

Abstract
Formation and accumulation of amyloid-beta (Aβ) plaques are associated with declined memory and other neurocognitive function in Alzheimer's Disease (AD) patients. However, the effects of Aβ plaques on neural progenitor cells (NPCs) and neurogenesis from NPCs remain largely unknown. The existing data on neurogenesis in AD patients and AD-like animal models remain controversial. For this reason, we utilized the nestin second-intron enhancer controlled LacZ (pNes-LacZ) reporter transgenic mice (pNes-Tg) and Bi-transgenic mice (Bi-Tg) containing both pPDGF-APPSw,Ind and pNes-LacZ transgenes to investigate the effects of Aβ plaques on neurogenesis in the hippocampus and other brain regions of the AD–like mice. We chose transgenic mice at 2, 8 and 12 months of age, corresponding to the stages of Aβ plaque free, plaque onset and plaque progression to analyze the effects of Aβ plaques on the distribution and de novo neurogenesis of (from) NPCs. We demonstrated a slight increase in the number of NPCs in the hippocampal regions at the Aβ plaque free stage, while a significant decrease in the number of NPCs at Aβ plaque onset and progression stages. On the other hand, we showed that Aβ plaques increase neurogenesis, but not gliogenesis from post-mitotic NPCs in the hippocampus of Bi-Tg mice compared with age-matched control pNes-Tg mice. The neurogenic responses of NPCs to Aβ plaques suggest that experimental approaches to promote de novo neurogenesis may potentially improve neurocognitive function and provide an effective therapy for AD.
Keywords: Neural Progenitor Cells, Alzheimer's Disease, Regenerative Medicine, Neurogenesis, Nestin, Aβ plaque
Introduction
Alzheimer's disease (AD) is a degenerative disorder of the brain that is characterized by senile plaques containing amyloid beta (Aβ) aggregates, and neurofibrillary tangles containing hyperphosporylated tau-protein. The generation and progression of Aβ plaques in the brain are generally recognized as the principal cause of AD pathogenesis (1-3). The Aβ plaque-mediated neurotoxicity in the distinct anatomic regions of the brain including the hippocampus, frontal cortex and limbic system leads AD patients to loss of memory along with a progressive decline in other cognitive skills (2,3). Despite the advancements made in understanding the cellular and molecular mechanisms associated with the neurodegeneration by Aβ plaques, effective therapy to intervene disease onset and progression and to improve neurocognitive functions in AD patients is not available at the present time. To a large extent, significant numbers of neurons died or on the way of dying at or after clinical manifestation of AD. Neuronal loss leads to neurocircuit alteration and consequently neurocognitive dysfunction. At this stage, therapy toward neuronal protection may not be effective. On the other hand, therapy aiming at neuronal regeneration may be potentially delay or reverse the onset and progression of AD. Since neural stem cells and/or neural progenitors cells (NPCs) are largely present in the adult CNS, regenerative medicine aiming to improve neurocognitive function by increasing de novo neurogenesis may have great potential to the therapy of AD.
Neurogenesis has been demonstrated to contribute to maintaining normal neurological functions in animal models and has been implicated in humans (4-6). The persistent neurogenesis was originally thought to occur in only a few restricted areas, such as the subventricular zone (SVZ), and the subgranular zone of hippocampus (7-9). However, more recent studies have demonstrated the generation of new neurons in many other CNS regions, for example, the neocortex, substantia nigra and spinal cord (10-15). Increasing evidence indicates that neurogenesis is critical to maintain normal neurological function, while alteration of neurogenesis is associated with neurological dysfunction (16,17). Furthermore, neurogenesis has been demonstrated in the animal models of ischemic stroke (18), amyotrophic lateral sclerosis (ALS) (19), Alzheimer's disease (AD) (20), Parkinson's disease (PD) and epilepsy (21,22). Moreover, enhanced neurogenesis has been reported in human patients with AD (23) and Huntington's disease (24,25). Although substantial progress has been made in identifying and characterizing neurogenesis in the adult CNS, the effects of NPCs and de novo neurogenesis in response to Aβ plaque-mediated neuropathophysiology remain to be established. In fact, the few research reports on neurogenesis in AD animal models remain controversial. Several studies showed that Aβ plaques decrease, while others demonstrated an increase in neurogenesis in the hippocampus and other anatomical regions of AD transgenic mouse models (26-28). These studies primarily used BrdU labeling to identify neural stem cells and/or NPCs. On the other hand, neurogenesis could be derived from NPCs. To apply regenerative medicine, particularly promotion of de novo neurongenesis for therapy of AD, extensive knowledge is required on the distribution and organization of NPCs in the brain of AD patients. Because the CNS tissues from AD patients and age-matched normal persons are generally not available during disease onset and progression stages, research progress on the response of endogenous NPCs to neurodegeneration and neurocognitive dysfunction in human AD is substantially limited. Therefore, we utilized the Bi-Tg mice containing both PDGF-APPSw,Ind and pNes-LacZ transgenes, and age-matched pNes-Tg mice to investigate the effects of Aβ plaques on neurogenesis in the hippocampus and other brain regions of the AD–like mice. We demonstrated the formation and progression of Aβ plaques increases neurogenesis, but not gliogenesis from NPCs in the hippocampus of Bi-Tg mice compared with that of age-matched pNes-LacZ controls. The neurogenic responses of NPCs to Aβ plaques suggest that experimental approaches to promote de novo neurogenesis may provide an effective therapy to improve neurocognitive dysfunction in AD.
