

Abstract
Epidermal growth factor (EGF) receptor (EGFR) modulates mitosis and apoptosis through signaling by its high-affinity (HA) and low-affinity (LA) EGF-binding states. The prevailing model of EGFR activation—derived from x-ray crystallography—involves the transition from tethered ectodomain monomers to extended back-to-back dimers and cannot explain these EGFR affinities or their different functions. Here, we use single-molecule Förster resonant energy transfer analysis in combination with ensemble fluorescence lifetime imaging microscopy to investigate the three-dimensional architecture of HA and LA EGFR-EGF complexes in cells by measuring the inter-EGF distances within discrete EGF pairs and the vertical distance from EGF to the plasma membrane. Our results show that EGFR ectodomains form interfaces resulting in two inter-EGF distances (∼8 nm and < 5.5 nm), different from the back-to-back EGFR ectodomain interface (∼11 nm). Distance measurements from EGF to the plasma membrane show that HA EGFR ectodomains are oriented flat on the membrane, whereas LA ectodomains stand proud from it. Their flat orientation confers on HA EGFR ectodomains the exclusive ability to interact via asymmetric interfaces, head-to-head with respect to the EGF-binding site, whereas LA EGFRs must interact only side-by-side. Our results support a structural model in which asymmetric EGFR head-to-head interfaces may be relevant for HA EGFR oligomerization.
Introduction
The epidermal growth factor (EGF) receptor (EGFR) belongs to the ErbB family of transmembrane receptor tyrosine kinases responsible for the signaling pathways leading to cell proliferation, differentiation, migration, and apoptosis (1–3). A long-standing puzzle regarding EGFR signaling is the underlying mechanism conferring distinct physiological roles on the high-affinity (HA) and low-affinity (LA) receptors detected by Scatchard analysis (4,5). HA EGFRs constitute <5% of cell surface EGFRs in most cell lines (6), and their affinity for EGF appears to be modulated by interactions occurring at the plasma membrane and/or the inner side of the membrane (7,8). HA EGFRs control all the early cell responses to EGF (4). These include inositol phosphate production, release of Ca2+ from intracellular stores, rise in intracellular pH, activation of protein kinase C, induction of the c-Fos protooncogene, and alterations in cell morphology. LA complexes constitute >95% of surface EGFRs and appear to have a role in hyperproliferation and apoptosis (9,10). EGFR therefore can behave as two different receptors—which both bind certain growth factors, such as EGF—but are also selectively activated by other ligands (11).
The bimodal function of EGFR in the cell is not yet understood. Expressed as a single translation product (2), wild-type EGFR is the prototype receptor tyrosine kinase molecule, with an ectodomain that binds EGF with a 1:1 stoichiometry (12,13), connected to its cytoplasmic tyrosine kinase domain by a single membrane-spanning sequence (2). EGFR activation is triggered by the binding of growth factor and is mediated by still poorly understood interreceptor ectodomain interactions, which ultimately bring neighboring receptor kinase domains together to allow allosteric transphosphorylation and the recruitment of cytoplasmic effector molecules (14). A better understanding of the interactions between ectodomains has arisen from x-ray crystal structures of solubilized ectodomain fragments. These have revealed an autoinhibited/tethered monomer, an extended “back-to-back” truncated dimer with two ligands bound at opposite outer sites (hence termed “back-to-back”) and a weaker, asymmetric “head-to-head” dimer with two ligands centrally located between ectodomains (12,13,15). Among the dimers, the back-to-back structure is favored as biologically relevant because it displays a much stronger interface between EGFR monomers (12).
The prevailing model of EGFR activation derived from the tethered and extended back-to-back structures, which involves ligand-mediated EGFR dimerization, has proven insufficient to explain the different EGFR affinities or their signaling functions. It was initially thought that these two structures could explain the high and low ligand-binding affinities of EGFR because in the tethered monomer only one contact is made—between ligand and subdomain I—in the receptor ectodomain, whereas in the dimer each ligand makes contact with both subdomains I and III (12,13). However, the model predicts EGF-binding kinetics that is inconsistent with a wealth of experimental data from cells and has been found to be consistent only with kinetic data from a single LA EGFR population (4,6,16–19). The kinetics of HA EGF-binding are not yet understood (20).
Cell-based evidence also suggests that additional EGFR ectodomain interfaces, other than the back-to-back arrangement, might be involved in EGFR activation. Förster resonance energy transfer (FRET) microscopy, a well-established spectroscopic ruler to measure intermolecular distances in the range 2–10 nm (21), has consistently shown ensemble-averaged FRET between fluorophores bound to the N-termini of EGF ligands bound to cell surface EGFR (22–26). The presence of ensemble-averaged FRET in cells was initially believed to represent the transition from LA EGFR monomers to HA dimers, interpreted in the context of a two-state non-FRET/FRET transition (22,23). However, after the realization that the preferred (back-to-back) crystallographic dimer would show inter-EGF distances of ∼11 nm, i.e., not detectable by FRET, two alternative models of EGFR activation in cells have since been proposed. In one, the presence of FRET in cells was explained by the presence of higher-order oligomers of back-to-back dimers that—in the context of crystallographic data—would be formed by asymmetric head-to-head EGFR ectodomain interactions. These head-to-head interactions would result in short distances between the N-termini of the two bound EGF molecules of <5 nm (25). In the second model, FRET was combined with submicron EGFR aggregation measurements to propose a dimer-to-tetramer transition incorporating an alternative EGFR ectodomain arrangement (24). The tetramers were generated through side-by-side contacts between two adjacent back-to-back dimers, for which there is no crystallographic evidence, with short (∼4 nm) inter-EGF distances. However, the relevance of these models to the activation mechanism of HA EGFRs needs to be investigated because the measurement detection limits required the use of high EGF concentrations (10–50 nM) that resulted in large, nonphysiological fractions (50%–97%) of the LA EGFR population—which constitute >90% of cell surface receptors—being occupied by EGF (24,25,27). Depending on the cell phenotype, environmental conditions, and receptor occupancy, EGFR can initiate different signals through selective activation of its two affinity states (10,11). Correlations have been shown between enhanced HA signaling and tumor invasiveness (28). A better understanding of the nature of the HA and LA EGFRs will therefore provide new insights into the mechanisms of EGFR signaling in normal and diseased cells.
In this study, we have investigated the differences in the structural arrangements of HA and LA EGFR-EGF complexes in A431 cells, an epithelial tumor cell line that expresses high levels of surface EGFRs (1–2 million receptors per cell) (29) of which 2%–12% show HA for EGF (KD < 1 nM) (4). A431 cells were chosen because they are one of the best characterized cell lines with a high level of EGFR expression. Comparative studies have shown that A431 shares early characteristics in signal transduction with other cell lines that express 1%–10% EGFR on the surface, including having HA and LA receptor populations (crucial to this study) (4), dynamic signaling amplification (30), degrees of oligomerization (31), localization to rafts (32), Ca2+ signaling (4), InsP3 and PLC-γ pathways (33), adaptor recruitment (34), and STAT (signal transducer and activator of transcription) activation (35).
Furthermore, A431 cells are ideal for studying EGFR signaling in disease states. Overexpression of EGFR is common in human squamous cell carcinomas, some of which express even more receptors than A431 cells, and amplification of the EGFR gene has also been observed in other tumor types and correlated with poor prognoses in breast and other cancers ((36) and references therein). In women, EGFR overexpression is thought to play a critical role in tumor etiology and progression. Furthermore, EGFR overexpression is associated with disease recurrence and decreased patient survival (37).
We first used single-molecule FRET analysis to survey the inter-EGF distances within pairs of EGF molecules that can be found on the surface of A431 cells. Single-pair FRET measurement has the advantage of reporting specific distances without the need to invoke a particular model and deconvoluting ensemble-averaged FRET data. It also has the advantage of allowing the use of low EGF concentrations (<1 nM) and hence small fractions of LA EGFR occupancy, relevant to physiological conditions. We then compared the three-dimensional (3D) architecture of HA and LA complexes using fluorescence lifetime imaging microscopy (FLIM) and FRET to measure the distance from EGF molecules to the plasma membrane. Starting from the known structures of solubilized EGFR monomers and dimers, we constructed models of EGFR oligomers consistent with the FRET-derived in vivo inter-EGF distances and ectodomain orientations with respect to the plasma membrane. This has revealed differences in the quaternary structures of HA and LA EGFRs.
Materials and methods
Cell culture
A431 cells were purchased from the European Collection of Animal Cell Cultures and cultured in Dulbecco's modified Eagles medium without phenol red, supplemented with 10% fetal bovine serum, 2 mM glutamine, and 1% penicillin/streptomycin (all Invitrogen, Carlsbad, CA) at 37°C in the presence of 5% CO2 in air. A431R cell media were supplemented with PD 153035 to block EGFR tyrosine kinase activity (38).
Binding affinity of EGFCy3
A431 cells were seeded into dishes, cultured as described above, and, when the cultures reached 70%–80% confluency, deprived of serum overnight. Cells were chilled by incubation on ice at 4°C for 20 min. Duplicate samples of cells were treated with ice cold 20 nM EGFCy3 or with a 1:1 mixture of 10 nM EGFCy3 and 10 nM murine EGF (mEGF) for 2 h at 4°C. Excess EGF was removed, and cells were fixed with 1% paraformaldehyde at room temperature for 10 min. Cells were imaged with an inverted fluorescence microscope (Axiovert 25; Zeiss, Thornwood, NY) using a 12 bit digital camera (C7780 3CCD; Hamamatsu, Japan). For each treatment, total fluorescence intensities from 30 confluent areas were measured and the mean and standard deviation calculated.
Phosphorylation of EGFR by EGFCy3 and EGFCy5 at low temperatures
A431 cells were seeded into wells of a six-well plate, cultured as described above, and, when the cultures reached ∼50% confluency, deprived of serum for 5 h. Cells were then treated for 2 h with either phosphate buffer saline (PBS) or 20 nM mEGF (diluted in PBS; Peprotech, Rocky Hill, NJ) at room temperature and at 4°C, or with 20 nM mEGFCy3or mEGFCy5 (Cambridge Research Biochemicals, Cleveland, OH; using mEGF from Peprotech and CyDye monoreactive esters from Amersham Biosciences, Buckinghamshire, UK) at 4°C. Whole cell lysates were prepared by adding 50 μL of boiling 2× laemmeli sample buffer (12% sodium dodecylsulfate (SDS), 36% glycerol, 150 mM Tris at pH 6.8, 0.01% bromophenol blue) with 100 nM sodium fluoride, 1 mM sodium orthovanadate, and 100 mM dithiothreitol to each well. Cellular proteins were separated by SDS-polyacrylamide gel electrophoresis in a 6% acrylamide gel using the method by Schagger and von Jagow (39) and transferred onto Hybond-LFP (poly(vinylidene difluoride)) membrane (Amersham Biosciences) using semidry transblot apparatus (BioRad, Hercules, CA) according to the manufacturer's instructions. The membrane was blocked and probed with antiphosphotyrosine (clone 4G10; Upstate, Lake Placid, NY) and then stripped (62.5 mM Tris at pH 6.7, 2% SDS, 0.8% β-mercaptoethanol, incubated at 60°C for 1 h), blocked, and reprobed with antivinculin (a gift from Bipin Patel, University of Leicester, UK) according to the manufacturer's instructions. Primary antibodies were detected using horseradish peroxidase conjugated anti-mouse antibody (Jackson ImmunoResearch Laboratories, West Grove, PA). The membranes were developed using enhanced chemiluminescence (West Pico chemiluminescent substrate; Pierce, Rockford, IL) onto x-ray film (Kodak, Rochester, NY).
