

Summary
Blood transfusion therapy is life-saving for patients with β-thalassaemia and sickle cell disease (SCD), but often results in severe iron overload. This pilot study examined whether the biomarkers of tissue injury or inflammation differ in these two diseases. Plasma malondialdehyde (MDA) was significantly increased 1.8-fold in thalassaemia relative to control patients. In contrast, MDA in SCD was not significantly different from controls. In multivariate analysis, the strongest predictors of elevated MDA were liver iron concentration (P < 0.001) and specific diagnosis (P = 0.019). A significant 2-fold elevation of non-transferrin bound iron (NTBI) was observed in thalassaemia relative to SCD. NTBI was not a significant predictor of high MDA in multivariate analysis. SCD patients showed a significant 2.2-fold elevation of the inflammatory marker interleukin (IL)-6 relative to controls, and a 3.6- and 1.7-fold increase in IL-5 and IL-10 relative to thalassaemia. Although α-tocopherol was significantly decreased by at least 32% in both thalassaemia and SCD, indicating ongoing oxidant stress and antioxidant consumption, γ-tocopherol, a nitric oxide-selective antioxidant, was increased 36% in SCD relative to thalassaemia. These results demonstrate that thalassaemia patients have increased MDA and circulating NTBI relative to SCD patients and lower levels of some cytokines and γ-tocopherol. This supports the hypothesis that the biology of SCD may show increased inflammation and increased levels of protective antioxidants compared with thalassaemia.
Keywords: iron overload, thalassaemia, sickle cell disease, oxidative stress, inflammation, cytokines, non-transferrin bound iron, malondialdehyde, lipid peroxidation, vitamin E
Although the prognosis for patients with thalassaemia and sickle cell disease (SCD) has greatly improved in recent decades with the use of modern newborn screening, prophylactic penicillin (and the availability of newer antibiotics), blood transfusions and iron chelation therapy, patients continue to be at high risk for iron overload and iron-induced toxicities. Iron-related cardiac disease remains the most common cause of death in thalassaemia (Zurlo et al, 1989; Borgna-Pignatti et al, 1998). In addition, over 70% of patients with thalassaemia suffer from primary or secondary amenorrhoea, hypogonadism, osteoporosis and other endocrine disorders (Bronspiegel-Weintrob et al, 1990). However, transfused patients with SCD have a conspicuous absence of iron overload-related organ injury (Finch et al, 1982; Wood et al, 2004). In pilot data from a recent study, Vichinsky et al 2005a confirmed this finding and demonstrated that the strongest predictors of organ injury in thalassaemia were duration of iron exposure and the specific diagnosis.
Biomarkers of oxidative damage are increased in both thalassaemia and SCD (Rachmilewitz et al, 1976; Repka & Hebbel, 1991). In spite of the iron overload in both diseases, oxidants originate from sources other than the iron loaded tissues. For example, in β-thalassaemia the excess unpaired α-haemoglobin chains denature and autoxidise, contributing to increased oxidants, ineffective erythropoiesis, haemolysis and shortened erythrocyte survival (Scott et al, 1993). Malondialdehyde (MDA), a product of lipid peroxidation and protein carbonyls, representing oxidation of the circulating proteins, are elevated in thalassaemia (Graziano et al, 1976; Snyder et al, 1981; Ramenghi et al, 1989; Shalev et al, 1995; Cighetti et al, 2002). On the other hand, SCD haemoglobin can bind to the RBC membrane and act as a Fenton reagent, increasing the generation of oxidants such as superoxide and hydroxyl radical (Repka & Hebbel, 1991). Further sources of oxidants unique to SCD vasculature include xanthine oxidase released from the liver, nitric oxide and secondary oxides of nitrogen (Aslan et al, 2001, 2003; Aslan & Freeman, 2004, Aslan et al, 2000) and endothelial NADPH oxidase (Wood et al, 2005). These increased oxidants and intravascular haemoglobin from haemolysis (Reiter et al, 2002) may contribute to the consumption of nitric oxide in SCD (Aslan et al, 2001). In turn, low levels of nitric oxide may result in haemodynamic instability (Aslan et al, 2001) and reduction of antioxidant capacity (Gladwin et al, 2003). Additionally, some studies have shown increased levels of plasma MDA in SCD (Jain et al, 1990; Sess et al, 1992; Sertac et al, 1997). However, it is unknown whether MDA levels differ in these two iron overload diseases that share similar chelation treatments.
To protect against oxidative damage, the body has endogenous defence mechanisms that are supported by dietary antioxidants. The lipid-soluble vitamin E (tocopherols) antioxidants, including α- and γ-tocopherol, are an important front line defence (Sess et al, 1992; Christen et al, 1997; Jiang et al, 2002; Jiang & Ames, 2003; Kassab-Chekir et al, 2003; Ruhl & Everhart, 2003). These antioxidants are better known as oxygen radical scavengers. However, γ-tocopherol, the primary form of vitamin E in the USA diet, has recently been shown to have a unique role in preventing oxidant damage (Christen et al, 1997; Jiang & Ames, 2003). In addition to functioning as an oxygen radical scavenger, γ-tocopherol scavenges reactive nitric oxide species and inhibits prostaglandin E2-mediated inflammation (Jiang et al, 2000, 2001, 2002; Jiang & Ames, 2003). γ-Tocopherol, in contrast to α-tocopherol, is increased in response to inflammatory stimuli (Himmelfarb et al, 2003). It is unknown whether γ-tocopherol levels differ in these two diseases or contribute to differences in oxidative damage.
Sickle cell disease is well recognised as a chronic inflammatory disease (Belcher et al, 2000, 2003; Klings & Farber, 2001; Jison et al, 2004). Patients with SCD in crisis show elevated tumour necrosis factor-α (TNF-α) and interleukin-6 (IL-6) compared with steady state SCD. In addition, IL-1, IL-6 and interferon-γ (IFN-γ) are elevated in steady state SCD compared with controls. It is unknown from these studies how iron overload will affect the levels of chronic inflammation in SCD. Elevations of the pro-inflammatory cytokines TNF-α and IL-2 have also been demonstrated in iron overloaded patients with thalassaemia (Del Vecchio et al, 2002). These cytokines returned to normal after treatment with the iron chelator deferiprone (L1) (Del Vecchio et al, 2002). Although it is true that oxidant stress can stimulate pro-inflammatory responses in both diseases, different patterns of cytokine and immuno-modulator expression have been described and therefore it is unknown whether the two diseases develop similar inflammatory responses.
The present pilot study examined whether biomarkers of oxidant injury (such as MDA), evidence for inflammation [C-reactive protein (CRP) and cytokines] and antioxidant levels (α- and γ-tocopherol) differ in SCD and thalassaemia.
Methods
Study design
Patients with thalassaemia or SCD and a history of receiving chronic transfusion therapy were recruited from the hematology clinic at the Children’s Hospital and Research Center Oakland (CHRCO) at the time of a clinically indicated liver biopsy for evaluation of iron overload. Healthy controls were recruited from CHRCO staff and their families. The study received Institutional Review Board approval. Consent for adults and assent for children were obtained in accordance with the CHRCO Institutional Review Board guidelines. Twenty-eight chronically transfused haemoglobinopathy patients were studied (eight or more transfusions per year). Case subjects included 17 β-thalassaemia patients [7 male, age: 23.7 ± 8.6 years (mean ± standard deviation (SD), Table I)], 11 SCD patients [seven male, age: 12.7 ± 3.7 years (mean ± SD)] and nine healthy controls [six male, age: 28.2 ± 11.4 years (mean ± SD); serum ferritin <300 μg/l; eight Caucasian, one Asian]. Clinical information including demographics and history of transfusion, chelation, and therapy were obtained by interview and chart review.
