

Abstract
The role of nitric oxide (NO) in regulating neutrophil migration has been investigated. Human neutrophil migration to interleukin (IL)-8 (1 nmol/L) was measured after a 1-hour incubation using a 96-well chemotaxis plate assay. The NO synthase inhibitor NG-nitro-l-arginine methyl ester (L-NAME) significantly (P < 0.001) enhanced IL-8-induced migration by up to 45%. Anti-CD18 significantly (P < 0.001) inhibited both IL-8-induced and L-NAME enhanced migration. Antibodies to L-selectin or PSGL-1 had no effect on IL-8-induced migration but prevented the increased migration to IL-8 induced by L-NAME. L-NAME induced generation of neutrophil-derived microparticles that was significantly (P < 0.01) greater than untreated neutrophils or D-NAME. This microparticle formation was dependent on calpain activity and superoxide production. Only microparticles from L-NAME and not untreated or D-NAME-treated neutrophils induced a significant (P < 0.01) increase in IL-8-induced migration and transendothelial migration. Pretreatment of microparticles with antibodies to L-selectin (DREG-200) or PSGL-1 (PL-1) significantly (P < 0.001) inhibited this effect. The ability of L-NAME-induced microparticles to enhance migration was found to be dependent on the number of microparticles produced and not an increase in microparticle surface L-selectin or PSGL-1 expression. These data show that NO can modulate neutrophil migration by regulating microparticle formation.
Accumulation and activation of inflammatory cells is vital to host defense but can also cause pathology. Neutrophils, for example, are critical for clearance of various pathogens but also cause injury and death of host tissue if their activity is misdirected or exaggerated.1 Induction of inflammation is associated with increased expression or altered avidity of adhesion molecules on endothelial cells and leukocytes, which increases the likelihood of interaction between these cell types.2,3 Initial attachment and rolling of neutrophils on endothelium is principally mediated by the selectin family of adhesion molecules, whereas stable adhesion and transmigration out of vessels is controlled by agents such as chemoattractants, integrins, members of the immunoglobulin superfamily, and junctional adhesion molecules.4,5,6,7,8
Nitric oxide (NO), a short-lived small molecule produced by most cell types, has a variety of well defined physiological and pathophysiological roles. A strong case for anti-inflammatory effects of NO is provided by studies showing that pharmacological inhibition of NO or genetic deletion of its synthases elevates leukocyte-endothelial cell interaction in diverse organs and tissues.9,10 This case is supported by studies showing that NO-releasing compounds inhibit neutrophil migration.11,12 Early investigations using human umbilical vein endothelial cells suggested that NO regulates leukocyte recruitment by modulating adhesion molecule expression on endothelial cells,13,14 although more recent studies refute this showing only minimal effects on microvascular endothelial cell adhesion molecule expression in vitro15 and in vivo.10 This suggests that regulation of adhesion by NO may involve direct effects on leukocytes.
Regulation of neutrophil function by NO is not straightforward however. In apparent conflict to the above, the NO synthase (NOS) inhibitor NG-monomethyl-l-arginine (L-NMMA) and the NO scavenger carboxy-PTIO attenuate neutrophil chemotaxis in vitro,16 and exogenously generated NO has been reported to both induce17 and inhibit16 neutrophil chemotaxis, supporting both pro- and anti-inflammatory activities of this mediator. The complexities of the effects of NO on inflammatory responses were highlighted in a recent investigation using inducible (i)NOS-deficient mice.18 Neutrophil sequestration in the pulmonary microvasculature in response to cecal ligation and puncture was reduced in iNOS−/− mice compared with wild types. In contrast, accumulation of neutrophils in bronchoalveolar lavage fluid was enhanced in the iNOS−/− animals, suggesting that NO can have differential effects on different stages of leukocyte recruitment.
Human neutrophils are reported to contain all three NOS enzymes.19,20,21,22,23 However, it is generally accepted that only endothelial (e)NOS is constitutively expressed in unstimulated peripheral blood neutrophils. Depending on levels produced, NO may be released to influence nearby cells or may act as an intracellular messenger regulating the activity of the cell responsible for its generation. Inside cells, NO stimulates guanylyl cyclase to increase cGMP formation and may influence a number of factors including chemotaxis,24 superoxide anion release,16 integrin expression,25 and F-actin polymerization.26 These and other functional responses of neutrophils are highly polarized suggesting that the messenger systems supporting them are restricted to localized intracellular compartments. Discrepancies between experiments dealing with inhibition of endogenous NOS versus exogenous application of NO-generating compounds may be partially explained by localized versus widespread activity of NO.
Given the complexity surrounding regulation of inflammation by NO, we used an in vitro transmigration assay to investigate direct effects of NO inhibition on human neutrophils. We find that the broad-spectrum NOS inhibitor NG-nitro-l-arginine methyl ester (L-NAME) enhances neutrophil migration in response to the chemokine interleukin (IL)-8 via a mechanism that is dependent on adhesion between L-selectin and P-selectin glycoprotein ligand-1 (PSGL-1). We have also identified a mechanism for this enhancement, finding that L-NAME-treated neutrophils generate L-selectin- and PSGL-1-expressing microparticles and that these coat the surface of artificial migration chambers or endothelial cells to support enhanced migration of subsequently added neutrophils.
Materials and Methods
Antibodies
Anti-human CD18 (6.5E) was a gift from M. Robinson, SLH Celltech Group, Slough, UK. Anti-human PSGL-1 (blocking PL-1 and nonblocking PL-2) were gifts from Professor R. McEver, University of Oklahoma, Norman, OK. Purified and phycoerythrin-conjugated anti-human L-selectin (DREG-200) and isotype control (MOPC-21) were purchased from Becton Dickinson (Oxford, UK).
Neutrophil Isolation
Venous blood was drawn from healthy adult volunteers and immediately transferred to tubes containing EDTA (1.6 mg/ml; Sarstedt Ltd., Beaumount Leys, Leicester, UK). Neutrophils were isolated from whole blood using a two-step density gradient. Briefly, 2.5 ml of high-density histopaque (1.119 g/ml; Sigma, Dorset, UK) was placed in a round-bottom 10-ml tube, and 2.5 ml of low-density histopaque (1.077 g/ml, Sigma) was carefully layered on top. Whole blood (5 ml) was then layered above the histopaque gradient and centrifuged for 30 minutes (700 × g at 20°C) to allow separation of the blood into its components. The granulocyte layer was harvested, resuspended in buffer [phosphate-buffered saline (PBS) containing 1 mmol/L Ca2+, 0.5 mmol/L Mg2, and supplemented with 0.1% low-endotoxin bovine serum albumin (BSA); Sigma], washed by centrifugation (350 × g for 6 minutes) and red blood cells lysed. Neutrophils were washed, counted using a hemocytometer, and centrifuged (350 × g for 6 minutes). Finally, neutrophils were diluted to the required concentration in RPMI (Invitrogen Ltd., Paisley, UK) supplemented with 0.1% BSA. Differential counts showed that preparations were consistently >97% neutrophils, which were >95% viable (as measured by Trypan blue dye exclusion).