Preparation of samples for single-molecule microscopy measurements
Measurements in fixed cells
A431 cells were seeded onto glass-bottomed dishes, cultured as described above, and, when the cultures reached 60%–70% confluency, deprived of serum for 16 h. Cells were incubated in PBS supplemented with 1 mM HEPES at pH 7.4 for 2.5 h at 37°C in the presence of 5% CO2 in air. Cells to be fixed before EGF binding were treated with 1% paraformaldehyde for 10 min at 18°C, washed, and stored at 4°C before labeling 16–24 h later. Cells to be fixed after EGF binding were chilled to 4°C. EGFCy3 and EGFCy5 (Cambridge Research Biochemicals, using mEGF from Peprotech and CyDye monoreactive esters from Amersham Biosciences) solutions were prepared in PBS supplemented with 1 mM HEPES at pH 7.4 and 0.1% bovine serum albumin (BSA) chilled to 4°C. MEGF has only one reactive amino residue (at the N-terminus) and was labeled at a ratio of exactly 1:1 protein/dye, as confirmed by high performance liquid chromatography and mass spectrometry. Cells were labeled with 1:1 EGFCy3/EGFCy5 solutions, at concentrations of 0.1–1.0 nM, for 2 h at 4°C. Cells to be fixed after EGF binding were treated with paraformaldehyde as described above, washed, and stored at 4°C. EGFR phosphorylation was confirmed by Western blot (Fig. 1 B). Cells were imaged on the same day as EGF labeling.
Figure 1.
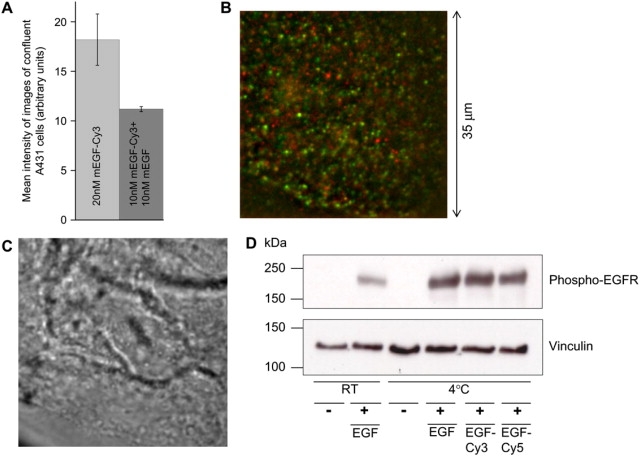
(A) Affinity of labeled and unlabeled EGF for binding to EGFR (see Materials and Methods). Since the intensity approximately halves when the proportion of EGF labeled is also halved, labeling does not significantly affect the EGF binding affinity to EGFR. (B) Merged time-integrated donor (EGFCy3; green) and acceptor (EGFCy5; red) images of a sample of A431 cells exposed to 0.25 nM EGFCy3 and 0.5 nM EGFCy5. (C) White light transmission image corresponding to the same area as in B. (D) Western blots of whole cell lysates to show phosphorylation of EGFR in resting and EGF-stimulated A431 cells. A431 cells were treated for 2 h with either PBS or 20 nM mEGF (mEGF, diluted in PBS) at room temperature or with PBS, 20 nM mEGF, mEGFCy3, or mEGFCy5 at 4°C. Probing with an antiphosphotyrosine antibody shows EGFR is phosphorylated when A431 cells are stimulated with EGF, EGFCy3, or EGFCy5at 4°C. The blot was subsequently probed with antivinculin as a loading control showing an approximately equal amount of total protein in each lane.
Measurements on LA receptors in fixed cells
A431 cells were seeded and serum-starved as described above. Cells were incubated in PBS supplemented with 1 mM HEPES at pH 7.4, 0.1% BSA, and 200 nM anti-EGFR clone 108 (gift from Kathryn M. Ferguson) for 4 h at 4°C, to block EGF binding to HA receptors (40). The cells were then fixed and treated with EGF, or vice versa, according to the protocol described above.
Measurements in live cells
A431 cells were seeded onto glass coverslips and cultured as described above until ∼60%–70% confluent. On the day of the experiment cells were incubated in antiinternalization buffer (PBS supplemented with 1 mM HEPES at pH 7.4, 10 mM sodium azide, 10 mM deoxy-d-glucose, and 0.1% BSA (23)) at 37°C for 20 min in the presence of 5% CO2 in air. Cells were labeled with 0.2 nM EGFCy3 and 1 nM EGFCy5 in antiinternalization buffer at 18°C and imaged ∼10 min later using the single-molecule microscope.
Preparation of samples for confocal microscopy and FLIM
Measurement of distances from EGF to the cell surface for HA receptors
A431 cells, treated as described above, were incubated with 5 μM DiI (1,1′-dioctadecyl-3,3,3′,3′- tetramethylindocarbocyanine perchlorate) (Vybrant cell-labeling solution, Invitrogen) diluted in serum-free culture media at 37°C for 15 min to label the plasma membrane. Cells were then incubated in PBS supplemented with 1 mM HEPES at pH 7.4, 0.1% BSA, and 200 nM Anti-EGFR clone 2E9 (Caltag-MedSystems, Carlsbad, CA) for 4 h at 4°C to block EGF binding to LA receptors (4). Cells to be fixed before EGF binding were treated with 1% paraformaldehyde for 10 min at 18°C. Cells were labeled with 100 nM EGFFluorescein (mEGF conjugated at its N-terminus with fluorescein, Invitrogen) diluted in PBS supplemented with 1 mM HEPES at pH 7.4 and 0.1% BSA for 2 h at 4°C. Cells to be fixed after EGF binding were treated with paraformaldehyde as described above, washed, and stored at 4°C. Cells were imaged on the same day.
Measurement of distances from EGF to the cell surface for LA receptors
The above protocol was followed to prepare these samples, except that the cells were not treated with anti-EGFR clone 2E9 but were chilled on ice at 4°C for 20 min before treatment with EGF.
Measurement of mean inter-EGF distances for HA and LA receptors
The protocol used to prepare these samples has been previously reported (23).
Preparation of samples to measure the mean FRET efficiency of noninternalized receptor populations using FLIM
A431 cells were labeled with 100 nM of unlabeled EGF, allowed to internalize EGF-EGFR for 15 min at 37°C, then cooled to 4°C to block EGFR recycling to the cell surface. This was followed by the removal of unlabeled EGF remaining on the cell surface by an acid wash (41). Noninternalized EGFRs were labeled with a 1:2 mixture of EGFAlexa488 and EGFErythrosin at 4°C.
Single-molecule fluorescence microscopy
The single-molecule total internal reflection fluorescence (TIRF) microscope used was as previously reported (42). Briefly, samples were placed in a custom-built TIRF microscope and excited with 1 mW at 532 nm. The resultant fluorescence was divided into orthogonal polarizations and two wavelength bands corresponding to Cy3 and Cy5 emission (Quad View, Roper Scientific, Tucson, AZ), giving four images on the charge-coupled device (CCD) (iXon; Andor, South Windsor, CT) of the same field of view, but differing in spectral and polarization content. Images were acquired at ∼3 Hz with an integration time of 277 ms.
Single-molecule data analysis
Fluorescent spot locations in time-integrated image series were identified using custom-written IDL routines (incorporating some from Crocker and Grier (43) and Crocker and Weeks (44)). The average image for a data set was calculated and band-pass filtered to reduce noise on the scale of 1 pixel and remove smooth variations on scales greater than the spot size. Peaks in this filtered image were then used as potential fluorescent spot positions. The background for each frame was calculated by smoothing it on a scale larger than the spot size while masking out the region around all spot positions. Intensity time traces for the two wavelengths were calculated by integrating the unfiltered background-subtracted images in each channel around each spot position throughout the time series. FRET efficiencies were calculated allowing for bleed through (3%) and following calibration as reported previously (42) during selected time intervals. Extensive manual filtering of the obtained traces ensured that only reliably measured features were retained.
Random noise in the images (e.g., detector readout noise, Poisson counting statistics) means that individual background-subtracted intensity measurements from the images can be negative (see Fig. 3 B). Negative intensities are clearly unphysical but are correctly handled by modeling the underlying intensity correctly over a series of frames. The simplest case, where the intensity is assumed to be constant, is to take the mean and standard deviation of intensities. We did this to measure the donor and acceptor intensity distributions in the selected frame ranges. From these we calculated the FRET efficiency.
Figure 3.
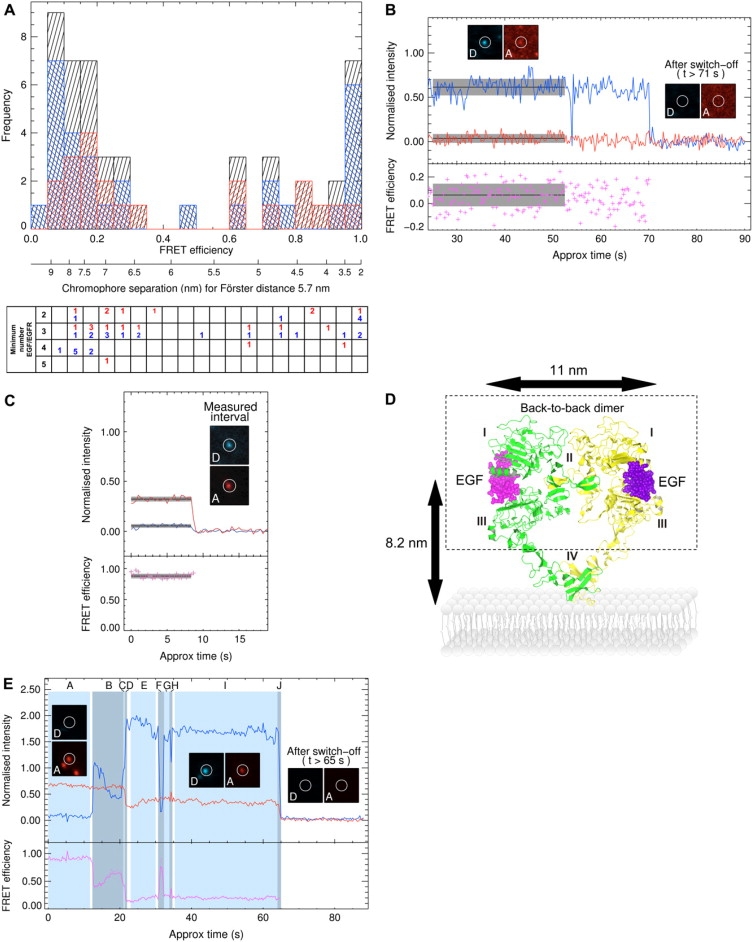
(A) (Upper) Distribution of FRET efficiencies and equivalent separations in A431 cells exposed to EGF concentrations (0.1–1 nM) of a 1:1 mixture of EGFCy3 and EGFCy5. (Red) Cells pre-fixed with 1% paraformaldehyde and then exposed to EGF. (Blue) Cells first exposed to EGF at 4°C and then fixed; the combined data are shown in black. (Lower) The table specifies for each FRET efficiency value in the histogram the minimum number of EGFRs in the associated spot (determined from the number of EGFCy3 and/or EGFCy5 photobleaching steps). (B and C) Traces shown as described in Fig. 2. (D) Model of the crystal structure of the ectodomains of an EGFR dimer standing proud (15), based on coordinates of the EGF-EGFR dimer from Ogiso et al. (13). The ectodomain is divided into four subdomains. Domains I and III (or L1 and L2, large homologous domains) bind to peptide ligands (e.g., EGF, transforming growth factor-α, amphiregulin, betacellulin, epigen, epiregulin, and heparin binding EGF-like growth factor) (74,75). Domain II (or CR1, cysteine-rich domain 1) includes a dimerization arm, two of which interact to form the back-to-back EGFR-EGFR dimer (12,13). Interactions between domain IVs (or CR2, cysteine-rich domain 2) of EGFR may also be involved in the back-to-back EGFR-EGFR interface ((76) reviewed in Burgess et al. (77) and Saxon and Lee (78). (E) Trace showing three donors and two acceptors in one spot.