Materials and Methods
Transgenic mouse lines
Nestin promoter (enhancer) driven LacZ reporter transgenic mice (pNes-Tg) (29,30) and pPDGF-APPSw,Ind (K670N/M671L and V717F) (31) mimicking human AD transgenic mice (Jackson Laboratory, Bar Harbor, ME) were used to generate Bi-Tg mice containing both pPDGF-APPSw,Ind and pNes-LacZ transgenes through heterozygous breeding. Transgenic progeny were identified by regular PCR amplification of tail DNA using specific primers. Age-matched littermates of pNes-Tg mice were used as controls. The experimental protocols for animal breeding and neurogenic analyses were approved by the Institutional Animal Use and Care Committee at University of North Dakota, and are in close agreement with the National Institutes of Health guideline for the care and use of laboratory animals.
In vivo 5-bromodeoxyuridine (BrdU) labeling
BrdU at 50 mg/kg (Sigma, St. Louis, MO) was intraperitoneally (IP) administrated for three days to adult Bi-Tg and age-matched pNes-Tg mice, twice daily at 8 hour intervals. Brains were dissected out 3 days after the last injection of BrdU and processed for BrdU immunohistochemical staining as described in the following section.
LacZ staining, immunohistochemical staining, image analysis and quantification
The prefrontal cortex, hippocampus and lateral ventricle of the mouse brain were used to analyze the organization and distribution of NPCs in response to neuron degeneration. For LacZ staining, sections (12 μm) were incubated in 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) solution for 16 hours at room temperature as previously described (14,15). For immunohistochemical staining, sections were first antigen unmasked in 0.01 M citric acid, pH 6.0, 95°C, 10 minutes, followed by incubating in blocking buffer (10% goat serum/0.4% triton X-100 in 1×PBS, pH 7.5) for 1 hour at room temperature. Primary antibody was then added to the blocking buffer (1:250) and sections were incubated at 4 °C overnight. The next day, sections were washed 5 times (5 min each) in 1×PBST (pH 7.5, containing 0.2% tween-20), followed by incubation with specific fluorescein-conjugated secondary antibody for 2 hours at room temperature. After extensive washes, sections were covered with anti-fade medium and sealed for fluorescent microscopic analysis. For negative control staining, sections were incubated without primary antibody.
All images were collected and analyzed with a Nikon fluorescent microscope 85I (Nikon, Japan) equipped with the Spot digital camera (Diagnostic Instruments, Sterling Heights, MI) and Photoshop software (Adobe Systems, San Jose, CA).
Quantifications and Statistical Analysis
Quantifications of LacZ positive, LacZ and NeuN positive, LacZ and BrdU positive cells in the hippocampus were manually counted. A total of 8 sections (every tenth section) between Bregma −1.70mm and −2.66mm of the mouse brain stereotaxic coordinates were counted with fluorescent microscope, and the image was displayed on a computer monitor. This method of quantification for LacZ (NPCs) analysis eliminates the potential false positive effect that may be caused by the background. The total number of cells in the specific volume of the hippocampal regions was calculated using the following formula: T = (Sum N1-8) ×10; where T represents the total number of cells in the specific volume; Sum represents the summation; N1-8 represents sections 1 through 8 of every 10th section (Average ± SD; n=3 mice/group). For analysis of the distribution of NPCs and neurogenesis in the prefrontal cortex, sections at Bregma 2.80mm regions were made. The number of cells per section is the average of 10 consecutive sections in the regions (Average ± SD; n=3 mice/group). For quantification of NPC distribution in the lateral ventricle, Image J was used to derive the relative LacZ staining intensity. The relative intensity is the average of 10 consecutive sections at Bregma −1.82 mm regions (Average ± SD; n=3 mice/group).
Statistical analysis of NPC distribution and neurogenesis in the specific anatomical regions of the brain in the Bi-Tg mice compared with age-matched littermate control pNes-Tg mice (Average ± SD; n= 3 mice/group) was performed using the paired Student t test. P < 0.05 was considered significant.
Results
Aβ plaque onset and progression in the brain of AD-like (Bi-Tg) mice
Similar to pPDGF-APPSw,Ind Tg mice, Bi-Tg mice have age-dependent Aβ plaque onset and progression in the hippocampus and neocortex, which is associated with neurocognitive decline (32,33). No Aβ plaques were detected in the CNS of the control pNes-Tg mice and in the 2 months of age Bi-Tg mice (Figure 1). The onset of Aβ plaques begins at 8 months of age in the Bi-Tg mice (Figure 1). By 12 months of age, large numbers of Aβ plaques were accumulated in the hippocampus (Figure 1) and neocortex (data not shown) in the Bi-Tg mice (33,35). Based on kinetic formation of Aβ plaques, we defined Bi-Tg mice at 2, 8, and 12 months of age as the stages of Aβ plaque free, Aβ plaque onset, and Aβ plaque progression respectively.
Figure 1.
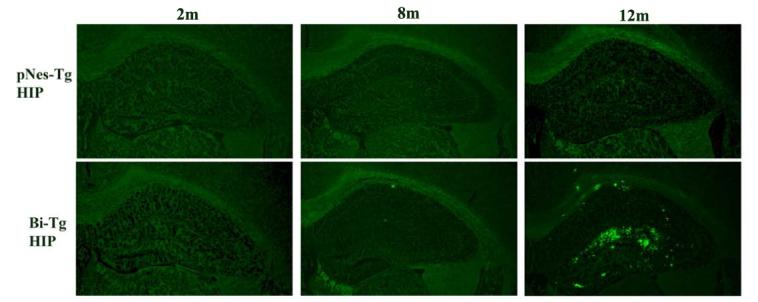
Aβ plaque formation and progression in the AD-like transgenic mice. Representative IHC images demonstrating Bi-Tg mice have age-dependent Aβ plaque onset and progression in the hippocampus compared with age-matched pNes-Tg mice.
