

Abstract
Numerous studies have revealed that the BCR-ABL oncoprotein abnormally engages a multitude of signaling pathways, some of which may be important for its leukemogenic properties. Central to this has been the determination that the tyrosine kinase function of BCR-ABL is mainly responsible for its transforming potential, and can be targeted with small molecule inhibitors, such as imatinib mesylate (Gleevec, STI-571). Despite this apparent success, the development of clinical resistance to imatinib therapy, and the inability of imatinib to eradicate BCR-ABL-positive malignant hematopoietic progenitors demand detailed investigations of additional effector pathways that can be targeted for CML treatment. The promotion of cellular survival via the suppression of apoptotic pathways is a fundamental characteristic of tumor cells that enables resistance to anti-cancer therapies. As substrates of survival kinases such as Akt, the FoxO family of transcription factors, particularly FoxO3a, has emerged as playing an important role in the cell cycle arrest and apoptosis of hematopoietic cells. This review will discuss our current understanding of BCR-ABL signaling with a focus on apoptotic suppressive mechanisms and alternative approaches to CML therapy, as well as the potential for FoxO transcription factors as novel therapeutic targets.
Keywords: Apoptosis, BCR-ABL, CML, FoxO, Imatinib Resistance, Proteasomal Degradation
Introduction
While our understanding of the molecular underpinnings of tumorigenesis is far from complete, it has become clear that a fundamental property of cancer cells is the ability to circumvent the apoptotic cellular death program [1]. Investigating the mechanisms underlying resistance of tumor cells to apoptosis has been of significant interest in the field since a desired goal of anti-cancer therapies is to selectively unleash the apoptotic potential that remains inhibited in tumor cells.
In the normal cellular context, proliferation and death programs are tightly linked. Given this, cells harboring a single oncogenic mutation driving proliferation undergo a protective growth inhibitory response, appropriately resulting in apoptosis of the pre-neoplastic cell. In contrast, such as in the evolution of cancers, oncogenes must overcome such protective cellular responses, by taking advantage of cooperating mutations in apoptosis signaling molecules, resulting in the abnormal proliferation and suppression of apoptosis in the tumor cell [2–4]. In a classic example, over-expression of the wild type c-MYC oncoprotein can induce apoptosis as well as sensitize cells towards a host of apoptotic stimuli in certain cell types. However, events such as inactivation of p53, over-expression of BCL-2, or loss of BIM, are able to cooperate with MYC in inducing tumorigenesis[5]. In another mechanism for the promotion of tumorigenesis, oncogenes such as BCR-ABL can simultaneously activate multiple pathways including those involved in cellular proliferation, as well as in the promotion of survival and suppression of apoptosis. The dissection of signaling pathways critical for BCR-ABL-mediated leukemogenesis is essential towards the discovery and deve lopment of rational and successful treatments for BCR-ABL positive chronic myeloid leukemia (CML) and will be the focus of this review.
BCR-ABL and Chronic Myeloid Leukemia (CML)
The Philadelphia (Ph) chromosome, first identified by Nowell and Hungerford in 1960, is the cytogenetic hallmark of chronic myeloid leukemia (CML)[6]. The Ph chromosome is a shortened chromosome 22 that is a by-product of a reciprocal chromosomal translocation between the long arms of chromosomes 9 and 22 t(9;22)(q34;q11) [7]. A consequence of this chromosomal translocation is the replacement of the first exon of the cellular ABL non-receptor tyrosine kinase gene with sequences from the cellular BCR (break point cluster) gene [8, 9], resulting in a chimeric BCR-ABL oncoprotein with highly dysregulated, constitutive tyrosine kinase activity [10]. Three major forms of the BCR-ABL oncogene have been reported based on the break point occurring in the BCR gene. The most commonly occurring form of BCR-ABL is a 210kDa oncoprotein that is found in most cases of CML and 5 to 10% of adults with acute leukemia. The other two forms of BCR-ABL include 230kDa and 185kDa proteins that are associated with chronic neutrophilic leukemia and acute lymphocytic leukemia, respectively [11].
CML is a hematopoietic stem cell malignancy that progresses in several defined stages. In the initial stage of the disease, known as the chronic phase, the BCR-ABL-transformed clone is a progenitor for the granulocytic, monocytic, erythroid, megakaryocytic and lymphoid lineages, but only results in enhanced proliferation of maturing granulocytes. This genetically unstable chronic phase of the disease is inevitably followed by clonal evolution of the neoplastic cells resulting in the more aggressive stages of the disease, known as the accelerated and blast phases. During these phases, which may involve transformation to either acute myeloid or lymphoid leukemia, hematopoiesis is severely compromised because the leukemic clone loses its capacity to differentiate, leading to the accumulation of abnormally differentiated cells or blasts in the bone marrow and blood [12–15]. Indeed, a recent study demonstrated that BCR-ABL-dependent transcriptional upregulation of the Id-1 (inhibitor of differentiation) transcription factor is a critical determinant in the differentiation block that exists in BCR-ABL-transformed K562 cells [16].
Importantly, various studies have established that the BCR-ABL p210kDa protein is oncogenic, and is essential for the pathogenesis of CML. By far, the most recent and convincing evidence for the importance of BCR-ABL in CML includes the ability of the ABL tyrosine kinase inhibitor, imatinib mesylate (Gleevec, STI-571, Novartis Pharmaceuticals), to selectively induce apoptosis in BCR-ABL-transformed leukemic cells [17, 18] and to produce molecular and cytogenetic remissions in chronic phase CML patients [19–21]. A further revelation that BCR-ABL is critical in CML comes from the determination that clinical resistance to imatinib can arise either through BCR-ABL gene amplification or point mutations within BCR-ABL [22].
Earlier studies aimed at investigating the oncogenic potential of BCR-ABL were performed in various systems in vitro and in vivo, some of which have emerged as useful tools in elucidating the molecular mechanisms behind BCR-ABL-induced leukemogenesis. BCR-ABL can transform Rat-1 fibroblasts [23], primary bone marrow B-lymphoid cells [24], and growth factor-dependent hematopoietic cells [25]. The latter was first demonstrated in the interleukin-3 (IL-3)-dependent murine bone marrow-derived cell line BaF3. While untransformed BaF3 cells fail to proliferate and undergo apoptosis in the absence of IL-3 stimulation, BCR-ABL expression protects BaF3 cells from apoptosis, enabling their growth independently of IL-3. Interestingly, BCR-ABL-positive progenitors isolated from CML patients do not sustain growth in the absence of growth factors, but are partially dependent on growth factors, indicating possible differences between growth-factor-dependent cell line models and primary cell culture. Overall, growth factor-dependent hematopoietic cells, while not free of disadvantages [26], offer a convenient in vitro model to study the effects of BCR-ABL transformation and allows for direct comparisons between non-transformed parental and BCR-ABL-transformed cells [27]. On the other hand, such comparisons are not possible in CML patient-derived BCR-ABL-positive cell lines, such as K562 and BV173. These cell lines have been useful, but results need to be interpreted cautiously since they originate from blast crisis CML, in which case mutations in addition to BCR-ABL could be present [28].
The ability of BCR-ABL to induce leukemia in vivo has been tested using various murine models. Transplantation of BCR-ABL-transformed cell lines into syngeneic mice results in the rapid development of acute leukemias [29]. Chronic phase and blast crisis CML cells are also able to produce leukemias in varying capacities in NOD/SCID mice [30]. Efforts in generating transgenic mice with constitutive expression of BCR-ABL failed due to embryonic lethality [31]. These studies suggested that the target cell for BCR-ABL expression is extremely important for determining a leukemic outcome. Nevertheless, inducible expression of BCR-ABL in transgenic mice did reveal the development of leukemias, although these were mostly acute B and T lymphoid leukemias [32, 33]. Consequently, one of the challenges was to develop an in vivo murine model that would more accurately represent the myeloproliferative disease with which BCR-ABL is associated in human CML. As a major advance in the development of a suitable mouse model for CML, the murine bone marrow transduction-transplantation model has so far been the most accurate in producing a myeloproliferative disease in mice that resembles human CML [34–38]. This involves using high titer BCR-ABL retroviral stocks to transduce primary bone marrow cells, and subsequent transplantation of BCR-ABL-transduced bone marrow cells into irradiated syngeneic recipients. Mice receiving BCR-ABL-transduced bone marrow effectively develop a fatal CML-like myeloproliferative disease in three to four weeks and show an expansion of cells belonging to the myeloid lineage (i.e. neutrophils), with infiltration of these cells in the spleen, liver and lungs. This murine model has so far been used to evaluate novel therapies, determine which domains of the BCR-ABL oncogene are important for its leukemogenic effect, and investigating which signaling pathways may be critical for the induction of leukemogenesis.
The Tyrosine Kinase Activity of BCR-ABL and Avenues for Therapy
Remarkably, one of the greatest leaps towards molecular targeted therapy in cancer has been made in the field of CML, with the development of imatinib mesylate (Gleevec, STI-571-Novartis Pharmaceuticals), a small molecule inhibitor of the constitutive tyrosine kinase activity of BCR-ABL [39]. Prior to the development of tyrosine kinase inhibition therapy for CML, earlier studies provided strong evidence that constitutive ABL tyrosine kinase activity is required for BCR-ABL transformation. In particular, the tyrosine kinase activity of BCR-ABL is increased relative to that of c-ABL [10], and kinase-deficient BCR-ABL (K1176R) is unable to transform either growth factor-dependent hematopoietic cells or murine bone marrow cells [40], or to induce myeloproliferative disease in the murine bone marrow-transduction-transplantation model in vivo [38]. BCR-ABL tyrosine kinase activity contributes to the phosphorylation of various cellular substrates such as Crk-L[41], but also results in BCR-ABL autophosphorylation [10], which creates a platform for the binding of additional adaptor molecules. BCR-ABL-mediated phosphorylation of various substrates leads to activation of signaling pathways that promote cellular proliferation and survival [11, 42] (Figure 1).
Figure 1. BCR-ABL Activates Multiple Signaling Pathways.
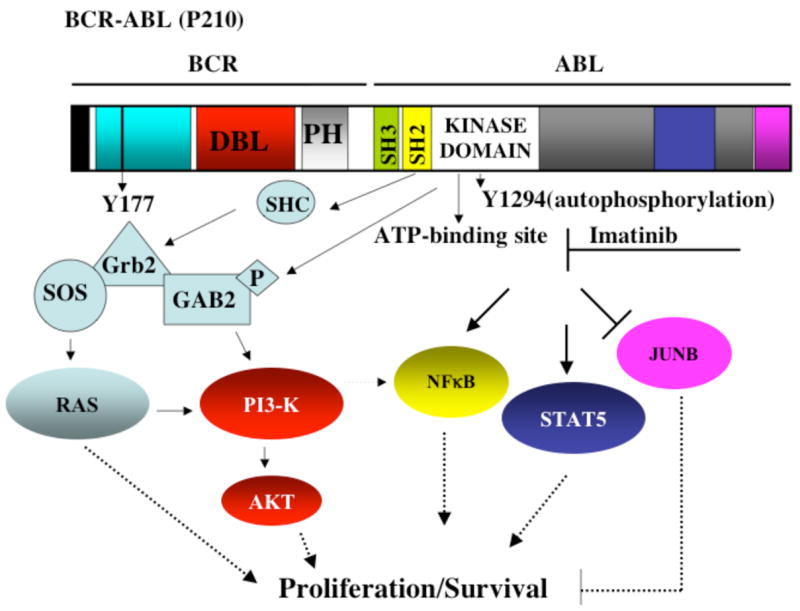
BCR-ABL kinase activity and autophosphorylation create platforms for the activation of various pathways involved in cellular proliferation and survival, such as the Ras, PI3-K/Akt, NF-κB, and STAT5, among others. The small molecule inhibitor imatinib mesylate (imatinib) inhibits BCR-ABL’s kinase activity by competing for the ATP binding site within the kinase domain. See text for details.
Imatinib is a 2-phenylaminopyrimidine derivative designed according to the structure of the ATP binding site of protein kinases. Hence, imatinib functions by competitive inhibition of the ATP binding site of BCR-ABL, thereby preventing tyrosine kinase activity and the subsequent phosphorylation of substrates involved in BCR-ABL signaling. Crystal structure studies have revealed that imatinib binds to the kinase domain of ABL in its inactive conformation and therefore locks it in an inactive state [43, 44]. Imatinib inhibits not only ABL but also the receptor for platelet-derived growth factor A and B (PDGFR-α, β), stem cell factor receptor (KIT) and ARG (abl-related gene) tyrosine kinases, making it useful in attenuating oncogenic events triggered upon activation of these kinases [45–47]. Imatinib appears to selectively induce apoptosis in numerous BCR-ABL-positive leukemic cell lines and BCR-ABL-transduced hematopoietic cells (BaF3 and 32D), but not in BCR-ABL-negative hematopoietic cancer cell lines (HL60, Jurkat and U937), or non-transformed growth factor-dependent hematopoietic cells. Similarly, at concentrations of 1μM, imatinib can selectively inhibit colony formation of CML progenitors over that of normal progenitors. However, in these studies, it is important to note that despite the presence of imatinib, a minor percentage of BCR-ABL-positive colonies persist, suggesting that imatinib does not completely inhibit BCR-ABL-induced growth and transformation [17, 48].
The activity of imatinib against BCR-ABL-induced tumor growth has also been evaluated in murine models. Imatinib causes a dose-dependent inhibition of tumor growth in mice injected with BCR-ABL-transformed 32D cells [17], with similar results observed in nude mice injected with human BCR-ABL-positive KU812 cells [49]. Finally, when tested in the murine bone marrow transduction-transplantation model, imatinib attenuated symptoms of myeloproliferative disease, as evidenced by a reduction in white blood cell counts and spleen size and prolonged survival. However, this positive response was not uniform, since 25% of the animals in this study remained refractory to treatment [50].
Imatinib: The Success Story
In the clinical setting, promising results from Phase I, II, and III studies have led to the approval of imatinib as the front-line therapy for CML. Overall, imatinib is effective at producing durable hematologic and cytogenetic remissions in most chronic phase CML patients. The results and details relating to these clinical trials have been extensively reviewed in [51, 52]. Briefly, phase I trials of imatinib mesylate on CML in chronic phase (CP) conducted by Druker et al. (2001) employed a dose-escalating study, which was administered to patients with CML in CP in whom treatment with IFN-α had failed [20]. Complete hematologic responses were observed in 98% (53/54) of patients treated with daily doses of 300mg or more, and typically occurred within the first month of therapy. Of the 54 patients treated with 300mg or more, cytogenetic responses occurred in 29, including 17 (31%) with major cytogenetic responses; 7 of these patients had complete cytogentic remissions. Thirty-eight CML patients with blast crisis (BC) were also treated with doses ranging from 300 to 1000mg. Responses occurred in 21 of 38 patients (55%); 4 of these 21 patients had a complete hematologic response. Seven patients with a myeloid BC continued to receive treatment and remained in remission from 101 to 349 days after starting treatment [21, 53]. The results provided evidence of the essential role of BCR-ABL tyrosine kinase activity in CML, and demonstrated the potential for the devlopment of anticancer drugs based on the specific molecular abnormality present in human cancer. Kantarjian et al. (2002) conducted a phase II study of imatinib on CML in CP [21]. A total of 532 patients with CML in late CP in whom previous therapy with IFN-α had failed were treated with 400mg of imatinib daily. Imatinib induced major cytogenetic responses in 60% of the 454 patients with confirmed CP CML and complete hematologic responses in 95%. After 18 months, CML had not progressed to accelerated phase (AP) or BC in about 89% of patients, and 95% of the patients were alive. Thus, imatinib induced high rates of cytogenetic and hematologic responses in patients with CML in CP in whom previous INF–α therapy had failed. Talpaz et al. (2002) conducted a phase II study of imatinib on CML in AP. A total of 235 CML patients were enrolled in this study, with 181 having a confirmed diagnosis of AP. Patients were treated with 400 or 600mg daily. Imatinib induced a hematologic response in 82% patients and sustained hematologic responses lasting at least 4 weeks in 69% (complete in 34%). The rate of major cytogenetic response was 24% (complete in 17%). Estimated 12-month progression free and overall survival rates were 59% and 74%, respectively. In comparison with 400mg, imatinib doses of 600mg daily led to more cytogentic responses (28% compared with 16%), longer duration of response (79% compared with 57% at 12 months), time to disease progression (67% compared with 44% at 12 months), and overall survival (78% compared with 65% at 12 months), with no clinically relevant increase in toxicity. Thus, imatinib was an effective and well-tolerated treatment for patients with CML in AP. Sawyers et al. (2002) conducted a phase II study of imatinib on CML in myeloid BC [19]. A total of 260 patients with CML were enrolled, of whom 229 had a confirmed diagnosis of CML in BC. Patients were treated daily with doses of 400 or 600mg. Imatinib induced hematologic responses in 52% of patients and sustained hematologic responses lasting at least 4 weeks in 31% of patients including complete hematologic responses in 8%. For patients with a sustained response, the estimated median response duration was 10 months. Imatinib induced major cytogenetic responses in 16% of patients, with 7% of the responses being complete. Thus, imatinib had substantial activity when used as a single agent in patients with CML in BC. The best observed results at five years in patients remaining on first-line imatinib therapy from the phase III IRIS trial (International Randomized Study of Interferon and STI-571) [54] are summarized in Table 1.
Table 1.