Table I.
Demographics.
| Parameter | Thalassaemia | SCD | Control | P value |
|---|---|---|---|---|
| Age (years) | 23.6 ± 8.6 (17) (a) | 12.7 ± 3.7 (11) (b) | 26.5 ± 12.3 (9) (a) | 0.002* |
| Height (cm) | 154.2 ± 9.1 (16) (a) | 150.4 ± 23.8 (10) (a) | 164.8 ± 21.8 (9) (a) | 0.207* |
| Weight (kg) | 52.3 ± 8.9 (17) (a) | 45.4 ± 20.2 (11) (a) | 62.2 ± 26.1 (9) (a) | 0.125* |
| BMI (kg/m2) | 22.3 ± 4.7 (16) (a) | 19.1 ± 3.8 (10) (a) | 21.7 ± 4.6 (9) (a) | 0.201* |
| Sex M/F | 7/10 (a) | 7/4 (a) | 4/5 (a) | 0.489† |
| Splenectomy, n | 12/17 (67%) (a) | 3/11 (27%) (b) | – | 0.025† |
| Chelation, n¶ | 16/17 (94%) (a) | 7/11 (64%) (a) | – | 0.062‡ |
| Transfusion, years | 16.1 ± 8.6 (17) (a) | 4.6 ± 2.9 (11) (b) | – | <0.001§ |
| HCV, n | 5/17 (29%) (a) | 0 (0%) (a) | – | 0.125‡ |
Values are means ± SD. Number of patients analysed are shown in parentheses (n) to the right of the SD. Within a row groups sharing the same lowercase letter are not significantly different from each other (5% procedure wise error rate). BMI, body mass index; HCV, hepatitis C virus; SCD, sickle cell disease.
The P value was obtained from an anova.
The P value was obtained from a chi-square test.
The P value was obtained from a Fisher’s exact test.
The P value was obtained from a two-sample t-test.
Chelation was by deferoxamine.
Laboratory analyses
A blood sample (45 ml) was collected from subjects in the morning at least 2 h postprandial; subjects were advised to abstain from taking any medications (including chelation)/vitamin or mineral supplements for the previous 24 h. The study design specified that blood from transfused subjects be collected as long as possible after transfusion (i.e. in the period just proceeding the next transfusion). The mean days between transfusion and phlebotomy were 20.7 ± 10.4 d after the last transfusion. Blood was transported at room temperature to the analytical laboratory within 20 min of venepuncture for cell isolation and oxidation analysis. Serum, plasma and cells were separated by centrifugation.
Malondialdehyde (MDA)
Malondialdehyde was assayed in duplicate 250 μl plasma samples, using gas chromatography-mass spectrometry (GC–MS, model 5888; Agilent Technologies, Palo Alto, CA, USA), as previously described (Yeo et al, 1994, 1999). Two hundred micro molar diethylenetriaminepentaacetate and 2.5 mmol/l butylated hydroxytoluene (BHT) were added to prevent sample oxidation during analysis. Samples were spiked with 1 μmol/l 2H2-MDA as an internal standard. To hydrolyse protein-bound MDA, 10 μl of 6.6 N H2SO4 was added for 10 min at room temperature. MDA was derivatised to pentafluorophenylhydrazine at room temperature for 1 h. Derivatised MDA was extracted with isooctane, and 70 μl was injected into the GC–MS for detection.
Vitamin E
Plasma α-tocopherol and γ-tocopherol were analysed as previously described (Jiang et al, 2002; Jiang & Ames, 2003). Briefly, tocopherols were extracted using a mixture of methanol/hexane (2:5, v/v) in the presence of 0.8 mmol/l BHT added to prevent oxidative losses during work-up. Tocopherols were separated on a 150 × 4.6 mm, 5 μm Supelcosil™ LC-18-DB column (Supelco, Bellefonte, PA, USA) and monitored by coulometric detection (Model Coulochem II; ESA Inc., Chelmsford, MA, USA) using a Model 5011 analytical cell (Jiang et al, 2002; Jiang & Ames, 2003).
Non-transferrin bound iron (NTBI)
Non-transferrin bound iron was determined by nitrilotriacetic acid (NTA) capture of NTBI and quantified by high performance liquid chromatography (HPLC) (Singh et al, 1990). This assay uses a large excess of a low affinity ligand NTA, which removes and complexes low molecular mass iron and iron non-specifically bound to serum proteins. Iron bound to transferrin, ferritin, desferrioxamine, and its metabolites is not accessed by the NTA ligand. The Fe–NTA complex present in the ultrafiltrate represents the NTBI and is then quantified using an HPLC procedure including on-column derivatisation with a high affinity iron chelator (3-hydroxy-1-propyl-2-methyl-pyridin-4-one). Using this method, normal individuals had negative NTBI values. A negative NTBI value, a common finding by several groups independent of the method used to measure NTBI (Singh et al, 1990; Porter et al, 1996; Bradley et al, 1997; Gosriwatana et al, 1999; Wickramasinghe et al, 1999; Jacobs et al, 2005), arises when the transferrin saturation in samples is less than 50% (Jacobs et al, 2005). Negative NTBI values can be caused when transferrin is less than 50% saturated and iron NTA loads rapidly onto transferrin. Hence, normal control samples, which contain around 30% saturated transferrin, bind iron bound to NTA and thus appear negative with respect to the experimental zero iron controls used with standards to generate calibration curves where no transferrin is present.
C-reactive protein
Plasma high sensitivity C-reactive protein (hsCRP) was determined in plasma samples by a nephelometric method utilising latex particles coated with (CRP) monoclonal antibodies by Quest Diagnostics Inc., San Juan Capistrano, CA, USA.
Cytokines
Plasma levels of 10 cytokines (IL-1β, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, granulocyte–macrophage colony-stimulating factor (GM-CSF), IFN-γ and TNF-α) were determined in samples by a multiplex antibody bead assay by BioSource International, Camarillo, CA, USA.
Liver iron
Tissue iron concentration was determined by inductively coupled plasma mass spectrometry (ICP-MS, Mayo, Rochester, NY, USA) as described previously (Vichinsky et al, 2005a). Liver biopsies were collected within an average of 15 d (mean 14.5 ± 20.5) of the phlebotomy for the study. Three samples were excluded from these analyses because they were collected more than 2 months from the blood sample.
Blood and liver function tests
Alanine transaminase (ALT) and ferritin were measured in the clinical lab at CHRCO. Serum ferritin concentrations were measured by a one step immunoassay (FERR Flex®; Dade Behring Inc., Newark, DE, USA). Serum ALT concentrations were measured on the Dimension® clinical chemistry system (Dade Behring Inc.). The manufacturer’s reference range for the normal general population is 30–65 U/l (n = 244 adults). Ferritin and ALT are presented as the mean values obtained during the 6 month prior to the study phlebotomy. Hepatitis C RNA was identified by polymerase chain reaction.
Percentage transferrin saturation
This was determined on serum samples using the urea gel method of Evans and Williams (1978). Twenty-five micro litres of serum were treated with an excess of rivanol to remove most of the serum proteins excluding transferrin. After centrifugation, samples of the supernatants were run on polyacrylamide gels containing 6 mol/l urea. Under these partially denaturating conditions, the four different species of transferrin, namely apo-, diferric-, C- and N-terminal monoferric, denatured to differing extents and were therefore separated on the gel, which enabled quantification using densitometry from which the per cent saturation was calculated.
Protein carbonyls
Oxidative damage to plasma proteins were assayed as previously described by measuring carbonyl groups on plasma proteins by reaction with 2,4-dinitrophenylhydrazine to form a hydrazine complex, which was measured spectrophotometrically (Houglum et al, 1990, 1997).