Effects of Aβ plaques on alteration of NPCs in the hippocampus and other brain regions
We focused on the three defined stages, i.e., Aβ plaque free, Aβ plaque onset and Aβ plaque progression, to study the effects of Aβ plaques on the alteration of NPCs in the Bi-Tg mouse model mimicking AD. We demonstrated that the number of NPCs in the dentate gyrus of Bi-Tg mice was slightly increased at 2 months of age, compared to the age-matched control pNes-Tg mice (Figure 2A and 2B). However, the number of NPCs in the dentate gyrus of Bi-Tg mice was significantly decreased at 8 and 12 months of ages compared to that of the age-matched control pNes-Tg mice (Figure 2A and 2B).
Figure 2.
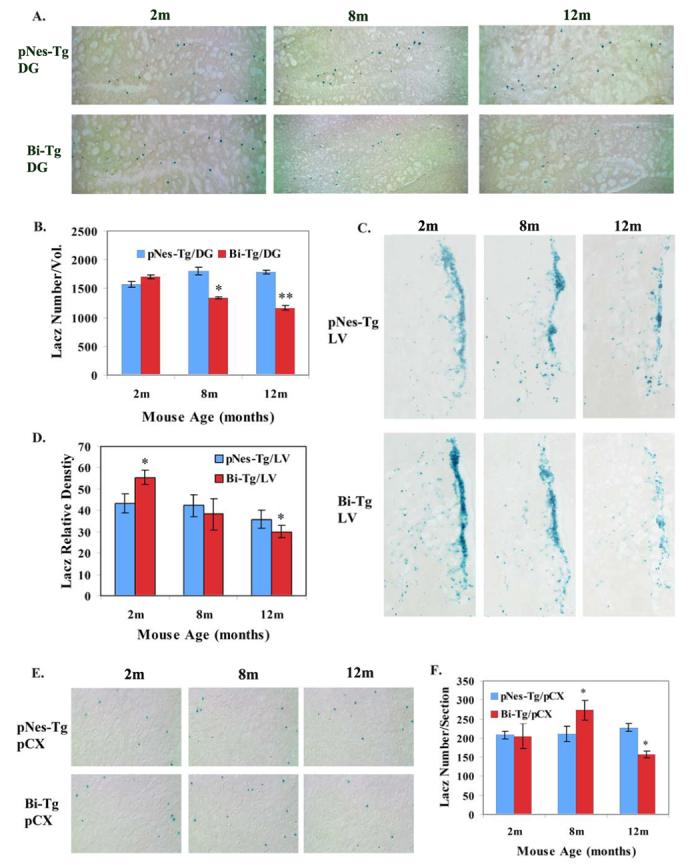
The effects of Aβ plaques on the distribution and organization of NPCs in the brain of AD-like transgenic mice. A. Representative LacZ staining images demonstrating the number of NPCs in the dentate gyrus (DG) of Bi-Tg mice at 2, 8 and 12 months of age (m) compared to that of the age-matched control pNes-Tg mice. B. The total number of NPCs in the DG (−1.70 to −2.66 from Bregma) of the Bi-Tg mice at 2, 8 and 12 months of age compared to that of the age-matched control pNes-Tg mice (* p<0.05; ** p<0.005). C. Representative LacZ staining images demonstrating the distribution of NPCs in the lateral ventricle regions in the Bi-Tg mice at 2, 8 and 12 months of age compared to age-matched normal control (pNes-Tg) mice. D. The relative staining intensity in the lateral ventricle regions of Bi-Tg mice at 2, 8 and 12 months of age (m) compared to age-matched pNes-Tg mice (*p< 0.05). E. Representative LacZ staining images demonstrating the distribution of NPCs in the prefrontal cortex of Bi-Tg mice at 2, 8 and 12 months of age compared to age-matched normal control (pNes-Tg) mice. F. The total number of NPCs in the prefrontal cortex of Bi-Tg mice at 2, 8 and 12 months of age compared to age-matched pNes-Tg mice (*p< 0.05).
We took the same approach to analyze the response of NPCs to Aβ plaques in the lateral ventricles and prefrontal cortex. We demonstrated an increase of NPCs at the Aβ plaque free stage (2 months of age mice), and a decrease of NPCs at the Aβ plaque progression stage (12 months of age) (Figure 2C and 2D). As mice are aging, NPCs in the lateral ventricles are reduced in both normal and AD-like mice (Figure 2C and 2D). In the prefrontal cortex, we showed that an increase of NPCs at Aβ plaque onset stage, while a decrease of NPCs at Aβ plaque progression stage (Figure 2E and 2F).
Effects of Aβ plaques on the survival, proliferation and differentiation of NPCs
We carried out several experiments to study the effects of Aβ plaques on the fate of NPCs in the hippocampus. Since the number of NPCs is reduced at the stages of Aβ plaque onset and progression in the hippocampus, we first performed double staining with LacZ and active caspase 3 (Figure 3), LacZ and TUNEL (data not shown) respectively to examine whether the formation of Aβ plaques is associated with apoptotic death of NPCs. We did not detect significant co-localization of apoptotic markers with NPCs in the hippocampus (Figure 3), prefrontal cortex (data not shown) and lateral ventricle (data not shown) of the 8 and 12 months of age Bi-Tg mice (Figure 3), suggesting that Aβ plaque onset and progression do not induce apoptotic death of NPCs (Figure 3).
Figure 3.