FLIM microscopy
For FRET between EGF and the membrane, fluorescence decays were recorded using a frequency-doubled laser (Coherent, Santa Clara, CA; λ = 460 nm, 76 MHz repetition rate). Fluorescence decays were collected between λ = 480 nm and λ = 520 nm, where there was no bleed through from the acceptor. FRET efficiencies and distances were calculated as described previously (45). For the mean FRET efficiency of low- and high-affinity receptors, the method was as reported previously (23).
Anisotropy measurements
The steady-state anisotropies of the fluorophores conjugated to EGF were determined using a Jobin Yvon (Longjumeau, France) Fluorlog-3 steady-state fluorimeter. Solutions of EGFCy3, EGFCy5, and EGFFluorescein were prepared in 99% glycerol and PBS at concentrations of 0.1–1 μM and excited with polarized light at 552 nm, 640 nm, and 445 nm, respectively. Time-resolved anisotropy decays were collected as previously described (23).
Calculation of Förster radius for EGF derivatives in solution
We evaluated Förster radius (R0) from EGFCy3 fluorescence and EGFCy5 absorption solution measurements at the 649 nm Cy5 absorption maximum, using an absorption coefficient of 250,000 M−1cm−1 and an orientation factor of . This error in κ2 was taken to be the same as that calculated by Yasuda et al. (46) because the semicone angles of the subnanosecond wobble (φc) of each fluorophore were very similar to those reported therein. φc was calculated to be ∼28° for Cy3 and ∼38° for Cy5, using , where r∞ and r0 are the experimentally determined residual and limiting anisotropies, respectively. The refractive index was assumed to be 1.33. The error in R0 was mainly from κ2 and was estimated using (46). R0 was thus calculated to be 5.7 nm ± 14%, consistent with previous results (47).
For dyes with a fluorescence lifetime τ > 1 ns, such as fluorescein, the anisotropy decay when bound to protein in solution is typically multiexponential (23), i.e., . φc was hence calculated from r∞, preexponential terms β1, β2, and correlation times φ1, φ2 for EGFFluorescein in PBS (Table 1). The equivalent residual anisotropy after depolarization due to rotation of the dye with respect to EGF is , leading to φc = 53°. This is equivalent to a donor depolarization factor of 0.04 and an error in . (48).
Table 1.
Experimental steady-state anisotropy values and time-resolved anisotropy decay parameters
| A* | Free Cy3 | Free Cy5 | EGFCy3 | EGFCy5 | EGFFluorescein |
|---|---|---|---|---|---|
| PBS | 0.230 | 0.194 | 0.271 | 0.210 | 0.077 |
| 99% Glycerol |
0.382 |
0.388 |
0.313‡ |
||
| B† | β1 | φ1 | β2 | φ2 | r∞ |
| EGFFluorescein | 0.20 | 0.11 | 0.06 | 4.58 | 0.04 |
The measurement errors are ∼1%.
Experimental steady-state anisotropy values for free fluorophores and EGF derivatives in solution used to calculate the errors in FRET-derived inter-EGF distances.
Time-resolved anisotropy decay parameters of EGFFluorescein in PBS.
Excited away from absorption maximum at 445 nm.
Analysis of crystal structures
Crystal structures were manipulated and analyzed using programs from Collaborative Computational Project No. 4 (49). Coordinates were downloaded from the Protein Data Bank (50). The structures were analyzed and distances calculated using Coot (51), and figures were prepared with CCP4mg (52). The PISA web service (53) was used to analyze interfaces between EGFR monomers. The normal modes of the EGFR dimer were estimated using the elNemo web service (54).
Results
Single-pair FRET analysis of EGF-EGF distances in EGFR complexes
EGF labeled at its N-termini with Cy3 and Cy5 dyes (denoted EGFCy3 and EGFCy5) was added to the medium of A431 cells in culture as FRET donor-acceptor pairs. Addition of dye to the N-terminus of EGF did not modify the affinity of EGF for EGFR (Fig. 1 A). Cy3 and Cy5 are ideal for single-pair FRET measurements as they possess well-separated emission spectra, a significant overlap between donor emission and acceptor absorption, comparable quantum yields that can result in easily identifiable anticorrelated donor and acceptor signal fluctuations, and good photostability (55). We used final concentrations of a 1:1 mixture of EGFCy3 and EGFCy5 of 0.1–1 nM that are close to physiological conditions for EGFR occupancy (i.e., ≤50% HA and ≤10% LA EGFR bound to EGF) and optimal for single-molecule microscopy.
The occupancy fraction F was calculated using F = [EGF]/[EGF] + KD, where KD is the dissociation constant. Given that the Förster radius of the Cy3/Cy5 pair is 5.7 nm, FRET will only occur if EGFCy3 and EGFCy5 are within <10 nm of each other. Fig. 1 B is a time-integrated fluorescence image showing an example of the fluorescent spots that appear on the cell surface when Cy3 was selectively excited at 532 nm under total internal reflection conditions and emissions from Cy3 and Cy5 were simultaneously imaged onto different areas of a CCD camera (42); Fig. 1 C is a white light transmission image of the same area. The number of spots is lower than the nominal density because, for example, many receptors are not excited due to cell surface ruffling placing them outside the evanescent field and photobleaching before data acquisition started. On excitation of donor EGFCy3 the occurrence of FRET is shown by a decrease in donor fluorescence intensity and the simultaneous appearance of sensitized acceptor EGFCy5 fluorescence. The fluorescence intensity versus time traces associated with the spots displayed typical characteristics of single-molecule fluorescence data (47), including discrete bleaching steps and blinking in the donor and acceptor channels.
To investigate the FRET efficiencies that can be found in the EGFR-EGF complexes that form in A431 cells with physiological EGF concentrations, we used 12 samples of A431 cells. Six were first lightly fixed to trap EGFR in inactive configurations and then exposed to EGF. The other six samples were first exposed to EGF while held at 4°C to inhibit ligand-induced EGFR internalization (23), then fixed to trap EGFR complexes in active configurations. Given that the FRET efficiency determined by FLIM measurement is identical in the absence and presence of fixative (Supplementary Fig. 1), we conclude that FRET values at the plasma membrane are not affected by the light fixation. The low temperature did not inhibit receptor phosphorylation, as determined by Western blot (Fig. 1 D). We could not detect any difference in FRET at the single-molecule level between cells exposed to EGF exposure at 4°C or at room temperature (42). There is a wealth of evidence that signaling by EGFR can still proceed at 4°C—e.g., incubation with EGF at 4°C does not affect the lateral diffusion dynamics of complexes at the cell surface (56,57), EGFR signaling to the level of Ras activation is also efficient in the cold (58), and immunoblots using antibodies against phosphotyrosine demonstrated that EGF treatment of intact cells increases the phosphotyrosine content of at least six EGFR substrates within 5 s at 4°C (59)—which suggests that ligand receptor complexes form as normal at the cell surface at this temperature. Fig. 1 D also shows that EGFCy3 and EGFCy5 activate EGFR as efficiently as native EGFR.
The features identified on the cell surface were classified into three categories: i) spots that displayed clear FRET, defined as having sensitized fluorescence intensity distinctly above the bleed through from the donor channel (∼3%); ii) spots that displayed very low FRET and/or donor-only fluorescence; and iii) overlapping, out-of-focus, weak intensity and otherwise unreliable spots (all rejected). From category (i) we selected spots showing clear abrupt bleaching steps that were well separated from other features, within a diffraction-limited size and had FRET efficiency traces with a signal/noise ratio greater than ∼3. For the selected spots, examples of which are shown in Fig. 2, A–F, we built a histogram of FRET efficiency values for intervals between bleaching steps in individual spots (Fig. 3 A). According to Förster theory, the FRET efficiency increases with the inverse sixth power of the donor-acceptor separation (21). From the anisotropy data in Table 1 and assuming , the errors in the distances shown are ±14% for single pair FRET (see Materials and Methods).
Figure 2.
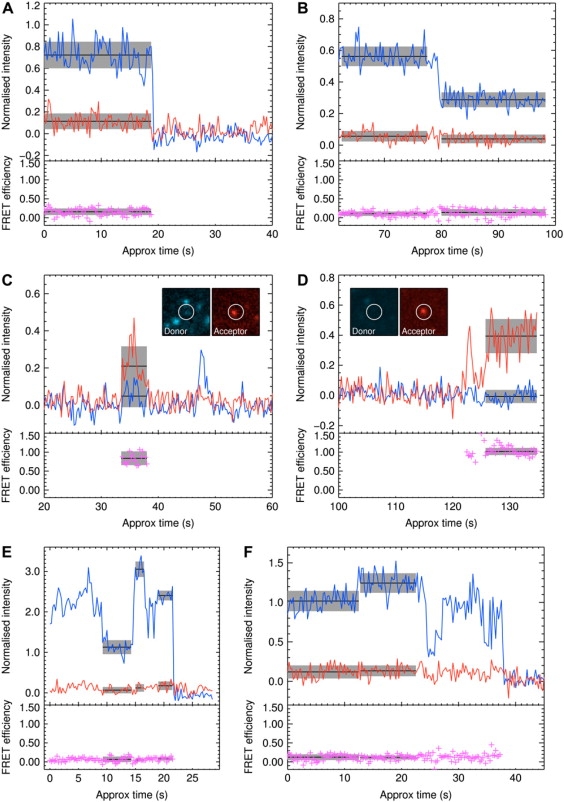
Examples of single-pair FRET traces. (A) A single-donor photobleaching step. FRET efficiency 0.16, standard deviation 0.09. (B) One of two donors photobleaching. The low intensity suggests the molecule is out of focus (away from the peak of the evanescent field). FRET efficiencies 0.11 and 0.14, standard deviations 0.06 and 0.09, respectively. (C) Single strong FRET interaction. FRET 0.84, standard deviation 0.18. (D) Single interaction with completely quenched donor. (E and F) Traces showing multiple steps. Fluorescence intensity versus time traces of donor (blue) and acceptor (red) fluorophores and temporal variation FRET efficiency (purple). Black lines and gray error bars show mean ± SD in the marked region. Images show mean donor and acceptor fluorescence (background-subtracted) during time intervals as marked.