Diet
The Block Food Frequency Questionnaire (Block, 1998) was completed by a majority of subjects to obtain a semi-quantitative assessment of usual food intake over the previous 12 months. The Block Food Frequency Questionnaire has been validated in adult (Sinha et al, 1993) and paediatric populations (Block et al, 1995) and is used commonly in epidemiological trials of oxidative stress.
Statistics
Data are reported as mean ± SD. Comparisons among groups were made using one-way analysis of variation (anova) analyses, where P < 0.05 was considered statistically significant. Post hoc tests were done by Tukey’s analysis at a 5% procedure-wise error rate. As MDA and ferritin values were right skewed, the analyses on these two variables were performed on log-transformed data. Logistic regression was used to create multivariate models to describe relationships between oxidative injury (MDA, ALT) and factors that may influence their development (i.e. diagnosis, duration of chronic transfusion, liver iron concentration, NTBI or age). Other variables were also examined as potential confounders, such as serum ferritin and hepatitis C virus (HCV) and were found to be non-significant in the model.
Results
Demographics
The demographics of the study populations are shown in Table I. No significant differences were seen in sex, height, weight, body mass index, chelation or percentage of HCV infection, among the study groups. Duration of chronic transfusion was significantly longer in thalassaemia patients (P < 0.001) and they were significantly older than the SCD group (P = 0.002). Splenectomy (P = 0.025) was more common in thalassaemia patients, but there was no significant difference in chelation treatment (deferioxamine, P = 0.062) compared with SCD patients.
The caloric intake was not different among the three groups (approximately 2000 ± 1000 kcal) and no difference was observed in total fat, protein, carbohydrate, calcium, iron, vitamin A, C, E and D intake between groups (including supplements).
Comparison of iron burden
Parameters of iron burden are shown in Table II. Although both thalassaemia and SCD groups had evidence of severe haemosiderosis by quantitative liver iron measurements, these were higher in SCD than thalassaemia patients in this study population (15.0 ± 5.6 vs. 9.2 ± 7.2 mg/g dry tissue weight, P = 0.033, Table II). Serum ferritin and transferrin saturation showed similar high elevation in both thalassaemia and SCD patients relative to controls. Despite higher tissue iron burden, SCD patients had significantly lower NTBI than thalassaemia patients (Table II).
Table II.
Measurements of iron status.
| Parameter | Thalassaemia | SCD | Control | anovaP value |
|---|---|---|---|---|
| Liver iron (mg/g dw) | 9.2 ± 7.2 (16) (b) | 15.0 ± 5.6 (11) (a) | 0.033 | |
| Ferritin (μg/l) | 1868 ± 1042 (17) (a) | 2515 ± 1153 (11) (a) | 65.0 ± 70.9 (9)(b) | <0.001 |
| Transferrin saturation (%) | 82.5 ± 21.7 (6) (a) | 64.5 ± 23.3 (4) (a,b) | 40.2 ± 13.1 (5) (b) | 0.014 |
| NTBI (μmol/l) | 4.0 ± 1.6 (15) (a) | 1.9 ± 2.1 (11) (b) | −1.0 ± 0.4 (9) (c) | <0.001 |
Values are means ± SD. Number of patients analysed are shown in parentheses (n) to the right of the SD. Within a row, groups not sharing the same letter are significantly different from each other (5% procedure wise error rate).
dw, dry weight; NTBI, non-transferrin bound iron; SCD, sickle cell disease.
Oxidative injury and inflammation
In thalassaemia patients, MDA levels were significantly higher than controls. This difference was not observed for SCD patients (MDA values were log-transformed prior to analysis; Table III). Further analysis of MDA comparing it with liver iron concentration in both disease patient groups showed a positive correlation between liver iron concentration and plasma MDA (Fig 1; thalassaemia, R2 = 0.695, P = 0.003 and SCD, R2 = 0.644, P = 0.032). Plasma α-tocopherol was significantly reduced in both thalassaemia and SCD patients relative to controls. In contrast, γ-tocopherol levels were significantly higher in SCD compared with thalassaemia patients as well as controls [Table III, the control group was in the range found for normal healthy controls used in other studies (Himmelfarb et al, 2003)]. The dietary questionnaire found no difference in intake of γ- or α-tocopherol. Protein carbonyls, a measure of oxidative damage to amino acid side chains, were not significantly changed in either group in comparison to controls.
Table III.
Measurements of oxidative injury and inflammation.
| Parameter | Thalassaemia | SCD | Control | anovaP value |
|---|---|---|---|---|
| MDA, nmol/l* | 35.3 ± 20.0 (17) (a) | 26.1 ± 9.7 (11) (a,b) | 19.3 ± 13.4 (9) (b) | 0.006 |
| α-tocopherol, μmol/l | 14.8 ± 4.6 (17) (b) | 15.7 ± 4.1 (11) (b) | 22.7 ± 3.4 (9) (a) | <0.001 |
| γ-tocopherol, μmol/l | 3.3 ± 2.1 (17) (b) | 5.5 ± 1.4 (11) (a) | 1.8 ± 0.7 (9) (c) | <0.001 |
| Protein carbonyls, nmol/mg | 0.7 ± 0.3 (16) (a) | 0.6 ± 0.1 (11) (a) | 0.6 ± 0.1 (9) (a) | 0.454 |
| ALT, U/l | 57.5 ± 31.1 (17) (a) | 36.1 ± 11.0 (11) (b) | 34.9 ± 7.7 (9) (b) | 0.020 |
| hsCRP, mg/l | 1.0 ± 1.2 (16) (a) | 2.7 ± 3.2 (10) (a) | 0.9 ± 0.7 (9) (a) | 0.061 |
| Cytokines: pg/ml | ||||
| IL-5 | 0.3 ± 0.3 (13) (b) | 1.0 ± 0.6 (10) (a) | 0.6 ± 0.6 (8) (a,b) | 0.010 |
| IL-6 | 3.1 ± 1.7 (15) (a,b) | 4.5 ± 2.2 (11) (a) | 2.0 ± 1.0 (8) (b) | 0.014 |
| IL-10 | 2.1 ± 1.2 (17) (b) | 3.5 ± 1.6 (11) (a) | 1.8 ± 1.0 (9) (b) | 0.007 |
Values are means ± SD. Number of patients analysed are shown in parentheses (n) to the right of the SD. Within a row, groups not sharing the same letter are significantly different from each other (5% procedure wise error rate).
MDA values were log-transformed prior to analysis.
ALT, alanine transaminase; IL, interleukin; hsCRP, high sensitivity C-reactive protein; MDA, malondialdehyde; SCD, sickle cell disease.
Fig 1.
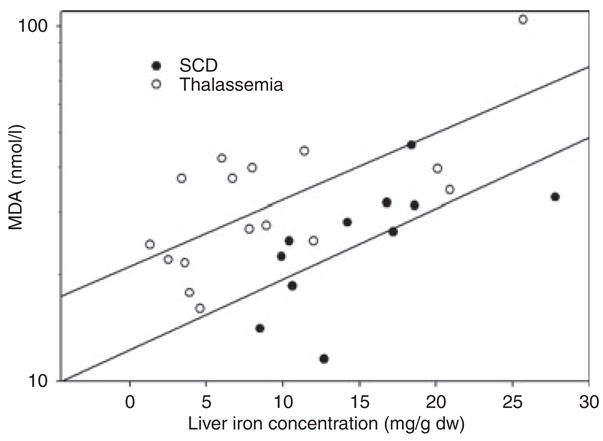
Regression analysis comparing plasma malondialdehyde (MDA) (nmol/l, log scale) with liver iron concentration [mg/g dry weight (dw)]. Regressions for thalassaemia (top; R = 0.695, P = 0.003) and sickle cell disease (bottom; R = 0.644, P = 0.032), show that, in both diseases, higher liver iron concentrations are associated with more plasma MDA.