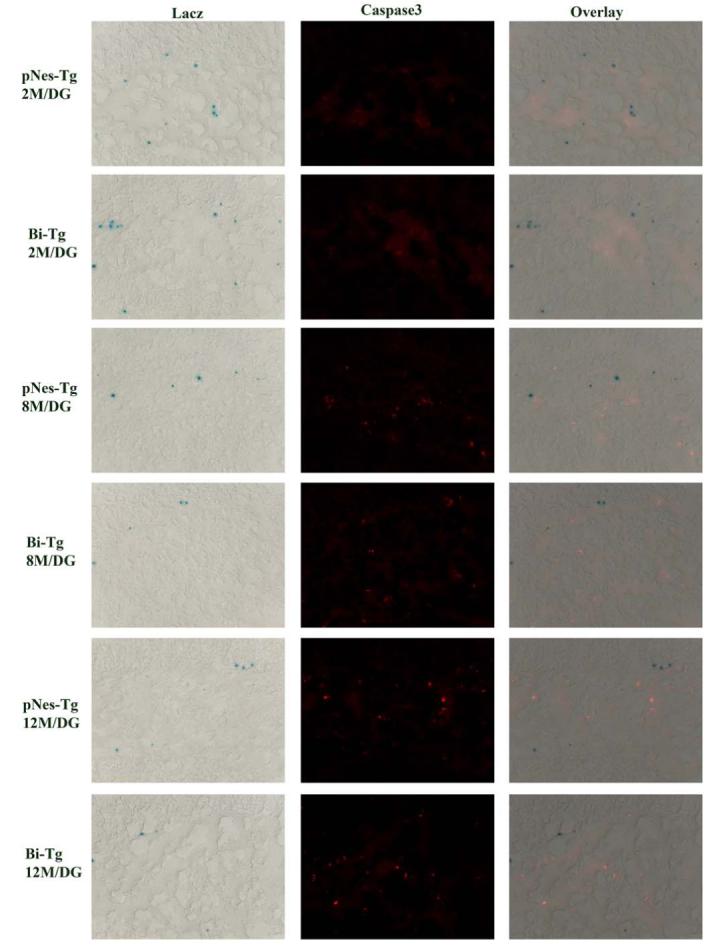
Analysis of caspase 3-mediated apoptosis of NPCs in the DG of AD-like transgenic mice. Representative IHC images showing that there is no co-localization of NPCs with active caspase 3.
We next examined whether Aβ plaques affect the proliferative capacity of NPCs in situ. We administered BrdU at 50 mg/kg through intraperitoneal (IP) injection twice a day for 3 days. Three days after the last BrdU administration, we processed the brain samples to analyze the effects of Aβ plaques on NPC proliferation. We showed that NPCs identified by LacZ-positive cells in the dentate gyrus of the hippocampal regions are not proliferative, since LacZ staining and BrdU-labeling are not co-localized (Figure 4A). Using the BrdU labeling approach, we also demonstrated that Aβ plaques alter cell proliferative status in the hippocampal regions. Compared with the age-matched controls, we observed a slight increase in the number of BrdU-positive cells at Aβ plaque free stage, a significant increase in the number of BrdU-positive cells at the Aβ plaque onset stage, and a decrease in the number of BrdU-positive cells at Aβ plaque progression stage in the dentate gyrus (Figure 4A and 4B). The effects of Aβ plaque on cell proliferation in the subgranular zone and the outer portion of the granular cell layer of the dentate gyrus were shown in Figure 4C. Again, we did not detect significant numbers of proliferative NPCs in the regions of the hippocampus (Figure A, and data not shown). In contrast, we detected that some of the NPCs in lateral ventricle are actively proliferative (Figure 4D and 4E).
Figure 4.
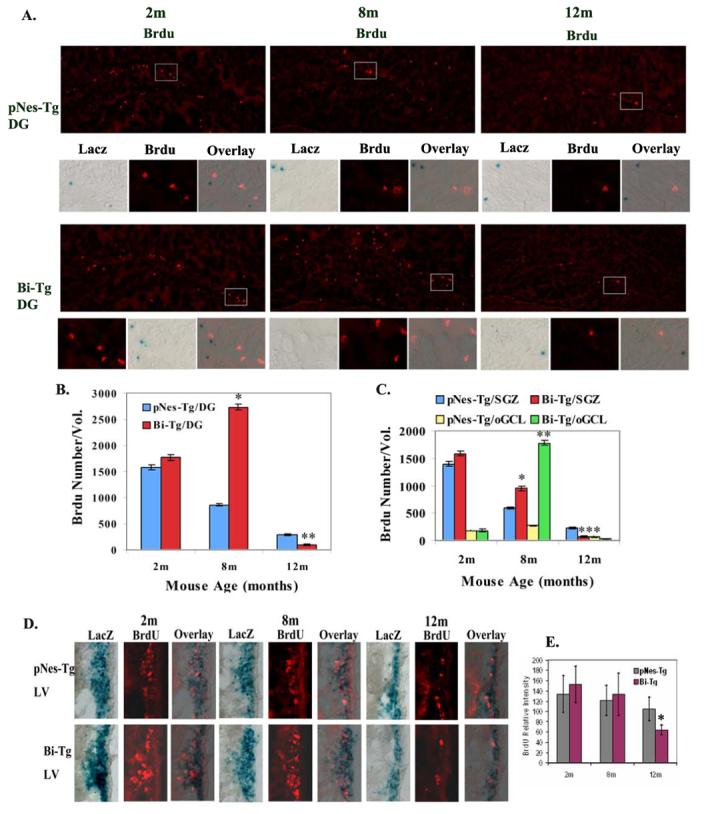
Proliferation of NPCs in the brain of AD-like transgenic mice. A. Representative BrdU and LacZ staining images demonstrating that the majority of NPCs in the dentate gyrus (DG) of Bi-Tg and age-matched control pNes-Tg mice at 2, 8 and 12 months of age (m) are not co-localized. B. The total number of BrdU-positive cells in the DG (−1.70 to −2.66 from Bregma) of the Bi-Tg mice at 2, 8 and 12 months of age compared to that of the age-matched control pNes-Tg mice (* p<0.01; ** p<0.05;). C. The total number of BrdU-positive cells in the subgranular zone (SGZ) and the outer portion of the granular cell layer (oGCL) (−1.70 to −2.66 from Bregma) of the Bi-Tg mice at 2, 8 and 12 months of age compared to that of the age-matched control pNes-Tg mice (* p<0.01; ** p<0.05; ***p<0.05). D. Representative BrdU and LacZ staining images demonstrating that some of NPCs in the lateral ventricle (LV) of Bi-Tg and age-matched control pNes-Tg mice at 2, 8 and 12 months of age (m) are co-localized with BrdU, suggesting NPCs in the LV are proliferative. E. The relative BrdU staining intensity in the lateral ventricle regions of Bi-Tg mice at 2, 8 and 12 months of age (m) compared to age-matched pNes-Tg mice (*p< 0.05).