Two broad clusters of low and high FRET efficiencies were observed in the EGFR complexes, whether cells were fixed before exposure to EGF (Fig. 3 A, red) or after (Fig. 3 A, blue). Given that there are 50–1,000 EGFRs per μm2 on the cell surface of A431 cells (26,56), we checked whether the interactions shown in Fig. 3 A could also be found in HeLa cells, which have an estimated density of <10 receptors per μm2 (assuming ∼50,000 EGFRs per cell and a cell diameter of ∼20 μm). Since we could find examples of spots showing high FRET and low FRET values on the surface of HeLa cells (Supplementary Fig. 2), we conclude that random colocalization of receptors due to EGFR overexpression cannot explain the FRET results shown in Fig. 3 A.
Fig. 3, B and C, shows examples of long (9 nm) and short (4 nm) inter-EGF distances. Besides the broadening introduced by the inherent ±14% error in the distance calculation from uncertainties in the value of the orientation factor, some additional broadening of the FRET efficiency distribution can also be expected from the breakdown of the point-dipole approximation at distances comparable to the size of the dyes (2 nm) (60). However the FRET efficiency clusters in Fig. 3 A are so far apart that they must report on at least two structurally different ectodomain interfaces, which place EGF-binding sites long (∼8 nm) and short (<5.5 nm) distances apart. The data in Fig. 3 A provide the first direct evidence of high-FRET interfaces between single EGF pairs in EGFR-EGF complexes in cells. We were unable to explain these distances in light of the back-to-back EGFR dimer (Fig. 3 D) even after applying extreme deformations to this crystal structure—for example, by changing the angle between domains I and III and domain II and/or applying extreme perturbations along low frequency normal modes—as the predicted distances between N-termini always remain >9 nm (M. D. Winn, unpublished observations). The occurrence of long and short distances in single-pair interactions (Fig. 3 A, lower), therefore, shows that EGFR ectodomains can form at least two interfaces different from the crystallographic back-to-back interface, not just the one assumed by simple FRET/non-FRET models (22,24,25). Significant FRET was observed independent of whether cells were fixed before exposure to EGF (inactive states) or vice versa (active states), consistent with some unliganded EGFRs being constitutively oligomerized on the cell surface, as previously suggested (23,26,47).
Multiple fluorescence bleaching steps by the donor and/or acceptor were also observed alongside many examples of single-step photobleaching (Fig. 3 A, lower). Examples are shown in Fig. 2, E and F, and have been collated in Table 2, the latter including additional data from live samples. These multiple steps report on groups of EGFRs colocalized within spots, i.e., within the point spread function (PSF) of the microscope (full width at half-maximum ∼500 nm). Given that spots can also contain any number of nonfluorescent, unliganded EGFRs and/or unpaired EGFR-EGFCy5 units, the number of bleaching steps actually reports on the minimum number of receptors colocalized in a sub-500 nm diameter area on the cell surface (Table 2). This finding is consistent with previous reports which showed that EGFR can aggregate within microclusters (24,26). Considering the low density of spots formed at the low EGF concentrations employed, the probability of receptor groups colocalizing by chance within the PSF of the microscope is small. Given that spots remain intact while slowly diffusing in the plasma membrane (Supplementary Fig. 3), our findings suggest that receptors are stably held together by interactions at the plasma membrane, such as by oligomerization or lipid rafts smaller than the PSF of the microscope (8).
Table 2.
Number of donor and acceptor molecules and hence the minimum number of EGFRs per spot determined from the number of photobleaching steps in the associated donor and acceptor fluorescence versus time traces
| Minimum number of receptors | In pre-fixed cells | In cells stimulated with EGF |
|---|---|---|
| 2 | 7 | 12 |
| 3 | 7 | 9 |
| 4 | 1 | 4 |
| 5 | 1 | 2 |
We illustrate many of the features of the different modes of interactions between EGFR-EGF complexes described by Fig. 3 A in an example from live cells (Fig. 3 E). First, we see several receptors colocalized. The two acceptor fluorescence steps (D and J in Fig. 3 E) report the presence of two acceptor EGFCy5 molecules. Therefore, at least one donor EGFCy3 must be transferring energy to the two acceptors during A. Second, high FRET is demonstrated by the increase in donor intensity upon the bleaching of one acceptor (D), which is equivalent to that observed in single unquenched EGFCy3 molecules; the large magnitude of these changes demonstrates a short inter-EGF separation had existed in this pair (61). High FRET is observed again as this acceptor fluorescence briefly recovers and bleaches for a second time in H. During B, a further EGFR-EGFCy3 unit diffuses and becomes colocalized with the complex at C. Neither colocalization at C nor the transient dissociation (or blinking) of two of the three donor EGFR-EGFCy3 units during F changes the intensity of any of the acceptors, indicating distances >10 nm between these groups. Since the donor fluorescence intensity that disappears during F (whereas an acceptor continues fluorescing) is equivalent to that expected from two “donor units” (one unit being the increase at C), there are at least three donors present. We conclude that this spot contains at least three donors and two acceptors.
FRET analysis of inter-EGF distances in HA and LA EGFR complexes
Depending on the conditions, such as the donor/acceptor ratio employed, HA EGFRs have been shown by FLIM measurement to display mean FRET efficiency values between 0.22 and 0.40 (22,23). Selective EGF saturation-binding of the HA EGFR population was achieved using a monoclonal anti-EGFR antibody 2E9 (mAb 2E9) to block EGF binding to the LA population (4). (The 2E9 antibody does not disturb binding to HA EGFRs (4).) Our ensemble FRET measurements reproduce these results (Fig. 4 A). We conjugated EGF to Alexa 488 (donor) and Erythrosin (acceptor) (EGFAlexa488 and EGFErythrosin) and exposed A431 cells to saturating concentrations of mAb 2E9 to block EGF binding to the LA population (4) followed by a 1:2 mixture of EGFAlexa488 and EGFErythrosin at a final EGF concentration of 100 nM at 4°C. At this EGF concentration, most HA EGFRs are bound to EGF (23). The Alexa 488/Erythrosin FRET pair is ideal for ensemble FLIM-FRET measurements because the fluorescence lifetime of Alexa 488 is ∼10 times longer than that of Cy3, so larger changes in lifetime are expected for Alexa 488 for the same FRET efficiency values. In addition, the Förster radius (5.7 nm) (23) has the same value as the Cy3/Cy5 pair, allowing direct comparison of ensemble and single-molecule FRET results. We found the FRET efficiency between donor and acceptor EGF ligands bound to the HA EGFR population to be 0.24 ± 0.05 (Fig. 4 A, blue bar). This, therefore, shows that EGFR ectodomain interfaces with inter-EGF distances of <10 nm are involved in the formation of HA EGFR complexes, potentially explaining some of the interactions contributing to Fig. 3 A.
Figure 4.
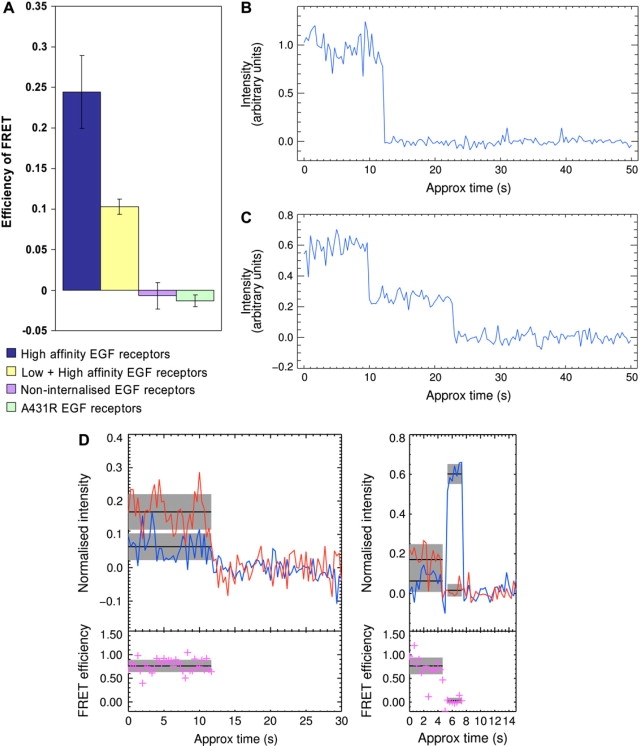
(A) Histograms of ensemble FRET efficiencies of HA EGFR populations alone and with LA EGFR in A431 cells, plus the noninternalized EGFR population in A431 cells and in tyrosine kinase-inhibited A431R cells, using EGFAlexa488, EGFErythrosin, and FLIM. Fluorescence traces showing one step (B) and two steps (C) in the donor channel. (D) Traces showing high and low FRET efficiencies in LA EGFR complexes in A431 cells labeled using a 2 nM 1:1 mixture of EGFCy3 and EGFCy5.
When the binding of EGF to HA EGFRs is not blocked by mAb 2E9, previous ensemble FRET measurements have returned lower mean FRET efficiencies (between 0.05 and 0.18) than in the conditions when HA EGFRs were selectively labeled (22,24). In the absence of mAb 2E9, using a 1:2 mixture of EGFAlexa488 and EGFErythrosin at a final EGF concentration of 100 nM at 4°C, we found a mean FRET efficiency value of 0.1 ± 0.01 (Fig. 4 A, yellow bar). However, in cells allowed to internalize EGFR-EGF for 15 min at 37°C and then cooled to 4°C to block EGFR recycling to the cell surface, there was no evidence of FRET between donor- and acceptor-labeled EGFR-EGF complexes that were not internalized (∼60% of the total number) (Fig. 4 A, purple bar). This shows that receptors displaying FRET on the cell surface are targeted for downregulation. We also found that FRET is abolished at the surface of A431 cells by exposing the cells to the tyrosine kinase inhibitor PD 153035 (i.e., A431R cells) (38) (Fig. 4 A, green bar) even though the cells still express the same number of receptors. We therefore conclude that the interactions at the plasma membrane of A431 cells described by FRET are biologically relevant.