Multivariate analysis using log MDA as the dependent variable representing oxidant injury was employed to examine the pattern of several of the following relationships simultaneously (Table IV). This analysis examined as co-variants (i.e. predictors): diagnosis, liver iron concentration, duration of chronic transfusion and age. Two significant predictors of MDA were found, diagnosis of thalassaemia and quantitative liver iron (P < 0.001 and =0.019 respectively). Age was not a significant predictor in multivariate analysis (Table IV). ALT, another marker of tissue injury was significantly increased (1.7-fold, P = 0.020) in the thalassaemia group relative to the SCD group (Table III). Further multivariate analysis, with ALT as the dependent variable, and liver iron concentration and diagnosis as independent variables in the model, found that diagnosis of thalassaemia was a predictor of higher ALT (P = 0.025, data not shown).
Table IV.
Oxidant injury analysis model using two groups (thalassaemia and SCD).
| Bivariate (crude)
|
Multivariate (adjusted)
|
|||
|---|---|---|---|---|
| P-value | P-value | Regression coefficient | 95% confidence intervals | |
| MDA (dependent variable, nmol/l) R2 for model =0.55, n = 26 | ||||
| Diagnosis [thalassaemia (reference) versus SCD] | 0.185 | 0.019 | −0.481 | −0.870, −0.091 |
| Liver iron concentration (mg/g) | 0.012 | <0.001 | +0.045 | 0.024, 0.067 |
| Duration of chronic transfusion (years) | 0.199 | 0.641 | +0.005 | −0.016, 0.026 |
| Age (years) | 0.041 | 0.697 | +0.004 | −0.017, 0.025 |
MDA values were log transformed prior to analysis.
MDA, malondialdehyde; SCD, sickle cell disease.
Markers of inflammation including hsCRP and 10 cytokines were compared with each of the groups (Table III). A 2.5-fold higher hsCRP level was observed in SCD compared with thalassaemia patients, which showed a trend toward statistical significance (P = 0.061; Table III). The inflammatory cytokines IL-5, IL-6 and IL-10 were highest in SCD patients, with IL-6 and IL-10 being significantly different from controls, while IL-5 and IL-10 were also significantly elevated in SCD compared with thalassaemia patients. There was no significant difference between the levels of IL-5, IL-6 and IL-10 in thalassaemia and control patients (Table III). There were no significant differences among groups for the other cytokines examined (GM-CSF, IFN-γ, IL-1β, IL-2, IL-4, IL-8, and TNF-α, data not shown).
Discussion
Emerging clinical data suggest that iron overloaded SCD patients are at less risk for organ injury than patients with thalassaemia. Wood et al 2004 recently reported increased cardiac iron deposition in chronically transfused haemoglobinopathy patients. Decreased cardiac function only occurred in thalassaemia patients, not in iron overloaded SCD patients (Wood et al, 2004). In a recent study involving 30 thalassaemia and 43 SCD patients with severe haemosiderosis, we confirmed that greater cardiac and endocrine disease occurred in the thalassaemia patients (Vichinsky et al, 2005a). Although the specific role of iron overload in mediating injury is the primary focus of this paper, the unique pathophysiologies of SCD and thalassaemia certainly play a critical role in promoting the observed pathology. Specifically, in SCD the tissue injury related to sickling of RBCs and in thalassaemia the surplus of α-globin chains and intramedullary ineffective erythropoiesis are certainly important factors. Also important is the enhanced gastrointestinal iron absorption seen thalassaemia patients that may initiate different iron trafficking compared with SCD. Clearly, disease diagnosis is important; in fact, multivariate analysis showed disease diagnosis of thalassaemia to be a significant predictor of MDA and this variable may be closely related to these distinct underlying fundamental pathophysiologies. Furthermore, concerning the ethnicity difference between SCD and thalassaemia; we cannot discount ethnicity being a factor. However, while this is true, the incidence and prevalence of iron-induced organ injury was not influenced by the ethnicity of the patients in multiple large thalassaemia trials (Cunningham et al, 2004; Vichinsky et al, 2005b). In order to investigate the relationship of iron overload and specific disease to injury, we examined biomarkers of oxidative stress, inflammation and tissue injury. However, while each class of biomarker is traditionally thought to represent one of these processes, it is important to note that they are not distinct entities and have considerable interaction, i.e. oxidative stress can initiate tissue injury and/or inflammation.
Plasma malondialdehyde was studied as a marker of tissue injury and oxidative stress. MDA is a well-recognised biomarker of lipid peroxidation, but its measurement has been limited by the specificity of the methodology. The present study utilises GC–MS, a technique that avoids problems of oxidation during processing (as may be seen in thiobarbituric acid reactive substance analysis) and specifically measures MDA (Liu et al, 1997). Another factor we have taken into account is artifactual oxidation during the measurement of MDA because of the high NTBI in our samples. We took great care to prevent any possible enhanced MDA during the preparation of our samples by adding chelators and antioxidants. Regression analysis revealed that higher liver iron concentrations are associated with more plasma MDA in both disease groups. Further multivariate analysis identified specific disease (thalassaemia) and liver iron concentration as significant predictors of high MDA. In animal models, high liver iron levels induce elevation of lipid peroxides and oxidants (Knutson et al, 2000; Walter et al, 2002) presumably through iron-initiated Fenton chemistry. Increased lipid peroxidation markers have previously been observed in thalassaemia patients (Livrea et al, 1998; Cighetti et al, 2002; Laksmitawati et al, 2003). However, MDA studies in SCD have been largely focused on in vitro studies of sickle-cell erythrocyte membranes (Buchanan & Holtkamp, 1981; Hebbel & Miller, 1984; Jain & Shohet, 1984; Jain et al, 1989, 1990; Sertac et al, 1997). This is the first study to compare oxidative injury with iron overloaded SCD and thalassaemia patients.
Duration of chronic transfusion and NTBI seem to be likely candidates as predictors of MDA, because duration of transfusion indicates the time the iron stress has been endured and NTBI estimates the possible availability of free iron. However, neither was a predictor of MDA in multivariate analysis. Concerning duration of chronic transfusion, elevated MDA is probably a real-time marker of oxidative injury (Kadiiska et al, 2005a,b) and indicative of current liver iron concentration. Duration of chronic transfusion correlates better with cumulative tissue injury (Vichinsky et al, 2005a). We suggest that this may explain why duration of transfusion is not a predictor of MDA in multivariate analysis. NTBI is also not a significant predictor of MDA. This is surprising, because with high levels of both plasma MDA and NTBI, it is the potential free iron in NTBI that could be initiating Fenton chemistry in vivo, which induces the lipid peroxidation that gives increased MDA. However, this is not the case. We suggest that the increased levels of plasma MDA in thalassaemia may result from several mechanisms. First, plasma MDA could be enhanced in thalassaemia patients because it may be dependent on the amount of circulating erythroid precursors and peripheral blood erythrocytes that have a high density of unpaired α-haemoglobin chains (Scott et al, 1993). The excess α-chains in thalassemic red blood cells are unstable and prone to denaturation and oxidation (Scott et al, 1993) and, in the present work, may have contributed more to enhanced plasma MDA than the SCD haemoglobin, which is also capable of producing oxidants (Repka & Hebbel, 1991). The α-chains in thalassemic red blood cells can autoxidise, release haem and generate superoxide (Scott et al, 1993), which can then increase lipid peroxidation and thus enhance MDA levels. Furthermore, plasma MDA may be increased in thalassaemia because of peroxidation of tissues that leak MDA into the plasma. For example, plasma ALT, another marker of tissue injury, was also increased in thalassaemia relative to SCD and is indicative of liver dysfunction and leakage of liver metabolites into the plasma. Parallel to ALT, plasma MDA may also rise partly as a result of possible liver lipid peroxidation and leakage into the plasma. It is an attractive suggestion that MDA may leak from the liver because of its strong correlation to liver iron concentration in multivariate analysis.