Neutrophil Transmigration Assay
Neutrophil chemotaxis was measured in a 96-well chemotaxis chamber (Neuroprobe, Inc., Gaithersburg, MD) using a modification of the method described by Frevert and colleagues.27 Wells were filled with 25 μl of human IL-8 (PeproTech, Rocky Hill, NJ), RPMI, or neutrophils (5 × 104) resuspended in RPMI. A filter membrane was positioned over the loaded wells, and 25 μl of neutrophils (2 × 106/ml) were placed directly onto 3.0-μm filter sites. The chamber was incubated for 1 hour (37°C in 5% CO2), and any nonmigrated neutrophils were removed from the upper surface of the filter by wiping and washing with 25-μl aliquots of RPMI. Both neutrophils that had migrated to the underside of the filter and those that had migrated into the lower wells were counted. To dislodge any migrated cells adherent to the underside of the filter membrane, the plate with the filter attached was centrifuged (350 × g for 10 minutes). The filter was removed, and neutrophils in the wells of the chemotaxis plate were resuspended and counted using a hemocytometer. To correct for chemokinesis, equal concentrations of chemoattractant were placed above and below the filter in control wells. To assess the role of adhesion molecules, neutrophils were resuspended in saturating doses of antibodies: anti-CD18 (6.5E, 6 μg/106), anti-L-selectin (DREG-200, 15 μg/106), anti-PSGL-1 (PL-1, 10 μg/106), or the appropriate control antibody. Aliquots (25 μl) were then deposited directly onto the filter membranes, and the transmigration assay performed as above.
Generation of Neutrophil-Derived Microparticles
Human neutrophils (5 × 106/ml for each treatment, unless otherwise stated) were incubated with L-NAME (30 μmol/L; Tocris Cookson, Bristol, UK), its inactive enantiomer D-NAME (30 μmol/L, Sigma), N-formyl-Met-Leu-Phe (fMLP, 10 μmol/L; Sigma), or RPMI for 1 hour (37°C in 5% CO2) to stimulate microparticle formation. After incubation, microparticle-containing suspensions were cleared of large cellular fragments by centrifugation (350 × g for 6 minutes) and supernatants transferred to microcentrifuge tubes. After ultracentrifugation (100,000 × g for 2 hours),28 the microparticle pellets were resuspended in 100 μl of RPMI and 0.1% BSA.
To investigate the effects of microparticles on neutrophil migration, neutrophils were incubated with L-NAME and the resulting microparticles isolated by centrifugation. This was followed by dialysis to remove any remaining L-NAME. Dialyzed microparticles (5-μl aliquots) were deposited onto the filter membranes of chemotaxis chambers and incubated for 30 minutes (37°C, 5% CO2) before addition of neutrophils and chemoattractants.
Flow Cytometric Investigation of Microparticles
Neutrophils were labeled with the membrane-intercalating red fluorescent dye PKH26 using a cell linker kit according to the manufacturer’s instructions (Sigma). Microparticle formation was stimulated as above, and neutrophil suspensions, microparticle-containing supernatants, and filtered (0.2 μm) supernatants were compared using a FACScan flow cytometer (Becton Dickinson). Microparticles were defined as positive for red fluorescence and were identified and analytically separated from whole cells by their smaller forward and side scatter parameters and lower fluorescent intensities. A gate was positioned around the microparticle population and events analyzed for phosphatidylserine externalization using fluorescein isothiocyanate (FITC)-conjugated Annexin V (5 μl of Annexin 5 per 1 × 105 neutrophils; Becton Dickinson), which binds with high affinity to negatively charged phospholipids in the presence of calcium ions. The role of calpain and superoxide dismutase in microparticle generation was determined by adding the selective calpain inhibitor PD150606 or negative control PD145305 (1 to 3 μmol/L; Merck, Nottingham, UK)29 or superoxide dismutase (10 to 30 μg, Sigma) to isolated neutrophils together with the stimulus for microparticle formation. The number of microparticles formed was analyzed using flow cytometry as above.
To determine adhesion molecule expression on the surface of microparticles, neutrophils were labeled with PKH26 as above and incubated with D-NAME (30 μmol/L) or L-NAME (30 μmol/L) for 1 hour (37°C, 5% CO2). The resulting microparticles were incubated for 30 minutes at 4°C with saturating concentrations of FITC-conjugated antibodies directed against L-selectin (DREG-200) and PSGL-1 (PL-1), with isotype control (mouse IgG1), or with binding nonblocking anti-PSGL-1 (PL-2).
Measurement of Neutrophil Shape Change Using the Gated Autofluorescence/Forward Scatter Assay
The effects of stimuli on neutrophil shape change were assessed using a method based on that described by Sabroe and colleagues.30 Rested isolated human neutrophils (5 × 105) were placed in 1.2-ml polypropylene cluster tubes and incubated with 100 μl of PBS, PMA (10−9 mol/L), fMLP (10−5 mol/L), D-NAME (3 × 10−5 mol/L), or L-NAME (3 × 10−5 mol/L) for 6 minutes in a shaking water bath set at 37°C. Tubes were then removed and 250 μl of ice-cold optimized fixative added to terminate the reaction. Effects on neutrophil shape change were analyzed immediately using flow cytometry as described by Sabroe and colleagues.30
Measurement of Intracellular Calcium Mobilization Using Flow Cytometry
Fluo 3-acetoxymethyl ester (Fluo 3-AM) was used to measure intracellular calcium mobilization in response to stimuli according to the technique described by Storey and colleagues.31 Briefly, isolated neutrophils (5 × 106/ml) were resuspended in HEPES/Tyrode’s buffer (10 mmol/L HEPES, 137 mmol/L NaCl, 2.68 mmol/L KCl, 0.42 mmol/L Na2PO4, 11.9 mmol/L NaHCO3, 5 mmol/L glucose without CaCl2 and MgCl2), pH 7.4. To prevent leakage, probenecid (2.5 mmol/L) was added to all buffers.32 Neutrophils were incubated for 30 minutes (37°C, 5% CO2) with Fluo 3-AM (5 μmol/L, Invitrogen) and washed twice in buffer. Neutrophils were transferred to 1.2-ml polypropylene cluster tubes and CaCl2 (1 mmol/L) or EGTA (100 μmol/L) added. Baseline fluorescence was measured using flow cytometry. In selected samples, RPMI, D-NAME (30 μmol/L), L-NAME (30 μmol/L), or the calcium ionophore A23187 (10 μmol/L) was added, and fluorescence was immediately measured. The median fluorescence value (Ftest) was recorded every 15 seconds after stimulation for 2 minutes. Median fluorescence was also measured in an unlabeled sample (Fmin) and a sample containing A23187 (Fmax). Intracellular calcium concentration was calculated according to Storey and colleagues.31
Electron Microscopy
Microparticle formation was induced as described above. Microparticle-containing pellets were fixed using 3% glutaraldehyde (Sigma) in 0.1 mol/L phosphate buffer. Secondary fixation was performed using 2% osmium tetroxide (Sigma). The samples were embedded in Araldite resin for 48 to 72 hours at 60°C. Ultra-thin sections (70 to 90 nm) were cut using a Reichert Ultracut E ultramicrotome (Reichert, Depew, NY). The sections were stained using 3% uranyl acetate in 50% ethanol (Sigma) followed by staining with Reynolds lead citrate for 25 minutes (Polysciences Inc., Eppelheim, Germany). The sections were analyzed under transmission electron microscopy (Philips CM10; Philips, Eindhoven, Holland), and micrographs were recorded on Kodak electron microscope film (Sigma, Dorset, UK).