Summary of five years response in patients remaining on first-line imatinib therapy from the phase III IRIS trial
| Parameter | % of patients |
|---|---|
| Complete hematologic response | 97 |
| Major cytogenetic response | 89 |
| Complete cytogenetic response | 82 |
| Survival
Overall Excluding non-CML deaths |
89 95 |
| Survival without accelerated or blastic phase | 93 |
| Event-free survival | 83 |
Clinical Resistance to Imatinib and The Need for Alternative Therapies
The emergence of resistance to imatinib therapy is of great concern and mechanisms explaining resistance have been urgently investigated in the last few years. The most prominent finding from clinical samples is that 50–90% of cases in which resistance develops after imatinib therapy involve point mutations in the BCR-ABL kinase domain. Another mechanism of resistance may involve increased expression of BCR-ABL, resulting from the amplification of the BCR-ABL gene sequence [55–57].
The kinase domain mutations can be grouped according to their location, effect on kinase conformation, and facilitation of imatinib binding [55–57]. These include mutations within the P-loop of the kinase domain, which are thought to alter the kinase towards an active conformation and therefore preclude imatinib binding. In another class, amino acid 315, which forms a critical hydrogen bond with imatinib, is mutated to an isoleucine residue (T315I) and thereby abrogates binding of imatinib. Other classes of mutations involve those at residue 351, and A-loop mutations, which also shift the conformation to an active kinase state and therefore prevent imatinib binding. The T315I BCR-ABL mutation is one of the most frequently occurring and highly resistant to imatinib as well as the newer generation of kinase inhibitors such as BMS-354825 (Dasatinib) and AMN107 (Nilotinib) [58, 59]. Another aspect of imatinib resistance is that some of these mutations may be present even before imatinib therapy. These mutations have been postulated to offer a growth advantage and may dictate disease progression [51].
In a related clinical problem with imatinib treatment, it has become apparent that a minor percentage of BCR-ABL-positive progenitors persist in chronic phase CML patients despite imatinib treatment [60, 61]. Disturbingly, BCR-ABL transcripts coding for mutations in the kinase domain have been detected even in some patients who have achieved a complete cytogenetic response, suggesting a link between resistance and disease persistence [62, 63]. Together with mathematical models that take into account the bi-phasic response to imatinib as well as a continued need for imatinib therapy [64], all these observations have led to the conclusion that imatinib is unable to target the quiescent BCR-ABL-positive stem cell population, and thus cannot completely eliminate the disease [65].
Overall, it is clear that while imatinib and the newer generation kinase inhibitors show promise in the management of CML, a detailed understanding of the signaling pathways emanating from and in addition to BCR-ABL kinase activity is necessary to decipher key downstream events that govern BCR-ABL-induced leukemogenesis. Such studies will yield potential insights into novel therapeutic strategies that can then be applied independently or combined with tyrosine kinase inhibition therapy to cure CML.
Signaling Pathways Activated by BCR-ABL and the Suppression of Apoptosis
A well-known aspect of BCR-ABL transformation is its ability to activate multiple signaling pathways that lead to proliferation, reduced growth factor-dependence and apoptosis, and abnormal interaction with extracellular matrix and stroma. Accumulating evidence suggests that the suppression of apoptosis constitutes an important mechanism by which BCR-ABL drives the expansion of myeloid cells. Notably, the primary consequence of tyrosine kinase inhibition with imatinib in BCR-ABL-transformed cells is the induction of apoptosis [17, 18]. In growth factor-dependent hematopoietic cells, BCR-ABL induces the survival and proliferation of cells that would otherwise undergo apoptotic cell death in response to growth factor withdrawal. Furthermore, antisense oligonucleotide-mediated inhibition of BCR-ABL in these growth factor-independent transformed cells results in apoptosis without altering their cell cycle [66]. In other studies using antisense oligonucleotides, it was shown that BCR-ABL-positive cells are highly resistant to various apoptotic stimuli and become sensitized to drug treatment upon inhibition of BCR-ABL [67]. Further support for the anti-apoptotic effects of BCR-ABL comes from experiments with temperature-sensitive BCR-ABL kinase mutants, in which induction of BCR-ABL kinase activity at the permissive temperature led to a significant decrease in apoptosis in the absence of growth factors [68, 69]. In fact, investigations using primary cells have revealed that CML progenitors show a normal proliferative response to growth factors and do not have a greater proliferative potential than normal progenitors [70]. Furthermore, in the absence of serum and growth factors, neither normal nor CML progenitors proliferated, yet the latter maintained higher cell viability [66]. Altogether, these observations detail the important role that BCR-ABL inhibition of apoptosis plays in myeloid cell expansion, tumor progression, and resistance to cytotoxic therapy.
As a result of its elevated tyrosine kinase activity, BCR-ABL activates a multitude of signaling pathways, some of which may be crucial for its leukemogenic activity. The Ras [71], PI3-K/Akt [72, 73], JAK/STAT [74–76], and NF-κB [77] signaling pathways are among those activated by BCR-ABL (Figure 1). In accordance with the ability of BCR-ABL to substitute for the requirement of cytokines, many of these pathways are also activated by hematopoietic cytokines upon binding to their respective cytokine receptors. Furthermore, it has been shown that the serine/threonine protein phosphatase, PP2A, is also functionally inactivated by BCR-ABL [78]. A functional consequence of the activation of these pathways involves changes in the activity and gene expression of key molecules, which have a direct impact on cellular survival, growth, and behavior. In particular, the Ras, PI3-K/Akt, JAK/STAT, and NF-κB pathways are capable of transmitting anti-apoptotic signals, and it has become of interest to determine which anti-apoptotic signals play a role in BCR-ABL-mediated leukemogenesis.
BCR-ABL Activation of STAT
The signal transduction and activators of transcription (STAT) transcription factors have been extensively studied for their potential role in leukemogenesis. The STAT family of transcription factors participates in diverse processes, including cell growth, differentiation, apoptosis, fetal development, inflammation, and immune response. Ligand binding to cytokine or growth factor receptors initiates a series of signaling events that results in STAT phosphorylation, dimerization, and subsequent translocation to the nucleus. Some STAT target genes include Bcl-xL and Mcl-1, substantiating an anti-apoptotic role for the activity of STAT transcription factors [79].
BCR-ABL-positive CML cell lines display constitutive phosphorylation and activation of STAT-1 and STAT-5. STAT-5 activation induces up-regulation of the serine/threonine kinase Pim-1 and the anti-apoptotic genes of the Bcl-2 family, A1 and Bcl-xL, [80–82]. Inhibition of STAT-5 signaling with dominant negative mutants impairs BCR-ABL transformation in vitro [83, 84]. Confoundingly, BCR-ABL-transduced bone marrow cells from STAT-5a/b double null mice efficiently induce a fatal myeloproliferative disease in the transduction-transplantation model [85]. Therefore, it remains possible that while STAT-5 does induce an anti-apoptotic effect in BCR-ABL transformation, other pathways substitute for this effect in its absence.
BCR-ABL Activation of NF-κB
The Nuclear Factor-κB (NF-κB) family of pleiotropic transcription factors function as dimers and are activated by a broad range of stimuli including cytokines, physical and oxidative stresses, viruses and viral products. The IκB (inhibitor of κB) proteins negatively regulate NF-κB by sequestering it to the cytoplasm. Phosphorylation and subsequent degradation of IκB relieves NF-κB to translocate to the nucleus. Upon their activation, NF-κB proteins promote the transcription of numerous genes involved in diverse cellular processes, including inflammation, cell cycle, survival/anti-apoptosis, and angiogenesis [86]. The constitutive activation of NF-κB is frequently observed in various cancers, and correlates with resistance of tumor cells to apoptosis. The NF-κB anti-apoptotic target genes include those from the Bcl-2 family (Bcl-xL, BFL1) and the inhibitors of apoptosis proteins, IAP1, IAP2, and XIAP [87].
Earlier studies demonstrated that BCR-ABL-expressing hematopoietic cells showed increased DNA binding activity of the p65 (RelA) subunit of NF-κB [88]. Using a mutated form of IκB that cannot be phosphorylated and therefore constitutively inhibits NF-κB (the super repressor form of IκBα), it was determined that NF-κB activation is not required for BCR-ABL to protect cells from growth factor withdrawal or exposure to DNA-damaging agents. On the other hand, NF-κB activity was found to be required for BCR-ABL-transformed hematopoietic cells to form tumors in nude mice, and for the transformation of primary bone marrow cells [77]. The exact mechanism of NF-κB activation in BCR-ABL-transformed cells, and its relevance to BCR-ABL-induced CML, however, remains to be elucidated.
BCR-ABL Activation of the Ras Pathway
Within the Ras superfamily of low molecular weight GTP-binding proteins is the Ras subfamily, consisting of H-Ras, N-Ras and K-Ras. The Ras pathway regulates various aspects of cellular growth and has been one of the most extensively studied both in the context of normal and cancer cells [89–91]. Activating mutations in Ras, or changes in molecular components that comprise Ras signaling, are found in most human cancers including leukemias, and result in increased cellular proliferation and survival [92].
Indeed, the Ras pathway is constitutively activated in BCR-ABL-expressing cells. Inhibition of Ras activity with dominant negative mutants impairs the ability of BCR-ABL to transform murine bone marrow cells and rodent fibroblasts [71]. A major link between BCR-ABL and the Ras pathway is provided through the adaptor protein Grb2. Grb2 associates with the guanine nucleotide exchange factor Sos, which forms a platform for the recruitment of Ras, enabling the activation of Ras from the GDP- to GTP-bound state. Via its SH2 domain, Grb2 binds to the phosphorylated tyrosine residue 177 within the BCR region of BCR-ABL. A mutation in the tyrosine 177 position of BCR-ABL to phenylalanine (Y177F) abrogates Grb2 binding and subsequent Ras activation, resulting in reduced foci formation in rodent fibroblasts [93, 94]. However, BCR-ABL Y177F maintains its ability to activate Ras in cytokine-dependent hematopoietic cells, rendering them cytokine-independent [95]. Given conflicting results on the role of Y177 in BCR-ABL transformation in vitro, Million et al. investigated a role for Y177 in vivo using the murine transduction-transplantation model. In this model, BCR-ABL (Y177F) significantly attenuates the development of a myeloproliferative disease, and results in delayed occurrence of B and T lymphoid leukemias, indicating that this site is important in determining the lineage and severity of leukemic disease [96]. Efforts to characterize the signaling events from Y177 have revealed that the scaffolding adaptor protein Gab2 is phosphorylated in BCR-ABL-expressing cells through its recruitment to phosphorylated Y177 in a complex with Grb2. BaF3 cells expressing BCR-ABL Y177F show decreased phosphorylation and association with Gab2 and reduced activation of the Ras/Erk and PI3-K/Akt pathways, resulting in decreased cellular proliferation and migration. Interestingly, BCR-ABL cannot transform primary myeloid progenitors from Gab2-deficient mice. Similar to cells expressing BCR-ABL Y177F, BCR-ABL-expressing Gab2-deficient myeloid cells show defects in Ras/Erk and PI3-K/Akt activation. Altogether, these studies identify Gab2 and its downstream activation of the Ras and PI3-K/Akt pathways as playing an important role in BCR-ABL-mediated leukemogenesis [97].
In terms of the anti-apoptotic effects of Ras activation in BCR-ABL transformation, the anti-apoptotic member of the BCL-2 family, Mcl-1, is constitutively expressed in a Ras/Raf/MEK-dependent manner in primary CML cells. Antisense inhibition of Mcl-1 led to decreased survival and apoptosis of imatinib-sensitive as well as -resistant BCR-ABL-positive K562 cells, and produced synergistic effects with imatinib [98].
BCR-ABL Inhibition of the PP2A Phosphatase
A recent study by Neviani et al. [78] has revealed that the tumor suppressor serine/threonine protein phosphatase, PP2A, is functionally inactivated in blast crisis CML through BCR-ABL-mediated transcriptional upregulation of the PP2A inhibitor, SET. The inactivation of PP2A then allows for hyperphosphorylation and inactivation of proapoptotic PP2A substrates such as phospho-BAD. Similarly, hyperphosphorylation of PP2A substrate kinases such as phospho-Akt and phospho-ERK leads to their prolonged activation and ability to drive survival and proliferative signaling pathways. In imatinib-sensitive and –resistant (T315I) BCR-ABL positive cell lines and CML blast crisis progenitors, restoration of PP2A activity promotes dephosphorylation of key regulators of cell survival and proliferation, suppresses BCR-ABL activity, and induces BCR-ABL degradation. Ultimately, this leads to growth suppression, enhanced apoptosis, restored differentiation, and decreased in vivo leukemogenesis of imatinib-sensitive and –resistant BCR-ABL cells. Thus, functional inactivation of PP2A is essential for BCR-ABL-mediated leukemogenesis and, perhaps, required for blastic transformation. These findings underscore the possibility that pharmacological enhancement of PP2A activity represents a possible therapeutic strategy for blast crisis and imatinib-resistant cases of CML.
BCR-ABL Activation of the PI3-K/Akt Pathway
Signal transduction pathways that emanate from the activation of phosphoinositide-3 kinase (PI3-K) have been intensively investigated in mammalian systems, and play a central role in survival, proliferation, differentiation, adhesion, metabolism, and motility [99]. While there are several families and classes of phosphoinositide kinases (PIKs), the class IA PI3-Ks within the PI3-K family, become activated when recruited to the cell surface by growth factor receptor tyrosine kinases. PI3-K is a lipid kinase that functions as a heterodimer consisting of a p110 catalytic subunit and a p85 regulatory subunit. Upon its activation by growth factor tyrosine kinase receptors, PI3-K phosphorylates phosphatidylinositol bisphosphate (PIP2) to form phosphatidylinositol triphosphate (PIP3) [100]. The formation of PIP3 can be reversed by the phosphatase and tensin homolog deleted on chromosome 10 (PTEN) [101]. On the other hand, PIP3 provides a platform for the recruitment of kinases, such as the serine/threonine kinases Akt, 3-phosphoinositide-dependent protein kinase-1 (PDK1), and perhaps others via their pleckstrin homology (PH) domains. In this setting, Akt is phosphorylated at distinct residues, namely at threonine 308 within the activation loop, most probably by PDK1, and at serine 473 within the hydrophobic motif by a mechanism that involves PDK2 (mTORC2). Activated Akt regulates numerous cellular substrates, resulting in cell growth, survival, and suppression of apoptosis, among other effects [102].
The constitutive activation of the PI3-K/Akt pathway is common in various cancers, and is emerging as a crucial event in tumorigenesis [103, 104]. The activation of the PI3-K/Akt pathway also occurs in BCR-ABL transformation [73], with mounting evidence for its importance in mediating BCR-ABL’s leukemogenic effects. Original studies carried out by Skorski and colleagues showed that treatment of chronic, acute, and blast crisis primary CD34+ CML cells, as well as BCR-ABL-infected murine bone marrow cells with a PI3-K inhibitor, wortmannin, led to decreased colony formation [105]. Subsequent investigations corroborated the involvement of Akt as a downstream effector of PI3-K activation in BCR-ABL transformation. Co-expression of BCR-ABL with a dominant negative form of Akt (K179M) in primary murine bone marrow cells resulted in reduced colony formation. The significance of Akt activation was substantiated in experiments that showed the ability of a constitutively active Akt (E40K) to rescue the transformation defect of a BCR-ABL mutant lacking the SH2 domain. These in vitro studies were confirmed with in vivo studies that indicated the importance of Akt activation in BCR-ABL-induced leukemia. Injection of marrow cells co-expressing BCR-ABL and dominant negative Akt into SCID mice greatly attenuated the development of leukemia as evidenced by milder splenomegaly, and lack of tumors in other organs such as lungs, liver and kidney [72].
Notably, pharmacological inhibitors of PI3-K (LY294002 and Wortmannin) synergize with imatinib in inducing apoptosis of both chronic and blast crisis CML cells [106]. Combination of a PDK-1 inhibitor (OSU-03012), which essentially inhibits Akt activation, with imatinib resulted in apoptosis even in cells expressing the BCR-ABL T315I imatinib-resistant mutant [107]. Besides substantiating a role for PI3-K/Akt signaling in BCR-ABL-mediated transformation and leukemogenesis, some of these observations also indicate that PI3-K/Akt activation is potentially a crucial event in BCR-ABL-mediated resistance to imatinib.
While the precise mechanism(s) by which BCR-ABL activates PI3-K is not completely understood, several possibilities exist, with stronger evidence for some than others. For one, the direct activation of PI3-K by BCR-ABL is not likely. Even though earlier studies showed that BCR-ABL directly binds to the p85 regulatory subunit of PI3-K [105], further investigations revealed that this interaction was not required for PI3-K activation [108]. In addition, an optimal YXXM motif for recognition of the PI3-K regulatory subunit by SH2 domains was likewise found not to be required for PI3-K activation [108].
On the other hand, BCR-ABL-induced tyrosine auto-phosphorylation, and/or phosphorylation of its substrates, can provide the platform for PI3-K activation. As mentioned earlier, recent evidence indicates a strong role for tyrosine 177 of BCR-ABL in PI3-K/Akt activation. Central to the effects of signaling via this site in BCR-ABL is the adaptor protein Gab2, which is indirectly recruited to BCR-ABL tyrosine 177 through a Grb2/Gab2 complex. Consequent Gab2 tyrosine phosphorylation provides binding sites for the SH2 domain of the p85 regulatory subunit of PI3-K, thereby causing activation of PI3-K. BCR-ABL Y177F-expressing BaF3 cells show reduced activation of PI3-K and Akt and slightly decreased proliferation in the absence of cytokines compared to wild type BCR-ABL. In a related manner, BCR-ABL-expressing Gab2-deficient myeloid cells and lymphoblasts also display drastic reductions in PI3-K and Akt activation, and remain refractory to BCR-ABL transformation [97].