The larger FRET efficiency value observed when only HA EGFR are allowed to bind EGF (Fig. 4 A) shows that EGFR ectodomain interfaces yielding high FRET efficiencies are more prevalent in HA complexes. The mean FRET efficiency when both HA and LA EGFR are allowed to bind EGF must largely reflect the FRET value from the LA EGFR population alone, as the contribution from the HA population (<10%) is smaller than the error of the measurement (10%) (Fig. 4 A). However, unlike ensemble FRET measurements for HA EGFRs, in which the number of EGFRs occupied by EGF (∼7.5 × 104) was comparable to that in the single-molecule FRET experiments (1–4 × 104 HA EGFR occupied at 0.1–1 nM (EGF)), the ensemble FRET efficiency value from the LA population (Fig. 4 A) was averaged over 1–2 × 106 EGF-bound receptors, which is 10–100× larger.
To investigate whether FRET could still be found in LA EGFRs at low fractions of EGF occupancy, we treated two samples of A431 cells with saturating concentrations of monoclonal anti-EGFR 108 (mAb 108) that blocks EGF binding to the HA population (40) before exposing the cells to a 1:1 mixture of EGFCy3 and EGFCy5 at a total EGF concentration of 2 nM. Fig. 4, B and C, shows two examples of donor-only fluorescence intensity versus time traces from LA complexes showing one- and two-step photobleaching, the latter showing that LA EGFR-EGF in A431 cells also colocalize within the PSF of the microscope consistent with previous results (26). Although spots showing high FRET efficiency values were relatively harder to find, Fig. 4 D shows two clear examples of high-FRET interfaces in two LA EGFR complexes. The FRET efficiency values correspond to inter-EGF distances of ∼4.5 nm. These observations suggest that LA EGFR ectodomains can also interact at low EGF occupancy in an arrangement that results in short inter-EGF distances.
FLIM-FRET measurements of the distance from EGF to the plasma membrane
To gain insight into the 3D arrangement of the EGFR ectodomain interfaces shown in Fig. 3 A, we investigated the distance between EGF conjugated at its N-terminus to donor fluorescein (EGFFluorescein) and the membrane-labeling acceptor chromophore DiI in HA and LA EGFR-EGF complexes. The fluorescein-DiI FRET pair was chosen for these measurements because its Förster radius (3.5 nm) has been very well characterized in a similar geometry (62). We used ensemble-averaged FLIM-based FRET measurement because the concentration of DiI needed to ensure colocalization of the membrane probe with EGFR-bound EGF precluded single-molecule measurement. The DiI was observed to be uniformly distributed across the cell. To study HA EGFRs, three samples of A431 cells were incubated with DiI, exposed to a saturating concentration of mAb 2E9 to block EGF binding to LA receptors (4), and then fixed and exposed to a saturating concentration of EGF (100 nM). Three further mAb 2E9-labeled samples were first exposed to EGF at 4°C and then fixed. We found that fluorescence lifetime distributions of EGFFluorescein bound to HA receptors peaked at much shorter values in samples colabeled with the DiI acceptor than in samples without acceptor, which did not show evidence of donor lifetime quenching (Fig. 5 A). We determined that local membrane acidification effects could not contribute in any way to the observed fluorescence lifetime shortening (for example, via a change in EGF conformation that alters the interactions between EGF and the probe) (Fig. 5 B). Given that the donor fluorescence was recovered on acceptor photobleaching (data not shown), we conclude that the shortening of the donor lifetime in the presence of acceptor is due to FRET.
Figure 5.
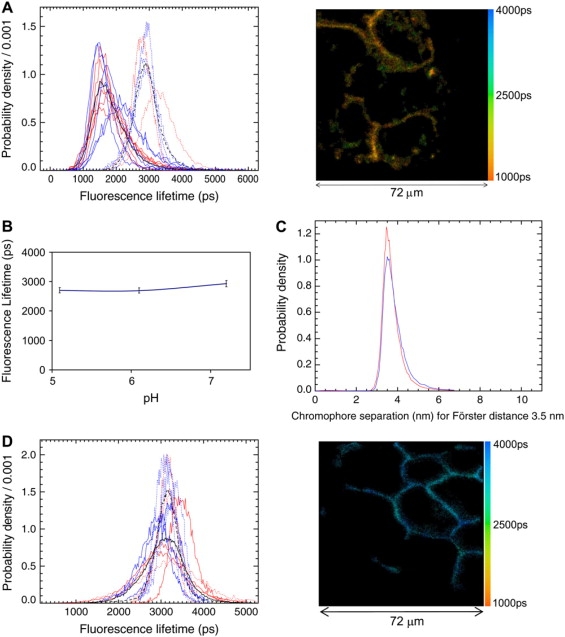
(A) Mean fluorescence lifetime distributions of EGFFluorescein bound to HA EGFRs in A431 cells whose plasma membrane was labeled with DiI, for cells fixed with 1% paraformaldehyde before (red) and after (blue) binding EGF. Control FLIM data in the absence of DiI (acceptor) are shown as dotted lines. Curves correspond to different fields of view, an example of which is shown to the right, and are normalized as photon-weighted fluorescence lifetime distributions. The black curves are the normalized sums of the corresponding unnormalized photon-weighted fluorescence lifetime distributions. (B) Fluorescence lifetime of EGFFluorescein in buffer solution measured at the range of pH shown. (C) FRET-derived separation between EGFFluorescein (donor), bound to HA EGFRs, and the cell surface decorated with DiI (acceptor). (D) Fluorescence lifetime distributions of EGFFluorescein bound to LA EGFRs (EGF binding to LA EGFR was blocked using mAb 2E9 (S3)) before (red) and after (blue) fixation with 1% paraformaldehyde.
The efficiency of FRET was determined from the mean fluorescence lifetime of donor EGFFluorescein bound to HA EGFR in the presence and absence of acceptor DiI. From the anisotropy data in Table 1, the errors in the distances using the inverse sixth power of the donor-acceptor separation and the randomized orientation factor are ±10% (see Materials and Methods). The observed FRET efficiency of 0.5, therefore, shows a peak distance from EGF to the cell surface of ∼3.5 nm ± 10% (Fig. 5 C). This distance is substantially smaller than that predicted by the back-to-back dimer model in Fig. 3 D, which, based on the configuration in the autoinhibited monomer (15), is usually represented fused with domain IV and standing proud from the membrane (63), giving a distance between the N-terminus of EGF and the outer surface of the plasma membrane of ∼8.2 nm (Fig. 3 D). This distance comprises 4.3 nm from the EGF N-termini to a plane containing the EGFR ectodomain C-termini of the back-to-back dimer crystal structure (from 1ivo, Table 3), plus the length of missing residues 513–621 of domain IV (added by Ogiso et al. (13)). Our FLIM-FRET measurement shows that ectodomains of HA receptors must instead lie flat on the plasma membrane, not perpendicularly as previously thought. For this to occur the hinge region between domain IV and the transmembrane domain must be very flexible, consistent with x-ray crystallography predictions of the high degree of disorder in this region (12,13,15,64). Since the same distance from EGF to the membrane was found in pre-fixed cells (Fig. 5 C), we conclude that HA EGFRs are constitutively oriented flat on the plasma membrane.
Table 3.
Distances between EGF molecules and interface parameters in models of possible EGFR oligomers generated from available EGFR crystal structures
| EGFR interface | Crystal structure used | Distance between ligands* (Å) | Effect of addition of dye to distance† | Surface area (Å2)‡ | ΔG§ (kcal/mol) (p-value) |
|---|---|---|---|---|---|
| Back-to-back | 1mox (12) | 111 | Increase | 1107 | −17.3 (0.043) |
| Head-to-head | 1mox (12) | 50 | Decrease (min 27 Å) | 440 | −3.1 (0.430) |
| Back-to-back | 1ivo (13) | 111 | Increase | 1315 | −14.0 (0.132) |
| Weak A:A | 1ivo (13) | 64 | Increase | 455 | 0.3 (0.559) |
| Weak B:B | 1ivo (13) | 46 | Decrease | 284 | 1.0 (0.749) |
| Head-to-back§ | 1nql (15) | 53 | Similar | 956 | −9.9 (0.263) |
| Domain II/IV interactions¶ | 1nql (15) | 68 | Similar | 655 | −5.7 (0.366) |
| Perpendicular | 1yy9 (73) | 816 | −5.0 (0.417) | ||
| HER3 conformation‖ | 1m6b (64) | 1877 | −21.0 (0.061) |
Distances between EGF or TGFα molecules are from the first determined amino acid residues (residues 2/3 in 1mox and residue 5 in 1ivo).
Size of Cy3 or Cy5 dye used is ∼20 Å.
Calculated by PISA (53).
ΔG is the solvation free energy gain upon formation of the interface, as calculated by PISA. It does not include the effect of satisfied hydrogen bonds and salt bridges across the interface. The p-value is the probability of getting a lower than obtained ΔG, if interface atoms are picked randomly from the protein surface.
Inactive crystallographic dimer.
Crystal of unliganded HER3 dimer.
Similarly, we measured the distance between LA EGFR-EGFFluorescein and DiI probes on the cell surface by incubating A431 cells with DiI and then exposing them to a saturating concentration of EGF (100 nM), which, in the absence of mAb 2E9, labels most HA and LA EGFR binding sites (23). As described before, the contribution to the measured FRET efficiency from the HA EGFR population (<10% of surface EGFRs) is negligible. Unlike the HA case, we did not find any evidence of FRET, either in pre-fixed cells or cells first activated by EGF. The absence of FRET is unlikely to be solely due to preferential labeling by DiI of lipid rafts, which has been reported in the literature to occur in artificial membrane preparations (65) because EGFRs colocalize with lipid rafts (32). Given the magnitude of the fluorescence lifetime quenching observed for HA EGFRs, a substantial shortening of the lifetime would therefore have been observed if LA receptors were similarly oriented flat on the plasma membrane. However, given that the Förster radius for this pair is 3.5 nm, zero FRET efficiencies correlate with distances from EGF to the cell surface >7 nm (Fig. 5 D), showing LA ectodomains stand proud from the plasma membrane. FLIM-FRET measurement therefore shows that HA and LA EGFRs constitutively have different orientations with respect to the plasma membrane, consistent with the notion that EGFR affinity is regulated by the formation of ternary complexes between HA EGFR and intracellular substrates, a finding that could also explain the equilibrium kinetics of EGF binding (66,67).
Crystal structure analysis of ectodomain interfaces in HA and LA complexes
To investigate possible 3D quaternary structures consistent with the inter-EGF distances we observed (Fig. 3 A) and the distances from EGF to the membrane (Fig. 5 C), we surveyed the EGFR interectodomain interfaces so far described by x-ray crystallography (Table 3). The back-to-back EGFR dimer (Fig. 3 D) (12,13) deposited with Protein Data Bank (PDB) ID codes 1mox and 1ivo provides an interface between the two EGFR monomers with a large surface area with a negative solvation Gibbs free energy difference (ΔG) and a p-value very close to zero. Alternative weaker interfaces between EGFR monomers have also been described (12,13,15) and are compared here. The weak A:A and weak B:B conformations contain very weak interfaces, shown by the small surface area at the contact points. These contacts are not favorable as they have positive ΔG values and p-values of greater than 0.5 and therefore will not be considered further here. Interfaces in an inactive crystallographic dimer (15), denoted here as head-to-back and domain II/IV interactions and deposited with PDB ID code 1nql, show that these contact points create a reasonable surface area and a negative ΔG value.