Furthermore, we analyzed the effects of Aβ plaques on the differentiation of NPCs in the hippocampal regions. For analysis of neurogenesis, we performed the double staining with LacZ and NeuN (Figure 5A). We demonstrated a slight increase of neurogenesis at Aβ plaque free stage, and a significant increase of neurogenesis at Aβ plaque onset and progression stages in the dentate gyrus and CA1 regions of the Bi-Tg mice compared with age-matched control pNes-Tg mice (Figure 5A-5C).
Figure 5.
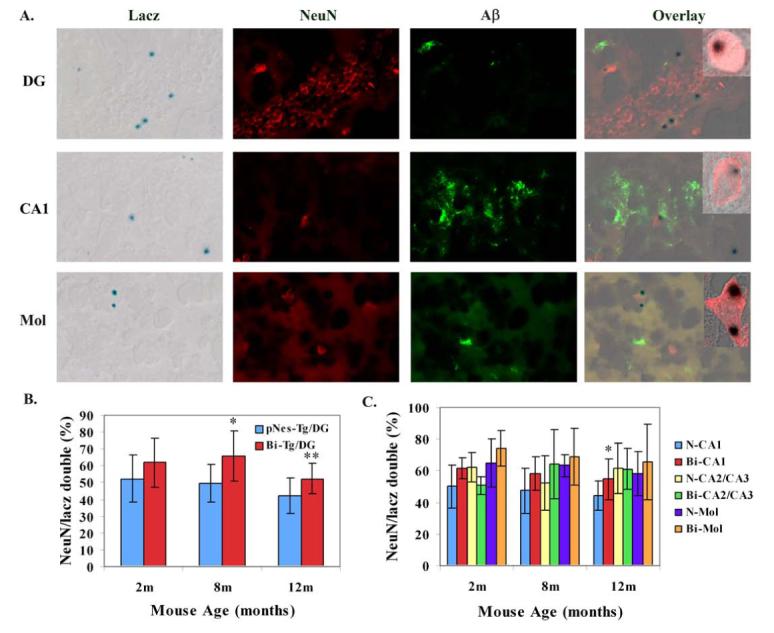
The effects of Aβ plaques on differentiation of NPCs in the hippocampus of AD-like transgenic mice. A. Representative LacZ, NeuN, Aβ staining and overlay images demonstrating some of the NPCs are co-localized with NeuN in the DG, CA1 and molecular layer (Mol) of Bi-Tg mice (inserts were confocal images showing the co-localization of LacZ and NeuN). B. The percentage of NeuN-positive cells out of NPCs in the DG in the Bi-Tg mice at 2, 8 and 12 months of age compared to control pNes-Tg mice (* p<0.05; ** p<0.01). C. The percentage of NeuN-positive cells out of NPCs in the CA1, CA2/CA3, and Molecular layer in the Bi-Tg mice at 2, 8 and 12 months of age compared to control age-matched control pNes-Tg mice (*p<0.05).
For analysis of gliogenesis from NPCs in the hippocampus, we performed a double staining using LacZ and GFAP. We did not detect significant gliogenesis from NPCs in the hippocampal regions (Figure 6A). On the other hand, we did detect gliogenesis from NPCs at the lateral ventricles (Figure 6B).
Figure 6.
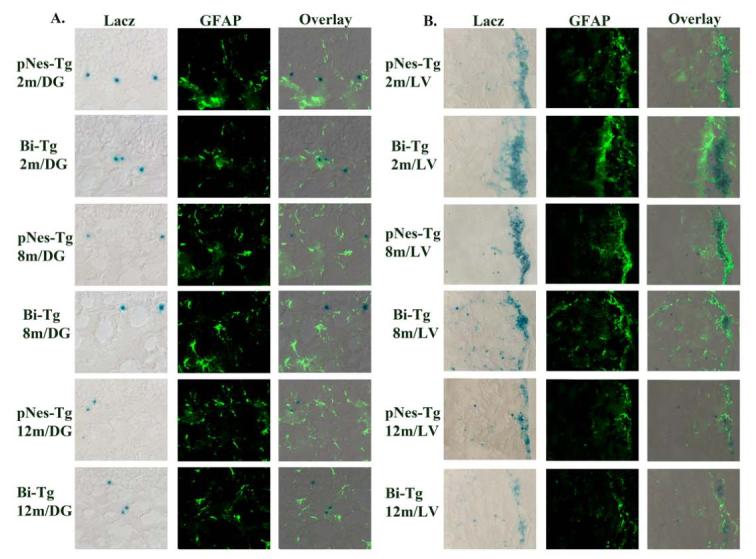
The effects of Aβ plaques on gliogenesis of NPCs in the hippocampus and lateral ventricle of AD-like transgenic mice. A. Representative LacZ, GFAP staining and overlay images in the DG of Bi-Tg mice compared with age-matched pNes-Tg mice controls. No overlay of LacZ and GFAP staining was detected in the hippocampal regions. B. Representative LacZ, GFAP staining and overlay images in the lateral ventricles of Bi-Tg mice compared with age-matched pNes-Tg mice controls. Overlay of LacZ and GFAP staining was detected in the lateral ventricle regions.