We also examined inflammation, a known regulator of iron transport and storage, as another possible explanation for the observed differences in MDA levels, and to determine the important inflammatory differences that may exist between these two diseases. SCD is characterised by microvascular hypoxia-reperfusion leading to inflammation. Human and animal models have found elevated TNF-α, IL-6, IL-10 and hsCRP as markers of inflammation in SCD (Hedo et al, 1993; Singhal et al, 1993; Bank et al, 1998; Bourantas et al, 1998; Dale & Alberio, 1998; Sess et al, 1998; Belcher et al, 2000, 2003; Makis et al, 2000; Barbeau et al, 2001; Ludwiczek et al, 2003; Banerjee & Kuypers, 2004). Our results support these observations. Inflammation (Fillet et al, 1989) and cytokines (IL-10) stimulate the uptake and retention of iron into monocytes and reticuloendothelial (RE) cells (Ludwiczek et al, 2003). Recently, hepcidin that is increased in inflammation, has been identified as a critical participant in this cellular regulation of iron storage (Papanikolaou et al, 2005). In transfused thalassaemia or SCD patients the hepicidin level may vary depending on the severity of anaemia as suggested in a recent study (Kearney et al, 2005). Increased cytokines (such as IL-10) may explain the lower NTBI levels in SCD patients as compared with thalassaemia patients (Ludwiczek et al, 2003). We and others suggest that the NTBI of SCD patients may be taken up by the RE system, sparing the parenchymal tissues of injury (Pippard, 1994). In contrast, the high NTBI in thalassaemia may also contribute to the increased cardiac iron and organ dysfunction observed in thalassaemia patients.
Antioxidant capacity is an important determinant of tissue injury, especially in patients with increased oxidant stress. We have found significant differences in antioxidant capacity in thalassaemia and SCD patients in terms of plasma tocopherol levels. Plasma α-tocopherol was decreased in both thalassaemia and SCD patients in ours and other studies (Rachmilewitz et al, 1976; Sess et al, 1998; De Luca et al, 1999; Kassab-Chekir et al, 2003). Our observed changes in α-tocopherol were not because of variation in dietary intake and are probably related to its consumption as a scavenger of oxidants. These low levels probably contribute to increased MDA levels. In contrast to the decreased α-tocopherol, γ-tocopherol levels were increased in both groups, but levels were significantly higher in the SCD patients. Inflammation has been shown to decrease degradation of γ-tocopherol through inhibition of a cytochrome P450 3A–dependent process (Parker et al, 2000; Sontag & Parker, 2002) and may explain our observed difference. We suggest that the higher levels of γ-tocopherol in SCD may be important in reducing tissue injury related to inflammation and oxidant stress.
Conclusions
Malondialdehyde levels were higher in thalassaemia compared with SCD (conclusions summarised in Table V). The strongest predictors of elevated MDA, by multivariate analysis, were diagnosis and liver iron concentration. In contrast, several inflammatory markers appeared higher in SCD. This inflammation in SCD may generate increased levels of γ-tocopherol, leading to decreased tissue peroxidation and injury. Inflammation may also be contributing to the lower NTBI levels observed in SCD, further protecting against tissue injury by removing a source of circulating labile iron. In summary, we suggest that the unique inflammatory physiology of SCD may protect against organ injury by restricting iron to protected areas within the RE system (Fillet et al, 1989; Ludwiczek et al, 2003) and maintaining protective antioxidants.
Table V.
Model of transfusional iron overload in thalassaemia and SCD (shown are the relative differences).
| Thalassaemia | SCD | |
|---|---|---|
| General tissue changes: | ||
| More organ dysfunction | ||
|
| ||
| Changes in specific biomarkers: | ||
| More MDA (adjusted), ALT, NTBI | More IL-5 and IL-10 | |
| More γ-tocopherol | ||
|
| ||
| Hypothesis: | ||
| Transfusional iron load stored in the liver, heart and endocrine organs. | Transfusional iron load is taken up by activated RE cells keeping iron away from vital organs. | |
| Tissue iron load leads to dysfunction. | γ-tocopherol is higher which may protect. | |
ALT, alanine transaminase; MDA, malondialdehyde; NTBI, non-transferrin bound iron; RE, reticuloendothelial; SCD, sickle cell disease.
Acknowledgments
We gratefully acknowledge Roland Fischer for helping in data analysis, Esther Roitman for assisting in the GC–MS experiments and Annie Lui and Gladys Warr for help in sample preparation and storage. This work has been supported in part by the following: NIH grants R01-DK057778, M01 RR01271, U54HL070583, R01-AT001821, the National Center for Minority Health and Health Disparities Grant P60MD000222 and the National Center for Complementary and Alternative Medicine Research Scientist Award K05 AT001323.
References
- Aslan M, Freeman BA. Oxidant-mediated impairment of nitric oxide signaling in sickle cell disease – mechanisms and consequences. Cellular and Molecular Biology. 2004;50:95–105. [PubMed] [Google Scholar]
- Aslan M, Thornley-Brown D, Freeman BA. Reactive species in sickle cell disease. Annals of the New York Academy of Sciences. 2000;899:375–391. doi: 10.1111/j.1749-6632.2000.tb06201.x. [DOI] [PubMed] [Google Scholar]
- Aslan M, Ryan TM, Adler B, Townes TM, Parks DA, Thompson JA, Tousson A, Gladwin MT, Patel RP, Tarpey MM, Batinic-Haberle I, White CR, Freeman BA. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proceedings of the National Academy of Sciences of the Unites States of America. 2001;98:15215–15220. doi: 10.1073/pnas.221292098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslan M, Ryan TM, Townes TM, Coward L, Kirk MC, Barnes S, Alexander CB, Rosenfeld SS, Freeman BA. Nitric oxide-dependent generation of reactive species in sickle cell disease. Actin tyrosine induces defective cytoskeletal polymerization. Journal of Biological Chemistry. 2003;278:4194–4204. doi: 10.1074/jbc.M208916200. [DOI] [PubMed] [Google Scholar]
- Banerjee T, Kuypers FA. Reactive oxygen species and phosphatidylserine externalization in murine sickle red cells. British Journal of Haematology. 2004;124:391–402. doi: 10.1046/j.1365-2141.2003.04781.x. [DOI] [PubMed] [Google Scholar]
- Bank N, Kiroycheva M, Ahmed F, Anthony GM, Fabry ME, Nagel RL, Singhal PC. Peroxynitrite formation and apoptosis in transgenic sickle cell mouse kidneys. Kidney International. 1998;54:1520–1528. doi: 10.1046/j.1523-1755.1998.00148.x. [DOI] [PubMed] [Google Scholar]