Neutrophil Transendothelial Migration
The methods used to investigate transendothelial migration were based on those described by Dunzendorfer and colleagues.33 Briefly, gelatin-coated Transwell filter inserts (Corning, Surrey, UK) were seeded with 1 × 105 human umbilical vein endothelial cells (HUVECs) and cultured in complete growth media in 24-well plates. Media were replaced every 2 days until a confluent monolayer was established (∼4 to 6 days after seeding). Monolayer permeability was checked using FITC-BSA (Sigma) leakage for 10, 30, 60, and 120 minutes. The monolayers were found to retain >93% FITC-BSA in the upper chamber of the plate. Once a confluent monolayer was established, neutrophil-derived microparticles were produced as described above. Media from the upper chamber was removed, the cells washed twice with PBS, and 20 μl of microparticles (derived from 5 × 106 neutrophils) were added to the filter. The plates were incubated for 30 minutes (37°C, 5% CO2) to allow the microparticles to coat the HUVECs. The Transwell filters were then moved to new plates and 600 μl of RPMI or IL-8 10−9 mol/L added to the lower wells. Neutrophils (5 × 105 in 100 μl of RPMI) were then added to the upper chamber and the plates incubated for 1 hour (37°C, 5% CO2) to allow migration to occur. The upper chambers were then carefully washed twice with fresh RPMI to remove any remaining neutrophils. To dislodge any migrated cells adherent to the underside of the filter membrane, the plate with the filter attached was centrifuged (350 × g for 10 minutes). The filter was removed and neutrophils in the wells of the chemotaxis plate were resuspended and counted using a hemocytometer. One set of wells was used to correct for chemokinesis by placing IL-8 (10−9 mol/L) in both the upper chambers and lower wells.
Statistical Analyses
Results are presented as mean ± SEM. Statistical analyses were performed using GraphPad Instat version 3.01 (GraphPad Software, San Diego, CA). One-way analysis of variance followed by Bonferroni’s or Dunnett’s test for multiple comparisons were performed on appropriate data sets. P values less than 0.05 were considered significant.
Results
Effect of L-NAME on Neutrophil Migration to IL-8
IL-8 caused dose-dependent neutrophil migration that was maximal at 10−8 mol/L (Figure 1A). Vehicle (RPMI) did not induce significant neutrophil chemotaxis in this and subsequent experiments (chemokinesis control = 4.1 ± 0.8%, n = 3; RPMI = 7.4 ± 0.5%, n = 3). A submaximal concentration of 10−9 mol/L was chosen for subsequent experiments so that increases or decreases in IL-8-induced migration might be investigated. L-NAME (30 μmol/L) significantly enhanced IL-8-induced neutrophil migration, whereas the inactive enantiomer, D-NAME, did not (Figure 1B). Higher concentrations of L-NAME (100 μmol/L and 300 μmol/L) and D-NAME significantly reduced neutrophil viability (data not shown). The effect of L-NAME (30 μmol/L) on IL-8-induced migration was reversed by L-Arginine (10 mmol/L) but not the nonmetabolized enantiomer, D-Arginine (10 mmol/L, Figure 1B). These data suggest that enhanced neutrophil migration to IL-8 in the presence of L-NAME was a direct consequence of reduced NO production.
Figure 1.
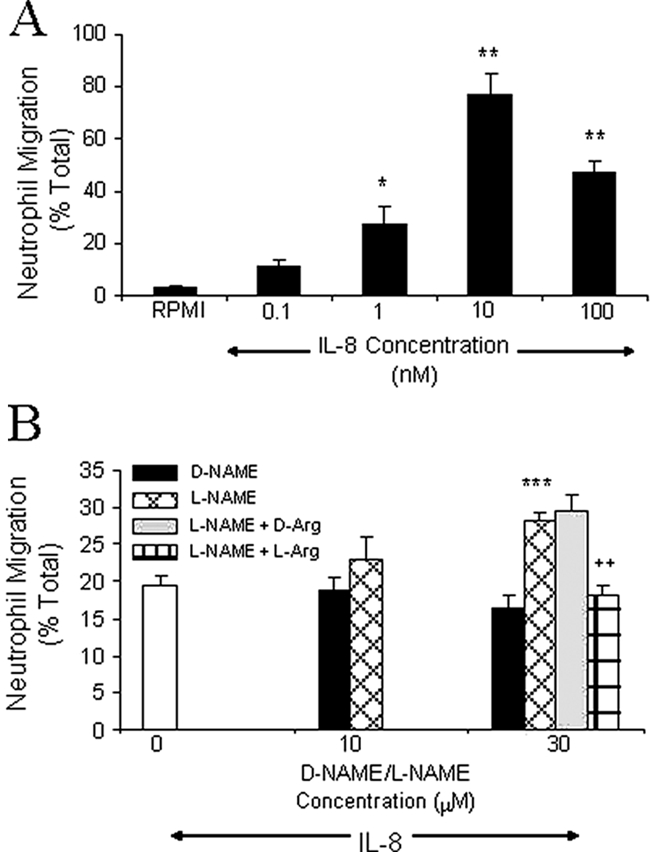
Effect of L-NAME on neutrophil migration to IL-8. A: The number of neutrophils that migrated for 1 hour to RPMI or IL-8 (0.1 to 100 nmol/L) was counted and results expressed as a percentage of the total number of neutrophils added to filter membranes of chemotaxis chambers. B: Isolated human neutrophils were resuspended in vehicle (RPMI, open bar), D-NAME, L-NAME, L-NAME with d-arginine (10 mmol/L), or L-NAME with l-arginine (10 mmol/L) and migration to IL-8 (1 nmol/L) assessed after 1 hour. Results are corrected for spontaneous migration by subtracting the percentage of neutrophils migrating to the chemokinesis control. Results are presented as mean ± SEM (n = 3 to 4) and analyzed for statistical significance using one-way analysis of variance followed by Dunnett’s t-test (A) or Bonferroni’s test (B) for multiple comparisons. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to vehicle. ++P < 0.01 compared to the IL-8 response of L-NAME-treated neutrophils.
Adhesion Mechanisms Involved in IL-8-Induced Migration in the Presence of L-NAME
Neutrophil migration toward IL-8 was significantly reduced by anti-CD18 (6.5E) antibody whether L-NAME was present or not (Figure 2A). The results demonstrate that CD18 is required for neutrophil migration toward IL-8, but do not identify the mechanism for enhanced migration induced by NO inhibition. Neutrophil-neutrophil and neutrophil-microparticle interactions, which may explain enhanced migration in the presence of L-NAME, may be L-selectin- and PSGL-1-dependent.34,35,36 We therefore investigated the role of L-selectin and PSGL-1 in enhanced IL-8-induced neutrophil migration in the presence of L-NAME (Figure 2, B and C). Interestingly, antibodies to L-selectin (DREG-200) or PSGL-1 (PL-1) had no effect on migration of untreated neutrophils to IL-8 but prevented the increased migration to IL-8 induced by L-NAME.