Discussion
Though it has been well established that neurogenesis is present in the adult mammalian CNS (4-9), the effects of Aβ plaques on NPCs and on de novo neurogenesis (and/or gliogenesis) from NPCs in AD patients during disease onset and progression remain largely unknown. This may be largely due to the unavailability of tissues at disease free, onset and progression stages in AD patients along with normal controls. For this reason, we utilized Bi-Tg mice that contain both pPDGF-APPSw,Ind and pNes-LacZ transgenes to investigate the effects of Aβ plaques on neurogenesis. Selection of the Bi-Tg transgenic mouse model for this study was primarily based on the following two major characteristics: 1). Bi-Tg mice, like pPDGF-APPSw,Ind Tg mice, develop an age-dependent Aβ plaque formation and accumulation that are associated with neurocognitive dysfunction, which recapitulates some etiological and pathological features of AD patients (31); 2). Bi-Tg mice, like pNes-Tg mice carrying LacZ reporter gene under the control of the nestin second-intron enhancer, are the well-established animal model for identification and characterization of NPCs in the CNS (14,15,29,30). Together, the advantages of the Bi-Tg mouse model system allowed us to define the temporal responses of NPCs and neurogenesis/gliogenesis from NPCs to Aβ plaques in relation to AD-like disease onset and progression. The present study using the Bi-Tg mice with the age-matched control pNes-Tg mice demonstrated two major findings: 1). Aβ plaques modulate the tempospatial distribution of NPCs in the hippocampus and other anatomical regions of the adult mouse brain; 2). Aβ plaques enhance de novo neurogenesis, but not gliogenesis from NPCs in the hippocampus. In addition, the previous and current studies also established that the NPCs identified by the LacZ reporter in the subventricular zone of the adult CNS are proliferative, while the NPCs presented outside of the subventricular zone are not at the actively proliferative state (Figure 4) (34). To the best of our knowledge, this is the first report defining the temporal responses of the post-mitotic NPCs in the hippocampus to Aβ plaques in the mouse model mimicking human AD. However, whether the organization of NPCs and neurogenesis from NPCs in the AD-like mouse model truly reflect the activity of NPCs in the CNS of human AD patients remains to be established. Nevertheless, the results from this study provide compelling evidence to suggest that NPCs are good targets for neurogenic therapy of AD to improve neurocognitive dysfunction induced by Aβ plaques.
In light of the current findings with the Bi-Tg mice, we demonstrated an increase of neurogenesis in the hippocampus, particularly at the Aβ plaque onset and progression stages. We reason that the increased neurogenesis is a compensatory response to Aβ plaque-mediated neurocognitive dysfunction. Changes in neurogenesis have been observed in specific anatomical regions of the adult brain in the animal models associated with neurological and psychiatric disorders, such as stroke (16,18), PD(34), and epilepsy (22). An increase of neurogenesis in the hippocampus of human AD patients was reported with immature neuronal staining (23). Similarly, an increase of neurogenesis was shown in transgenic mice mimicking human AD (20). On the other hand, a decrease of hippocampal neurogenesis was reported in several transgenic mouse models of AD (28). Apparently, the differences reported on the neurogenesis in the transgenic mouse models of AD may be due to the methodological approaches used in identification and characterization of neurogenesis. For example, the BrdU labeling dose and duration, and the animal ages were demonstrated to attribute to the different outcomes of neurogenesis observed (35,36). For this reason, we took both the BrdU-dependent and BrdU-independent approaches to analyze the effects of Aβ aggregation on NPC distribution and neurogenesis in the adult mouse CNS. Our study in contrast with previous reports focused on neurogenesis from post-mitotic NPCs which are present throughout the CNS. In addition, we chose three stages corresponding to Aβ plaque free, plaque onset and plaque progression to define the effects of Aβ plaques on neurogenesis. We demonstrated diverse responses of NPCs to Aβ plaques, depending on age of the Aβ-borne animals and the anatomical regions of the brain. For example, Aβ plaque formation and accumulation significantly decrease the number of NPCs in the dentate gyrus of the hippocampus (Figure 2A and 2B). On the other hand, Aβ plaque formation increases, while Aβ plaque accumulation decreases the numbers of NPCs in the prefrontal cortex (Figure 2E and 2F). The changes of NPCs and neurogenesis in the anatomical regions of the Bi-Tg mouse model compared to age-matched controls might reflect the clinical and pathological manifestation in AD, since the different brain regions were disproportionally affected (37,38). In addition, the changes of NPCs and neurogenesis in the hippocampus to Aβ plaques may also associate with the alterations of neuropathophysiology (31-33). Nevertheless, the cause/effect of the alterations on NPCs and neurogenesis in relation to neuropathophysiology by Aβ plaques remain to be established.
The increase of neurogenesis from NPCs at Aβ plaque onset and progression stages suggests Aβ plaques can promote differentiation of NPCs. More recently, an in vivo study showed that Aβ influences the fate of NPCs, driving their differentiation toward a neuronal direction (39). We observed a decrease of NPCs in the hippocampus at the Aβ plaque onset and progression stages. Since we did not detect significant cell death of NPCs (Figure 2) and gliogenesis from NPCs in the hippocampus (Figure 6), we reason that the decreased NPCs are most likely due to the elevation of migration, which leads to NPC distribution and re-organization, and the increase of neurogenesis, which turns NPCs into neuron-like cells (Figure 5A-5C).