The Gab2 adaptor protein, however, may not provide the only mechanism for PI3-K/Akt activation in BCR-ABL-transformed cells. The adaptor protein CrkL can bind via its SH3 domain to BCR-ABL. The resulting phosphorylation of CrkL provides a docking site for c-Cbl. The phosphorylation of c-Cbl in turn creates a binding site for the p85 regulatory subunit of PI3-K [109]. The importance of c-Cbl in BCR-ABL transformation, however, is not clear, as c-Cbl-deficient bone marrow cells expressing BCR-ABL can effectively generate CML-like disease in vivo [110].
Collectively, in vitro and in vivo studies have substantiated a role for PI3-K/Akt activation in BCR-ABL transformation and leukemogenesis. Furthermore, multiple mechanisms exist for PI3-K and subsequent Akt activation in BCR-ABL-transformed cells.
Signaling Downstream of the Serine/Threonine Kinase Akt
An important set of questions, given the findings on BCR-ABL-induced PI3-K/Akt activation, has been to determine which molecules downstream of activated Akt play an important role in mediating the leukemogenic effects of BCR-ABL. It is well known that activated Akt can phosphorylate and therefore functionally regulate the activity of numerous cellular substrates in order to promote survival. The range of Akt substrates include, but are not limited to the pro-apoptotic BCL-2 family member BAD [111, 112], caspase-9 [113], Mdm2 [114], mTOR and the FoxO subclass of forkhead transcription factors (FoxO1, FoxO3a, and FoxO4) (reviewed in [115]). A thorough appraisal of which Akt substrates are relevant in BCR-ABL transformation has not yet been accomplished, and only a handful of studies have focused on some of these substrates as potentially playing an important role in mediating the leukemogenic effects of BCR-ABL.
Since the suppression of apoptosis is a significant mechanism that contributes to the oncogenic effects of BCR-ABL, BAD initially presented itself as an attractive and a potentially important substrate regulated by activated Akt during BCR-ABL transformation. BAD exerts its pro-apoptotic effects by binding to, and preventing the function of anti-apoptotic BCL-2 proteins, such as BCL-XL [116]. Akt-mediated phosphorylation of BAD results in sequestration of BAD in the cytoplasm through binding to the 14-3-3 family of phospho-serine interacting proteins [111, 112, 117]. In vitro studies revealed that even though BCR-ABL promotes the phosphorylation and inactivation of BAD, BAD phosphorylation is not essential for governing BCR-ABL-induced survival [118]. In these studies a sub-population of BCR-ABL-expressing cells continued to survive in the absence of BAD phosphorylation, yet remained dependent upon PI3-K for their survival. The presence of BAD-independent routes to BCR-ABL-induced survival argues for the involvement of other substrates downstream of PI3-K/Akt activation that contribute to the survival or anti-apoptotic signaling induced by BCR-ABL.
While caspase-9 is an Akt substrate [113], it is unclear if its phosphorylation-dependent inhibition in BCR-ABL-transformed cells [119] is a prominent mechanism for apoptotic evasion downstream of Akt activation. It has been demonstrated, however, that downstream of cytochrome c release, BCR-ABL interferes with the recruitment of caspase-9 to the apoptosome suggesting that BCR-ABL employs multiple mechanisms to inhibit apoptosome function [119]. Another target of Akt, Mdm2, is a regulator of the p53 tumor suppressor that when phosphorylated by Akt leads to p53 inactivation [114]. While there is no direct evidence at present to show that Akt-dependent Mdm2 phosphorylation is critical for BCR-ABL-induced transformation, a few studies have indicated that BCR-ABL can down-regulate p53 by increasing the expression of Mdm2 [120, 121].
The serine/threonine kinase mammalian target of rapamycin (mTOR) is an Akt substrate that does not play a direct role in apoptosis, but functions in the translational control of proteins involved in regulating cellular growth, size, and the cell cycle [122]. An interesting line of investigation has revealed that mTOR is constitutively active in BCR-ABL expressing cells, and that rapamycin, an mTOR inhibitor, inhibits the growth of both imatinib-sensitive and resistant BCR-ABL transformed cells [123, 124]. Rapamycin induces cell cycle arrest and apoptosis in primary CML cells from untreated chronic phase and imatinib resistant patients [125]. In in-vivo studies, rapamycin suppresses the growth of CML cells when given to an imatinib resistant CML patient [125]. Current efforts on BCR-ABL and mTOR research are aimed at determining the mechanism by which rapamycin inhibits growth of BCR-ABL positive leukemic cells.
The FoxO Sub-Class of Forkhead Transcription Factors
Yet another prominent class of molecules regulated by Akt consists of the FoxO sub-family of forkhead transcription factors [115]. A highly conserved 100 amino acid forkhead DNA binding domain that engages in sequence-specific contacts with DNA regulatory elements defines the eukaryotic family of forkhead transcription factors. The forkhead transcription factors are also referred to as the winged helix transcription factors, since the structure of their DNA binding domain consists of 2 large loops, or wings, and three α helices. Numerous forkhead genes have been identified in a broad range of organisms from yeast and worms to humans, with the first member identified in Drosophila melanogaster. Given the large numbers and diversity of forkhead family members, the forkhead transcription factors display a variety of functions in development, differentiation, and even tumorigenesis, with this latter function attracting great attention in recent years [126].
According to the revised nomenclature [127], all vertebrate forkhead transcription factors have been assigned the symbol Fox (Forkhead Box), and are further divided into sub-classes based on phylogenetic analysis of their DNA binding domains. The FoxO (Forkhead Box, class O) transcription factors are a sub-class of the Fox forkhead transcription factors. The FoxO sub-class is evolutionarily conserved and shares a unique amino acid sequence (GDSNS) within the DNA binding domain that is not present in other forkhead proteins. The mammalian members of the FoxO sub-class include FoxO1 (also known as FKHR), FoxO3a (also known as FKHRL1), FoxO4 (also known as AFX), and the recently characterized FoxO6 [128, 129].
FoxO Regulation in Mammalian Systems
Initial insight into the functions of the FoxO transcription factors in mammalian cells was largely derived from genetic studies on the regulation of the FoxO orthologue, daf-16, in the nematode C. elegans. In C. elegans, the insulin receptor daf-2, the phosphatidylinositol 3-kinase, daf-23, and the Akt related kinases, Akt-1/2, were found to oppose the function of daf-16 [130–132]. Together with these studies were reports that the forkhead related transcription factor HNF-3 can bind to the insulin response DNA sequence of the Insulin-like Growth Factor-binding Protein-1 (IGFBP-1) promoter [133], and that Akt mediates the effects of insulin on the expression of IGFBP-1 via the insulin response DNA sequence [134]. These findings from various model systems, along with others, suggested that the FoxO transcription factors are important targets of insulin signaling via growth factor receptors, and supported a role for the negative regulation of FoxO transcription by the PI3-K/Akt signal transduction pathway.
Following these clues, an elegant set of in vitro studies clearly revealed a role for the negative regulation of the mammalian FoxO transcription factors during Akt-mediated cellular survival. FoxO transcription factors are direct targets of activated Akt, with three evolutionarily conserved Akt phosphorylation sites identified within each of the FoxO proteins, FoxO3a [135], FoxO1 [136–138] and FoxO4 [139, 140]. In response to growth factors such as IGF-1, EGF and PDGF, Akt-mediated phosphorylation of the FoxO factors within the nucleus causes their re-localization to the cytoplasm, thereby preventing FoxO-dependent transcription (Figure 2). Akt-directed phosphorylation is crucial in determining FoxO sub-cellular localization, as mutation of all three Akt-phosphorylation sites to alanine (as in the FoxO triple mutant) results in the exclusive localization of FoxO proteins in the nucleus, even in the presence of growth factor-induced PI3-K/Akt activation. In many cellular systems, expression of the FoxO triple mutant results in cell cycle arrest and/or apoptosis, indicating an important role for FoxO transcription factors in regulating the expression of genes that determine survival. The recent exception to the nucleo-cytoplasmic shuttling of FoxO proteins is FoxO6, which is constitutively nuclear even in the presence of survival signals, with Akt-mediated phosphorylation preventing its activity in the nucleus [141]. The precise mechanisms through which Akt directed phosphorylation of FoxO (FoxO1, FoxO3a, and FoxO4) dictates their sub-cellular localization and activity are complex. In terms of FoxO sub-cellular localization, the emerging model drawn from various studies is that the exclusion of FoxO from the nucleus in response to growth factors is a dynamic process involving FoxO interaction with various cellular components that results in its effective net nuclear export [135, 138, 142, 143]. The adaptor protein 14-3-3 distinctly recognizes serine phosphorylated FoxO, and potentially disrupts FoxO association with DNA, as well as with FoxO co-factors such as CBP/p300. 14-3-3 binding and the presence of a nuclear export signal (NES) on FoxO are essential for FoxO nuclear export. FoxO nuclear export is dependent on the Crm-1 export receptor machinery, as well as phosphorylation of FoxO at the casein kinase 1 (CK1) phosphorylation sites serines 322 and 325 [144, 145]. A functional nuclear localization signal (NLS) has been identified and overlaps with one of the Akt phosphorylation sites, suggesting that Akt-mediated phosphorylation also interferes with the function of the FoxO NLS. Similarly, 14-3-3 binding may also occlude the FoxO NLS and prevent FoxO re-importation to the nucleus. Together with the net promotion of nuclear export and the inhibition of nuclear import, FoxO transcription factors are rapidly re-localized to the cytoplasm in the presence of growth-promoting signals.
Figure 2. Survival signaling through the PI3-K/Akt pathway prevents FOXO3a transcriptional activity.
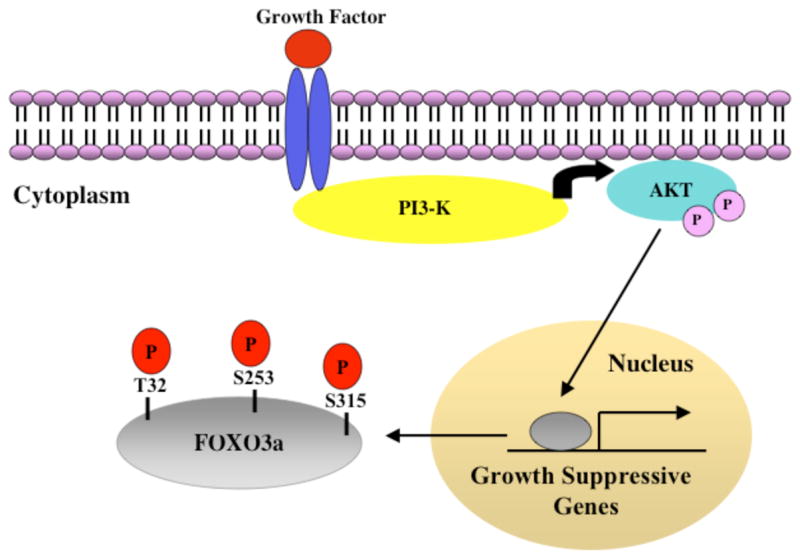
During survival such as in the presence of growth factors, growth factor stimulation of cognate receptors results in the activation of PI3-K, and the subsequent recruitment and activation of the Serine/Threonine kinase Akt through phosphorylation at Threonine 308 and Serine 473. Activated Akt phosphorylates FOXO3a at Threonine 32, Serine 253, and Serine 315, thereby resulting in the exclusion of FOXO3a from the nucleus and its inability to target the expression of genes that antagonize cell survival. On the contrary, in the absence of growth factors, FOXO3a is uninhibited by Akt, and is therefore not phosphorylated, and in the nucleus. See text for details.
Whereas survival signaling-dependent phosphorylation of FoxO transcription factors, such as through PI3-K/Akt, constitutes an important mechanism for FoxO regulation involving sub-cellular localization, additional post-translational modifications on FoxO proteins have been recently characterized, clearly revealing the complexity of FoxO regulation. FoxO transcription factors can be acetylated by the c-AMP response element binding (CREB)-binding protein (CBP) and/or p300, and deacetylated by the NAD-dependent mammalian Sir2 homolog, SIRT1 [146–151]. It has been proposed that CBP/p300, which serves to make DNA more accessible, is important for FoxO transcriptional activity. However, acetylation can repress FoxO-dependent gene transcription, while SIRT1 deacetylase activity promotes FoxO-dependent transcription in certain cellular responses, such as to oxidative stress [147]. The effects of FoxO acetylation, however, are not uniform, varying based on cell type and target gene, since FoxO acetylation shifts cells towards apoptosis, whereas FoxO deacetylation promotes target gene expression involved in cell cycle arrest and protection against oxidative stress [152].
In a related mechanism for FoxO regulation, Akt-induced phosphorylation not only affects FoxO sub-cellular localization, but also targets FoxO proteins, such as FoxO1 and FoxO3a, for ubiquitination and subsequent degradation via the 26S proteasome [153, 154]. The ubiquitination of FoxO1 by the E3 ubiquitin ligase, Skp2, requires Akt-mediated phosphorylation at serine 256 [155]. The IκB kinase (IKK) can also phosphorylate FoxO3a at a distinct residue (serine 644) in an Akt-independent manner, which not only promotes its nuclear exclusion but also targets FoxO3a for ubiquitination and proteasomal degradation [156]. Proteasomal degradation of FoxO proteins could prevent their recycling to the nucleus and thereby prevent their activation and thus is another key mechanism for the negative regulation of these tumor suppressors.
The ability of FoxO transcription factors to be regulated by various modifications, such as phosphorylation, ubiquitination, acetylation, and as yet unidentified mechanisms, indicate that FoxO transcription factors carry out central cellular functions that need to be precisely controlled.
Multiple Functions of the FoxO Transcription Factors: A Focus on Apoptosis
By regulating the transcription of genes involved in differentiation, DNA damage repair, cell cycle control, glucose metabolism, and apoptosis, FoxO transcription factors function in a variety of cellular processes that determine cell fate [157, 158]. The repertoire of FoxO gene targets has not been completely elucidated, but continues to increase. Given the diverse set of FoxO functions in regulating cellular survival, the effect of FoxO activity appears to be largely dependent on the cellular context, which includes the particular extra-cellular signal, cell type, and possibly the FoxO member. With respect to the latter, it is possible that given their important role in regulating cellular survival, FoxO family members possess distinct as well as overlapping functions. This is supported by recent evidence from mice singly deficient in the FoxO members FoxO1, FoxO3a, or FoxO4 [159]. FoxO1 deficiency resulted in severe vascular defects that led to embryonic lethality, but FoxO3a- and FoxO4-deficient mice were viable. FoxO3a-deficient female mice, however, display abnormal ovarian follicular development that results in an age-dependent infertility [160]. The determination of whether the various functions ascribed to FoxO activity in vitro translate in vivo, awaits the generation of mice that are multiply deficient in FoxO members. In addition, the examination of FoxO-deficient mice in the context of cancer may yield interesting insights regarding the role of FoxO in tumorigenesis. Recently, two groups have reported the consequences of the conditional and simultaneous disruption of both alleles of the three principal FoxO genes in mice [161, 162]. Tothova et al. [162], demonstrate that FoxOs are critical for long-term maintenance of hematopoietic stem cells. Paik et al. [161], show that FoxOs suppress the development of hemangiomas and lymphomas in mice. Although these two reports make a significant contribution to our understanding of the importance of FoxOs at the organismal level many questions remain to be addressed. Why does disruption of FoxOs produce such a restricted tumor phenotype? Furthermore, what other factors contribute to the context-dependent effects of FoxO deficiency?
As mentioned earlier, most of the functions of FoxO have been described in vitro. In accordance with the negative regulation of FoxO during growth factor-induced survival, the ability of FoxO proteins to initiate apoptosis was the first function associated with this group of transcription factors. A constitutively active FoxO3a protein that cannot be phosphorylated by Akt (also known as the triple mutant) and therefore localized to the nucleus overrides growth factor-induced survival and induces apoptosis in several cell types such as fibroblastoid, neuronal, and hematopoietic [135]. One of FoxO3a’s pro-apoptotic target genes is the death-inducing Fas Ligand (FasL) and in some cell types such as cerebellar granule neuronal cells and the acute T lymphoid leukemic Jurkat cells, apoptosis induced by the FoxO3a triple mutant is partially dependent on FasL signaling [135].