Figure 2.

The effect of adhesion molecule inhibition on L-NAME-induced enhanced migration to IL-8. Neutrophils were resuspended in RPMI, isotype control antibodies, anti-CD18 (6.5E) (A), anti-L-selectin (DREG-200) (B), or blocking and nonblocking anti-PSGL-1 (PL-1, PL-2), with or without L-NAME (C), and added to the filter membrane. Migration to IL-8 was assessed after 1 hour. Data are presented as mean ± SEM (n = 3 to 4) and analyzed for statistical significance using one-way analysis of variance followed by Bonferroni’s test for multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001 compared to the IL-8 response of neutrophils treated with isotype control antibody. +P < 0.05, +++P < 0.001 compared to the IL-8 response of neutrophils co-incubated with L-NAME and isotype control.
Regulation of Microparticle Formation by NO
Neutrophils incubated with L-NAME (Figure 3B) generated more microparticles than resting neutrophils (Figure 3A). The microparticles were analytically separated from intact neutrophils by their smaller forward (size) and side (granularity) scatter parameters and were positive for the membrane-binding dye PKH26GL. These microparticles were still present in large numbers after removal of intact neutrophils by centrifugation at 350 × g for 6 minutes (Figure 3C) but were removed by passage through a 0.2-μm filter (Figure 3D). The number of microparticles (as assessed by the number of events/minute counted in the microparticle gate) formed in response to L-NAME (3816 ± 370, n = 8) was comparable to that seen with the positive control, fMLP (3799 ± 288, n = 13), and was significantly (P < 0.05) different from vehicle-treated (2495 ± 210, n = 13) and D-NAME-treated (2510 ± 242, n = 8) cells.
Figure 3.
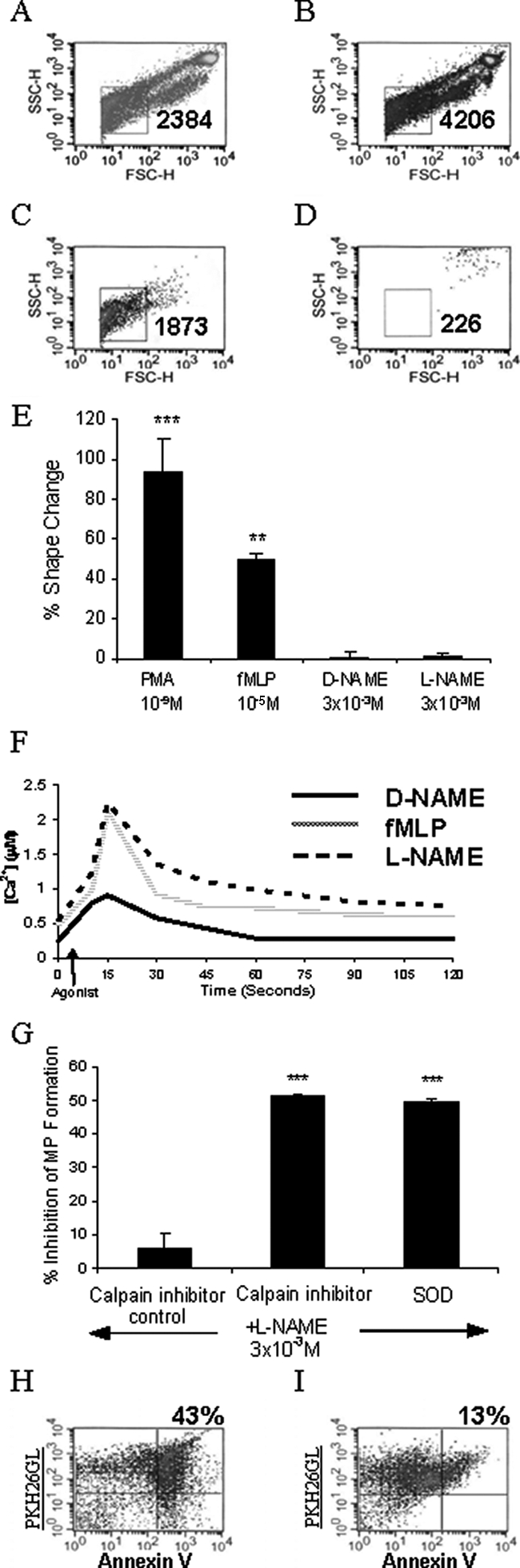
Analysis of L-NAME-induced microparticle formation. Neutrophils were labeled with PKH26 lipid-intercalating dye (4 μmol/L). Representative flow cytometry density plots showing PKH26-positive events from resting neutrophils (A), neutrophils incubated with L-NAME for 1 hour (B), cell-free supernatant from neutrophils incubated with L-NAME (C), and filtered (0.2 μm) supernatants (D) are shown. Microparticles were identified (square gate) by their smaller forward and side scatter parameters, and events counted for 60 seconds are shown. Density plots are representative of 8 to 13 experiments. E: Gated autofluorescence/forward scatter analysis is shown as mean ± SEM percent change, n = 4. Data were analyzed using one-way analysis of variance followed by Dunnett’s t-test. **P < 0.01, ***P < 0.001 compared to buffer control. F: Flow cytometry analysis of D-NAME, L-NAME, and fMLP effects on intracellular calcium in fluo-3 AM-labeled neutrophils for 2 minutes. Histogram is representative of four experiments. G: Events counted in the microparticle gate for 60 seconds were recorded and the percent inhibition calculated. Results are shown as mean ± SEM, n = 3. Data were analyzed using one-way analysis of variance followed by Dunnett’s t-test. ***P < 0.001 compared to L-NAME-stimulated control. H: Events in the microparticle gate were analyzed for phosphatidylserine externalization by labeling with FITC-conjugated Annexin-V in a calcium-containing buffer. I: As a negative control Annexin-V was added in the presence of EDTA. Microparticles were identified as Annexin V (FL-1)- and PKH26 (FL-2)-positive events located in the top right quadrant on the density plots (percent events shown). The data shown is representative of three experiments.
Chemotactic factors such as C5a and IL-8 are known to induce changes in the shape of human neutrophils.30 To investigate whether L-NAME induced a similar response, a gated autofluorescence/forward scatter assay was performed. PMA and fMLP induced a significant change in shape compared to unstimulated neutrophils whereas D-NAME and L-NAME did not (Figure 3E). However, using a calcium probe (fluo-3), it was found that L-NAME induced a rapid increase in intracellular calcium concentration that peaked 10 seconds after stimulation (Figure 3F). This was followed by a rapid decline and a sustained phase. The response was very similar to that seen after fMLP stimulation, but D-NAME was unable to elicit a pronounced increase in intracellular calcium.
Increased levels of intracellular calcium are known to activate the enzyme calpain, which in turn degrades membrane proteins.37 The selective calpain inhibitor PD150606 was used to investigate the effects of calpain inhibition on L-NAME-induced microparticle formation. PD150606 significantly (P < 0.001) inhibited microparticle formation and reduced the numbers produced down to those of unstimulated/resting cells. In the same experiment, the superoxide (O2−) scavenger superoxide dismutase was co-administered with L-NAME. Again the levels of microparticles derived from these neutrophils were significantly (P < 0.001) reduced to those of unstimulated cells.