One interesting but unresolved issue in the current study is the lineage relationships between LacZ-positive NPCs and BrdU-positive cells in the hippocampus. The majority of the NPCs defined by LacZ staining that are present outside of the subventricular zone are not at an actively proliferative state (Figure 4A-4C), but at highly differentiating state (Figure 5). About 60% NPCs are co-localized with NeuN in the hippocampal region of AD-like (Bi-Tg) mice (Figure 5B). Aβ plaques enhance the differentiation of NPCs toward neuronal direction (Figure 5B-5C). Previous reports have shown that BrdU positive cells can differentiate into neuron-like cells (5,20,28). More significantly, Aβ plaques elicit alterations of the neurogenic status from BrdU-positive cells (20,28). It is likely that post-mitotic NPCs and mitotically active (BrdU-positive) cells represent two different lineages, both of which can differentiate into neurons in response to Aβ plaques. Thus, neurogenesis in the hippocampus consists of at least two components, one is derived from post-mitotic NPCs which may mobilize an early and acute response (Figure 5). On the other hand, another neurogenic component is derived from BrdU-positive cells (20,28), which provide the long-lasting response (source) for regeneration. In the later event, some of proliferative NPCs may never reach the maturity in the journey of proliferation, migration and differentiation (40). Together, the extensive existence of NPCs in the CNS makes them good targets for neuronal regeneration. To a large extent, experimental approaches to enhance neurogenesis aiming to improve neurocognitive function would contribute significantly to the effective therapy of AD.
Acknowledgements
This study was supported in part by NIH NS45829, and HL75034 Grants and ND EPSCORE Grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Metsaars WP, Hauw JJ, van Welsem ME, Duyckaerts C. A grading system of Alzheimer disease lesions in neocortical areas. Neurobiol Aging. 2003;24:563–572. doi: 10.1016/s0197-4580(02)00134-3. [DOI] [PubMed] [Google Scholar]
- 2.Hayashi H, Kimura N, Yamaguchi H, et al. A seed for Alzheimer amyloid in the brain. J Neurosci. 2004;24:4894–4902. doi: 10.1523/JNEUROSCI.0861-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selkoe DJ. Alzheimer's disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J Alzheimers Dis. 2001;3:75–80. doi: 10.3233/jad-2001-3111. [DOI] [PubMed] [Google Scholar]
- 4.Temple S. The development of neural stem cells. Nature. 2001;414:112–117. doi: 10.1038/35102174. [DOI] [PubMed] [Google Scholar]
- 5.van PH, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature. 2002;415:1030–1034. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bjorklund A, Lindvall O. Cell replacement therapies for central nervous system disorders. Nat Neurosci. 2000;3:537–544. doi: 10.1038/75705. [DOI] [PubMed] [Google Scholar]
- 7.Cameron HA, McKay R. Stem cells and neurogenesis in the adult brain. Curr Opin Neurobiol. 1998;8:677–680. doi: 10.1016/s0959-4388(98)80099-8. [DOI] [PubMed] [Google Scholar]
- 8.Horner PJ, Gage FH. Regeneration in the adult and aging brain. Arch Neurol. 2002;59:1717–1720. doi: 10.1001/archneur.59.11.1717. [DOI] [PubMed] [Google Scholar]
- 9.Gage FH. Neurogenesis in the adult brain. J Neurosci. 2002;22:612–613. doi: 10.1523/JNEUROSCI.22-03-00612.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rietze R, Poulin P, Weiss S. Mitotically active cells that generate neurons and astrocytes are present in multiple regions of the adult mouse hippocampus. J Comp Neurol. 2000;424:397–408. [PubMed] [Google Scholar]
- 11.Greenberg DA, Jin K. Regenerating the brain. Int Rev Neurobiol. 2007;77:1–29. doi: 10.1016/S0074-7742(06)77001-5. [DOI] [PubMed] [Google Scholar]
- 12.Gould E, Reeves AJ, Graziano MS, Gross CG. Neurogenesis in the neocortex of adult primates. Science. 1999;286:548–552. doi: 10.1126/science.286.5439.548. [DOI] [PubMed] [Google Scholar]
- 13.Lie DC, Dziewczapolski G, Willhoite AR, Kaspar BK, Shults CW, Gage FH. The adult substantia nigra contains progenitor cells with neurogenic potential. J Neurosci. 2002;22:6639–6649. doi: 10.1523/JNEUROSCI.22-15-06639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ke Y, Chi L, Xu R, Luo C, Gozal D, Liu R. Early response of endogenous adult neural progenitor cells to acute spinal cord injury in mice. Stem Cells. 2006;24:1011–1019. doi: 10.1634/stemcells.2005-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chi L, Ke Y, Luo C, et al. Motor neuron degeneration promotes neural progenitor cell proliferation, migration, and neurogenesis in the spinal cords of amyotrophic lateral sclerosis mice. Stem Cells. 2006;24:34–43. doi: 10.1634/stemcells.2005-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakatomi H, Kuriu T, Okabe S, et al. Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell. 2002;110:429–441. doi: 10.1016/s0092-8674(02)00862-0. [DOI] [PubMed] [Google Scholar]
- 17.Monje ML, Mizumatsu S, Fike JR, Palmer TD. Irradiation induces neural precursor-cell dysfunction. Nat Med. 2002;8:955–962. doi: 10.1038/nm749. [DOI] [PubMed] [Google Scholar]
- 18.Jin K, Minami M, Xie L, et al. Ischemia-induced neurogenesis is preserved but reduced in the aged rodent brain. Aging Cell. 2004;3:373–377. doi: 10.1111/j.1474-9728.2004.00131.x. [DOI] [PubMed] [Google Scholar]