- Barbeau P, Woods KF, Ramsey LT, Litaker MS, Pollock DM, Pollock JS, Callahan LA, Kutlar A, Mensah GA, Gutin B. Exercise in sickle cell anemia: effect on inflammatory and vasoactive mediators. Endothelium. 2001;8:147–155. doi: 10.3109/10623320109165323. [DOI] [PubMed] [Google Scholar]
- Belcher JD, Marker PH, Weber JP, Hebbel RP, Vercellotti GM. Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood. 2000;96:2451–2459. [PubMed] [Google Scholar]
- Belcher JD, Bryant CJ, Nguyen J, Bowlin PR, Kielbik MC, Bischof JC, Hebbel RP, Vercellotti GM. Transgenic sickle mice have vascular inflammation. Blood. 2003;101:3953–3959. doi: 10.1182/blood-2002-10-3313. [DOI] [PubMed] [Google Scholar]
- Block G. Invited commentary: comparison of the Block and the Willett food frequency questionnaires. American Journal of Epidemiology. 1998;148:1160–1161. doi: 10.1093/oxfordjournals.aje.a009601. discussion, 1162–1165. [DOI] [PubMed] [Google Scholar]
- Block G, Norris JC, Mandel RM, DiSogra C. Sources of energy and six nutrients in diets of low-income Hispanic-American women and their children: quantitative data from HHANES, 1982–1984. Journal of the American Dietetic Association. 1995;95:195–208. doi: 10.1016/S0002-8223(95)00048-8. [DOI] [PubMed] [Google Scholar]
- Borgna-Pignatti C, Rugolotto S, De Stefano P, Piga A, Di Gregorio F, Gamberini MR, Sabato V, Melevendi C, Cappellini MD, Verlato G. Survival and disease complications in thalassemia major. Annals of the New York Academy of Sciences. 1998;850:227–231. doi: 10.1111/j.1749-6632.1998.tb10479.x. [DOI] [PubMed] [Google Scholar]
- Bourantas KL, Dalekos GN, Makis A, Chaidos A, Tsiara S, Mavridis A. Acute phase proteins and interleukins in steady state sickle cell disease. European Journal of Haematology. 1998;61:49–54. doi: 10.1111/j.1600-0609.1998.tb01060.x. [DOI] [PubMed] [Google Scholar]
- Bradley SJ, Gosriwitana I, Srichairatanakool S, Hider RC, Porter JB. Non-transferrin-bound iron induced by myeloablative chemotherapy. British Journal of Haematology. 1997;99:337–343. doi: 10.1046/j.1365-2141.1997.4143221.x. [DOI] [PubMed] [Google Scholar]
- Bronspiegel-Weintrob N, Olivieri NF, Tyler B, Andrews DF, Freedman MH, Holland FJ. Effect of age at the start of iron chelation therapy on gonadal function in beta-thalassaemia major. New England Journal of Medicine. 1990;323:713–719. doi: 10.1056/NEJM199009133231104. [DOI] [PubMed] [Google Scholar]
- Buchanan GR, Holtkamp CA. Platelet aggregation, malondialdehyde generation and production time in children with sickle cell anaemia. Thrombosis and Haemostasis. 1981;46:690–693. [PubMed] [Google Scholar]
- Christen S, Woodall AA, Shigenaga MK, Southwell-Keely PT, Duncan MW, Ames BN. Gamma-tocopherol traps mutagenic electrophiles such as NO(X) and complements alphatocopherol: physiological implications. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:3217–3222. doi: 10.1073/pnas.94.7.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cighetti G, Duca L, Bortone L, Sala S, Nava I, Fiorelli G, Cappellini MD. Oxidative status and malondialdehyde in beta-thalassaemia patients. European Journal of Clinical Investigation. 2002;32:55–60. doi: 10.1046/j.1365-2362.2002.0320s1055.x. [DOI] [PubMed] [Google Scholar]
- Cunningham MJ, MacKlin EA, Neufeld EJ, Cohen AR. Complications of beta-thalassemia major in North America. Blood. 2004;104:34–39. doi: 10.1182/blood-2003-09-3167. [DOI] [PubMed] [Google Scholar]
- Dale GL, Alberio L. Is there a correlation between raised erythropoietin and thrombotic events in sickle-cell anaemia? Lancet. 1998;352:566–567. doi: 10.1016/S0140-6736(97)11506-9. [DOI] [PubMed] [Google Scholar]
- De Luca C, Filosa A, Grandinetti M, Maggio F, Lamba M, Passi S. Blood antioxidant status and urinary levels of catecholamine metabolites in beta-thalassaemia. Free Radical Research. 1999;30:453–462. doi: 10.1080/10715769900300491. [DOI] [PubMed] [Google Scholar]
- Del Vecchio GC, Schettini F, Piacente L, De Santis A, Giordano P, De Mattia D. Effects of deferiprone on immune status and cytokine pattern in thalassaemia major. Acta Haematologica. 2002;108:144–149. doi: 10.1159/000064705. [DOI] [PubMed] [Google Scholar]
- Evans RW, Williams J. Studies of the binding of different iron donors to human serum transferrin and isolation of ironbinding fragments from the N- and C-terminal regions of the protein. Biochemical Journal. 1978;173:543–552. doi: 10.1042/bj1730543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillet G, Beguin Y, Baldelli L. Model of reticuloendothelial iron metabolism in humans: abnormal behavior in idiopathic hemochromatosis and in inflammation. Blood. 1989;74:844–851. [PubMed] [Google Scholar]
- Finch CA, Lee MY, Leonard JM. Continuous RBC transfusions in a patient with sickle cell disease. Archives of Internal Medicine. 1982;142:279–282. [PubMed] [Google Scholar]
- Gladwin MT, Schechter AN, Ognibene FP, Coles WA, Reiter CD, Schenke WH, Csako G, Waclawiw MA, Panza JA, Cannon RO., III Divergent nitric oxide bioavailability in men and women with sickle cell disease. Circulation. 2003;107:271–278. doi: 10.1161/01.cir.0000044943.12533.a8. [DOI] [PubMed] [Google Scholar]
- Gosriwatana I, Loreal O, Lu S, Brissot P, Porter J, Hider RC. Quantification of non-transferrin-bound iron in the presence of unsaturated transferrin. Analytical Biochemistry. 1999;273:212–220. doi: 10.1006/abio.1999.4216. [DOI] [PubMed] [Google Scholar]
- Graziano JH, Miller DR, Grady RW, Cerami A. Inhibition of membrane peroxidation in thalassaemic erythrocytes by 2,3-dihydroxybenzoic acid. British Journal of Haematology. 1976;32:351–356. doi: 10.1111/j.1365-2141.1976.tb00938.x. [DOI] [PubMed] [Google Scholar]
- Hebbel RP, Miller WJ. Phagocytosis of sickle erythrocytes: immunologic and oxidative determinants of hemolytic anemia. Blood. 1984;64:733–741. [PubMed] [Google Scholar]
- Hedo CC, Aken’ova YA, Okpala IE, Durojaiye AO, Salimonu LS. Acute phase reactants and severity of homozygous sickle cell disease. Journal of Internal Medicine. 1993;233:467–470. doi: 10.1111/j.1365-2796.1993.tb01000.x. [DOI] [PubMed] [Google Scholar]
- Himmelfarb J, Kane J, McMonagle E, Zaltas E, Bobzin S, Boddupalli S, Phinney S, Miller G. Alpha and gamma tocopherol metabolism in healthy subjects and patients with endstage renal disease. Kidney International. 2003;64:978–991. doi: 10.1046/j.1523-1755.2003.00151.x. [DOI] [PubMed] [Google Scholar]
- Houglum K, Filip M, Witztum JL, Chojkier M. Malondialdehyde and 4-hydroxynonenal protein adducts in plasma and liver of rats with iron overload. Journal of Clinical Investigation. 1990;86:1991–1998. doi: 10.1172/JCI114934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houglum K, Ramm GA, Crawford DH, Witztum JL, Powell LW, Chojkier M. Excess iron induces hepatic oxidative stress and transforming growth factor beta 1 in genetic hemochromatosis. Hepatology. 1997;26:605–610. doi: 10.1002/hep.510260311. [DOI] [PubMed] [Google Scholar]