Previous studies have shown that ectosomes released from human neutrophils in response to fMLP or PMA stimulation bind Annexin V.38 We therefore investigated binding of Annexin V by microparticles released from neutrophils treated with L-NAME. Microparticles were identified as Annexin V and PKH26GL-positive (Figure 3H, top right quadrant). Most of this binding was calcium-dependent as shown by the reduced number of events in the upper right quadrant in the presence of EDTA (Figure 3I). Formation of microparticles in response to L-NAME was also analyzed by electron microscopy (Figure 4). Neutrophil membrane blebs and microparticles are evident on those neutrophils stimulated with fMLP or L-NAME.
Figure 4.

Electron micrographs showing the formation of microparticles from human neutrophil membranes. Isolated neutrophils were resuspended in RPMI (A), fMLP (B), or L-NAME (C) and incubated for 1 hour. Neutrophils were then fixed in 3% glutaraldehyde-containing buffer and analyzed for microparticle formation.
Microparticles Enhance Neutrophil Migration
Having shown by flow cytometry that inhibition of endogenous NO production by L-NAME induced the formation of microparticles from the membrane of human neutrophils, the contribution of microparticles to the elevated migratory response toward IL-8 induced by L-NAME was investigated. Microparticles from untreated, D-NAME- and L-NAME-treated neutrophils were dispensed onto the filter of the chemotaxis plate and incubated for 30 minutes. Only microparticles derived from L-NAME-treated neutrophils enhanced migration to IL-8 (Figure 5A). Pretreating the microparticles with antibodies against either L-selectin or PSGL-1 reduced migration (Figure 5B). Levels of L-selectin and PSGL-1 expression per microparticle were not altered by different treatments (Figure 5C), suggesting that enhanced migration occurs in response to a higher number of microparticles rather than a change in their function. This was confirmed by adding microparticles derived from different numbers of neutrophils. At least 0.5 × 106 neutrophils were required to produce enough microparticles to significantly enhance migration (Figure 5D). To determine whether the effects of L-NAME-induced neutrophil-derived microparticles on neutrophil migration to IL-8 were altered in the presence of endothelial cells, transendothelial migration experiments were performed (Figure 5E). As in Figure 5A, coating HUVECs with microparticles derived from L-NAME-treated neutrophils enhanced neutrophil migration toward IL-8. Neutrophils did not migrate in the absence of IL-8.
Figure 5.

Microparticles enhance migration to IL-8. Microparticles derived from neutrophils (5 × 106) incubated for 1 hour with RPMI, D-NAME, or L-NAME were resuspended in RPMI (A) or isotype control, nonblocking anti-PSGL-1, anti-L-selectin, or blocking anti-PSGL-1 (B), and migration to IL-8 assessed after 1 hour. C: Microparticle adhesion molecule expression was assessed using PKH26-labeled neutrophils incubated with D-NAME (top) or L-NAME (bottom) for 1 hour. Microparticles were labeled with FITC-anti-L-selectin, FITC-anti-PSGL-1 (shown by the superimposable dark and light gray lines, respectively), or FITC-isotype control (solid area) for 30 minutes at 4°C and analyzed by flow cytometry. Histograms are representative of three experiments. D: The influence of microparticle density was determined by using 0.05 to 1.25 × 106 neutrophils stimulated with L-NAME as a source of microparticles. Migration to IL-8 was measured after 1 hour. Results are presented as mean ± SEM (n = 4). Data were analyzed for statistical significance using one-way analysis of variance followed by Dunnett’s t-test. *P < 0.05, **P < 0.01 compared to the response in the absence of added microparticles, +++P < 0.001 compared to isotype control. E: Migration experiments in A were repeated in the presence of HUVEC monolayer cultured on filter inserts. Results are shown as mean ± SEM from two experiments using two separate blood donors and two separate HUVEC preparations. Each experiment contained four replicates of each treatment.
Discussion
The current paradigm for the involvement of NO in inflammation is that low levels of NO produced by constitutive NOS enzymes are anti-inflammatory, whereas higher, sustained levels produced by inducible NOS are proinflammatory. The evidence from this study further supports a role of NOS in regulating inflammation through modulation of neutrophil migration. We found that incubation of unstimulated human peripheral blood neutrophils with the nonspecific NOS inhibitor, L-NAME, significantly increased the percentage of neutrophils migrating toward IL-8. L-Arginine reversed this effect, showing the dependence on inhibition of NO production. Studies using neutrophils isolated from iNOS-deficient mice showed significant reduction in the enhanced migration to KC (murine equivalent of IL-8) induced by L-NAME (see supplementary Figure S1 at http://ajp.amjpathol.org). However, this inhibition was not total, suggesting involvement of another NOS isoform.
The migration of neutrophils in response to IL-8, regardless of the presence of L-NAME, occurred via a predominantly integrin-dependent pathway because saturating concentrations of anti-CD18 function-blocking antibody almost completely abolished migration. These results supported previous findings that chemoattractant-induced human neutrophil migration on artificial surfaces occurs through an integrin-dependent mechanism39,40 but did not identify the mechanism by which L-NAME increases migration.
Inhibition of L-selectin and PSGL-1 attenuated the enhanced migration to IL-8 induced by L-NAME. These same antibodies, however, had no effect on neutrophil migration to IL-8 in the absence of L-NAME. This finding and data published in previous studies showing the dependence of neutrophil-neutrophil and neutrophil-microparticle interactions on L-selectin and PSGL-134,35,36,41,42 led us to investigate the role of neutrophil microparticles in the response to L-NAME. Microparticles are formed from the membrane of activated cells (eg, leukocytes, platelets, endothelial cells) and express adhesion molecules that enable them to influence cell-cell interactions43 and cell recruitment.44 Data presented here demonstrate that inhibition of endogenous NO production by treatment with the nonselective NOS inhibitor L-NAME is a sufficient stimulus for the formation of microparticles from neutrophils.
Flow cytometric analysis of the microparticles produced by neutrophils stimulated with L-NAME showed the presence of L-selectin and PSGL-1 on their surface. Gasser and colleagues38 have also reported that L-selectin is expressed on neutrophil-derived microparticles after stimulation with fMLP (1 μmol/L) or PMA (10 nmol/L). However, the levels of adhesion molecules expressed by neutrophil-derived microparticles were the same irrespective of the stimulus. We determined that the significantly increased number of microparticles derived from neutrophils in the presence of L-NAME was the major contributing factor to the enhanced migration to IL-8 observed. Initial experiments showed that the smaller numbers of microparticles produced in response to D-NAME had no significant effect on migration whereas the larger quantities of microparticles formed in response to L-NAME treatment significantly enhanced the migratory response toward IL-8. To reinforce this hypothesis, increasing numbers of neutrophils were stimulated with L-NAME to prepare increasing numbers of microparticles. It was found that the amount of migration was directly proportional to the number of neutrophils from which microparticles were derived. Therefore it was the number rather than an alteration in the adhesive properties of microparticles generated in response to L-NAME stimulation that enhanced neutrophil migration.