- 19.Klein SM, Behrstock S, McHugh J, et al. GDNF delivery using human neural progenitor cells in a rat model of ALS. Hum Gene Ther. 2005;16:509–521. doi: 10.1089/hum.2005.16.509. [DOI] [PubMed] [Google Scholar]
- 20.Jin K, Galvan V, Xie L, et al. Enhanced neurogenesis in Alzheimer's disease transgenic (PDGF-APPSw,Ind) mice. Proc Natl Acad Sci U S A. 2004;101:13363–13367. doi: 10.1073/pnas.0403678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crespel A, Rigau V, Coubes P, et al. Increased number of neural progenitors in human temporal lobe epilepsy. Neurobiol Dis. 2005;19:436–450. doi: 10.1016/j.nbd.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 22.Mazarati A, Lu X, Shinmei S, Badie-Mahdavi H, Bartfai T. Patterns of seizures, hippocampal injury and neurogenesis in three models of status epilepticus in galanin receptor type 1 (GalR1) knockout mice. Neuroscience. 2004;128:431–441. doi: 10.1016/j.neuroscience.2004.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin K, Peel AL, Mao XO, et al. Increased hippocampal neurogenesis in Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:343–347. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Curtis MA, Penney EB, Pearson AG, et al. Increased cell proliferation and neurogenesis in the adult human Huntington's disease brain. Proc Natl Acad Sci U S A. 2003;100:9023–9027. doi: 10.1073/pnas.1532244100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curtis MA, Penney EB, Pearson J, Dragunow M, Connor B, Faull RL. The distribution of progenitor cells in the subependymal layer of the lateral ventricle in the normal and Huntington's disease human brain. Neuroscience. 2005;132:777–788. doi: 10.1016/j.neuroscience.2004.12.051. [DOI] [PubMed] [Google Scholar]
- 26.Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS, Mattson MP. Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer's disease. J Neurochem. 2002;83:1509–1524. doi: 10.1046/j.1471-4159.2002.01267.x. [DOI] [PubMed] [Google Scholar]
- 27.Haughey NJ, Liu D, Nath A, Borchard AC, Mattson MP. Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid beta-peptide: implications for the pathogenesis of Alzheimer's disease. Neuromolecular Med. 2002;1:125–135. doi: 10.1385/NMM:1:2:125. [DOI] [PubMed] [Google Scholar]
- 28.Donovan MH, Yazdani U, Norris RD, Games D, German DC, Eisch AJ. Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer's disease. J Comp Neurol. 2006;495:70–83. doi: 10.1002/cne.20840. [DOI] [PubMed] [Google Scholar]
- 29.Aoki Y, Huang Z, Thomas SS, et al. Increased susceptibility to ischemia-induced brain damage in transgenic mice overexpressing a dominant negative form of SHP2. FASEB J. 2000;14:1965–1973. doi: 10.1096/fj.00-0105com. [DOI] [PubMed] [Google Scholar]
- 30.Mitsuhashi T, Aoki Y, Eksioglu YZ, et al. Overexpression of p27Kip1 lengthens the G1 phase in a mouse model that targets inducible gene expression to central nervous system progenitor cells. Proc Natl Acad Sci U S A. 2001;98:6435–6440. doi: 10.1073/pnas.111051398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsia AY, Masliah E, McConlogue L, et al. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.German DC, Yazdani U, Speciale SG, Pasbakhsh P, Games D, Liang CL. Cholinergic neuropathology in a mouse model of Alzheimer's disease. J Comp Neurol. 2003;462:371–381. doi: 10.1002/cne.10737. [DOI] [PubMed] [Google Scholar]
- 33.Reilly JF, Games D, Rydel RE, et al. Amyloid deposition in the hippocampus and entorhinal cortex: quantitative analysis of a transgenic mouse model. Proc Natl Acad Sci U S A. 2003;100:4837–4842. doi: 10.1073/pnas.0330745100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shan X, Chi L, Bishop M, et al. Enhanced de novo neurogenesis and dopaminergic neurogenesis in the substantia nigra of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson's disease-like mice. Stem Cells. 2006;24:1280–1287. doi: 10.1634/stemcells.2005-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao M, Momma S, Delfani K, et al. Evidence for neurogenesis in the adult mammalian substantia nigra. Proc Natl Acad Sci U S A. 2003;100:7925–7930. doi: 10.1073/pnas.1131955100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frielingsdorf H, Schwarz K, Brundin P, Mohapel P. No evidence for new dopaminergic neurons in the adult mammalian substantia nigra. Proc Natl Acad Sci U S A. 2004;101:10177–10182. doi: 10.1073/pnas.0401229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imbimbo BP, Lombard J, Pomara N. Pathophysiology of Alzheimer's disease. Neuroimaging Clin N Am. 2005;15:727–53. doi: 10.1016/j.nic.2005.09.009. ix. [DOI] [PubMed] [Google Scholar]
- 38.Gotz J, Streffer JR, David D, et al. Transgenic animal models of Alzheimer's disease and related disorders: histopathology, behavior and therapy. Mol Psychiatry. 2004;9:664–683. doi: 10.1038/sj.mp.4001508. [DOI] [PubMed] [Google Scholar]
- 39.Calafiore M, Battaglia G, Zappala A, et al. Progenitor cells from the adult mouse brain acquire a neuronal phenotype in response to beta-amyloid. Neurobiol Aging. 2006;27:606–613. doi: 10.1016/j.neurobiolaging.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 40.Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ. Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci. 1999;2:260–265. doi: 10.1038/6365. [DOI] [PubMed] [Google Scholar]