FoxO transcription factors are subject to negative regulation not only in the presence of growth factors such as insulin and IGF-1, but also in response to hematopoietic cytokines, such as IL-2, IL-3, and Erythropoietin. During an immune response, signaling initiated by the cytokine IL-2 is crucial for the survival and proliferation of activated T cells, with PI3-K/Akt activation playing a central role in this response. When activated T cells are no longer required, the gradual depletion of IL-2 signaling results in apoptosis of the activated T cell pool [163]. FoxO3a appears to play an important role in mediating this process, as it is highly phosphorylated in a PI3-K/Akt-dependent manner during the growth of IL-2-dependent murine cytotoxic T lymphocytes. IL-2 withdrawal causes the rapid de-phosphorylation of FoxO3a concomitant with the up-regulation of the cyclin-dependent kinase inhibitor p27Kip1and the pro-apoptotic BCL-2 family member BIM, and results in cell cycle arrest followed by apoptosis [164]. Indeed, the ability of FoxO to directly regulate p27Kip1 and Bim has been investigated in several cellular backgrounds, with these genes identified as FoxO targets [165–169]. The regulation of p27Kip1 expression plays an important role in hematopoietic cell survival, as bone marrow cells derived from p27Kip1-deficient mice show resistance towards apoptosis induced by cytokine withdrawal. Intriguingly, in some cellular contexts, FoxO3a activity imposes multiple effects, such as changes in cell cycle and the up-regulation of pro-apoptotic factors, that cooperate to shift the cellular threshold towards apoptosis [164, 166].
FoxO inhibition downstream of PI3-K/Akt constitutes an important mechanism for the survival of IL-3-dependent hematopoietic cells, such as BaF3 [167, 168]. The apoptosis induced in BaF3 cells upon either IL-3 withdrawal or expression of a FoxO3a triple mutant is not mediated through FasL signaling, however [170]. The FoxO3a target, BIM, is one mediator of apoptosis in response to IL-3 withdrawal or FoxO3a triple mutant expression. Indeed, BIM expression is sufficient to induce apoptosis in BaF3 cells, and bone marrow cells from BIM-deficient mice display decreased apoptosis in the absence of cytokines. The suppression of BIM as a consequence of FoxO inhibition is an important, but not exclusive pathway governing hematopoietic cell survival. The TNF-related apoptosis inducing ligand, (TRAIL) which induces apoptosis via the extrinsic death-receptor mediated pathway is yet another target of FoxO3a that is suppressed during hematopoietic cell survival promoted by the cytokines IL-3, GM-CSF, and Epo [170].
FoxO transcription factors function both in the activation and the repression of gene transcription. In order to promote cell cycle arrest, FoxO transcription factors can repress the transcription of the D type cyclins, D1 and D2 [171, 172]. Furthermore, the inhibitor of differentiation Id1 was found to be transcriptionally repressed by FoxO3a and sustained signaling through the FoxO arm of the PI3-K/Akt pathway driven by BCR-ABL removed this transcriptional block to sustain leukemogenesis [173]. FoxO4 can suppress the transcription of the anti-apoptotic Bcl-2 member, Bcl-xL, through an indirect mechanism involving transcriptional activation of Bcl-6, which is a repressor of Bcl-xL transcription [174].
These studies collectively provide evidence for FoxO transcription factor functions in cell cycle arrest and apoptosis, which occurs through the transcriptional regulation genes such as p27Kip1, cyclin D, FasL, TRAIL, Bim, and indirectly Bcl-xL. Intriguingly, the aberrant expression of these very same target genes is witnessed in various cancers, thus providing a compelling rationale to investigate the role of FoxO transcription factors in tumorigenesis, and the potential for developing therapeutic strategies focused on the activation of FoxO transcription factors.
FoxO Transcription Factors and Tumorigenesis
Interestingly, some of the human FoxO transcription factors were first identified in fusion genes from chromosomal translocations associated with certain types of cancers such as leukemias and the soft-tissue tumor known as alveolar rhabdomyosarcoma (ARMS). In particular, FoxO4 is fused to the MLL transcription factor as a result of the t(X; 11) chromosomal translocation found in acute lymphoblastic leukemia [175]. Another chromosomal translocation, t(6; 11), found in a few cases of acute myeloid leukemia, involves a fusion between MLL and FoxO2, which may be identical to FoxO3a [176]. Finally, fusions between the FoxO1 and PAX transcription factors, t(2;13), or t(1;13), have been detected in ARMS [177]. While the precise role and mechanism of these FoxO fusions in promoting tumorigenesis is not completely understood, these rearrangements involve fusions between the transactivation domain of FoxO and the DNA binding domain of the other fusion partner. Such fusion events have been proposed to be tumorigenic through the gain of function of the fusion protein, and/or the loss of endogenous FoxO function. For example, the MLL-FoxO4 fusion is thought to transform by interfering with the function of endogenous forkhead transcription factors [178, 179]. The loss of FoxO function as an important consequence of tumorigenesis associated with the PAX-FoxO fusions in ARMS has been substantiated by studies showing that the PAX3-FoxO1 fusion itelf is not sufficient to induce tumorigenesis in mice [180]. In addition, it has been recently discovered that ARMS tumors display drastic loss of FoxO1 expression, and that restoration of FoxO1 expression in an ARMS tumor-derived cell line results in cell cycle arrest and apoptosis [181]. These findings thereby support a role for FoxO1 as a tumor suppressor, and the overall involvement of the human FoxO transcription factors in tumor-associated chromosomal aberrations indeed raises interesting questions regarding their relevance in various cancers. Moreover, the tumor-suppressive activity of FoxO genes has been further demonstrated by somatic deletion of FoxO1, 3a, and 4 in mice. The deletion of these genes produced a cancer-prone phenotype in mice resulting mostly in thymic lymphomas and hemangiomas [161].
A crucial line of investigation into FoxO transcription factor involvement in tumorigenesis has stemmed not only from the discovery of FoxO’s transcriptional targets, but also its regulation by survival-promoting pathways such as the PI3-K/Akt pathway. Indeed, the constitutive activation of Akt is observed in many cancers, and can result from activating mutations in growth factor receptor tyrosine kinases, creation of constitutively active tyrosine kinases (i.e. BCR-ABL), activating mutations in PI3-K subunits, amplification of the PIK-3CA, AKT1, and AKT2 genes, or loss of the tumor suppressor phosphatase, PTEN [103, 104].
The catalytic subunit of the class1A PI3-K, PIK3CA, is frequently mutated in colorectal, gastric, brain, and breast tumors [182]. Until recently, the consequences of Akt activation resulting from activating mutations in PIK3CA remained unknown. Strikingly, human colon carcinoma cells engineered to express constitutively active PIK3CA showed a specific effect on the regulation of the FoxO transcription factors FoxO3a and FoxO1, but not other Akt substrates such as mTOR, 4EBP-1, p70S6K, GSK3β, Tuberin, and even FoxO4. FoxO3a and FoxO1 were highly phosphorylated, and FoxO1 levels were suppressed in mutant PIK3CA-transformed cells. Silencing of FoxO1 attenuated apoptosis in control colon carcinoma cells grown under low serum conditions, but did not reduce apoptosis in mutant PIK3CA-expressing cells, supporting a role for FoxO1 suppression in the increased tumorigenicity of mutant PIK3CA-expressing colon carcinoma cells [183].
Somatic mutations that result in the inactivation of the tumor suppressor PTEN, a phosphatase that antagonizes PI3-K/Akt signaling, have been observed in cancers such as prostate and endometrial, as well as in glioblastomas and melanomas [104]. In prostate carcinoma cells lacking PTEN, FoxO1 and FoxO3a are retained in the cytoplasm and remain transcriptionally inactive. Similar to the re-introduction of PTEN expression, the FoxO1 triple mutant is able to restore either cell cycle arrest or cell death in cells lacking PTEN, suggesting that FoxO1 is an important effector of PTEN tumor suppressor function [184, 185].
The involvement of FoxO1 in prostate cancer appears to extend beyond its ability to be regulated by the PI3-K/Akt pathway. Androgens prevent FoxO1 transcriptional activity by promoting complex formation between the androgen receptor and FoxO1, and inhibit cell cycle arrest and apoptosis induced by the FoxO1 triple mutant. In the coming years, it will be interesting to determine whether this mechanism of FoxO inhibition, which does not involve its sub-cellular localization, is present in other cellular contexts [186].
The amplification or over-expression of the Her2/Neu receptor tyrosine kinase is observed in various epithelial tumors and often results in the activation of survival pathways such as PI3-K/Akt [187]. Expression of a FoxO4 triple mutant reduces survival and transformation of Her2/Neu over-expressing NIH3T3 cells in vitro and their tumorigenicity in vivo [188]. The specific involvement of FoxO transcription factors in this class of cancers, however, needs to be further examined by investigation of endogenous FoxO proteins in Her2/Neu over-expressing tumors.
Fundamental studies detailing the impact of the IκB Kinase (IKK) pathway on the function of the FoxO member FoxO3a have been recently described in breast cancer [156]. The constitutive activation of IKK has been implicated as an important mechanism by which breast carcinomas are able to sustain cellular proliferation and evade apoptosis. In many primary breast tumor specimens, cytoplasmic FoxO3a correlated not only with an increase in phosphorylated Akt but also increased IKKβ expression. However, in a fraction of primary breast tumor specimens (23/131) examined, FoxO3a cytoplasmic localization was observed even in the absence of phosphorylated Akt, and instead correlated with increased IKKβ expression, suggesting a possible link between IKKβ activity and FoxO3a inhibition. Indeed, in an Akt-independent fashion, IKK interacts with and phosphorylates FoxO3a at serine 644, resulting in FoxO3a cytoplasmic retention as well as ubiquitination and degradation by the 26S proteasome. IKK-imposed inhibition of FoxO3a is functionally relevant since expression of FoxO3a in cells constitutively expressing IKKβ, impairs IKKβ-induced cellular proliferation in vitro and tumor formation in nude mice in vivo.
Moreover, the tumor-suppressive activity of FoxO genes has been further demonstrated by somatic deletion of FoxO1, 3a and 4 in mice. The deletion of these genes produced a cancer-prone phenotype in mice resulting mostly in thymic lymphomas and hemangiomas [161].
Therefore, the growth-suppressive functions of the FoxO transcription factors are intricately inhibited by several inputs, through the aberrant activation of PI3-K/Akt, IKK and as yet to be identified central regulators, and in this manner potentially constitute a significant mechanism in promoting tumorigenesis.
FoxO Transcription Factors in Leukemia
Recent studies have revealed that the inhibition of FoxO function is a potentially important event even in hematological malignancies such as leukemias. BCR-ABL transformation inhibits FoxO3a activity by maintaining PI3-K- dependent constitutive phosphorylation and cytoplasmic retention of FoxO3a [170, 189]. Sustained activation of PI3-K/Akt by BCR-ABL in both BCR-ABL-transformed cells (Mo7e-p210 and BaF3-p210) and primary CML CD34+ cells leads to Skp2 transcriptional upregulation and likely contributes to leukemogenesis through enhanced proteasomal degradation and downregulation of FoxO protein levels [190]. One of the consequences of FoxO3a inhibition by BCR-ABL is the suppression of the downstream pro-apoptotic target TRAIL [170]. FoxO3a inhibition plays an important role in governing BCR-ABL-induced transformation as expression of a constitutively active FoxO3a triple mutant in BCR-ABL-transformed cells overrides growth factor-independent survival and induces apoptosis [169, 170]. The BH3-only pro-apoptotic protein BIM is also suppressed in a FoxO3a dependent manner in BCR-ABL-transformed cells, and FoxO3a-mediated regulation of BIM contributes to apoptosis induced by imatinib in BCR-ABL-transformed cell lines [169]. Therefore, FoxO3a inhibition in BCR-ABL-transformed cells can yield the suppression of genes that regulate the apoptotic program. Furthermore, FoxO3a inhibition by BCR-ABL not only affects the expression of apoptotic genes, but also cell cycle regulatory genes, such as cyclin D [191]. Therefore, BCR-ABL-mediated inhibition of FoxO3a potentially represents an important mechanism by which BCR-ABL dysregulates the expression of multiple genes involved in the cell cycle and apoptosis, thereby promoting tumorigenesis. Further assessment of BCR-ABL-induced inhibition of FoxO3a has revealed that in addition to promoting phosphorylation at Akt-dependent sites, BCR-ABL enhances the suppression of FoxO3a protein expression in a proteasome-dependent manner. The clinically relevant proteasome inhibitor bortezomib (VELCADE, PS-341) restores FOXO3a expression and induces apoptosis not only in imatinib-sensitive, but also in imatinib-resistant (T315I) BCR-ABL-transformed patient cells (our unpublished observations).
The negative regulation of FoxO has also been observed with other activated tyrosine kinases such as FLT3-ITD [192], and NPM-ALK [193] implicated in acute myeloid leukemia and anaplastic large cell lymphoma, respectively. In these studies, it was demonstrated that the FLT3-ITD and NPM-ALK oncogenes inhibit FoxO3a by promoting its PI3-K-dependent phosphorylation and cytoplasmic retention. Together, these studies indicate that FoxO transcription factor inactivation is a potentially important mechanism by which activated oncogenic tyrosine kinases such as BCR-ABL and FLT3-ITD operate in driving leukemogenesis, and provide insight into novel targeting strategies for the treatment of these cancers.
Current and Future Approaches for CML treatment
The emergence of clinical resistance to imatinib therapy, as well as the inability of imatinib to eradicate BCR-ABL leukemic disease, has prompted not only the search for more potent tyrosine kinase inhibitors, but also the testing of combinations of anti-cancer agents that inhibit classical growth pathways with imatinib (Table 2). In another category of anti-cancer agents, inhibitors of heat shock protein 90 (Hsp90), histone deacetylases, and the 26S proteasome may also prove to be equally valuable in preventing, as well as combating, drug-resistant mutants of BCR-ABL (Table 2).
Table 2.
Current clinical and experimental approaches for CML therapy
| Inhibitor Class | Target(s) | Clinical Trials for CML | Activity in Imatinib- Refractory BCR-ABL Variants | Comments | References |
|---|---|---|---|---|---|
| Tyrosine Kinase | |||||
| Imatinib mesylate (Gleevec, STI-571) | ABL, PDGF, Kit, Arg | Current CML Therapy | No | First generation tyrosine kinase inhibitor | Reviewed in [51] |
| AMN107 | ABL, PDGF, Kit | Phase I-II | Yes, Not T315I | Approx. 20 fold more potent than imatinib | [59], [194], [195] |
| BMS-354825 | ABL, SRC | Phase II | Yes, Not T315I | Approx. 325 fold more potent than imatinib | [58], [194] |
| SKI-606 | ABL, SRC | Phase I | Resistant mutants not tested | Effectiveness in CML xenografts of K562 cells suggests utility in blast crisis | [196] |
| AP23464 | ABL, SRC | No | Yes, not T315I | More potent than imatinib | [197] |
| Farnesyl protein transferase | |||||
| SCH66336 | Ras, other farnesylated proteins | Phase 1 | Yes | Single, or enhanced with imatinib | [198], [199], [200] |
| R115777 | Phase I | Yes, T315I not tested | Single, or synergistic with imatinib | [201], [202] | |
| MEK1/2 | |||||
| PD184352 | MEK1/2 | No | Yes, T315I not tested | Synergistic with imatinib, HDACi (SAHA) | [203], [204] |
| Phosphatidylinositol-3-Kinase (PI3-K) | |||||
| LY-294002 | PI3-K | No | Yes, T315I not tested | Demonstrates the importance of PI3-K activation in BCR-ABL- induced CML | [106], [200] |
| Wortmannin | PI3-K | No | Yes, T315I not tested | [106] | |
| 3-phosphoinositide-dependent kinase-1 (PDK-1) | |||||
| OSU-03012 | PDK-1, and thus inhibits Akt | No | Yes | Synergistic with imatinib | [107] |
| mTOR | |||||
| Rapamycin | MTOR | Yes | Yes | Synergistic with imatinib | [123], [124] |
| Aurora Kinase | |||||
| VX-680 | Aurora Kinases, ABL, FLT3 | No | Yes | In phase I development for solid tumors | [205] |
| P38 | |||||
| BIRB-796 | p38, ABL | No | Yes | In clinical trials for inflammatory disease | [205] |
| Histone Deacetylase (HDAC) | |||||
| LAQ824 | Histone deacetylases | Phase I | Conflicting results with T315I mutant reported | Single, synergistic with imatinib | [206] [207] |
| LBH589 | Histone deacetylases | Yes | T315I-sensitive with 17- AAG combination | Synergistic with Hsp90 inhibitor, 17-AAG | [208] |
| SAHA | Histone deacetylases | Phase I | Yes, T315I not tested | Single, synergistic with imatinib, MEK1/2 inhibitor, bortezomib, 17-AAG. | [209] [210] [204], [211], [212] |
| Heat Shock Protein 90 (Hsp90) | |||||
| 17-AAG | Hsp90 | Yes | Yes | Single, synergistic with imatinib or HDACis, SAHA, or LBH589 | [57], [200], [212], [208] |
| Proteasome | |||||
| Bortezomib (Velcade, 26S Proteasome PS-341) | Yes | Yes, T315I sensitive to Bortezomib alone but not tested in combinations | Single, or synergistic with flavopiridol or HDACi (SAHA) | [213], [214], [211] | |
Proteasome Inhibitors: A New Class of Anti-Cancer Agents
The ubiquitin-proteasome pathway is crucial not only for the proteolysis of old, damaged, or misfolded proteins, but also for the regulated degradation of molecules that control key cellular processes such as the cell cycle and apoptosis. The mammalian 26S proteasome consists of a regulatory 19S subunit, which binds and recognizes ubiquitinated substrates, and a 20S catalytic core that carries out the essential function of protein degradation [215–217]. In recent years, the development of a specific, potent, and reversible inhibitor of the proteasome, known as bortezomib (VELCADE, PS-341, Millennium Pharmaceuticals) which displays activity against the growth of various cancers in vitro and in vivo, has established the proteasome as a viable target, and thus heralded a novel strategy in cancer treatment [218–220]. In fact, bortezomib treatment has shown great promise in the clinic, and is currently used for the treatment of patients with relapsed and/or refractory multiple myeloma or mantle cell lymphoma [221–225]. Pre-clinical and clinical investigations with bortezomib alone or in combination with other chemotherapeutic agents are being currently performed for various solid and hematologic cancers. The anti-cancer effects of bortezomib have been shown in xenograft models not only of multiple myeloma, but also adult T-cell leukemia, lung, breast, prostate, pancreatic, head and neck, and colon cancers, and in melanoma [226].