- Jacobs EM, Hendriks JC, van Tits BL, Evans PJ, Breuer W, Liu DY, Jansen EH, Jauhiainen K, Sturm B, Porter JB, Scheiber-Mojdehkar B, von Bonsdorff L, Cabantchik ZI, Hider RC, Swinkels DW. Results of an international round robin for the quantification of serum non-transferrin-bound iron: need for defining standardization and a clinically relevant isoform. Analytical Biochemistry. 2005;341:241–250. doi: 10.1016/j.ab.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Jain SK, Shohet SB. A novel phospholipid in irreversibly sickled cells: evidence for in vivo peroxidative membrane damage in sickle cell disease. Blood. 1984;63:362–367. [PubMed] [Google Scholar]
- Jain SK, Ross JD, Levy GJ, Little RL, Duett J. The accumulation of malonyldialdehyde, an end product of membrane lipid peroxidation, can cause potassium leak in normal and sickle red blood cells. Biochemical Medicine and Metabolic Biology. 1989;42:60–65. doi: 10.1016/0885-4505(89)90041-8. [DOI] [PubMed] [Google Scholar]
- Jain SK, Ross JD, Levy GJ, Duett J. The effect of malonyldialdehyde on viscosity of normal and sickle red blood cells. Biochemical Medicine and Metabolic Biology. 1990;44:37–41. doi: 10.1016/0885-4505(90)90042-y. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Ames BN. Gamma-tocopherol, but not alphatocopherol, decreases proinflammatory eicosanoids and inflammation damage in rats. FASEB Journal. 2003;17:816–822. doi: 10.1096/fj.02-0877com. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Elson-Schwab I, Courtemanche C, Ames BN. Gamma-tocopherol and its major metabolite, in contrast to alphatocopherol, inhibit cyclooxygenase activity in macrophages and epithelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:11494–11499. doi: 10.1073/pnas.200357097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Christen S, Shigenaga MK, Ames BN. Gammatocopherol, the major form of vitamin E in the US diet, deserves more attention. American Journal of Clinical Nutrition. 2001;74:714–722. doi: 10.1093/ajcn/74.6.714. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lykkesfeldt J, Shigenaga MK, Shigeno ET, Christen S, Ames BN. Gamma-tocopherol supplementation inhibits protein nitration and ascorbate oxidation in rats with inflammation. Free Radical Biology and Medicine. 2002;33:1534–1542. doi: 10.1016/s0891-5849(02)01091-2. [DOI] [PubMed] [Google Scholar]
- Jison ML, Munson PJ, Barb JJ, Suffredini AF, Talwar S, Logun C, Raghavachari N, Beigel JH, Shelhamer JH, Danner RL, Gladwin MT. Blood mononuclear cell gene expression profiles characterize the oxidant, hemolytic, and inflammatory stress of sickle cell disease. Blood. 2004;104:270–280. doi: 10.1182/blood-2003-08-2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadiiska MB, Gladen BC, Baird DD, Germolec D, Graham LB, Parker CE, Nyska A, Wachsman JT, Ames BN, Basu S, Brot N, Fitzgerald GA, Floyd RA, George M, Heinecke JW, Hatch GE, Hensley K, Lawson JA, Marnett LJ, Morrow JD, Murray DM, Plastaras J, Roberts LJ, II, Rokach J, Shigenaga MK, Sohal RS, Sun J, Tice RR, Van Thiel DH, Wellner D, Walter PB, Tomer KB, Mason RP, Barrett JC. Biomarkers of oxidative stress study II: are oxidation products of lipids, proteins, and DNA markers of CCl4 poisoning? Free Radical Biology and Medicine. 2005a;38:698–710. doi: 10.1016/j.freeradbiomed.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Kadiiska MB, Gladen BC, Baird DD, Graham LB, Parker CE, Ames BN, Basu S, FitzGerald GA, Lawson JA, Marnett LJ, Morrow JD, Murray DM, Plastaras J, Roberts LJ, 2nd, Rokach J, Shigenaga MK, Sun J, Walter PB, Tomer KB, Barrett JC, Mason RP. Biomarkers of oxidative stress study: III. Effects of the nonsteroidal anti-inflammatory agents indomethacin and meclofenamic acid on measurements of oxidative products of lipids in CCl4 poisoning. Free Radical Biology and Medicine. 2005b;38:711–718. doi: 10.1016/j.freeradbiomed.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Kassab-Chekir A, Laradi S, Ferchichi S, Haj Khelil A, Feki M, Amri F, Selmi H, Bejaoui M, Miled A. Oxidant, antioxidant status and metabolic data in patients with beta-thalassemia. Clinica Chimica Acta. 2003;338:79–86. doi: 10.1016/j.cccn.2003.07.010. [DOI] [PubMed] [Google Scholar]
- Kearney SL, Nemeth E, Neufeld EJ, Thapa D, Ganz T, Weinstein DA, Cunningham MJ. Urinary hepcidin in congenital chronic anemias. Pediatric Blood & Cancer. 2005 doi: 10.1002/pbc.20616. in press. [DOI] [PubMed] [Google Scholar]
- Klings ES, Farber HW. Role of free radicals in the pathogenesis of acute chest syndrome in sickle cell disease. Respiratory Research. 2001;2:280–285. doi: 10.1186/rr70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson MD, Walter PB, Ames BN, Viteri FE. Both iron deficiency and daily iron supplements increase lipid peroxidation in rats. Journal of Nutrition. 2000;130:621–628. doi: 10.1093/jn/130.3.621. [DOI] [PubMed] [Google Scholar]
- Laksmitawati DR, Handayani S, Udyaningsih-Freisleben SK, Kurniati V, Adhiyanto C, Hidayat J, Kusnandar S, Dillon HS, Munthe BG, Wirawan R, Soegianto RR, Ramelan W, Freisleben HJ. Iron status and oxidative stress in beta-thalassaemia patients in Jakarta. Biofactors. 2003;19:53–62. doi: 10.1002/biof.5520190107. [DOI] [PubMed] [Google Scholar]
- Liu J, Yeo HC, Doniger SJ, Ames BN. Assay of aldehydes from lipid peroxidation: gas chromatography-mass spectrometry compared to thiobarbituric acid. Analytical Biochemistry. 1997;245:161–166. doi: 10.1006/abio.1996.9990. [DOI] [PubMed] [Google Scholar]
- Livrea MA, Tesoriere L, Maggio A, D’Arpa D, Pintaudi AM, Pedone E. Oxidative modification of low-density lipoprotein and atherogenetic risk in beta-thalassemia. Blood. 1998;92:3936–3942. [PubMed] [Google Scholar]
- Ludwiczek S, Aigner E, Theurl I, Weiss G. Cytokine-mediated regulation of iron transport in human monocytic cells. Blood. 2003;101:4148–4154. doi: 10.1182/blood-2002-08-2459. [DOI] [PubMed] [Google Scholar]
- Makis AC, Hatzimichael EC, Mavridis A, Bourantas KL. Alpha-2-macroglobulin and interleukin-6 levels in steady-state sickle cell disease patients. Acta Haematologica. 2000;104:164–168. doi: 10.1159/000046509. [DOI] [PubMed] [Google Scholar]
