S, et al. Post-transplant Kaposi sarcoma originates from the seeding of donor-derived progenitors.[see comment][erratum appears in Nat Med. 2003 Jul;9(7):975] Nature Medicine. 2003;9:554–561. doi: 10.1038/nm862. [DOI] [PubMed] [Google Scholar]
- Boshoff C, Endo Y, Collins PD, Takeuchi Y, Reeves JD, Schweickart VL, Siani MA, Sasaki T, Williams TJ, Gray PW, et al. Angiogenic and HIV-inhibitory functions of KSHV-encoded chemokines. Science. 1997;278:290–294. doi: 10.1126/science.278.5336.290. [DOI] [PubMed] [Google Scholar]
- Boshoff C, Weiss R. AIDS-related malignancies. [Review] [133 refs] Nature Reviews Cancer. 2002;2:373–382. doi: 10.1038/nrc797. [DOI] [PubMed] [Google Scholar]
- Boshoff C, Weiss RA. Kaposi's sarcoma-associated herpesvirus. Adv Cancer Res. 1998;75:57–86. doi: 10.1016/s0065-230x(08)60739-3. [DOI] [PubMed] [Google Scholar]
- Brown LF, Tognazzi K, Dvorak HF, Harrist TJ. Strong expression of kinase insert domain-containing receptor, a vascular permeability factor/vascular endothelial growth factor receptor in AIDS-associated Kaposi's sarcoma and cutaneous angiosarcoma. American Journal of Pathology. 1996;148:1065–1074. [PMC free article] [PubMed] [Google Scholar]
- Browning PJ, Sechler JMG, Kaplan M, Washington RH, Gendelman R, Yarchoan R, Ensoli B, Gallo RC. Identification and Culture of Kaposi's Sarcoma-Like Spindle Cells from the Peripheral Blood of Human Immunodeficiency Virus-1 Individualas and Normal Controls. Blood. 1994;84:2711–2720. [PubMed] [Google Scholar]
- Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi's Sarcoma-Associated Herpes Virus-like DNA sequences are present in AIDS-Related Body cavity B-cell lymphomas. N Engl J Med. 1995;332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles D, Moore P. Identification of Herpesvirus-Like DNA sequences in AIDS-associated Kaposis's Sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- Cherqui S, Kurian SM, Schussler O, Hewel JA, Yates JR, 3rd, Salomon DR. Isolation and angiogenesis by endothelial progenitors in the fetal liver. Stem Cells. 2006;24:44–54. doi: 10.1634/stemcells.2005-0070. [DOI] [PubMed] [Google Scholar]
- Ciufo DM, Cannon JS, Poole LJ, Wu FY, Murray P, Ambinder RF, Hayward GS. Spindle cell conversion by Kaposi's sarcoma-associated herpesvirus: formation of colonies and plaques with mixed lytic and latent gene expression in infected primary dermal microvascular endothelial cell cultures. J Virol. 2001;75:5614–5626. doi: 10.1128/JVI.75.12.5614-5626.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng JH, Zhang YJ, Wang XP, Gao SJ. Lytic replication-defective Kaposi's sarcoma-associated herpesvirus: potential role in infection and malignant transformation. J Virol. 2004;78:11108–11120. doi: 10.1128/JVI.78.20.11108-11120.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dictor M, Rambech E, Way D, Witte M, Bendsoe N. Human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus) DNA in Kaposi's sarcoma lesions, AIDS Kaposi's sarcoma cell lines, endothelial Kaposi's sarcoma simulators, and the skin of immunosuppressed patients. Am J Pathol. 1996;148:2009–2016. [PMC free article] [PubMed] [Google Scholar]
- Dittmer D, Stoddart C, Renne R, Linquist-Stepps V, Moreno ME, Bare C, McCune JM, Ganem D. Experimental transmission of Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) to SCID-hu Thy/Liv mice. Journal of Experimental Medicine. 1999;190:1857–1868. doi: 10.1084/jem.190.12.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer DP. Transcription profile of Kaposi's sarcoma-associated herpesvirus in primary Kaposi's sarcoma lesions as determined by real-time PCR arrays. Cancer Research. 2003;63:2010–2015. [PubMed] [Google Scholar]
- Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, van Marck E, Salmon D, Gorin I, Escande JP, et al. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:4546–4551. doi: 10.1073/pnas.96.8.4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran EM, Shapshak P, Worley J, Minagar A, Ziegler F, Haliko S, Moleon-Borodowsky I, Haslett PA. Presenilin-1 detection in brain neurons and FOXP3 in peripheral blood mononuclear cells: normalizer gene selection for real time reverse transcriptase pcr using the deltadeltaCt method. Front Biosci. 2005;10:2955–2965. doi: 10.2741/1751. [DOI] [PubMed] [Google Scholar]
- Fakhari FD, Dittmer DP. Charting latency transcripts in Kaposi's sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. Journal of Virology. 2002;76:6213–6223. doi: 10.1128/JVI.76.12.6213-6223.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flore O, Rafii S, Ely S, O'Leary JJ, Hyjek EM, Cesarman E. Transformation of primary human endothelial cells by Kaposi's sarcoma- associated herpesvirus. Nature. 1998;394:588–592. doi: 10.1038/29093. [DOI] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis and angiogenesis inhibition: an overview. [Review] [39 refs] Exs. 1997;79:1–8. doi: 10.1007/978-3-0348-9006-9_1. [DOI] [PubMed] [Google Scholar]
- Gallo RC. The enigmas of Kaposi's sarcoma. Science. 1998;282:1837–1839. doi: 10.1126/science.282.5395.1837. [DOI] [PubMed] [Google Scholar]
- Ganem D. KSHV Infection and the Pathogenesis of Kaposi’s Sarcoma. Annu Rev Pathol Mech Dis. 2006;1:273–296. doi: 10.1146/annurev.pathol.1.110304.100133. [DOI] [PubMed] [Google Scholar]
- Gao SJ, Boshoff C, Jayachandra S, Weiss RA, Chang Y, Moore PS. KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene. 1997;15:1979–1985. doi: 10.1038/sj.onc.1201571. [DOI] [PubMed] [Google Scholar]
- Gill PS, Tsai YC, Rao AP, Spruck CH, 3rd, Zheng T, Harrington WA, Jr, Cheung T, Nathwani B, Jones PA. Evidence for multiclonality in multicentric Kaposi's sarcoma. Proc Natl Acad Sci U S A. 1998;95:8257–8261. doi: 10.1073/pnas.95.14.8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisotto MG, Garin A, Martin AP, Jensen KK, Chan P, Sealfon SC, Lira SA. The human herpesvirus 8 chemokine receptor vGPCR triggers autonomous proliferation of endothelial cells. J Clin Invest. 2006;116:1264–1273. doi: 10.1172/JCI26666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundhoff A, Ganem D. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis.[see comment] Journal of Clinical Investigation. 2004;113:124–136. doi: 10.1172/JCI200417803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo HG, Sadowska M, Reid W, Tschachler E, Hayward G, Reitz M. Kaposi's sarcoma-like tumors in a human herpesvirus 8 ORF74 transgenic mouse. J Virol. 2003;77:2631–2639. doi: 10.1128/JVI.77.4.2631-2639.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza MS, Reyes RA, Izumiya Y, Wisdom R, Kung HJ, Luciw PA. ORF36 protein kinase of Kaposi's sarcoma herpesvirus activates the c-Jun N-terminal kinase signaling pathway. J Biol Chem. 2004;279:38325–38330. doi: 10.1074/jbc.M400964200. [DOI] [PubMed] [Google Scholar]
- Hong YK, Foreman K, Shin JW, Hirakawa S, Curry CL, Sage DR, Libermann T, Dezube BJ, Fingeroth JD, Detmar M. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nature Genetics. 2004;36:683–685. doi: 10.1038/ng1383. [DOI] [PubMed] [Google Scholar]