Finally, the head-to-head conformation contains weak interfaces made up of contacts that constitute a small surface area. The ΔG value, however, is negative and the p-value slightly less than 0.5. The distance between the N-termini of bound ligands is similar for the head-to-head and head-to-back dimer conformations, 5.0 nm and 5.3 nm, respectively. By modeling the possible positions of the dye molecules attached to the N-termini of these ligands, the distance between the chromophores is reduced for the head-to-head conformation and is predicted to remain similar for the head-to-back conformation. Similarly, the conformation involving domain II/IV interfaces gives a distance of 6.8 nm between the N-termini of EGF. The observed weak crystal interfaces illustrate possible interectodomain arrangements that would explain the single-molecule low-FRET and high-FRET clusters (Fig. 3 A). There may well be other interreceptor interfaces in cells that are not observed in the available crystal structures.
Discussion
Using single-pair FRET analysis of inter-EGF distances in EGFR-EGF complexes, we have demonstrated for the first time to our knowledge that—in cells—EGFR ectodomains can form at least two interfaces, both structurally different from the back-to-back dimer arrangement which would not show any detectable FRET. One of these interfaces places two EGF molecules at short inter-EGF distances (<5.5 nm), like those previously invoked by simple no-FRET/high-FRET models employed to interpret ensemble FRET data (22,24–26). The other interface shows EGF pairs positioned in EGFR-EGF complexes with inter-EGF distances of ∼8 nm and has not been predicted by simple no-FRET/FRET models. The ∼8 nm distances could not solely be accounted for by a mixture of FRET states (i.e., colocalization of quenched and unquenched donors) because they were clearly observed for single EGF pairs, as reported by the single-step photobleaching in donor and acceptor channels (Fig. 3, A, lower, B, and C). Distances of ∼8 nm, therefore, need to be taken into consideration in interpretations of ensemble FRET data. We propose that such distances represent a mixture of conformations in the EGFR-EGF complex that were trapped by the cell fixation procedure and/or the kinetic intermediates previously proposed (68).
We have also provided the first evidence of the much sought structural differences between HA and LA EGFR complexes. The distance measured from EGF to the plasma membrane for HA EGFR complexes is ∼3.5 nm, whereas this distance is >7 nm for LA EGFR complexes, implying two different orientations for HA and LA ectodomains with respect to the plasma membrane. Different orientations were also found for HA and LA ectodomains in cells pre-fixed before exposure to EGF, showing the two different orientations are independent of EGF binding. This suggests that the EGFR ectodomain orientation must instead be regulated by interactions between EGFR and other cellular components (e.g., cytoskeleton (69), endocytic machinery (7), and lipid rafts (8)). The different constitutive orientations of HA and LA EGFR ectodomains also suggest that the ability to selectively block EGF binding to the HA and LA EGFR populations shown by different monoclonal anti-EGFR antibodies is due to steric constraints. We speculate that the different ectodomain orientations could give rise to the two affinities of EGFR for EGF binding because the electrostatic environment of the EGF-binding site is very different for the two orientations. The HA binding site would be much closer to the plasma membrane and therefore affected by the surface and dipole membrane potentials, which are significant up to ∼4 nm from the cell surface (70). HA EGF binding might therefore be regulated at the cell surface by different electrostatic and van der Waals forces.
Our single-molecule FRET results do not provide any clear evidence for the formation of stoichiometric oligomers larger than dimers at the sub-10-nm scale. However, because of the “resolution gap” between FRET measurements (<10 nm) and the optical resolution of the microscope (∼500 nm), we cannot quantify inter-EGFR distances between 10 nm and 500 nm in complexes observed to contain more than two receptors (Fig. 3 A and Table 2). As fluorescence spots were observed to remain intact while slowly diffusing laterally and given the low density of spots per image, we conclude that EGFR groups larger than dimers must be held together by additional interactions between these receptors and/or with other cell structures, for example, within lipid rafts smaller than the PSF of the microscope (8). We cannot, therefore, rule out the possibility that the observed groups of EGFRs are stoichiometric oligomers, as has previously been suggested (24). If this is the case, our data suggest that EGFR ectodomain interfaces showing FRET in dimers (Fig. 3 A) are kept separated by >10 nm by their 3D ectodomain arrangement within the oligomers, i.e., preventing FRET interactions. Considering that the back-to-back dimer structure—generally believed to be involved in the formation of EGFR oligomers—would provide such a >10 nm “spacer” between pairs of high-FRET ectodomains, our data would, therefore, be consistent with previous models of EGFR oligomerization (24,25).
In light of the growing evidence for EGFR stoichiometric oligomerization in the form of trimers (25,31,71), tetramers (24,26), previous models of oligomers (24,25), and the current crystallographic evidence (Table 3), we explored the complexes we could build that are consistent with the inter-EGF distances and EGF/plasma membrane separation that we measured by FRET. Fig. 6, A and B, shows a model of a HA EGFR-EGF tetramer that incorporates a strong back-to-back interface and two weak head-to-head interfaces. These interfaces are both present as dimers in the crystallographic asymmetric unit of a ligand-bound EGFR complex (12), and the latter is consistent with the <5.5 nm inter-EGF distances we have measured. Unlike other weak interfaces, the head-to-head interface has a negative solvation free energy difference (Table 3) and is particularly interesting in the context of our FRET results because it has the antiparallel symmetry that allows four receptors to lie flat on the membrane, as shown for HA EGFRs, while still interacting via both interfaces. Furthermore, the back-to-back interface provides the required >10 nm “spacer” between the two high-FRET head-to-head ectodomain interfaces. We note that this arrangement may, in principle, also contain any number of EGFRs, including the trimers recently identified by gel electrophoresis (25,71), rather than solely consisting of dimers as proposed elsewhere (24). Fig. 3 E may be an example of five receptors in such a complex. We therefore propose that the combination of parallel and antiparallel interfaces in Fig. 6, A and B, reflects some of the ectodomain associations that could generate EGFR oligomers in vivo but only in HA EGFR complexes.
Figure 6.
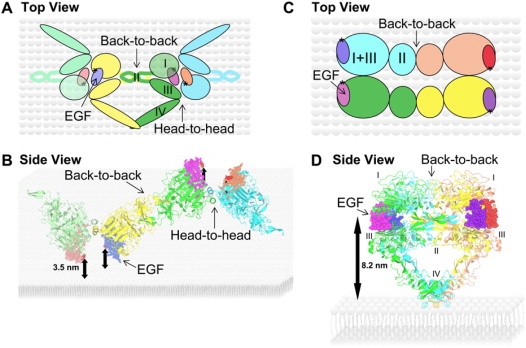
Model of HA and LA EGFR complexes. (A and B) Top and side views of a tetramer complex created through a combination of back-to-back and head-to-head interactions. The ectodomains need to lie on the cell surface for the two interactions to be able to occur simultaneously. Each receptor is represented by a color, and EGFR domains I–IV are labeled. Four EGF molecules are bound, one to each receptor in the tetramer. Dye molecules are represented by an asterisk. (C and D) Top and side views of an EGFR tetramer generated by placing two back-to-back dimers one behind the other. The side views of the tetramers (B and D) show the predicted distances between bound EGF and the cell surface (3.5 and 8.2 nm, respectively).
To model LA EGFR complexes, we placed two back-to-back EGFR dimers side-by-side so that there are four EGFRs in a rectangle, each aligned perpendicularly to the membrane (Fig. 6, C and D), as previously suggested (24). Simply placing two rigid back-to-back crystal structures side-by-side does not produce a good interface, because it does not take into account any conformational changes that would have to take place on interface formation. However, it does suggest that if such a tetramer did exist, the distance between the EGF N-termini along the short side of the rectangle formed would be ∼3.5 nm, comparable with the single-molecule FRET results (Fig. 3, A and D). The requirement inferred from FRET measurements (Fig. 5 D) that the distance from EGF to the plasma membrane be >7 nm would also be satisfied. We propose that the combination of interfaces depicted reflects some of the ectodomain associations that could generate LA EGFR tetramers in vivo.
Apart from domain II/IV interactions (Table 3), which are only observed in inactive EGFR states, there is no crystallographic evidence or models proposed that can explain the ∼8 nm inter-EGF distances we first (to our knowledge) report here. We have suggested above that these distances could result from conformational and/or kinetic intermediates. Work is currently underway to develop models that can explain how these interfaces might participate in the formation of EGFR-EGF complexes.
Based on our model for HA EGFR complexes (Fig. 6, A and B), we have produced a cartoon suggesting a possible mechanism for HA EGFR signal transduction (Fig. 7). We note that the resulting differences in the symmetries and orientations of the intracellular domains, which arise from the ectodomain interfaces and orientations identified here, provide a structural basis by which HA and LA signaling specificity can be achieved. In the tetramer shown, the alternation of back-to-back and head-to-head (parallel and antiparallel) ectodomains leads to an inherent asymmetry by which nearest ectodomain neighbors belong to a different receptor pair to that of the nearest kinase neighbors. Kinase activation would therefore involve interactions between three receptors arranged to allow optimal phosphorylation of the C-terminal tails as previously described (72). This may explain the mismatch between the twofold screw axis symmetry between ectodomains of HA EGFR and threefold screw axis asymmetry of the kinase domains, why constitutive dimers are inactive, and how signals can be regulated by oligomerization. The current model of active EGFR tetramers (24) is only consistent in A431 cells with the configuration of the LA EGFR population. Their different configuration, extracellularly and intracellularly, to HA receptors must therefore play a role in the exclusive ability of LA EGFRs to bind the EGF-like ligand Epigen and in the control of evasion of ubiquitination and downregulation (10,11).
Figure 7.
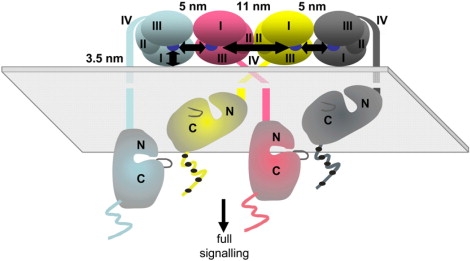
Model of HA EGFR signaling complexes. EGF bound to HA receptors in a tetramer involving a combination of two distinct configurations, giving distances between EGF ligands and between EGF and the plasma membrane that are consistent with our FRET data. This configuration results in a different intracellular kinase domain arrangement, which may explain how EGFR oligomers achieve multiple levels of signaling.