The potential for using highly specific inhibitors of the proteasome in anti-cancer therapies was realized from several discoveries, including the seminal finding that proteasome activity is necessary for the degradation of the NF-κB inhibitor IκB, in turn indirectly facilitating NF-κB transcription factor activity. The various studies reported behind this pathway have been extensively reviewed by Karin and Ben-Neriah [227]. Constitutive NF-κB activation is observed in, and proposed to contribute to the pathogenesis of various cancers [87]. This has initially provided a sound rationale for investigating the use of bortezomib in cancers with constitutive NF-κB activation, such as multiple myeloma. In retrospect, the proteasome presents itself as an appealing anti-cancer target, since it is a conduit for numerous proteins that are aberrantly regulated in cancer. In fact, it has become apparent that bortezomib’s anti-cancer activity must involve multiple mechanisms besides inhibition of NF-κB, with the details of these additional mechanisms remaining to be identified. Generally, proteasome inhibition by bortezomib causes changes in expression of various regulatory proteins, thereby inducing cell cycle arrest at the G1-S and G2-M phases and apoptosis. For example, bortezomib treatment of drug-resistant multiple myeloma cells, besides inhibiting NF-κB, leads to multiple effects that also explain the induction of apoptosis, including increased p53 expression, activation of JNK, and of caspases, such as -3 and –8 [228]. Most studied in multiple myeloma, these apoptosis inducing effects are coupled with the ability of bortezomib to alter the tumor microenvironment, inhibit angiogenesis, down-regulate growth factors, sensitize tumor cells to lower doses of chemotherapeutic agents, and re-sensitize tumor cell lines previously resistant to chemotherapeutic agents [218, 229]. Overall, the underlying mechanisms for bortezomib’s anti-tumor activity are still not completely understood, and may vary among different cancers.
Earlier studies that investigated the potential for proteasome inhibition to induce apoptosis in leukemic cells showed that proteasome inhibitors that are not as potent, specific, and clinically suitable (e.g. lactacystin and tripeptide aldehydes) as bortezomib, induce apoptosis in varying capacities in different leukemic cell lines, including ones that are BCR-ABL-positive, such as AR230 and K562. In fact, proteasome inhibition resulted in decreased BCR-ABL expression, providing one mechanism by which proteasome inhibitors can exert their growth inhibitory effects in this context [230, 231].
Whether bortezomib can inhibit BCR-ABL-induced leukemia, and the underlying mechanisms behind this have remained poorly understood. Indeed, bortezomib treatment clearly diminishes symptoms of CML-like illness in BCR-ABL-transduced mice, and prolongs their survival (our unpublished observations). Bortezomib can induce apoptosis in imatinib-sensitive and -resistant BCR-ABL-expressing leukemic cell lines derived from CML patients at the myeloid blast crisis phase (Table 2) [213]. Bortezomib treatment only resulted in a moderate and transient decrease in NF-κB DNA binding activity that did not correlate with apoptosis [213]. Therefore, such results suggest the presence of other mechanisms for the apoptosis-inducing effects of bortezomib in BCR-ABL-expressing cells and our studies indicate a contribution of FoxO3a activation towards the effects of bortezomib (our unpublished observations).
In accordance with its ability to affect the growth of BCR-ABL-expressing cells and synergize with anti-cancer agents at sub-toxic concentrations, bortezomib and the histone deacetylase inhibitor SAHA, or the cyclin-dependent kinase inhibitor flavopiridol, induce apoptosis in imatinib-sensitive and -resistant BCR-ABL-positive leukemic cells (Table 2) [211, 214].
FoxO Transcription Factors as Potential Targets in Cancer Therapy
Given the accumulating evidence for a role of FoxO inactivation in the pathogenesis of cancers, the ability to correct this deficiency with novel agents that reactivate FoxO may be invaluable in countering tumorigenesis. The application of proteasome inhibitors presents one approach for recovering FoxO3a expression and activity. In another strategy, Kau and colleagues have taken advantage of FoxO’s dynamic nucleo-cytoplasmic shuttling and employed a cell-based, chemical genetics approach to search for compounds that trap FoxO1 in the nucleus [232]. Their screen revealed two categories of compounds, consisting of general CRM-1-targeting export inhibitors, and another class that does not interfere with the CRM-1-dependent export of the HIV Rev protein, but that does affect FoxO1 nuclear export. Most inhibitors in the latter class resulted in FoxO entrapment by affecting Akt phosphorylation, while a few inhibitors functioned downstream of Akt. Examination of the structure of these compounds suggests that some may function as kinase inhibitors, even though their exact targets have yet to be identified. Importantly, some of these inhibitors were able to inhibit cellular proliferation of PTEN-deficient 786-0 cells, even though they displayed IC50 values in the micromolar range. Analyzing these FoxO1 nuclear export inhibitors may not only yield a new class of anti-cancer therapies, but may also shed light on novel regulators of the PI3-K, AKT, and FoxO pathways.
Targeting the “Right” Population of Tumor Cells in Anti-Cancer Therapy
An emerging concept in cancer biology and therapy is that cancers can arise from a small subset of a typically heterogeneous tumor cell population. This rare population of cells has been referred to as tumor-initiating cells or cancer “stem” cells [233, 234]. The cancer stem cell hypothesis has been substantiated and is exemplified in many types of leukemia, in which a stem or progenitor cell origin has been characterized [235, 236]. In the case of BCR-ABL, accumulating evidence suggests that the target cell for transformation is a hematopoietic stem cell rather than a committed progenitor cell [65, 237, 238]. Interestingly, a recent study by Tothova et al., (2007) reported an elevated level of reactive oxygen species (ROS) restricted to the hematopoietic stem cell (HSC) compartment of mice deficient in three members of the FoxO subfamily (FoxO1, FoxO3 and FoxO4) [162]. These genetic data show that FoxOs are essential and functionally redundant for the integrity of the HSC compartment. Furthermore, these data demonstrate a genetic link between FoxOs and protection from physiologic oxidative stress in the HSC compartment through the regulation of a subset of ROS genes such as MnSOD, calalase, and GADD45 [157, 239, 240] that serve to detoxify ROS and their deleterious effects on HSC survival and function. Indeed, a recent study by Lehtinen et al. (2006) reported that cytoplasmic FoxO3a could be reactivated in response to oxidative stress in a phosphorylation-dependent manner mediated by MST kinases [240]. Furthermore van der Horst et al. (2006) demonstrated that cytoplasmic FoxO4 is monoubiquitinated in response to oxidative stress resulting in its relocalization to the nucleus and enhancement in transcriptional activity [241]. Therefore, BCR-ABL-mediated inhibition of FoxOs in the HSC compartment and the consequent dysregulation of ROS levels likely contribute to genetic instability in CML leading to cellular transformation [242]. Additionally, upregulation of DNA polymerase-β, the most inaccurate of mammalian DNA polymerases, by BCR-ABL [243] is likely to replicate ROS-induced mutations thus promoting the accumulation of multiple genetic lesions allowing for cellular transformation and disease progression.
Unfortunately, it has been observed that while imatinib can eradicate the majority of proliferating CML progenitors and differentiated granulocytes, it is unable to target the minute population of quiescent CML stem cells [60, 65, 244]. In order to assess whether anti-neoplastics such as proteasome inhibitors will have long-term benefits in curing CML, it will be important to determine whether such compounds can target even those CML stem cells that persist during imatinib therapy. Combined treatment with the proteasome inhibitor MG-132 and idarubicin selectively induces apoptosis in acute myeloid leukemia (AML) stem cells [245], suggesting the potential for proteasome inhibition therapy in targeting leukemic stem cells. Crucial to the development of therapies that are tailored at targeting leukemic stem cells is the determination of which signaling pathways are crucial in this cellular context.
Perspectives and Conclusion
In over four decades of work since the identification of the Philadelphia chromosome in CML patients, we have witnessed great advances such as the identification of BCR-ABL oncoprotein as the target in CML, and the development of kinase inhibitors against BCR-ABL. Despite such progress, it has become apparent that BCR-ABL signaling is highly complex, and has numerous outputs that promote leukemogenesis. In addition the requirement for BCR-ABL, and BCR-ABL signaling, may vary among primitive quiescent stem cells, committed progenitors cells, and terminally differentiated cells. Since their initial discovery as downstream targets of PI3-K/Akt-mediated survival signaling, diverse roles of mammalian FoxO transcription factors in metabolism, stress responses, differentiation, cell cycle, and apoptosis have emerged [157, 158]. The theme arising from studies with BCR-ABL-induced leukemia and other cancers is that FoxO transcription factors are often inactivated in tumorigenesis. More recently, comparisons between FoxO and p53 have examined their similar modes of regulation and cellular functions, and their common deficiency in cancers [246]. Excitingly, the discovery that FoxO transcription factors represent an important node of negative regulation in cancers lends further opportunity for the discovery of therapies that are aimed at re-activating FoxO. Finally, as FoxO, NF-κB, and p53 appear to be the most centrally affected molecules in tumorigenesis, the determination of therapies aimed at restoring normal function of all these molecules may be ideal in the treatment of cancers.
Acknowledgments
We regret that due to space limitation the invaluable work of many colleagues could not be referenced. We would like to thank Dr. Susan Glueck for editing this manuscript. Our work is supported by NIH grants CA105306 and HL080192 to RKF and NIH training grant 5T32HL007623-22 to Amrik Singh. RKF is an American Cancer Society Scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 3.Abe K, Kurakin A, Mohseni-Maybodi M, Kay B, Khosravi-Far R. The complexity of TNF-related apoptosis-inducing ligand. Ann N Y Acad Sci. 2000;926:52–63. doi: 10.1111/j.1749-6632.2000.tb05598.x. [DOI] [PubMed] [Google Scholar]
- 4.Khosravi-Far R, Esposti MD. Death receptor signals to mitochondria. Cancer Biol Ther. 2004;3:1051–1057. doi: 10.4161/cbt.3.11.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2:764–776. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- 6.Nowell PC, Hungerford DA. Chromosome studies on normal and leukemic human leukocytes. J Natl Cancer Inst. 1960;25:85–109. [PubMed] [Google Scholar]
- 7.Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 8.Bartram CR, de Klein A, Hagemeijer A, van Agthoven T, Geurts van Kessel A, Bootsma D, Grosveld G, Ferguson-Smith MA, Davies T, Stone M, et al. Translocation of c-ab1 oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1983;306:277–280. doi: 10.1038/306277a0. [DOI] [PubMed] [Google Scholar]
- 9.Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984;36:93–99. doi: 10.1016/0092-8674(84)90077-1. [DOI] [PubMed] [Google Scholar]
- 10.Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 1990;247:1079–1082. doi: 10.1126/science.2408149. [DOI] [PubMed] [Google Scholar]
- 11.Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96:3343–3356. [PubMed] [Google Scholar]
- 12.Faderl S, Talpaz M, Estrov Z, O’Brien S, Kurzrock R, Kantarjian HM. The biology of chronic myeloid leukemia. N Engl J Med. 1999;341:164–172. doi: 10.1056/NEJM199907153410306. [DOI] [PubMed] [Google Scholar]
- 13.Sawyers CL. Chronic myeloid leukemia. N Engl J Med. 1999;340:1330–1340. doi: 10.1056/NEJM199904293401706. [DOI] [PubMed] [Google Scholar]
- 14.Melo JV. The molecular biology of chronic myeloid leukaemia. Leukemia. 1996;10:751–756. [PubMed] [Google Scholar]
- 15.Rosenthal S, Canellos GP, DeVita VT, Jr, Gralnick HR. Characteristics of blast crisis in chronic granulocytic leukemia. Blood. 1977;49:705–714. [PubMed] [Google Scholar]
- 16.Two new agents effective in Gleevec-resistant CML. Cancer Biol Ther. 2004;3:1198–1199. [PubMed] [Google Scholar]
- 17.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 18.Gambacorti-Passerini C, le Coutre P, Mologni L, Fanelli M, Bertazzoli C, Marchesi E, Di Nicola M, Biondi A, Corneo GM, Belotti D, Pogliani E, Lydon NB. Inhibition of the ABL kinase activity blocks the proliferation of BCR/ABL+ leukemic cells and induces apoptosis. Blood Cells Mol Dis. 1997;23:380–394. doi: 10.1006/bcmd.1997.0155. [DOI] [PubMed] [Google Scholar]
- 19.Sawyers CL, Hochhaus A, Feldman E, Goldman JM, Miller CB, Ottmann OG, Schiffer CA, Talpaz M, Guilhot F, Deininger MW, Fischer T, O’Brien SG, Stone RM, Gambacorti-Passerini CB, Russell NH, Reiffers JJ, Shea TC, Chapuis B, Coutre S, Tura S, Morra E, Larson RA, Saven A, Peschel C, Gratwohl A, Mandelli F, Ben-Am M, Gathmann I, Capdeville R, Paquette RL, Druker BJ. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood. 2002;99:3530–3539. doi: 10.1182/blood.v99.10.3530. [DOI] [PubMed] [Google Scholar]
- 20.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 21.Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, Niederwieser D, Resta D, Capdeville R, Zoellner U, Talpaz M, Druker B, Goldman J, O’Brien SG, Russell N, Fischer T, Ottmann O, Cony-Makhoul P, Facon T, Stone R, Miller C, Tallman M, Brown R, Schuster M, Loughran T, Gratwohl A, Mandelli F, Saglio G, Lazzarino M, Russo D, Baccarani M, Morra E. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346:645–652. doi: 10.1056/NEJMoa011573. [DOI] [PubMed] [Google Scholar]
- 22.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 23.Lugo TG, Witte ON. The BCR-ABL oncogene transforms Rat-1 cells and cooperates with v-myc. Mol Cell Biol. 1989;9:1263–1270. doi: 10.1128/mcb.9.3.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McLaughlin J, Chianese E, Witte ON. In vitro transformation of immature hematopoietic cells by the P210 BCR/ABL oncogene product of the Philadelphia chromosome. Proc Natl Acad Sci U S A. 1987;84:6558–6562. doi: 10.1073/pnas.84.18.6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daley GQ, Baltimore D. Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia-specific P210bcr/abl protein. Proc Natl Acad Sci U S A. 1988;85:9312–9316. doi: 10.1073/pnas.85.23.9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klucher KM, Lopez DV, Daley GQ. Secondary mutation maintains the transformed state in BaF3 cells with inducible BCR/ABL expression. Blood. 1998;91:3927–3934. [PubMed] [Google Scholar]
- 27.Ghaffari S, Daley GQ, Lodish HF. Growth factor independence and BCR/ABL transformation: promise and pitfalls of murine model systems and assays. Leukemia. 1999;13:1200–1206. doi: 10.1038/sj.leu.2401467. [DOI] [PubMed] [Google Scholar]
- 28.Drexler HG, MacLeod RA, Uphoff CC. Leukemia cell lines: in vitro models for the study of Philadelphia chromosome-positive leukemia. Leuk Res. 1999;23:207–215. doi: 10.1016/s0145-2126(98)00171-4. [DOI] [PubMed] [Google Scholar]
- 29.Ilaria RL, Jr, Van Etten RA. The SH2 domain of P210BCR/ABL is not required for the transformation of hematopoietic factor-dependent cells. Blood. 1995;86:3897–3904. [PubMed] [Google Scholar]
- 30.Dazzi F, Capelli D, Hasserjian R, Cotter F, Corbo M, Poletti A, Chinswangwatanakul W, Goldman JM, Gordon MY. The kinetics and extent of engraftment of chronic myelogenous leukemia cells in non-obese diabetic/severe combined immunodeficiency mice reflect the phase of the donor’s disease: an in vivo model of chronic myelogenous leukemia biology. Blood. 1998;92:1390–1396. [PubMed] [Google Scholar]