Although we have shown that small numbers of microparticles are present even in the absence of L-NAME, we only observed significant effects of L-selectin and PSGL-1 inhibition when L-NAME was present. These findings, together with the findings described above, suggest a threshold in the number of microparticles must be reached for migration to be enhanced by their presence. Therefore, migration was only enhanced when the number of microparticles present on the filter was significantly greater than those generated from unstimulated neutrophils, and this enhancement was dependent on L-selectin and PSGL-1.
Microparticles produced by different cell types in response to different stimuli have diverse properties including size, phospholipid content, and functional roles.45 However, previous studies have shown that placing platelet-derived microparticles on membrane filters enhances monocyte chemotaxis.46 Adhesion molecules expressed by neutrophil-derived microparticles may contribute to their adhesive properties and potentially enhance the migratory response toward IL-8 by promoting neutrophil-microparticle interactions. It is hypothesized that neutrophils suspended in L-NAME and dispensed onto the filter sites of the chemotaxis chamber release microparticles that coat the filter membrane. Neutrophil-derived microparticles express PSGL-1 and L-selectin and may capture and recruit intact neutrophils to the filter membrane through interactions between PSGL-1 on the microparticle and L-selectin on the neutrophil or vice versa. In the absence of P-selectin, leukocyte-expressed PSGL-1 can mediate L-selectin-dependent leukocyte rolling in vivo.36 Function-blocking antibodies that recognize PSGL-1 and L-selectin on the microparticles significantly attenuated neutrophil migration toward the chemoattractant in vitro. Once recruited to the membrane, exposure to the chemoattractant (IL-8) initiates the process of neutrophil migration.
In this study L-NAME has been shown to mediate neutrophil migration by inducing microparticle formation. The NO donor, sodium nitroprusside (SNP), has been shown to inhibit the catalytic activity of the calcium-dependent proteinase calpain47,48 that is normally responsible for the degradation of talin, an essential structural component of the neutrophil cytoskeleton and the shedding of membrane-derived vesicles.49 Using a calcium probe (fluo-3), we have found that L-NAME induces a rapid increase in intracellular calcium concentration in neutrophils that peaked 10 seconds after stimulation, similar to that seen after fMLP stimulation. This response was not evident in L-NAME-stimulated neutrophils in the presence of the calcium chelator EGTA. Also, the selective calpain inhibitor PD150606 significantly inhibited microparticle formation, suggesting that L-NAME increases activity of calpain leading to increased degradation of membrane proteins and an increase in microparticle generation.
It is known that there is a balance between NO and superoxide (O2−), and by reducing the levels of NO with L-NAME, the levels of O2− are increased.50,51 It is possible, therefore, that the effects of L-NAME on microparticle formation seen in this study may be attributable to O2−. Isolated neutrophils co-incubated with L-NAME and the O2− scavenger superoxide dismutase produced significantly fewer microparticles than cells treated with L-NAME alone. Because both calpain inhibition and O2− scavenging reduced microparticle numbers to those of unstimulated cells, it appears that L-NAME increases intracellular calcium and initiates a sequence of events within the cell involving O2− and calpain resulting in microparticle formation.
This study has focused on in vitro effects of L-NAME on neutrophils. The effects of release of neutrophil-derived microparticles in vivo, however, may be more far reaching. Neutrophil-derived microparticles have been shown to contain inflammatory mediators such as platelet-activating factor,52 CD11a, CD11b, and L-selectin38 and activate endothelial cells causing the release of the proinflammatory cytokines IL-6 and IL-8.53 It has also been found that severely injured patients have significantly higher levels of circulating neutrophil-derived microparticles ∼2 to 5 days after trauma.54 The absence of endothelial cells, and consequently endothelial-derived NO, in the in vitro system used in this study may make our findings more relevant to tissue migration of neutrophils. Hickey and colleagues55 demonstrated a role for L-selectin in neutrophil emigration and extravascular locomotion in the cremaster muscle of mice and showed that L-selectin expression is crucial for leukocytes to respond effectively to chemotactic stimuli. It is possible, therefore, that L-selectin expressed on microparticles derived from migrating neutrophils facilitates the subsequent migration of neutrophils not only in the vessel but also across the tissue.
We have identified a novel mechanism for regulation of neutrophil migration by NO. In our simplified in vitro system, enhancement of neutrophil migration is dependent on the concentration of L-NAME to which neutrophils are exposed and on a sufficient number of neutrophils being available to make microparticles. Apparently conflicting results proposing pro- and anti-inflammatory effects of NO in other studies may, therefore, be attributable to the differences in either of these factors. This study reiterates the need for highly selective iNOS inhibitors for use as anti-inflammatory therapies; inhibition of constitutive NO production may lead to increased neutrophil migration through microparticle formation and, therefore, propagation of the inflammatory response.
Supplementary Material
Acknowledgments
We thank Chris Hill from the Biomedical Centre for Light and Electron Microscopy, University of Sheffield, Sheffield, UK, for help with the electron microscopy.
Footnotes
Address reprint requests to Dr. Victoria Ridger, Cardiovascular Research Unit, University of Sheffield, School of Medicine and Biomedical Sciences, LU112 L Floor Royal Hallamshire Hospital, Glossop Rd., Sheffield S10 2JF UK. E-mail: v.c.ridger@sheffield.ac.uk.
Supported by the British Heart Foundation (studentship FS/2001007 and intermediate research fellowship FS/02/004).