Interestingly, in the crystal structure of the tethered ErbB3 (64), a closely related EGFR family member, the packing of molecules also reveals an antiparallel dimer (PDB ID code 1m6b) with a strong interface (Table 3) (64). The dimer is generated by crystal symmetry, with domain I of one molecule in contact with domain III of the other and vice versa. This interaction creates a strong interface, burying 1877 Å2 per receptor, and is highly favorable as shown by the large negative ΔG value and a p-value close to zero. Because the ErbB3 monomers are in an inactive conformation, this arrangement could point toward an antiparallel constitutive dimer. Although the details of this interface are different from those of the head-to-head structure of EGFR, it does suggest that ectodomain antiparallel arrangements are possible in other receptors. This new mode of oligomer formation, involving interfaces with different symmetries, may therefore be generally present in interactions between all four EGFR family members, regulate the selection of heterodimerization partners among members of the EGFR family, and explain how signals propagate in normal and cancerous cells.
Supplementary material
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Appendix A.
Supplement
Figure
Figure
{kind=link}
Movie
Acknowledgments
The authors are indebted to Louise N. Johnson and Tony Ng for helpful discussions and technical advice and to David Robinson for his help in the design and building of the instrumentation.
We gratefully acknowledge funding from the UK Biotechnology and Biological Sciences Research Council (BBSRC) grant numbers 719/B18301, BB/C51464X/1, BB/E000215/1.
Footnotes
Editor: Thomas Schmidt.
References
- 1.Carpenter G. Receptors for epidermal growth factor and other polypeptide mitogens. Annu. Rev. Biochem. 1987;56:881–914. doi: 10.1146/annurev.bi.56.070187.004313. [DOI] [PubMed] [Google Scholar]
- 2.Ullrich A., Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 3.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 4.Defize L.H., Boonstra J., Meisenhelder J., Kruijer W., Tertoolen L.G., Tilly B.C., Hunter T., van Bergen en Henegouwen P.M., Moolenaar W.H., de Laat S.W. Signal transduction by epidermal growth factor occurs through the subclass of high affinity receptors. J. Cell Biol. 1989;109:2495–2507. doi: 10.1083/jcb.109.5.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Defize L.H., Arndt-Jovin D.J., Jovin T.M., Boonstra J., Meisenhelder J., Hunter T., de Hey H.T., de Laat S.W. A431 cell variants lacking the blood group A antigen display increased high affinity epidermal growth factor-receptor number, protein-tyrosine kinase activity, and receptor turnover. J. Cell Biol. 1988;107:939–949. doi: 10.1083/jcb.107.3.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mattoon D., Klein P., Lemmon M.A., Lax I., Schlessinger J. The tethered configuration of the EGF receptor extracellular domain exerts only a limited control of receptor function. Proc. Natl. Acad. Sci. USA. 2004;101:923–928. doi: 10.1073/pnas.0307286101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ringerike T., Stang E., Johannessen L.E., Sandnes D., Levy F.O., Madshus I.H. High-affinity binding of epidermal growth factor (EGF) to EGF receptor is disrupted by overexpression of mutant dynamin (K44A) J. Biol. Chem. 1998;273:16639–16642. doi: 10.1074/jbc.273.27.16639. [DOI] [PubMed] [Google Scholar]
- 8.Pike L.J. Growth factor receptors, lipid rafts and caveolae: an evolving story. Biochim. Biophys. Acta. 2005;1746:260–273. doi: 10.1016/j.bbamcr.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 9.Brabyn C.J., Kleine L.P. EGF causes hyperproliferation and apoptosis in T51B cells: involvement of high and low affinity EGFR binding sites. Cell. Signal. 1995;7:139–150. doi: 10.1016/0898-6568(94)00073-k. [DOI] [PubMed] [Google Scholar]
- 10.Kamer A.R., Sacks P.G., Vladutiu A., Liebow C. EGF mediates multiple signals: dependence on the conditions. Int. J. Mol. Med. 2004;13:143–147. [PubMed] [Google Scholar]
- 11.Kochupurakkal B.S., Harari D., Di-Segni A., Maik-Rachline G., Lyass L., Gur G., Kerber G., Citri A., Lavi S., Eilam R., Chalifa-Caspi V., Eshhar Z., Pikarsky E., Pinkas-Kramarski R., Bacus S.S., Yarden Y. Epigen, the last ligand of ErbB receptors, reveals intricate relationships between affinity and mitogenicity. J. Biol. Chem. 2005;280:8503–8512. doi: 10.1074/jbc.M413919200. [DOI] [PubMed] [Google Scholar]
- 12.Garrett T.P., McKern N.M., Lou M., Elleman T.C., Adams T.E., Lovrecz G.O., Zhu H.J., Walker F., Frenkel M.J., Hoyne P.A., Jorissen R.N., Nice E.C., Burgess A.W., Ward C.W. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell. 2002;110:763–773. doi: 10.1016/s0092-8674(02)00940-6. [DOI] [PubMed] [Google Scholar]
- 13.Ogiso H., Ishitani R., Nureki O., Fukai S., Yamanaka M., Kim J.H., Saito K., Sakamoto A., Inoue M., Shirouzu M., Yokoyama S. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110:775–787. doi: 10.1016/s0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]
- 14.Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell. 2002;110:669–672. doi: 10.1016/s0092-8674(02)00966-2. [DOI] [PubMed] [Google Scholar]
- 15.Ferguson K.M., Berger M.B., Mendrola J.M., Cho H.-S., Leahy D.J., Lemmon M.A. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell. 2003;11:507–517. doi: 10.1016/s1097-2765(03)00047-9. [DOI] [PubMed] [Google Scholar]
- 16.Livneh E., Prywes R., Kashles O., Reiss N., Sasson I., Mory Y., Ullrich A., Schlessinger J. Reconstitution of human epidermal growth factor receptors and its deletion mutants in cultured hamster cells. J. Biol. Chem. 1986;261:12490–12497. [PubMed] [Google Scholar]
- 17.Klein P., Mattoon D., Lemmon M.A., Schlessinger J. A structure-based model for ligand binding and dimerization of EGF receptors. Proc. Natl. Acad. Sci. USA. 2004;101:929–934. doi: 10.1073/pnas.0307285101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lemmon M.A., Bu Z., Ladbury J.E., Zhou M., Pinchasi D., Lax I., Engelman D.M., Schlessinger J. Two EGF molecules contribute additively to stabilization of the EGFR dimer. EMBO J. 1997;16:281–294. doi: 10.1093/emboj/16.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wofsy C., Goldstein B., Lund K., Wiley H.S. Implications of epidermal growth factor (EGF) induced EGF receptor aggregation. Biophys. J. 1992;63:98–110. doi: 10.1016/S0006-3495(92)81572-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Özcan F., Klein P., Lemmon M.A., Lax I., Schlessinger J. On the nature of low- and high-affinity EGF receptors on living cells. Proc. Natl. Acad. Sci. USA. 2006;103:5735–5740. doi: 10.1073/pnas.0601469103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stryer L., Haugland R.P. Energy transfer: a spectroscopic ruler. Proc. Natl. Acad. Sci. USA. 1967;58:719–726. doi: 10.1073/pnas.58.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gadella T.W., Jovin T.M. Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. J. Cell Biol. 1995;129:1543–1558. doi: 10.1083/jcb.129.6.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin-Fernandez M., Clarke D.T., Tobin M.J., Jones S.V., Jones G.R. Preformed oligomeric epidermal growth factor receptors undergo an ectodomain structure change during signaling. Biophys. J. 2002;82:2415–2427. doi: 10.1016/S0006-3495(02)75585-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clayton A.H.A., Walker F., Orchard S.G., Henderson C., Fuchs D., Rothacker J., Nice E.C., Burgess A.W. Ligand-induced dimer-tetramer transition during the activation of the cell surface epidermal growth factor receptor—a multidimensional microscopy analysis. J. Biol. Chem. 2005;280:30392–30399. doi: 10.1074/jbc.M504770200. [DOI] [PubMed] [Google Scholar]
- 25.Whitson K.B., Beechem J.M., Beth A.H., Staros J.V. Preparation and characterization of Alexa Fluor 594-labeled epidermal growth factor for fluorescence resonance energy transfer studies: application to the epidermal growth factor receptor. Anal. Biochem. 2004;324:227–236. doi: 10.1016/j.ab.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 26.Clayton A.H., Tavarnesi M.L., Johns T.G. Unligated epidermal growth factor receptor forms higher order oligomers within microclusters on A431 cells that are sensitive to tyrosine kinase inhibitor binding. Biochemistry. 2007;46:4589–4597. doi: 10.1021/bi700002b. [DOI] [PubMed] [Google Scholar]
- 27.Walker F., Orchard S.G., Jorissen R.N., Hall N.E., Zhang H.-H., Hoyne P.A., Adams T.E., Johns T.G., Ward C., Garrett T.P.J., Zhu H.-J., Nerrie M., Scott A.M., Nice E.C., Burgess A.W. CR1/CR2 interactions modulate the functions of the cell surface epidermal growth factor receptor. J. Biol. Chem. 2004;279:22387–22398. doi: 10.1074/jbc.M401244200. [DOI] [PubMed] [Google Scholar]
- 28.Francoual M., Etienne-Grimaldi M.C., Formento J.L., Benchimol D., Bourgeon A., Chazal M., Letoublon C., Andre T., Gilly N., Delpero J.R., Lasser P., Spano J.P., Milano G. EGFR in colorectal cancer: more than a simple receptor. Ann. Oncol. 2006;17:962–967. doi: 10.1093/annonc/mdl037. [DOI] [PubMed] [Google Scholar]
- 29.Fabricant R.N., De Larco J.E., Todaro G.J. Binding and thermal dissociation of nerve growth factor and its receptor on human melanoma cells. Biochem. Biophys. Res. Commun. 1977;79:299–304. doi: 10.1016/0006-291x(77)90095-x. [DOI] [PubMed] [Google Scholar]
- 30.Ichinose J., Murata M., Yanagida T., Sako Y. EGF signalling amplification induced by dynamic clustering of EGFR. Biochem. Biophys. Res. Commun. 2004;324:1143–1149. doi: 10.1016/j.bbrc.2004.09.173. [DOI] [PubMed] [Google Scholar]
- 31.Furuuchi K., Berezov A., Kumagai T., Greene M.I. Targeted antireceptor therapy with monoclonal antibodies leads to the formation of inactivated tetrameric forms of ErbB receptors. J. Immunol. 2007;178:1021–1029. doi: 10.4049/jimmunol.178.2.1021. [DOI] [PubMed] [Google Scholar]
- 32.Ringerike T., Blystad F.D., Levy F.O., Madshus I.H., Stang E. Cholesterol is important in control of EGF receptor kinase activity but EGF receptors are not concentrated in caveolae. J. Cell Sci. 2002;115:1331–1340. doi: 10.1242/jcs.115.6.1331. [DOI] [PubMed] [Google Scholar]
- 33.Gusev K., Glouchankova L., Zubov A., Kaznacheyeva E., Wang Z., Bezprozvanny I., Mozhayeva G.N. The store-operated calcium entry pathways in human carcinoma A431 cells: functional properties and activation mechanisms. J. Gen. Physiol. 2003;122:81–94. doi: 10.1085/jgp.200308815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wheeler M., Domin J. Recruitment of the class II phosphoinositide 3-kinase C2β to the epidermal growth factor receptor: role of Grb2. Mol. Cell. Biol. 2001;21:6660–6667. doi: 10.1128/MCB.21.19.6660-6667.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sadowski H.B., Shuai K., Darnell J.E., Gilman M.Z. A common nuclear signal transduction pathway activated by growth factor and cytokine receptors. Science. 1993;261:1739–1744. doi: 10.1126/science.8397445. [DOI] [PubMed] [Google Scholar]
- 36.Reis-Filho J., Milanezi F., Carvalho S., Simpson P., Steele D., Savage K., Lambros M., Pereira E., Nesland J., Lakhani S., Schmitt F. Metaplastic breast carcinomas exhibit EGFR, but not HER2, gene amplification and overexpression: immunohistochemical and chromogenic in situ hybridization analysis. Breast Cancer Res. 2005;7:R1028–R1035. doi: 10.1186/bcr1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baron A.T., Lafky J.M., Boardman C.H., Balasubramaniam S., Suman V.J., Podratz K.C., Maihle N.J. Serum sErbB1 and epidermal growth factor levels as tumor biomarkers in women with stage III or IV epithelial ovarian cancer. Cancer Epidemiol. Biomarkers Prev. 1999;8:129–137. [PubMed] [Google Scholar]
- 38.Nelson J.M., Fry D.W. Cytoskeletal and morphological changes associated with the specific suppression of the epidermal growth factor receptor tyrosine kinase activity in A431 human epidermoid carcinoma. Exp. Cell Res. 1997;233:383–390. doi: 10.1006/excr.1997.3571. [DOI] [PubMed] [Google Scholar]
- 39.Schagger H., von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100kDa. Anal. Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 40.Bellot F., Moolenaar W., Kris R., Mirakhur B., Verlaan I., Ullrich A., Schlessinger J., Felder S. High-affinity epidermal growth factor binding is specifically reduced by a monoclonal antibody, and appears necessary for early responses. J. Cell Biol. 1990;110:491–502. doi: 10.1083/jcb.110.2.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin-Fernandez M.L., Clarke D.T., Tobin M.J., Jones G.R. Real-time studies of the interactions between epidermal growth factor and its receptor during endocytic trafficking. Cell Mol. Biol. (Noisy-le-grand) 2000;46:1103–1112. [PubMed] [Google Scholar]
- 42.Webb S.E.D., Needham S.R., Roberts S.K., Martin-Fernandez M.L. Multidimensional single-molecule imaging in live cells using total-internal-reflection fluorescence microscopy. Opt. Lett. 2006;31:2157–2159. doi: 10.1364/ol.31.002157. [DOI] [PubMed] [Google Scholar]
- 43.Crocker J.C., Grier D.G. Methods of digital video microscopy for colloidal studies. J. Colloid Interface Sci. 1996;179:298–310. [Google Scholar]
- 44.Crocker, J. C., and E. R. Weeks. Particle Tracking Using IDL. http://www.physics.emory.edu/∼weeks/idl/.