- Klein U, Tu Y, Stolovitzky GA, Mattioli M, Cattoretti G, Husson H, Freedman A, Inghirami G, Cro L, Baldini L, et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med. 2001;194:1625–1638. doi: 10.1084/jem.194.11.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight JS, Cotter MA, 2nd, Robertson ES. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus transactivates the telomerase reverse transcriptase promoter. J Biol Chem. 2001;276:22971–22978. doi: 10.1074/jbc.M101890200. [DOI] [PubMed] [Google Scholar]
- Lagunoff M, Bechtel J, Venetsanakos E, Roy AM, Abbey N, Herndier B, McMahon M, Ganem D. De novo infection and serial transmission of Kaposi's sarcoma-associated herpesvirus in cultured endothelial cells. J Virol. 2002;76:2440–2448. doi: 10.1128/jvi.76.5.2440-2448.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Veazey R, Williams K, Li M, Guo J, Neipel F, Fleckenstein B, Lackner A, Desrosiers RC, Jung JU. Deregulation of cell growth by the K1 gene of Kaposi's sarcoma-associated herpesvirus. Nature Medicine. 1998;4:435–440. doi: 10.1038/nm0498-435. [DOI] [PubMed] [Google Scholar]
- Montaner S, Sodhi A, Molinolo A, Bugge TH, Sawai ET, He Y, Li Y, Ray PE, Gutkind JS. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi's sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell. 2003;3:23–36. doi: 10.1016/s1535-6108(02)00237-4. [DOI] [PubMed] [Google Scholar]
- Montaner S, Sodhi A, Ramsdell AK, Martin D, Hu J, Sawai ET, Gutkind JS. The Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor as a therapeutic target for the treatment of Kaposi's sarcoma. Cancer Res. 2006;66:168–174. doi: 10.1158/0008-5472.CAN-05-1026. [DOI] [PubMed] [Google Scholar]
- Moore PS, Boshoff C, Weiss RA, Chang Y. Molecular mimicry of human cytokine and cytokine response pathway genes by kshv. Science. 1996;274:1739–1744. doi: 10.1126/science.274.5293.1739. [DOI] [PubMed] [Google Scholar]
- Moses AV, Fish KN, Ruhl R, Smith PP, Strussenberg JG, Zhu L, Chandran B, Nelson JA. Long-term infection and transformation of dermal microvascular endothelial cells by human herpesvirus 8. J Virol. 1999;73:6892–6902. doi: 10.1128/jvi.73.8.6892-6902.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidhar S, Pumfery AM, Hassani M, Sadaie MR, Azumi N, Kishishita M, Brady JN, Doniger J, Medveczky P, Rosenthal LJ. Identification of kaposin (open reading frame K12) as a human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus) transforming gene. Journal of Virology. 1998;72:4980–4988. doi: 10.1128/jvi.72.6.4980-4988.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H, Li M, Zarycki J, Jung JU. Inhibition of p53 tumor suppressor by viral interferon regulatory factor. J Virol. 2001;75:7572–7582. doi: 10.1128/JVI.75.16.7572-7582.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naranatt PP, Krishnan HH, Svojanovsky SR, Bloomer C, Mathur S, Chandran B. Host gene induction and transcriptional reprogramming in Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: insights into modulation events early during infection. Cancer Research. 2004;64:72–84. doi: 10.1158/0008-5472.can-03-2767. [DOI] [PubMed] [Google Scholar]
- Nicholas J, Ruvolo VR, Burns WH, Sandford G, Wan XY, Ciufo D, Hendrickson SB, Guo HG, Hayward GS, Reitz MS. Kaposis sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nature Medicine. 1997;3:287–292. doi: 10.1038/nm0397-287. [DOI] [PubMed] [Google Scholar]
- Nicolaides A, Huang YQ, Li JJ, Zhang WG, Friedman-Kien AE. Gene amplification and multiple mutations of the K-ras oncogene in Kaposi's sarcoma. Anticancer Res. 1994;14:921–926. [PubMed] [Google Scholar]
- Pantanowitz L, Dezube BJ. Advances in the pathobiology and treatment of Kaposi sarcoma. Current Opinion in Oncology. 2004;16:443–449. doi: 10.1097/00001622-200409000-00006. [DOI] [PubMed] [Google Scholar]