- 31.Heisterkamp N, Jenster G, Kioussis D, Pattengale PK, Groffen J. Human bcr-abl gene has a lethal effect on embryogenesis. Transgenic Res. 1991;1:45–53. doi: 10.1007/BF02512996. [DOI] [PubMed] [Google Scholar]
- 32.Huettner CS, Zhang P, Van Etten RA, Tenen DG. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nat Genet. 2000;24:57–60. doi: 10.1038/71691. [DOI] [PubMed] [Google Scholar]
- 33.Heisterkamp N, Jenster G, ten Hoeve J, Zovich D, Pattengale PK, Groffen J. Acute leukaemia in bcr/abl transgenic mice. Nature. 1990;344:251–253. doi: 10.1038/344251a0. [DOI] [PubMed] [Google Scholar]
- 34.Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 35.Elefanty AG, Hariharan IK, Cory S. bcr-abl, the hallmark of chronic myeloid leukaemia in man, induces multiple haemopoietic neoplasms in mice. Embo J. 1990;9:1069–1078. doi: 10.1002/j.1460-2075.1990.tb08212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kelliher MA, McLaughlin J, Witte ON, Rosenberg N. Induction of a chronic myelogenous leukemia-like syndrome in mice with v-abl and BCR/ABL. Proc Natl Acad Sci U S A. 1990;87:6649–6653. doi: 10.1073/pnas.87.17.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, Bronson R, Aster JC, Scott ML, Baltimore D. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92:3780–3792. [PubMed] [Google Scholar]
- 38.Zhang X, Ren R. Bcr-Abl efficiently induces a myeloproliferative disease and production of excess interleukin-3 and granulocyte-macrophage colony-stimulating factor in mice: a novel model for chronic myelogenous leukemia. Blood. 1998;92:3829–3840. [PubMed] [Google Scholar]
- 39.Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Druker BJ, Lydon NB. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996;56:100–104. [PubMed] [Google Scholar]
- 40.Pendergast AM, Gishizky ML, Havlik MH, Witte ON. SH1 domain autophosphorylation of P210 BCR/ABL is required for transformation but not growth factor independence. Mol Cell Biol. 1993;13:1728–1736. doi: 10.1128/mcb.13.3.1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oda T, Heaney C, Hagopian JR, Okuda K, Griffin JD, Druker BJ. Crkl is the major tyrosine-phosphorylated protein in neutrophils from patients with chronic myelogenous leukemia. J Biol Chem. 1994;269:22925–22928. [PubMed] [Google Scholar]
- 42.Sattler M, Griffin JD. Mechanisms of transformation by the BCR/ABL oncogene. Int J Hematol. 2001;73:278–291. doi: 10.1007/BF02981952. [DOI] [PubMed] [Google Scholar]
- 43.Nagar B, Bornmann WG, Pellicena P, Schindler T, Veach DR, Miller WT, Clarkson B, Kuriyan J. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571) Cancer Res. 2002;62:4236–4243. [PubMed] [Google Scholar]
- 44.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 45.Carroll M, Ohno-Jones S, Tamura S, Buchdunger E, Zimmermann J, Lydon NB, Gilliland DG, Druker BJ. CGP 57148, a tyrosine kinase inhibitor, inhibits the growth of cells expressing BCR-ABL, TEL-ABL, and TEL-PDGFR fusion proteins. Blood. 1997;90:4947–4952. [PubMed] [Google Scholar]
- 46.Okuda K, Weisberg E, Gilliland DG, Griffin JD. ARG tyrosine kinase activity is inhibited by STI571. Blood. 2001;97:2440–2448. doi: 10.1182/blood.v97.8.2440. [DOI] [PubMed] [Google Scholar]
- 47.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 48.Deininger MW, Goldman JM, Lydon N, Melo JV. The tyrosine kinase inhibitor CGP57148B selectively inhibits the growth of BCR-ABL-positive cells. Blood. 1997;90:3691–3698. [PubMed] [Google Scholar]
- 49.le Coutre P, Mologni L, Cleris L, Marchesi E, Buchdunger E, Giardini R, Formelli F, Gambacorti-Passerini C. In vivo eradication of human BCR/ABL-positive leukemia cells with an ABL kinase inhibitor. J Natl Cancer Inst. 1999;91:163–168. doi: 10.1093/jnci/91.2.163. [DOI] [PubMed] [Google Scholar]
- 50.Wolff NC, Ilaria RL., Jr Establishment of a murine model for therapy-treated chronic myelogenous leukemia using the tyrosine kinase inhibitor STI571. Blood. 2001;98:2808–2816. doi: 10.1182/blood.v98.9.2808. [DOI] [PubMed] [Google Scholar]
- 51.Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105:2640–2653. doi: 10.1182/blood-2004-08-3097. [DOI] [PubMed] [Google Scholar]
- 52.Hehlmann R, Berger U, Hochhaus A. Chronic myeloid leukemia: a model for oncology. Ann Hematol. 2005;84:487–497. doi: 10.1007/s00277-005-1039-z. [DOI] [PubMed] [Google Scholar]
- 53.Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville R, Talpaz M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 54.Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM, Cervantes F, Hochhaus A, Powell BL, Gabrilove JL, Rousselot P, Reiffers J, Cornelissen JJ, Hughes T, Agis H, Fischer T, Verhoef G, Shepherd J, Saglio G, Gratwohl A, Nielsen JL, Radich JP, Simonsson B, Taylor K, Baccarani M, So C, Letvak L, Larson RA. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 55.Shah NP, Sawyers CL. Mechanisms of resistance to STI571 in Philadelphia chromosome-associated leukemias. Oncogene. 2003;22:7389–7395. doi: 10.1038/sj.onc.1206942. [DOI] [PubMed] [Google Scholar]
- 56.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 57.Gorre ME, Ellwood-Yen K, Chiosis G, Rosen N, Sawyers CL. BCR-ABL point mutants isolated from patients with imatinib mesylate-resistant chronic myeloid leukemia remain sensitive to inhibitors of the BCR-ABL chaperone heat shock protein 90. Blood. 2002;100:3041–3044. doi: 10.1182/blood-2002-05-1361. [DOI] [PubMed] [Google Scholar]
- 58.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 59.Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Fendrich G, Hall-Meyers E, Kung AL, Mestan J, Daley GQ, Callahan L, Catley L, Cavazza C, Azam M, Neuberg D, Wright RD, Gilliland DG, Griffin JD. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer cell. 2005;7:129–141. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 60.Bhatia R, Holtz M, Niu N, Gray R, Snyder DS, Sawyers CL, Arber DA, Slovak ML, Forman SJ. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood. 2003;101:4701–4707. doi: 10.1182/blood-2002-09-2780. [DOI] [PubMed] [Google Scholar]
- 61.Hughes T, Branford S. Molecular monitoring of chronic myeloid leukemia. Semin Hematol. 2003;40:62–68. doi: 10.1053/shem.2003.50044. [DOI] [PubMed] [Google Scholar]
- 62.Chu S, Xu H, Shah NP, Snyder DS, Forman SJ, Sawyers CL, Bhatia R. Detection of BCR-ABL kinase mutations in CD34+ cells from chronic myelogenous leukemia patients in complete cytogenetic remission on imatinib mesylate treatment. Blood. 2005;105:2093–2098. doi: 10.1182/blood-2004-03-1114. [DOI] [PubMed] [Google Scholar]
- 63.Deininger MW, Holyoake TL. Can we afford to let sleeping dogs lie? Blood. 2005;105:1840–1841. doi: 10.1182/blood-2004-12-4764. [DOI] [PubMed] [Google Scholar]
- 64.Michor F, Hughes TP, Iwasa Y, Branford S, Shah NP, Sawyers CL, Nowak MA. Dynamics of chronic myeloid leukaemia. Nature. 2005;435:1267–1270. doi: 10.1038/nature03669. [DOI] [PubMed] [Google Scholar]
- 65.Elrick LJ, Jorgensen HG, Mountford JC, Holyoake TL. Punish the parent not the progeny. Blood. 2005;105:1862–1866. doi: 10.1182/blood-2004-08-3373. [DOI] [PubMed] [Google Scholar]
- 66.Bedi A, Zehnbauer BA, Barber JP, Sharkis SJ, Jones RJ. Inhibition of apoptosis by BCR-ABL in chronic myeloid leukemia. Blood. 1994;83:2038–2044. [PubMed] [Google Scholar]
- 67.McGahon A, Bissonnette R, Schmitt M, Cotter KM, Green DR, Cotter TG. BCR-ABL maintains resistance of chronic myelogenous leukemia cells to apoptotic cell death. Blood. 1994;83:1179–1187. [PubMed] [Google Scholar]
- 68.Carlesso N, Griffin JD, Druker BJ. Use of a temperature-sensitive mutant to define the biological effects of the p210BCR-ABL tyrosine kinase on proliferation of a factor-dependent murine myeloid cell line. Oncogene. 1994;9:149–156. [PubMed] [Google Scholar]
- 69.Kabarowski JH, Allen PB, Wiedemann LM. A temperature sensitive p210 BCR-ABL mutant defines the primary consequences of BCR-ABL tyrosine kinase expression in growth factor dependent cells. Embo J. 1994;13:5887–5895. doi: 10.1002/j.1460-2075.1994.tb06934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Emanuel PD, Bates LJ, Castleberry RP, Gualtieri RJ, Zuckerman KS. Selective hypersensitivity to granulocyte-macrophage colony-stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood. 1991;77:925–929. [PubMed] [Google Scholar]
- 71.Sawyers CL, McLaughlin J, Witte ON. Genetic requirement for Ras in the transformation of fibroblasts and hematopoietic cells by the Bcr-Abl oncogene. J Exp Med. 1995;181:307–313. doi: 10.1084/jem.181.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Skorski T, Bellacosa A, Nieborowska-Skorska M, Majewski M, Martinez R, Choi JK, Trotta R, Wlodarski P, Perrotti D, Chan TO, Wasik MA, Tsichlis PN, Calabretta B. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. Embo J. 1997;16:6151–6161. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Varticovski L, Daley GQ, Jackson P, Baltimore D, Cantley LC. Activation of phosphatidylinositol 3-kinase in cells expressing abl oncogene variants. Mol Cell Biol. 1991;11:1107–1113. doi: 10.1128/mcb.11.2.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carlesso N, Frank DA, Griffin JD. Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr/Abl. J Exp Med. 1996;183:811–820. doi: 10.1084/jem.183.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shuai K, Halpern J, ten Hoeve J, Rao X, Sawyers CL. Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene. 1996;13:247–254. [PubMed] [Google Scholar]
- 76.Ilaria RL, Jr, Van Etten RA. P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J Biol Chem. 1996;271:31704–31710. doi: 10.1074/jbc.271.49.31704. [DOI] [PubMed] [Google Scholar]
- 77.Reuther JY, Reuther GW, Cortez D, Pendergast AM, Baldwin AS., Jr A requirement for NF-kappaB activation in Bcr-Abl-mediated transformation. Genes Dev. 1998;12:968–981. doi: 10.1101/gad.12.7.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Neviani P, Santhanam R, Trotta R, Notari M, Blaser BW, Liu S, Mao H, Chang JS, Galietta A, Uttam A, Roy DC, Valtieri M, Bruner-Klisovic R, Caligiuri MA, Bloomfield CD, Marcucci G, Perrotti D. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer cell. 2005;8:355–368. doi: 10.1016/j.ccr.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 79.Sternberg DW, Gilliland DG. The role of signal transducer and activator of transcription factors in leukemogenesis. J Clin Oncol. 2004;22:361–371. doi: 10.1200/JCO.2004.10.124. [DOI] [PubMed] [Google Scholar]
- 80.Gesbert F, Griffin JD. Bcr/Abl activates transcription of the Bcl-X gene through STAT5. Blood. 2000;96:2269–2276. [PubMed] [Google Scholar]
- 81.Nieborowska-Skorska M, Hoser G, Kossev P, Wasik MA, Skorski T. Complementary functions of the antiapoptotic protein A1 and serine/threonine kinase pim-1 in the BCR/ABL-mediated leukemogenesis. Blood. 2002;99:4531–4539. doi: 10.1182/blood.v99.12.4531. [DOI] [PubMed] [Google Scholar]
- 82.Horita M, Andreu EJ, Benito A, Arbona C, Sanz C, Benet I, Prosper F, Fernandez-Luna JL. Blockade of the Bcr-Abl kinase activity induces apoptosis of chronic myelogenous leukemia cells by suppressing signal transducer and activator of transcription 5-dependent expression of Bcl-xL. J Exp Med. 2000;191:977–984. doi: 10.1084/jem.191.6.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sillaber C, Gesbert F, Frank DA, Sattler M, Griffin JD. STAT5 activation contributes to growth and viability in Bcr/Abl-transformed cells. Blood. 2000;95:2118–2125. [PubMed] [Google Scholar]
- 84.Nieborowska-Skorska M, Wasik MA, Slupianek A, Salomoni P, Kitamura T, Calabretta B, Skorski T. Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL and is required for leukemogenesis. J Exp Med. 1999;189:1229–1242. doi: 10.1084/jem.189.8.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sexl V, Piekorz R, Moriggl R, Rohrer J, Brown MP, Bunting KD, Rothammer K, Roussel MF, Ihle JN. Stat5a/b contribute to interleukin 7-induced B-cell precursor expansion, but abl- and bcr/abl-induced transformation are independent of stat5. Blood. 2000;96:2277–2283. [PubMed] [Google Scholar]
- 86.Karin M. The NF-kappa B activation pathway: its regulation and role in inflammation and cell survival. Cancer J Sci Am. 1998;4(Suppl 1):S92–99. [PubMed] [Google Scholar]
- 87.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 88.Hamdane M, David-Cordonnier MH, D’Halluin JC. Activation of p65 NF-kappaB protein by p210BCR-ABL in a myeloid cell line (P210BCR-ABL activates p65 NF-kappaB) Oncogene. 1997;15:2267–2275. doi: 10.1038/sj.onc.1201411. [DOI] [PubMed] [Google Scholar]
- 89.Khosravi-Far R, Der CJ. The Ras signal transduction pathway. Cancer Metastasis Rev. 1994;13:67–89. doi: 10.1007/BF00690419. [DOI] [PubMed] [Google Scholar]
- 90.Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ. Increasing complexity of Ras signaling. Oncogene. 1998;17:1395–1413. doi: 10.1038/sj.onc.1202174. [DOI] [PubMed] [Google Scholar]
- 91.Khosravi-Far R, Campbell S, Rossman KL, Der CJ. Increasing complexity of Ras signal transduction: involvement of Rho family proteins. Advances in cancer research. 1998;72:57–107. doi: 10.1016/s0065-230x(08)60700-9. [DOI] [PubMed] [Google Scholar]
- 92.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 93.Pendergast AM, Quilliam LA, Cripe LD, Bassing CH, Dai Z, Li N, Batzer A, Rabun KM, Der CJ, Schlessinger J, et al. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell. 1993;75:175–185. [PubMed] [Google Scholar]
- 94.Puil L, Liu J, Gish G, Mbamalu G, Bowtell D, Pelicci PG, Arlinghaus R, Pawson T. Bcr-Abl oncoproteins bind directly to activators of the Ras signalling pathway. Embo J. 1994;13:764–773. doi: 10.1002/j.1460-2075.1994.tb06319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cortez D, Kadlec L, Pendergast AM. Structural and signaling requirements for BCR-ABL-mediated transformation and inhibition of apoptosis. Mol Cell Biol. 1995;15:5531–5541. doi: 10.1128/mcb.15.10.5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Million RP, Van Etten RA. The Grb2 binding site is required for the induction of chronic myeloid leukemia-like disease in mice by the Bcr/Abl tyrosine kinase. Blood. 2000;96:664–670. [PubMed] [Google Scholar]
- 97.Sattler M, Mohi MG, Pride YB, Quinnan LR, Malouf NA, Podar K, Gesbert F, Iwasaki H, Li S, Van Etten RA, Gu H, Griffin JD, Neel BG. Critical role for Gab2 in transformation by BCR/ABL. Cancer cell. 2002;1:479–492. doi: 10.1016/s1535-6108(02)00074-0. [DOI] [PubMed] [Google Scholar]
- 98.Aichberger KJ, Mayerhofer M, Krauth MT, Skvara H, Florian S, Sonneck K, Akgul C, Derdak S, Pickl WF, Wacheck V, Selzer E, Monia BP, Moriggl R, Valent P, Sillaber C. Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood. 2005;105:3303–3311. doi: 10.1182/blood-2004-02-0749. [DOI] [PubMed] [Google Scholar]
- 99.Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- 100.Toker A, Cantley LC. Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature. 1997;387:673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- 101.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 103.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 104.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer cell. 2003;4:257–262. doi: 10.1016/s1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- 105.Skorski T, Kanakaraj P, Nieborowska-Skorska M, Ratajczak MZ, Wen SC, Zon G, Gewirtz AM, Perussia B, Calabretta B. Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood. 1995;86:726–736. [PubMed] [Google Scholar]
- 106.Klejman A, Rushen L, Morrione A, Slupianek A, Skorski T. Phosphatidylinositol-3 kinase inhibitors enhance the anti-leukemia effect of STI571. Oncogene. 2002;21:5868–5876. doi: 10.1038/sj.onc.1205724. [DOI] [PubMed] [Google Scholar]
- 107.Tseng PH, Lin HP, Zhu J, Chen KF, Hade EM, Young DC, Byrd JC, Grever M, Johnson K, Druker BJ, Chen CS. Synergistic interactions between imatinib mesylate and the novel phosphoinositide-dependent kinase-1 inhibitor OSU-03012 in overcoming imatinib mesylate resistance. Blood. 2005;105:4021–4027. doi: 10.1182/blood-2004-07-2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jain SK, Susa M, Keeler ML, Carlesso N, Druker B, Varticovski L. PI 3-kinase activation in BCR/abl-transformed hematopoietic cells does not require interaction of p85 SH2 domains with p210 BCR/abl. Blood. 1996;88:1542–1550. [PubMed] [Google Scholar]