- Papanikolaou G, Tzilianos M, Christakis JI, Bogdanos D, Tsimirika K, MacFarlane J, Goldberg YP, Sakellaropoulos N, Ganz T, Nemeth E. Hepcidin in iron overload disorders. Blood. 2005;105:4103–4105. doi: 10.1182/blood-2004-12-4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker RS, Sontag TJ, Swanson JE. Cytochrome P4503A-dependent metabolism of tocopherols and inhibition by sesamin. Biochemical and Biophysical Research Communications. 2000;277:531–534. doi: 10.1006/bbrc.2000.3706. [DOI] [PubMed] [Google Scholar]
- Pippard MJ. Secondary iron overload. In: Brock JH, Halliday JW, Pippard MJ, editors. Iron Metabolism in Health and Disease. W.B. Saunders Ltd; London: 1994. pp. 272–300. [Google Scholar]
- Porter JB, Abeysinghe RD, Marshall L, Hider RC, Singh S. Kinetics of removal and reappearance of non-transferrin-bound plasma iron with deferoxamine therapy. Blood. 1996;88:705–713. [PubMed] [Google Scholar]
- Rachmilewitz EA, Shohet SB, Lubin BH. Lipid membrane peroxidation in beta-thalassaemia major. Blood. 1976;47:495–505. [PubMed] [Google Scholar]
- Ramenghi U, David O, Dianzani I, Sacchetti L, Biasi F, Chiarpotto E, Poli G. Analysis of the carbonyl compounds produced in beta thalassaemic erythrocytes by oxidative stress. Haematologica. 1989;74:531–535. [PubMed] [Google Scholar]
- Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO, Jr, Schechter AN, Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nature Medicine. 2002;8:1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- Repka T, Hebbel RP. Hydroxyl radical formation by sickle erythrocyte membranes: role of pathologic iron deposits and cyto-plasmic reducing agents. Blood. 1991;78:2753–2758. [PubMed] [Google Scholar]
- Ruhl CE, Everhart JE. Relation of elevated serum alanine aminotransferase activity with iron and antioxidant levels in the United States. Gastroenterology. 2003;124:1821–1829. doi: 10.1016/s0016-5085(03)00395-0. [DOI] [PubMed] [Google Scholar]
- Scott MD, van den Berg JJ, Repka T, Rouyer-Fessard P, Hebbel RP, Beuzard Y, Lubin BH. Effect of excess alpha-hemoglobin chains on cellular and membrane oxidation in model beta-thalassemic erythrocytes. Journal of Clinical Investigation. 1993;91:1706–1712. doi: 10.1172/JCI116380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sertac A, Bingol F, Aydin S, Uslu A. Peroxidative damage in sickle-cell erythrocyte ghosts: protective effect of allopurinol. General Pharmacology. 1997;28:427–428. doi: 10.1016/s0306-3623(96)00297-2. [DOI] [PubMed] [Google Scholar]
- Sess D, Carbonneau MA, Thomas MJ, Dumon MF, Peuchant E, Perromat A, Le Bras M, Clerc M. First observations on the main plasma parameters of oxidative stress in homozygous sickle cell disease. Bulletin de la Societe de Pathologie Exotique. 1992;85:174–179. [PubMed] [Google Scholar]
- Sess ED, Carbonneau MA, Meite M, Peuchant E, Dumont MF, Receveur MC, Thomas MJ, Perromat A, Sangare A, Le Bras M, Clerc M. Markers of lipid peroxidation, inflammatory proteins and plasma tocopherols in homozygotic and heterozygotic sickle cell anemia. Bulletin de la Societe de Pathologie Exotique. 1998;91:238–241. [PubMed] [Google Scholar]
- Shalev O, Repka T, Goldfarb A, Grinberg L, Abrahamov A, Olivieri NF, Rachmilewitz EA, Hebbel RP. Deferiprone (L1) chelates pathologic iron deposits from membranes of intact thalassemic and sickle red blood cells both in vitro and in vivo. Blood. 1995;86:2008–2013. [PubMed] [Google Scholar]
- Singh S, Hider RC, Porter JB. A direct method for quantification of non-transferrin-bound iron. Analytical Biochemistry. 1990;186:320–323. doi: 10.1016/0003-2697(90)90088-q. [DOI] [PubMed] [Google Scholar]
- Singhal A, Doherty JF, Raynes JG, McAdam KP, Thomas PW, Serjeant BE, Serjeant GR. Is there an acute-phase response in steady-state sickle cell disease? Lancet. 1993;341:651–653. doi: 10.1016/0140-6736(93)90418-g. [DOI] [PubMed] [Google Scholar]
- Sinha R, Patterson BH, Mangels AR, Levander OA, Gibson T, Taylor PR, Block G. Determinants of plasma vitamin E in healthy males. Cancer Epidemiology, Biomarkers and Prevention. 1993;2:473–479. [PubMed] [Google Scholar]
- Snyder LM, Sauberman N, Condara H, Dolan J, Jacobs J, Szymanski I, Fortier NL. Red cell membrane response to hydrogen peroxide-sensitivity in hereditary xerocytosis and in other abnormal red cells. British Journal of Haematology. 1981;48:435–444. doi: 10.1111/j.1365-2141.1981.tb02735.x. [DOI] [PubMed] [Google Scholar]
- Sontag TJ, Parker RS. Cytochrome P450 omega-hydroxylase pathway of tocopherol catabolism. Novel mechanism of regulation of vitamin E status. Journal of Biological Chemistry. 2002;277:25290–25296. doi: 10.1074/jbc.M201466200. [DOI] [PubMed] [Google Scholar]
- Vichinsky EP, Buetensky E, Fung E, Hudes M, Theil E, Ferrell L, Williams R, Louie L, Lee P, Harmatz P. A review comparing organ dysfunction in transfused patients with SCD or B thalassemia. American Journal of Hematology. 2005a;80:70–74. doi: 10.1002/ajh.20402. [DOI] [PubMed] [Google Scholar]
- Vichinsky EP, MacKlin EA, Waye JS, Lorey F, Olivieri NF. Changes in the epidemiology of thalassaemia in North America: a new minority disease. Pediatrics. 2005b;116:e818–e825. doi: 10.1542/peds.2005-0843. [DOI] [PubMed] [Google Scholar]
- Walter PB, Knutson MD, Paler-Martinez A, Lee S, Xu Y, Viteri FE, Ames BN. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:2264–2269. doi: 10.1073/pnas.261708798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe SN, Thein SL, Srichairatanakool S, Porter JB. Determinants of iron status and bilirubin levels in congenital dyserythropoietic anaemia type I. British Journal of Haematology. 1999;107:522–525. doi: 10.1046/j.1365-2141.1999.01745.x. [DOI] [PubMed] [Google Scholar]
- Wood JC, Tyszka JM, Carson S, Nelson MD, Coates TD. Myocardial iron loading in transfusion-dependent thalassemia and sickle cell disease. Blood. 2004;103:1934–1936. doi: 10.1182/blood-2003-06-1919. [DOI] [PubMed] [Google Scholar]
- Wood KC, Hebbel RP, Granger DN. Endothelial cell NADPH oxidase mediates the cerebral microvascular dysfunction in sickle cell transgenic mice. FASEB J. 2005;19:989–991. doi: 10.1096/fj.04-3218fje. [DOI] [PubMed] [Google Scholar]
- Yeo HC, Helbock HJ, Chyu DW, Ames BN. Assay of malondialdehyde in biological fluids by gas chromatography-mass spectrometry. Analytical Biochemistry. 1994;220:391–396. doi: 10.1006/abio.1994.1355. [DOI] [PubMed] [Google Scholar]
- Yeo HC, Liu J, Helbock HJ, Ames BN. Assay of malondialdehyde and other alkanals in biological fluids by gas chromatography-mass spectrometry. Methods in Enzymology. 1999;300:70–78. doi: 10.1016/s0076-6879(99)00115-9. [DOI] [PubMed] [Google Scholar]
- Zurlo MG, De Stefano P, Borgna-Pignatti C, Di Palma A, Piga A, Melevendi C, Di Gregorio F, Burattini MG, Terzoli S. Survival and causes of death in thalassaemia major. Lancet. 1989;2:27–30. doi: 10.1016/s0140-6736(89)90264-x. [DOI] [PubMed] [Google Scholar]