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Smith JA. Neutrophils, host defense, and inflammation: a double-edged sword. J Leukoc Biol. 1994;56:672–686. doi: 10.1002/jlb.56.6.672. [DOI] [PubMed] [Google Scholar]
- Lukacs NW, Ward PA. Inflammatory mediators, cytokines, and adhesion molecules in pulmonary inflammation and injury. Adv Immunol. 1996;62:257–304. doi: 10.1016/s0065-2776(08)60432-0. [DOI] [PubMed] [Google Scholar]
- Malik AB, Lo SK. Vascular endothelial adhesion molecules and tissue inflammation. Pharmacol Rev. 1996;48:213–229. [PubMed] [Google Scholar]
- Simon SI, Green CE. Molecular mechanics and dynamics of leukocyte recruitment during inflammation. Annu Rev Biomed Eng. 2005;7:151–185. doi: 10.1146/annurev.bioeng.7.060804.100423. [DOI] [PubMed] [Google Scholar]
- Kakkar AK, Lefer DJ. Leukocyte and endothelial adhesion molecule studies in knockout mice. Curr Opin Pharmacol. 2004;4:154–158. doi: 10.1016/j.coph.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Liu L, Kubes P. Molecular mechanisms of leukocyte recruitment: organ-specific mechanisms of action. Thromb Haemost. 2003;89:213–220. [PubMed] [Google Scholar]
- Rao RM, Shaw SK, Kim M, Luscinskas FW. Emerging topics in the regulation of leukocyte transendothelial migration. Microcirculation. 2005;12:83–89. doi: 10.1080/10739680590896018. [DOI] [PubMed] [Google Scholar]
- Nourshargh S, Marelli-Berg FM. Transmigration through venular walls: a key regulator of leukocyte phenotype and function. Trends Immunol. 2005;26:157–165. doi: 10.1016/j.it.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Akimitsu T, Gute DC, Korthuis RJ. Leukocyte adhesion induced by inhibition of nitric oxide production in skeletal muscle. J Appl Physiol. 1995;78:1725–1732. doi: 10.1152/jappl.1995.78.5.1725. [DOI] [PubMed] [Google Scholar]
- Hickey MJ, Granger DN, Kubes P. Inducible nitric oxide synthase (iNOS) and regulation of leucocyte/endothelial cell interactions: studies in iNOS-deficient mice. Acta Physiol Scand. 2001;173:119–126. doi: 10.1046/j.1365-201X.2001.00892.x. [DOI] [PubMed] [Google Scholar]
- Moilanen E, Vuorinen P, Kankaanranta H, Metsa-Ketela T, Vapaatalo H. Inhibition by nitric oxide-donors of human polymorphonuclear leucocyte functions. Br J Pharmacol. 1993;109:852–858. doi: 10.1111/j.1476-5381.1993.tb13653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamim CF, Ferreira SH, Cunha FQ. Role of nitric oxide in the failure of neutrophil migration in sepsis. J Infect Dis. 2000;182:214–223. doi: 10.1086/315682. [DOI] [PubMed] [Google Scholar]
- De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA, Jr, Shin WS, Liao JK. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96:60–68. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan BV, Harrison DG, Olbrych MT, Alexander RW, Medford RM. Nitric oxide regulates vascular cell adhesion molecule 1 gene expression and redox-sensitive transcriptional events in human vascular endothelial cells. Proc Natl Acad Sci USA. 1996;93:9114–9119. doi: 10.1073/pnas.93.17.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binion DG, Fu S, Ramanujam KS, Chai YC, Dweik RA, Drazba JA, Wade JG, Ziats NP, Erzurum SC, Wilson KT. iNOS expression in human intestinal microvascular endothelial cells inhibits leukocyte adhesion. Am J Physiol. 1998;275:G592–G603. doi: 10.1152/ajpgi.1998.275.3.G592. [DOI] [PubMed] [Google Scholar]
- Wanikiat P, Woodward DF, Armstrong RA. Investigation of the role of nitric oxide and cyclic GMP in both the activation and inhibition of human neutrophils. Br J Pharmacol. 1997;122:1135–1145. doi: 10.1038/sj.bjp.0701477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauvais F, Michel L, Dubertret L. Exogenous nitric oxide elicits chemotaxis of neutrophils in vitro. J Cell Physiol. 1995;165:610–614. doi: 10.1002/jcp.1041650319. [DOI] [PubMed] [Google Scholar]
- Razavi HM, Wang LF, Weicker S, Rohan M, Law C, McCormack DG, Mehta S. Pulmonary neutrophil infiltration in murine sepsis: role of inducible nitric oxide synthase. Am J Respir Crit Care Med. 2004;170:227–233. doi: 10.1164/rccm.200306-846OC. [DOI] [PubMed] [Google Scholar]
- de Frutos T, Sanchez de Miguel L, Farre J, Gomez J, Romero J, Marcos-Alberca P, Nunez A, Rico L, Lopez-Farre A. Expression of an endothelial-type nitric oxide synthase isoform in human neutrophils: modification by tumor necrosis factor-alpha and during acute myocardial infarction. J Am Coll Cardiol. 2001;37:800–807. doi: 10.1016/s0735-1097(00)01185-2. [DOI] [PubMed] [Google Scholar]
- Wallerath T, Gath I, Aulitzky WE, Pollock JS, Kleinert H, Forstermann U. Identification of the NO synthase isoforms expressed in human neutrophil granulocytes, megakaryocytes and platelets. Thromb Haemost. 1997;77:163–167. [PubMed] [Google Scholar]
- Bryant JL, Jr, Mehta P, Von der Porten A, Mehta JL. Co-purification of 130 kD nitric oxide synthase and a 22 kD link protein from human neutrophils. Biochem Biophys Res Commun. 1992;189:558–564. doi: 10.1016/0006-291x(92)91594-g. [DOI] [PubMed] [Google Scholar]
- Evans TJ, Buttery LD, Carpenter A, Springall DR, Polak JM, Cohen J. Cytokine-treated human neutrophils contain inducible nitric oxide synthase that produces nitration of ingested bacteria. Proc Natl Acad Sci USA. 1996;93:9553–9558. doi: 10.1073/pnas.93.18.9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukahara Y, Morisaki T, Kojima M, Uchiyama A, Tanaka M. iNOS expression by activated neutrophils from patients with sepsis. ANZ J Surg. 2001;71:15–20. doi: 10.1046/j.1440-1622.2001.02025.x. [DOI] [PubMed] [Google Scholar]
- Kaplan SS, Billiar T, Curran RD, Zdziarski UE, Simmons RL, Basford RE. Inhibition of chemotaxis Ng-monomethyl-L-arginine: a role for cyclic GMP. Blood. 1989;74:1885–1887. [PubMed] [Google Scholar]
- Conran N, Gambero A, Ferreira HH, Antunes E, de Nucci G. Nitric oxide has a role in regulating VLA-4-integrin expression on the human neutrophil cell surface. Biochem Pharmacol. 2003;66:43–50. doi: 10.1016/s0006-2952(03)00243-0. [DOI] [PubMed] [Google Scholar]
- Clancy R, Leszczynska J, Amin A, Levartovsky D, Abramson SB. Nitric oxide stimulates ADP ribosylation of actin in association with the inhibition of actin polymerization in human neutrophils. J Leukoc Biol. 1995;58:196–202. doi: 10.1002/jlb.58.2.196. [DOI] [PubMed] [Google Scholar]
- Frevert CW, Wong VA, Goodman RB, Goodwin R, Martin TR. Rapid fluorescence-based measurement of neutrophil migration in vitro. J Immunol Methods. 1998;213:41–52. doi: 10.1016/s0022-1759(98)00016-7. [DOI] [PubMed] [Google Scholar]
- Brodsky SV, Zhang F, Nasjletti A, Goligorsky MS. Endothelium-derived microparticles impair endothelial function in vitro. Am J Physiol. 2004;286:H1910–H1915. doi: 10.1152/ajpheart.01172.2003. [DOI] [PubMed] [Google Scholar]