- 109.Sattler M, Salgia R, Okuda K, Uemura N, Durstin MA, Pisick E, Xu G, Li JL, Prasad KV, Griffin JD. The proto-oncogene product p120CBL and the adaptor proteins CRKL and c-CRK link c-ABL, p190BCR/ABL and p210BCR/ABL to the phosphatidylinositol-3′ kinase pathway. Oncogene. 1996;12:839–846. [PubMed] [Google Scholar]
- 110.Dinulescu DM, Wood LJ, Shen L, Loriaux M, Corless CL, Gross AW, Ren R, Deininger MW, Druker BJ. c-CBL is not required for leukemia induction by Bcr-Abl in mice. Oncogene. 2003;22:8852–8860. doi: 10.1038/sj.onc.1206892. [DOI] [PubMed] [Google Scholar]
- 111.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 112.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 113.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 114.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001;98:11598–11603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Burgering BM, Medema RH. Decisions on life and death: FOXO Forkhead transcription factors are in command when PKB/Akt is off duty. J Leukoc Biol. 2003;73:689–701. doi: 10.1189/jlb.1202629. [DOI] [PubMed] [Google Scholar]
- 116.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 117.Datta SR, Katsov A, Hu L, Petros A, Fesik SW, Yaffe MB, Greenberg ME. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell. 2000;6:41–51. [PubMed] [Google Scholar]
- 118.Neshat MS, Raitano AB, Wang HG, Reed JC, Sawyers CL. The survival function of the Bcr-Abl oncogene is mediated by Bad-dependent and -independent pathways: roles for phosphatidylinositol 3-kinase and Raf. Mol Cell Biol. 2000;20:1179–1186. doi: 10.1128/mcb.20.4.1179-1186.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Deming PB, Schafer ZT, Tashker JS, Potts MB, Deshmukh M, Kornbluth S. Bcr-Abl-mediated protection from apoptosis downstream of mitochondrial cytochrome c release. Mol Cell Biol. 2004;24:10289–10299. doi: 10.1128/MCB.24.23.10289-10299.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Trotta R, Vignudelli T, Candini O, Intine RV, Pecorari L, Guerzoni C, Santilli G, Byrom MW, Goldoni S, Ford LP, Caligiuri MA, Maraia RJ, Perrotti D, Calabretta B. BCR/ABL activates mdm2 mRNA translation via the La antigen. Cancer cell. 2003;3:145–160. doi: 10.1016/s1535-6108(03)00020-5. [DOI] [PubMed] [Google Scholar]
- 121.Goetz AW, van der Kuip H, Maya R, Oren M, Aulitzky WE. Requirement for Mdm2 in the survival effects of Bcr-Abl and interleukin 3 in hematopoietic cells. Cancer Res. 2001;61:7635–7641. [PubMed] [Google Scholar]
- 122.Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344(Pt 2):427–431. [PMC free article] [PubMed] [Google Scholar]
- 123.Mohi MG, Boulton C, Gu TL, Sternberg DW, Neuberg D, Griffin JD, Gilliland DG, Neel BG. Combination of rapamycin and protein tyrosine kinase (PTK) inhibitors for the treatment of leukemias caused by oncogenic PTKs. Proc Natl Acad Sci U S A. 2004;101:3130–3135. doi: 10.1073/pnas.0400063101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ly C, Arechiga AF, Melo JV, Walsh CM, Ong ST. Bcr-Abl kinase modulates the translation regulators ribosomal protein S6 and 4E-BP1 in chronic myelogenous leukemia cells via the mammalian target of rapamycin. Cancer Res. 2003;63:5716–5722. [PubMed] [Google Scholar]
- 125.Mayerhofer M, Aichberger KJ, Florian S, Krauth MT, Hauswirth AW, Derdak S, Sperr WR, Esterbauer H, Wagner O, Marosi C, Pickl WF, Deininger M, Weisberg E, Druker BJ, Griffin JD, Sillaber C, Valent P. Identification of mTOR as a novel bifunctional target in chronic myeloid leukemia: dissection of growth-inhibitory and VEGF-suppressive effects of rapamycin in leukemic cells. Faseb J. 2005;19:960–962. doi: 10.1096/fj.04-1973fje. [DOI] [PubMed] [Google Scholar]
- 126.Kaufmann E, Knochel W. Five years on the wings of fork head. Mech Dev. 1996;57:3–20. doi: 10.1016/0925-4773(96)00539-4. [DOI] [PubMed] [Google Scholar]
- 127.Kaestner KH, Knochel W, Martinez DE. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 2000;14:142–146. [PubMed] [Google Scholar]
- 128.Arden KC, Biggs WH., 3rd Regulation of the FoxO family of transcription factors by phosphatidylinositol-3 kinase-activated signaling. Arch Biochem Biophys. 2002;403:292–298. doi: 10.1016/s0003-9861(02)00207-2. [DOI] [PubMed] [Google Scholar]
- 129.Jacobs FM, van der Heide LP, Wijchers PJ, Burbach JP, Hoekman MF, Smidt MP. FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J Biol Chem. 2003;278:35959–35967. doi: 10.1074/jbc.M302804200. [DOI] [PubMed] [Google Scholar]
- 130.Wolkow CA, Kimura KD, Lee MS, Ruvkun G. Regulation of C. elegans lifespan by insulinlike signaling in the nervous system. Science. 2000;290:147–150. doi: 10.1126/science.290.5489.147. [DOI] [PubMed] [Google Scholar]
- 131.Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- 132.Gottlieb S, Ruvkun G. daf-2, daf-16 and daf-23: genetically interacting genes controlling Dauer formation in Caenorhabditis elegans. Genetics. 1994;137:107–120. doi: 10.1093/genetics/137.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Unterman TG, Fareeduddin A, Harris MA, Goswami RG, Porcella A, Costa RH, Lacson RG. Hepatocyte nuclear factor-3 (HNF-3) binds to the insulin response sequence in the IGF binding protein-1 (IGFBP-1) promoter and enhances promoter function. Biochem Biophys Res Commun. 1994;203:1835–1841. doi: 10.1006/bbrc.1994.2401. [DOI] [PubMed] [Google Scholar]
- 134.Cichy SB, Uddin S, Danilkovich A, Guo S, Klippel A, Unterman TG. Protein kinase B/Akt mediates effects of insulin on hepatic insulin-like growth factor-binding protein-1 gene expression through a conserved insulin response sequence. J Biol Chem. 1998;273:6482–6487. doi: 10.1074/jbc.273.11.6482. [DOI] [PubMed] [Google Scholar]
- 135.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 136.Tang ED, Nunez G, Barr FG, Guan KL. Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem. 1999;274:16741–16746. doi: 10.1074/jbc.274.24.16741. [DOI] [PubMed] [Google Scholar]
- 137.Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem. 1999;274:17179–17183. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- 138.Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A. 1999;96:7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kops GJ, de Ruiter ND, De Vries-Smits AM, Powell DR, Bos JL, Burgering BM. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 1999;398:630–634. doi: 10.1038/19328. [DOI] [PubMed] [Google Scholar]
- 140.Takaishi H, Konishi H, Matsuzaki H, Ono Y, Shirai Y, Saito N, Kitamura T, Ogawa W, Kasuga M, Kikkawa U, Nishizuka Y. Regulation of nuclear translocation of forkhead transcription factor AFX by protein kinase B. Proc Natl Acad Sci U S A. 1999;96:11836–11841. doi: 10.1073/pnas.96.21.11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.van der Heide LP, Jacobs FM, Burbach JP, Hoekman MM, Smidt MP. FoxO6 transcriptional activity is regulated by Thr26 and Ser184, independent of nucleo-cytoplasmic shuttling. Biochem J. 2005 doi: 10.1042/BJ20050525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Brownawell AM, Kops GJ, Macara IG, Burgering BM. Inhibition of nuclear import by protein kinase B (Akt) regulates the subcellular distribution and activity of the forkhead transcription factor AFX. Mol Cell Biol. 2001;21:3534–3546. doi: 10.1128/MCB.21.10.3534-3546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Brunet A, Kanai F, Stehn J, Xu J, Sarbassova D, Frangioni JV, Dalal SN, DeCaprio JA, Greenberg ME, Yaffe MB. 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J Cell Biol. 2002;156:817–828. doi: 10.1083/jcb.200112059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rena G, Bain J, Elliott M, Cohen P. D4476, a cell-permeant inhibitor of CK1, suppresses the site-specific phosphorylation and nuclear exclusion of FOXO1a. EMBO Rep. 2004;5:60–65. doi: 10.1038/sj.embor.7400048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rena G, Woods YL, Prescott AR, Peggie M, Unterman TG, Williams MR, Cohen P. Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. Embo J. 2002;21:2263–2271. doi: 10.1093/emboj/21.9.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci U S A. 2005;102:11278–11283. doi: 10.1073/pnas.0502738102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 148.Fukuoka M, Daitoku H, Hatta M, Matsuzaki H, Umemura S, Fukamizu A. Negative regulation of forkhead transcription factor AFX (Foxo4) by CBP-induced acetylation. Int J Mol Med. 2003;12:503–508. [PubMed] [Google Scholar]
- 149.Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 150.Daitoku H, Hatta M, Matsuzaki H, Aratani S, Ohshima T, Miyagishi M, Nakajima T, Fukamizu A. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci U S A. 2004;101:10042–10047. doi: 10.1073/pnas.0400593101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1) J Biol Chem. 2004;279:28873–28879. doi: 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- 152.van der Heide LP, Smidt MP. Regulation of FoxO activity by CBP/p300-mediated acetylation. Trends Biochem Sci. 2005;30:81–86. doi: 10.1016/j.tibs.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 153.Plas DR, Thompson CB. Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J Biol Chem. 2003;278:12361–12366. doi: 10.1074/jbc.M213069200. [DOI] [PubMed] [Google Scholar]
- 154.Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci U S A. 2003;100:11285–11290. doi: 10.1073/pnas.1934283100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Huang H, Regan KM, Wang F, Wang D, Smith DI, van Deursen JM, Tindall DJ. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc Natl Acad Sci U S A. 2005;102:1649–1654. doi: 10.1073/pnas.0406789102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, Kobayashi R, Hung MC. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- 157.Tran H, Brunet A, Griffith EC, Greenberg ME. The many forks in FOXO’s road. Sci STKE. 2003;2003:RE5. doi: 10.1126/stke.2003.172.re5. [DOI] [PubMed] [Google Scholar]
- 158.Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117:421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- 159.Hosaka T, Biggs WH, 3rd, Tieu D, Boyer AD, Varki NM, Cavenee WK, Arden KC. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci U S A. 2004;101:2975–2980. doi: 10.1073/pnas.0400093101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Castrillon DH, Miao L, Kollipara R, Horner JW, DePinho RA. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science. 2003;301:215–218. doi: 10.1126/science.1086336. [DOI] [PubMed] [Google Scholar]
- 161.Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR, Jiang S, Gilliland DG, Chin L, Wong WH, Castrillon DH, DePinho RA. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309–323. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, Armstrong SA, Passegue E, DePinho RA, Gilliland DG. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 163.Reif K, Burgering BM, Cantrell DA. Phosphatidylinositol 3-kinase links the interleukin-2 receptor to protein kinase B and p70 S6 kinase. J Biol Chem. 1997;272:14426–14433. doi: 10.1074/jbc.272.22.14426. [DOI] [PubMed] [Google Scholar]
- 164.Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, Burgering BM, Medema RH. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol. 2002;168:5024–5031. doi: 10.4049/jimmunol.168.10.5024. [DOI] [PubMed] [Google Scholar]
- 165.Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol. 2003;162:613–622. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dijkers PF, Medema RH, Pals C, Banerji L, Thomas NS, Lam EW, Burgering BM, Raaijmakers JA, Lammers JW, Koenderman L, Coffer PJ. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1) Mol Cell Biol. 2000;20:9138–9148. doi: 10.1128/mcb.20.24.9138-9148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201–1204. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 168.Dijkers PF, Birkenkamp KU, Lam EW, Thomas NS, Lammers JW, Koenderman L, Coffer PJ. FKHR-L1 can act as a critical effector of cell death induced by cytokine withdrawal: protein kinase B-enhanced cell survival through maintenance of mitochondrial integrity. J Cell Biol. 2002;156:531–542. doi: 10.1083/jcb.200108084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Essafi A, Fernandez de Mattos S, Hassen YA, Soeiro I, Mufti GJ, Thomas NS, Medema RH, Lam EW. Direct transcriptional regulation of Bim by FoxO3a mediates STI571-induced apoptosis in Bcr-Abl-expressing cells. Oncogene. 2005;24:2317–2329. doi: 10.1038/sj.onc.1208421. [DOI] [PubMed] [Google Scholar]
- 170.Ghaffari S, Jagani Z, Kitidis C, Lodish HF, Khosravi-Far R. Cytokines and BCR-ABL mediate suppression of TRAIL-induced apoptosis through inhibition of forkhead FOXO3a transcription factor. Proc Natl Acad Sci U S A. 2003;100:6523–6528. doi: 10.1073/pnas.0731871100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Schmidt M, Fernandez de Mattos S, van der Horst A, Klompmaker R, Kops GJ, Lam EW, Burgering BM, Medema RH. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol Cell Biol. 2002;22:7842–7852. doi: 10.1128/MCB.22.22.7842-7852.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ramaswamy S, Nakamura N, Sansal I, Bergeron L, Sellers WR. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell. 2002;2:81–91. doi: 10.1016/s1535-6108(02)00086-7. [DOI] [PubMed] [Google Scholar]
- 173.Birkenkamp KU, Essafi A, van der Vos KE, da Costa M, Hui RC, Holstege F, Koenderman L, Lam EW, Coffer PJ. FOXO3a induces differentiation of Bcr-Abl-transformed cells through transcriptional down-regulation of Id1. The Journal of biological chemistry. 2007;282:2211–2220. doi: 10.1074/jbc.M606669200. [DOI] [PubMed] [Google Scholar]
- 174.Tang TT, Dowbenko D, Jackson A, Toney L, Lewin DA, Dent AL, Lasky LA. The forkhead transcription factor AFX activates apoptosis by induction of the BCL-6 transcriptional repressor. J Biol Chem. 2002;277:14255–14265. doi: 10.1074/jbc.M110901200. [DOI] [PubMed] [Google Scholar]
- 175.Parry P, Wei Y, Evans G. Cloning and characterization of the t(X;11) breakpoint from a leukemic cell line identify a new member of the forkhead gene family. Genes Chromosomes Cancer. 1994;11:79–84. doi: 10.1002/gcc.2870110203. [DOI] [PubMed] [Google Scholar]
- 176.Hillion J, Le Coniat M, Jonveaux P, Berger R, Bernard OA. AF6q21, a novel partner of the MLL gene in t(6;11)(q21;q23), defines a forkhead transcriptional factor subfamily. Blood. 1997;90:3714–3719. [PubMed] [Google Scholar]
- 177.Barr FG. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene. 2001;20:5736–5746. doi: 10.1038/sj.onc.1204599. [DOI] [PubMed] [Google Scholar]
- 178.So CW, Cleary ML. Dimerization: a versatile switch for oncogenesis. Blood. 2004;104:919–922. doi: 10.1182/blood-2004-03-0992. [DOI] [PubMed] [Google Scholar]
- 179.So CW, Cleary ML. MLL-AFX requires the transcriptional effector domains of AFX to transform myeloid progenitors and transdominantly interfere with forkhead protein function. Mol Cell Biol. 2002;22:6542–6552. doi: 10.1128/MCB.22.18.6542-6552.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Anderson MJ, Shelton GD, Cavenee WK, Arden KC. Embryonic expression of the tumor-associated PAX3-FKHR fusion protein interferes with the developmental functions of Pax3. Proc Natl Acad Sci U S A. 2001;98:1589–1594. doi: 10.1073/pnas.98.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bois PR, Izeradjene K, Houghton PJ, Cleveland JL, Houghton JA, Grosveld GC. FOXO1a acts as a selective tumor suppressor in alveolar rhabdomyosarcoma. J Cell Biol. 2005;170:903–912. doi: 10.1083/jcb.200501040. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 182.Parsons DW, Wang TL, Samuels Y, Bardelli A, Cummins JM, DeLong L, Silliman N, Ptak J, Szabo S, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Lengauer C, Velculescu VE. Colorectal cancer: mutations in a signalling pathway. Nature. 2005;436:792. doi: 10.1038/436792a. [DOI] [PubMed] [Google Scholar]
- 183.Samuels Y, Diaz LA, Jr, Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, Rago C, Huso DL, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7:561–573. doi: 10.1016/j.ccr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 184.Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol. 2000;20:8969–8982. doi: 10.1128/mcb.20.23.8969-8982.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Modur V, Nagarajan R, Evers BM, Milbrandt J. FOXO proteins regulate tumor necrosis factor-related apoptosis inducing ligand expression. Implications for PTEN mutation in prostate cancer. J Biol Chem. 2002;277:47928–47937. doi: 10.1074/jbc.M207509200. [DOI] [PubMed] [Google Scholar]