- Parsons CH, Adang LA, Overdevest J, O'Connor CM, Taylor JR, Jr, Camerini D, Kedes DH. KSHV targets multiple leukocyte lineages during long-term productive infection in NOD/SCID mice. J Clin Invest. 2006;116:1963–1973. doi: 10.1172/JCI27249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillay P, Chetty R, Reddy R. Bcl-2 and p53 immunoprofile in Kaposi's sarcoma. Pathol Oncol Res. 1999;5:17–20. doi: 10.1053/paor.1999.0017. [DOI] [PubMed] [Google Scholar]
- Rabkin CS, Janz S, Lash A, Coleman AE, Musaba E, Liotta L, Biggar RJ, Zhuang Z. Monoclonal origin of multicentric Kaposi's sarcoma lesions. N Engl J Med. 1997;336:988–993. doi: 10.1056/NEJM199704033361403. [DOI] [PubMed] [Google Scholar]
- Rafii S, Lyden D. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nature Medicine. 2003;9:702–712. doi: 10.1038/nm0603-702. [DOI] [PubMed] [Google Scholar]
- Reiner A, Yekutieli D, Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 2003;19:368–375. doi: 10.1093/bioinformatics/btf877. [DOI] [PubMed] [Google Scholar]
- Safai B, Johnson KG, Myskowski PL, Koziner B, Yang SY, Winningham-Ruddles S, Godbold JH, Dupont B. The Natural History of Kaposi's Sarcoma in Acquired Immuno Deficiency Syndrome. Ann Int Med. 1985;103:744–750. doi: 10.7326/0003-4819-103-5-744. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, DePinho RA. Cellular senescence: mitotic clock or culture shock? Cell. 2000;102:407–410. doi: 10.1016/s0092-8674(00)00046-5. [DOI] [PubMed] [Google Scholar]
- Skobe M, Brown LF, Tognazzi K, Ganju RK, Dezube BJ, Alitalo K, Detmar M. Vascular endothelial growth factor-C (VEGF-C) and its receptors KDR and flt-4 are expressed in AIDS-associated Kaposi's sarcoma. Journal of Investigative Dermatology. 1999;113:1047–1053. doi: 10.1046/j.1523-1747.1999.00798.x. [DOI] [PubMed] [Google Scholar]
- Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. Journal of Virology. 1997;71:715–719. doi: 10.1128/jvi.71.1.715-719.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt MR, Kanan Y, Jeong JH, Papin JF, Hines-Boykin R, Dittmer DP. The tumor microenvironment controls primary effusion lymphoma growth in vivo. Cancer Research. 2004;64:4790–4799. doi: 10.1158/0008-5472.CAN-03-3835. [DOI] [PubMed] [Google Scholar]
- Wang HW, Trotter MW, Lagos D, Bourboulia D, Henderson S, Makinen T, Elliman S, Flanagan AM, Alitalo K, Boshoff C. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nature Genetics. 2004;36:687–693. doi: 10.1038/ng1384. [DOI] [PubMed] [Google Scholar]
- Wang L, Dittmer DP, Tomlinson CC, Fakhari FD, Damania B. Immortalization of primary endothelial cells by the K1 protein of Kaposi's sarcoma-associated herpesvirus. Cancer Res. 2006;66:3658–3666. doi: 10.1158/0008-5472.CAN-05-3680. [DOI] [PubMed] [Google Scholar]
- Yang T, Chen S, Leach MW, Manfra D, Homey B, Wiekowski M, Sullivan L, Jenh C, Narula S, Chensue S, Lira SA. Transgenic expression of the chemokine receptor encoded by HHV8 induces an angioprolierative disease resembling Kaposi' s sarcoma. J Exp Med . 2000;191 doi: 10.1084/jem.191.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SM, Zhou FC, Ye FC, Pan HY, Gao SJ. Early and sustained expression of latent and host modulating genes in coordinated transcriptional program of KSHV productive primary infection of human primary endothelial cells. Virology. 2005;343:47–64. doi: 10.1016/j.virol.2005.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FC, Zhang YJ, Deng JH, Wang XP, Pan HY, Hettler E, Gao SJ. Efficient infection by a recombinant Kaposi's sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J Virol. 2002;76:6185–6196. doi: 10.1128/JVI.76.12.6185-6196.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.