- 45.Zhang S., Wang G., Fernig D.G., Rudland P.S., Webb S.E., Barraclough R., Martin-Fernandez M. Interaction of metastasis-inducing S100A4 protein in vivo by fluorescence lifetime imaging microscopy. Eur. Biophys. J. 2005;34:19–27. doi: 10.1007/s00249-004-0428-x. [DOI] [PubMed] [Google Scholar]
- 46.Yasuda R., Masaike T., Adachi K., Noji H., Itoh H., Kinosita K. The ATP-waiting conformation of rotating F1-ATPase revealed by single-pair fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA. 2003;100:9314–9318. doi: 10.1073/pnas.1637860100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sako Y., Minoghchi S., Yanagida T. Single-molecule imaging of EGFR signalling on the surface of living cells. Nat. Cell Biol. 2000;2:168–172. doi: 10.1038/35004044. [DOI] [PubMed] [Google Scholar]
- 48.Dale R.E., Eisinger J., Blumberg W.E. The orientational freedom of molecular probes. The orientation factor in intramolecular energy transfer. Biophys. J. 1979;26:161–193. doi: 10.1016/S0006-3495(79)85243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.CCP4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763. [DOI] [PubMed]
- 50.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 52.Potterton L., McNicholas S., Krissinel E., Gruber J., Cowtan K., Emsley P., Murshudov G.N., Cohen S., Perrakis A., Noble M. Developments in the CCP4 molecular-graphics project. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2288–2294. doi: 10.1107/S0907444904023716. [DOI] [PubMed] [Google Scholar]
- 53.Krissinel E., Henrick K. Detection of protein assemblies in crystals. In: Berthold M.R., Glen R., Diederichs K., Kohlbacher O., Fischer I., editors. Computational Life Sciences. Springer; Berlin, Heidelberg, Germany: 2005. pp. 163–174. [Google Scholar]
- 54.Suhre K., Sanejouand Y.H. ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32:W610–W614. doi: 10.1093/nar/gkh368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hohng S., Joo C., Ha T. Single-molecule three-color FRET. Biophys. J. 2004;87:1328–1337. doi: 10.1529/biophysj.104.043935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Belzen N., Rijken P.J., Hage W.J., de Laat S.W., Verkleij A.J., Boonstra J. Direct visualization and quantitative analysis of epidermal growth factor-induced receptor clustering. J. Cell. Physiol. 1988;134:413–420. doi: 10.1002/jcp.1041340312. [DOI] [PubMed] [Google Scholar]
- 57.Rees A.R., Gregoriou M., Johnson P., Garland P.B. High affinity epidermal growth factor receptors on the surface of A431 cells have restricted lateral diffusion. EMBO J. 1984;3:1843–1847. doi: 10.1002/j.1460-2075.1984.tb02057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chan E.Y.W., Stang S.L., Bottorff D.A., Stone J.C. Hypothermic stress leads to activation of Ras-Erk signaling. J. Clin. Invest. 1999;103:1337–1344. doi: 10.1172/JCI5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McCune B.K., Earp H.S. The epidermal growth factor receptor tyrosine kinase in liver epithelial cells. The effect of ligand-dependent changes in cellular location. J. Biol. Chem. 1989;264:15501–15507. [PubMed] [Google Scholar]
- 60.Schuler B., Lipman E.A., Steinbach P.J., Kumke M., Eaton W.A. Polyproline and the “spectroscopic ruler” revisited with single-molecule fluorescence. Proc. Natl. Acad. Sci. USA. 2005;102:2754–2759. doi: 10.1073/pnas.0408164102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ha T., Ting A.Y., Liang J., Caldwell W.B., Deniz A.A., Chemla D.S., Schultz P.G., Weiss S. Single-molecule fluorescence spectroscopy of enzyme conformational dynamics and cleavage mechanism. Proc. Natl. Acad. Sci. USA. 1999;96:893–898. doi: 10.1073/pnas.96.3.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shahrokh Z., Verkman A., Shohet S. Distance between skeletal protein 4.1 and the erythrocyte membrane bilayer measured by resonance energy transfer. J. Biol. Chem. 1991;266:12082–12089. [PubMed] [Google Scholar]
- 63.Hubbard S.R. EGF receptor activation: push comes to shove. Cell. 2006;125:1029–1031. doi: 10.1016/j.cell.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 64.Cho H.-S., Leahy D.J. Structure of the extracellular region of HER3 reveals an interdomain tether. Science. 2002;297:1330–1333. doi: 10.1126/science.1074611. [DOI] [PubMed] [Google Scholar]
- 65.Klausner R.D., Wolf D.E. Selectivity of fluorescent lipid analogs for lipid domains. Biochemistry. 1980;19:6199–6203. doi: 10.1021/bi00567a039. [DOI] [PubMed] [Google Scholar]
- 66.Mayo K.H., Nunez M., Burke C., Starbuck C., Lauffenburger D., Savage C.R. Epidermal growth factor receptor binding is not a simple one-step process. J. Biol. Chem. 1989;264:17838–17844. [PubMed] [Google Scholar]
- 67.Wiegant F.A., Blok F.J., Defize L.H., Linnemans W.A., Verkley A.J., Boonstra J. Epidermal growth factor receptors associated to cytoskeletal elements of epidermoid carcinoma (A431) cells. J. Cell Biol. 1986;103:87–94. doi: 10.1083/jcb.103.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Teramura Y., Ichinose J., Takagi H., Nishida K., Yanagida T., Sako Y. Single-molecule analysis of epidermal growth factor binding on the surface of living cells. EMBO J. 2006;25:4215–4222. doi: 10.1038/sj.emboj.7601308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van Bergen en Henegouwen P.M., Defize L.H., de Kroon J., van Damme H., Verkleij A.J., Boonstra J. Ligand-induced association of epidermal growth factor receptor to the cytoskeleton of A431 cells. J. Cell. Biochem. 1989;39:455–465. doi: 10.1002/jcb.240390411. [DOI] [PubMed] [Google Scholar]
- 70.O'Shea P. Intermolecular interactions with/within cell membranes and the trinity of membrane potentials: kinetics and imaging. Biochem. Soc. Trans. 2003;31:990–996. doi: 10.1042/bst0310990. [DOI] [PubMed] [Google Scholar]
- 71.Gan H.K., Walker F., Burgess A.W., Rigopoulos A., Scott A.M., Johns T.G. The epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor AG1478 increases the formation of inactive untethered EGFR dimers. Implications for combination therapy with monoclonal antibody 806. J. Biol. Chem. 2007;822:2840–2850. doi: 10.1074/jbc.M605136200. [DOI] [PubMed] [Google Scholar]
- 72.Zhang X., Gureasko J., Shen K., Cole P.A., Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 73.Li S., Schmitz K.R., Jeffrey P.D., Wiltzius J.J.W., Kussie P., Ferguson K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 74.Groenen L.C., Nice E.C., Burgess A.W. Structure-function relationships for the EGF/TGF-alpha family of mitogens. Growth Factors. 1994;11:235–257. doi: 10.3109/08977199409010997. [DOI] [PubMed] [Google Scholar]
- 75.Harris R.C., Chung E., Coffey R.J. EGF receptor ligands. Exp. Cell Res. 2003;284:2–13. doi: 10.1016/s0014-4827(02)00105-2. [DOI] [PubMed] [Google Scholar]
- 76.Berezov A., Chen J., Liu Q., Zhang H.-T., Greene M.I., Murali R. Disabling receptor ensembles with rationally designed interface peptidomimetics. J. Biol. Chem. 2002;277:28330–28339. doi: 10.1074/jbc.M202880200. [DOI] [PubMed] [Google Scholar]
- 77.Burgess A.W., Cho H.-S., Eigenbrot C., Ferguson K.M., Garrett T.P.J., Leahy D.J., Lemmon M.A., Sliwkowski M.X., Ward C.W., Yokoyama S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol. Cell. 2003;12:541–552. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 78.Saxon M.L., Lee D.C. Mutagenesis reveals a role for epidermal growth factor receptor extracellular subdomain IV in ligand binding. J. Biol. Chem. 1999;274:28356–28362. doi: 10.1074/jbc.274.40.28356. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement
Figure
Figure
Movie