- 186.Li P, Lee H, Guo S, Unterman TG, Jenster G, Bai W. AKT-independent protection of prostate cancer cells from apoptosis mediated through complex formation between the androgen receptor and FKHR. Mol Cell Biol. 2003;23:104–118. doi: 10.1128/MCB.23.1.104-118.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 188.Yang H, Zhao R, Yang HY, Lee MH. Constitutively active FOXO4 inhibits Akt activity, regulates p27 Kip1 stability, and suppresses HER2-mediated tumorigenicity. Oncogene. 2005;24:1924–1935. doi: 10.1038/sj.onc.1208352. [DOI] [PubMed] [Google Scholar]
- 189.Komatsu N, Watanabe T, Uchida M, Mori M, Kirito K, Kikuchi S, Liu Q, Tauchi T, Miyazawa K, Endo H, Nagai T, Ozawa K. A member of Forkhead transcription factor FKHRL1 is a downstream effector of STI571-induced cell cycle arrest in BCR-ABL-expressing cells. J Biol Chem. 2003;278:6411–6419. doi: 10.1074/jbc.M211562200. [DOI] [PubMed] [Google Scholar]
- 190.Andreu EJ, Lledo E, Poch E, Ivorra C, Albero MP, Martinez-Climent JA, Montiel-Duarte C, Rifon J, Perez-Calvo J, Arbona C, Prosper F, Perez-Roger I. BCR-ABL induces the expression of Skp2 through the PI3K pathway to promote p27Kip1 degradation and proliferation of chronic myelogenous leukemia cells. Cancer Res. 2005;65:3264–3272. doi: 10.1158/0008-5472.CAN-04-1357. [DOI] [PubMed] [Google Scholar]
- 191.Fernandez de Mattos S, Essafi A, Soeiro I, Pietersen AM, Birkenkamp KU, Edwards CS, Martino A, Nelson BH, Francis JM, Jones MC, Brosens JJ, Coffer PJ, Lam EW. FoxO3a and BCR-ABL regulate cyclin D2 transcription through a STAT5/BCL6-dependent mechanism. Mol Cell Biol. 2004;24:10058–10071. doi: 10.1128/MCB.24.22.10058-10071.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Scheijen B, Ngo HT, Kang H, Griffin JD. FLT3 receptors with internal tandem duplications promote cell viability and proliferation by signaling through Foxo proteins. Oncogene. 2004;23:3338–3349. doi: 10.1038/sj.onc.1207456. [DOI] [PubMed] [Google Scholar]
- 193.Gu TL, Tothova Z, Scheijen B, Griffin JD, Gilliland DG, Sternberg DW. NPM-ALK fusion kinase of anaplastic large-cell lymphoma regulates survival and proliferative signaling through modulation of FOXO3a. Blood. 2004;103:4622–4629. doi: 10.1182/blood-2003-03-0820. [DOI] [PubMed] [Google Scholar]
- 194.O’Hare T, Walters DK, Stoffregen EP, Jia T, Manley PW, Mestan J, Cowan-Jacob SW, Lee FY, Heinrich MC, Deininger MW, Druker BJ. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65:4500–4505. doi: 10.1158/0008-5472.CAN-05-0259. [DOI] [PubMed] [Google Scholar]
- 195.Golemovic M, Verstovsek S, Giles F, Cortes J, Manshouri T, Manley PW, Mestan J, Dugan M, Alland L, Griffin JD, Arlinghaus RB, Sun T, Kantarjian H, Beran M. AMN107, a novel aminopyrimidine inhibitor of Bcr-Abl, has in vitro activity against imatinib-resistant chronic myeloid leukemia. Clin Cancer Res. 2005;11:4941–4947. doi: 10.1158/1078-0432.CCR-04-2601. [DOI] [PubMed] [Google Scholar]
- 196.Golas JM, Arndt K, Etienne C, Lucas J, Nardin D, Gibbons J, Frost P, Ye F, Boschelli DH, Boschelli F. SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 2003;63:375–381. [PubMed] [Google Scholar]
- 197.O’Hare T, Pollock R, Stoffregen EP, Keats JA, Abdullah OM, Moseson EM, Rivera VM, Tang H, Metcalf CA, 3rd, Bohacek RS, Wang Y, Sundaramoorthi R, Shakespeare WC, Dalgarno D, Clackson T, Sawyer TK, Deininger MW, Druker BJ. Inhibition of wild-type and mutant Bcr-Abl by AP23464, a potent ATP-based oncogenic protein kinase inhibitor: implications for CML. Blood. 2004;104:2532–2539. doi: 10.1182/blood-2004-05-1851. [DOI] [PubMed] [Google Scholar]
- 198.Peters DG, Hoover RR, Gerlach MJ, Koh EY, Zhang H, Choe K, Kirschmeier P, Bishop WR, Daley GQ. Activity of the farnesyl protein transferase inhibitor SCH66336 against BCR/ABL-induced murine leukemia and primary cells from patients with chronic myeloid leukemia. Blood. 2001;97:1404–1412. doi: 10.1182/blood.v97.5.1404. [DOI] [PubMed] [Google Scholar]
- 199.Hoover RR, Mahon FX, Melo JV, Daley GQ. Overcoming STI571 resistance with the farnesyl transferase inhibitor SCH66336. Blood. 2002;100:1068–1071. doi: 10.1182/blood.v100.3.1068. [DOI] [PubMed] [Google Scholar]
- 200.Jorgensen HG, Allan EK, Graham SM, Godden JL, Richmond L, Elliott MA, Mountford JC, Eaves CJ, Holyoake TL. Lonafarnib reduces the resistance of primitive quiescent CML cells to imatinib mesylate in vitro. Leukemia. 2005;19:1184–1191. doi: 10.1038/sj.leu.2403785. [DOI] [PubMed] [Google Scholar]
- 201.Karp JE, Lancet JE, Kaufmann SH, End DW, Wright JJ, Bol K, Horak I, Tidwell ML, Liesveld J, Kottke TJ, Ange D, Buddharaju L, Gojo I, Highsmith WE, Belly RT, Hohl RJ, Rybak ME, Thibault A, Rosenblatt J. Clinical and biologic activity of the farnesyltransferase inhibitor R115777 in adults with refractory and relapsed acute leukemias: a phase 1 clinical-laboratory correlative trial. Blood. 2001;97:3361–3369. doi: 10.1182/blood.v97.11.3361. [DOI] [PubMed] [Google Scholar]
- 202.Cortes J, Albitar M, Thomas D, Giles F, Kurzrock R, Thibault A, Rackoff W, Koller C, O’Brien S, Garcia-Manero G, Talpaz M, Kantarjian H. Efficacy of the farnesyl transferase inhibitor R115777 in chronic myeloid leukemia and other hematologic malignancies. Blood. 2003;101:1692–1697. doi: 10.1182/blood-2002-07-1973. [DOI] [PubMed] [Google Scholar]
- 203.Yu C, Krystal G, Varticovksi L, McKinstry R, Rahmani M, Dent P, Grant S. Pharmacologic mitogen-activated protein/extracellular signal-regulated kinase kinase/mitogen-activated protein kinase inhibitors interact synergistically with STI571 to induce apoptosis in Bcr/Abl-expressing human leukemia cells. Cancer Res. 2002;62:188–199. [PubMed] [Google Scholar]
- 204.Yu C, Dasmahapatra G, Dent P, Grant S. Synergistic interactions between MEK1/2 and histone deacetylase inhibitors in BCR/ABL+ human leukemia cells. Leukemia. 2005;19:1579–1589. doi: 10.1038/sj.leu.2403868. [DOI] [PubMed] [Google Scholar]
- 205.Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK, Milanov ZV, Atteridge CE, Biggs WH, 3rd, Edeen PT, Floyd M, Ford JM, Grotzfeld RM, Herrgard S, Insko DE, Mehta SA, Patel HK, Pao W, Sawyers CL, Varmus H, Zarrinkar PP, Lockhart DJ. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci U S A. 2005;102:11011–11016. doi: 10.1073/pnas.0504952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Weisberg E, Catley L, Kujawa J, Atadja P, Remiszewski S, Fuerst P, Cavazza C, Anderson K, Griffin JD. Histone deacetylase inhibitor NVP-LAQ824 has significant activity against myeloid leukemia cells in vitro and in vivo. Leukemia. 2004;18:1951–1963. doi: 10.1038/sj.leu.2403519. [DOI] [PubMed] [Google Scholar]
- 207.Nimmanapalli R, Fuino L, Bali P, Gasparetto M, Glozak M, Tao J, Moscinski L, Smith C, Wu J, Jove R, Atadja P, Bhalla K. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res. 2003;63:5126–5135. [PubMed] [Google Scholar]
- 208.George P, Bali P, Annavarapu S, Scuto A, Fiskus W, Guo F, Sigua C, Sondarva G, Moscinski L, Atadja P, Bhalla K. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against human CML-BC cells and AML cells with activating mutation of FLT-3. Blood. 2005;105:1768–1776. doi: 10.1182/blood-2004-09-3413. [DOI] [PubMed] [Google Scholar]
- 209.Nimmanapalli R, Fuino L, Stobaugh C, Richon V, Bhalla K. Cotreatment with the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) enhances imatinib-induced apoptosis of Bcr-Abl-positive human acute leukemia cells. Blood. 2003;101:3236–3239. doi: 10.1182/blood-2002-08-2675. [DOI] [PubMed] [Google Scholar]
- 210.Xu Y, Voelter-Mahlknecht S, Mahlknecht U. The histone deacetylase inhibitor suberoylanilide hydroxamic acid down-regulates expression levels of Bcr-abl, c-Myc and HDAC3 in chronic myeloid leukemia cell lines. Int J Mol Med. 2005;15:169–172. [PubMed] [Google Scholar]
- 211.Yu C, Rahmani M, Conrad D, Subler M, Dent P, Grant S. The proteasome inhibitor bortezomib interacts synergistically with histone deacetylase inhibitors to induce apoptosis in Bcr/Abl+ cells sensitive and resistant to STI571. Blood. 2003;102:3765–3774. doi: 10.1182/blood-2003-03-0737. [DOI] [PubMed] [Google Scholar]
- 212.Rahmani M, Reese E, Dai Y, Bauer C, Kramer LB, Huang M, Jove R, Dent P, Grant S. Cotreatment with suberanoylanilide hydroxamic acid and 17-allylamino 17-demethoxygeldanamycin synergistically induces apoptosis in Bcr-Abl+ Cells sensitive and resistant to STI571 (imatinib mesylate) in association with down-regulation of Bcr-Abl, abrogation of signal transducer and activator of transcription 5 activity, and Bax conformational change. Mol Pharmacol. 2005;67:1166–1176. doi: 10.1124/mol.104.007831. [DOI] [PubMed] [Google Scholar]
- 213.Gatto S, Scappini B, Pham L, Onida F, Milella M, Ball G, Ricci C, Divoky V, Verstovsek S, Kantarjian HM, Keating MJ, Cortes-Franco JE, Beran M. The proteasome inhibitor PS-341 inhibits growth and induces apoptosis in Bcr/Abl-positive cell lines sensitive and resistant to imatinib mesylate. Haematologica. 2003;88:853–863. [PubMed] [Google Scholar]
- 214.Dai Y, Rahmani M, Pei XY, Dent P, Grant S. Bortezomib and flavopiridol interact synergistically to induce apoptosis in chronic myeloid leukemia cells resistant to imatinib mesylate through both Bcr/Abl-dependent and -independent mechanisms. Blood. 2004;104:509–518. doi: 10.1182/blood-2003-12-4121. [DOI] [PubMed] [Google Scholar]
- 215.Wolf DH, Hilt W. The proteasome: a proteolytic nanomachine of cell regulation and waste disposal. Biochim Biophys Acta. 2004;1695:19–31. doi: 10.1016/j.bbamcr.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 216.Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 217.Adams J. The proteasome: structure, function, and role in the cell. Cancer Treat Rev. 2003;29(Suppl 1):3–9. doi: 10.1016/s0305-7372(03)00081-1. [DOI] [PubMed] [Google Scholar]
- 218.Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. Proteasome inhibition as a novel therapeutic target in human cancer. J Clin Oncol. 2005;23:630–639. doi: 10.1200/JCO.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 219.Adams J. Proteasome inhibition: a novel approach to cancer therapy. Trends Mol Med. 2002;8:S49–54. doi: 10.1016/s1471-4914(02)02315-8. [DOI] [PubMed] [Google Scholar]
- 220.Adams J. Proteasome inhibition in cancer: development of PS-341. Semin Oncol. 2001;28:613–619. doi: 10.1016/s0093-7754(01)90034-x. [DOI] [PubMed] [Google Scholar]
- 221.Chauhan D, Hideshima T, Mitsiades C, Richardson P, Anderson KC. Proteasome inhibitor therapy in multiple myeloma. Mol Cancer Ther. 2005;4:686–692. doi: 10.1158/1535-7163.MCT-04-0338. [DOI] [PubMed] [Google Scholar]
- 222.Richardson PG, Hideshima T, Anderson KC. Bortezomib (PS-341): a novel, first-in-class proteasome inhibitor for the treatment of multiple myeloma and other cancers. Cancer Control. 2003;10:361–369. doi: 10.1177/107327480301000502. [DOI] [PubMed] [Google Scholar]
- 223.Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, San-Miguel JF, Blade J, Boccadoro M, Cavenagh J, Dalton WS, Boral AL, Esseltine DL, Porter JB, Schenkein D, Anderson KC. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352:2487–2498. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- 224.Fisher RI, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, Epner E, Krishnan A, Leonard JP, Lonial S, Stadtmauer EA, O’Connor OA, Shi H, Boral AL, Goy A. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006;24:4867–4874. doi: 10.1200/JCO.2006.07.9665. [DOI] [PubMed] [Google Scholar]
- 225.Mitsiades CS, Mitsiades N, Hideshima T, Richardson PG, Anderson KC. Proteasome inhibition as a new therapeutic principle in hematological malignancies. Current drug targets. 2006;7:1341–1347. doi: 10.2174/138945006778559247. [DOI] [PubMed] [Google Scholar]
- 226.Adams J, Kauffman M. Development of the proteasome inhibitor Velcade (Bortezomib) Cancer Invest. 2004;22:304–311. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- 227.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 228.Hideshima T, Mitsiades C, Akiyama M, Hayashi T, Chauhan D, Richardson P, Schlossman R, Podar K, Munshi NC, Mitsiades N, Anderson KC. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood. 2003;101:1530–1534. doi: 10.1182/blood-2002-08-2543. [DOI] [PubMed] [Google Scholar]
- 229.Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer. 2004;4:349–360. doi: 10.1038/nrc1361. [DOI] [PubMed] [Google Scholar]
- 230.Soligo D, Servida F, Delia D, Fontanella E, Lamorte G, Caneva L, Fumiatti R, Lambertenghi Deliliers G. The apoptogenic response of human myeloid leukaemia cell lines and of normal and malignant haematopoietic progenitor cells to the proteasome inhibitor PSI. Br J Haematol. 2001;113:126–135. doi: 10.1046/j.1365-2141.2001.02683.x. [DOI] [PubMed] [Google Scholar]
- 231.Dou QP, McGuire TF, Peng Y, An B. Proteasome inhibition leads to significant reduction of Bcr-Abl expression and subsequent induction of apoptosis in K562 human chronic myelogenous leukemia cells. J Pharmacol Exp Ther. 1999;289:781–790. [PubMed] [Google Scholar]
- 232.Kau TR, Schroeder F, Ramaswamy S, Wojciechowski CL, Zhao JJ, Roberts TM, Clardy J, Sellers WR, Silver PA. A chemical genetic screen identifies inhibitors of regulated nuclear export of a Forkhead transcription factor in PTEN-deficient tumor cells. Cancer Cell. 2003;4:463–476. doi: 10.1016/s1535-6108(03)00303-9. [DOI] [PubMed] [Google Scholar]
- 233.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 234.Huntly BJ, Gilliland DG. Cancer biology: summing up cancer stem cells. Nature. 2005;435:1169–1170. doi: 10.1038/4351169a. [DOI] [PubMed] [Google Scholar]
- 235.Jordan CT, Guzman ML. Mechanisms controlling pathogenesis and survival of leukemic stem cells. Oncogene. 2004;23:7178–7187. doi: 10.1038/sj.onc.1207935. [DOI] [PubMed] [Google Scholar]
- 236.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 237.Huntly BJ, Gilliland DG. Blasts from the past: new lessons in stem cell biology from chronic myelogenous leukemia. Cancer cell. 2004;6:199–201. doi: 10.1016/j.ccr.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 238.Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, Rowan R, Amaral S, Curley D, Williams IR, Akashi K, Gilliland DG. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer cell. 2004;6:587–596. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 239.Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 240.Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villen J, Becker EB, DiBacco S, de la Iglesia N, Gygi S, Blackwell TK, Bonni A. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 241.van der Horst A, de Vries-Smits AM, Brenkman AB, van Triest MH, van den Broek N, Colland F, Maurice MM, Burgering BM. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nature cell biology. 2006;8:1064–1073. doi: 10.1038/ncb1469. [DOI] [PubMed] [Google Scholar]
- 242.Brummendorf TH, Balabanov S. Telomere length dynamics in normal hematopoiesis and in disease states characterized by increased stem cell turnover. Leukemia. 2006;20:1706–1716. doi: 10.1038/sj.leu.2404339. [DOI] [PubMed] [Google Scholar]
- 243.Canitrot Y, Lautier D, Laurent G, Frechet M, Ahmed A, Turhan AG, Salles B, Cazaux C, Hoffmann JS. Mutator phenotype of BCR--ABL transfected Ba/F3 cell lines and its association with enhanced expression of DNA polymerase beta. Oncogene. 1999;18:2676–2680. doi: 10.1038/sj.onc.1202619. [DOI] [PubMed] [Google Scholar]
- 244.Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, Holyoake TL. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99:319–325. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- 245.Guzman ML, Swiderski CF, Howard DS, Grimes BA, Rossi RM, Szilvassy SJ, Jordan CT. Preferential induction of apoptosis for primary human leukemic stem cells. Proc Natl Acad Sci U S A. 2002;99:16220–16225. doi: 10.1073/pnas.252462599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.You H, Mak TW. Crosstalk between p53 and FOXO transcription factors. Cell Cycle. 2005;4:37–38. doi: 10.4161/cc.4.1.1401. [DOI] [PubMed] [Google Scholar]