- Lokuta MA, Nuzzi PA, Huttenlocher A. Calpain regulates neutrophil chemotaxis. Proc Natl Acad Sci USA. 2003;100:4006–4011. doi: 10.1073/pnas.0636533100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabroe I, Hartnell A, Jopling LA, Bel S, Ponath PD, Pease JE, Collins PD, Williams TJ. Differential regulation of eosinophil chemokine signaling via CCR3 and non-CCR3 pathways. J Immunol. 1999;162:2946–2955. [PubMed] [Google Scholar]
- Storey RF, Sanderson HM, White AE, May JA, Cameron KE, Heptinstall S. The central role of the P(2T) receptor in amplification of human platelet activation, aggregation, secretion and procoagulant activity. Br J Haematol. 2000;110:925–934. doi: 10.1046/j.1365-2141.2000.02208.x. [DOI] [PubMed] [Google Scholar]
- Merritt JE, McCarthy SA, Davies MP, Moores KE. Use of fluo-3 to measure cytosolic Ca2+ in platelets and neutrophils. Loading cells with the dye, calibration of traces, measurements in the presence of plasma, and buffering of cytosolic Ca2+. Biochem J. 1990;269:513–519. doi: 10.1042/bj2690513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunzendorfer S, Rothbucher D, Schratzberger P, Reinisch N, Kahler CM, Wiedermann CJ. Mevalonate-dependent inhibition of transendothelial migration and chemotaxis of human peripheral blood neutrophils by pravastatin. Circ Res. 1997;81:963–969. doi: 10.1161/01.res.81.6.963. [DOI] [PubMed] [Google Scholar]
- Bargatze RF, Kurk S, Butcher EC, Jutila MA. Neutrophils roll on adherent neutrophils bound to cytokine-induced endothelial cells via L-selectin on the rolling cells. J Exp Med. 1994;180:1785–1792. doi: 10.1084/jem.180.5.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel EJ, Chomas JE, Ley K. Role of primary and secondary capture for leukocyte accumulation in vivo. Circ Res. 1998;82:30–38. doi: 10.1161/01.res.82.1.30. [DOI] [PubMed] [Google Scholar]
- Sperandio M, Smith ML, Forlow SB, Olson TS, Xia L, McEver RP, Ley K. P-selectin glycoprotein ligand-1 mediates l-selectin-dependent leukocyte rolling in venules. J Exp Med. 2003;197:1355–1363. doi: 10.1084/jem.20021854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquet JM, Dachary-Prigent J, Nurden AT. Calcium influx is a determining factor of calpain activation and microparticle formation in platelets. Eur J Biochem. 1996;239:647–654. doi: 10.1111/j.1432-1033.1996.0647u.x. [DOI] [PubMed] [Google Scholar]
- Gasser O, Hess C, Miot S, Deon C, Sanchez JC, Schifferli JA. Characterisation and properties of ectosomes released by human polymorphonuclear neutrophils. Exp Cell Res. 2003;285:243–257. doi: 10.1016/s0014-4827(03)00055-7. [DOI] [PubMed] [Google Scholar]
- Harler MB, Wakshull E, Filardo EJ, Albina JE, Reichner JS. Promotion of neutrophil chemotaxis through differential regulation of {beta}1 and {beta}2 integrins. J Immunol. 1999;162:6792–6799. [PubMed] [Google Scholar]
- Mackarel AJ, Russell KJ, Brady CS, FitzGerald MX, O’Connor CM. Interleukin-8 and leukotriene-B4, but not formylmethionyl leucylphenylalanine stimulate CD18-independent migration of neutrophils across human pulmonary endothelial cells in vitro. Am J Respir Cell Mol Biol. 2000;23:154–161. doi: 10.1165/ajrcmb.23.2.3853. [DOI] [PubMed] [Google Scholar]
- Walcheck B, Moore KL, McEver RP, Kishimoto TK. Neutrophil-neutrophil interactions under hydrodynamic shear stress involve L-selectin and PSGL-1. A mechanism that amplifies initial leukocyte accumulation of P-selectin in vitro. J Clin Invest. 1996;98:1081–1087. doi: 10.1172/JCI118888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furie B, Furie BC. Role of platelet P-selectin and microparticle PSGL-1 in thrombus formation. Trends Mol Med. 2004;10:171–178. doi: 10.1016/j.molmed.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Farlow SB, McEver RP, Nollert MU. Leukocyte-leukocyte interactions mediated by platelet microparticles under flow. Blood. 2000;95:1317–1323. [PubMed] [Google Scholar]
- Hrachovinova I, Cambien B, Hafezi-Moghadam A, Kappelmayer J, Camphausen RT, Widom A, Kazazian H, Schaub RG, McEver RP, Magner DD. Interaction of P-selectin and PSGL-1 generates microparticles that correct hemostasis in a mouse model of hemophilia A. Nat Med. 2003;9:1020–1025. doi: 10.1038/nm899. [DOI] [PubMed] [Google Scholar]
- VanWijk MJ, VanBavel E, Sturk A, Nieuwland R. Microparticles in cardiovascular diseases. Cardiovasc Res. 2003;59:277–287. doi: 10.1016/s0008-6363(03)00367-5. [DOI] [PubMed] [Google Scholar]
- Barry OP, Pratico D, Savani RC, FitzGerald GA. Modulation of monocyte-endothelial cell interactions by platelet microparticles. J Clin Invest. 1998;102:136–144. doi: 10.1172/JCI2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michetti M, Salamino F, Melloni E, Pontremoli S. Reversible inactivation of calpain isoforms by nitric oxide. Biochem Biophys Res Commun. 1995;207:1009–1014. doi: 10.1006/bbrc.1995.1285. [DOI] [PubMed] [Google Scholar]
- Koh TJ, Tidball JG. Nitric oxide inhibits calpain-mediated proteolysis of talin in skeletal muscle cells. Am J Physiol. 2000;279:C806–C812. doi: 10.1152/ajpcell.2000.279.3.C806. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, Umeshita K, Sakon M, Imajoh-Ohmi S, Fujitani K, Gotoh M, Oiki E, Kambayashi J, Monden M. Calpain activation in plasma membrane bleb formation during tert-butyl hydroperoxide-induced rat hepatocyte injury. Gastroenterology. 1996;110:1897–1904. doi: 10.1053/gast.1996.v110.pm8964416. [DOI] [PubMed] [Google Scholar]
- Niu XF, Smith CW, Kubes P. Intracellular oxidative stress induced by nitric oxide synthesis inhibition increases endothelial cell adhesion to neutrophils. Circ Res. 1994;74:1133–1140. doi: 10.1161/01.res.74.6.1133. [DOI] [PubMed] [Google Scholar]
- Niu XF, Ibbotson G, Kubes P. A balance between nitric oxide and oxidants regulates mast cell-dependent neutrophil-endothelial cell interactions. Circ Res. 1996;79:992–999. doi: 10.1161/01.res.79.5.992. [DOI] [PubMed] [Google Scholar]
- Watanabe J, Marathe GK, Neilsen PO, Weyrich AS, Harrison KA, Murphy RC, Zimmerman GA, McIntyre TM. Endotoxins stimulate neutrophil adhesion followed by synthesis and release of platelet-activating factor in microparticles. J Biol Chem. 2003;278:33161–33168. doi: 10.1074/jbc.M305321200. [DOI] [PubMed] [Google Scholar]
- Mesri M, Altieri DC. Endothelial cell activation by leukocyte microparticles. J Immunol. 1998;161:4382–4387. [PubMed] [Google Scholar]
- Fujimi S, Ogura H, Tanaka H, Koh T, Hosotsubo H, Nakamori Y, Kuwagata Y, Shimazu T, Sugimoto H. Increased production of leukocyte microparticles with enhanced expression of adhesion molecules from activated polymorphonuclear leukocytes in severely injured patients. J Trauma. 2003;54:114–120. doi: 10.1097/00005373-200301000-00014. [DOI] [PubMed] [Google Scholar]
- Hickey MJ, Forster M, Mitchell D, Kaur J, De Caigny C, Kubes P. L-Selectin facilitates emigration and extravascular locomotion of leukocytes during acute inflammatory responses in vivo. J Immunol. 2000;165:7164–7170. doi: 10.4049/jimmunol.165.12.7164. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.