

Abstract
MHC molecules are expressed at the surface of nucleated cells to present peptides to T cells. Structural information on MHC molecules has been gathered by x-ray crystallography techniques by using soluble proteins. Although relationships between MHC molecules and cell membranes have not been studied in detail, they are of critical importance for T cell recognition. Using a chemically modified lipid, we have been able to capture and orient histidine-tagged MHC molecules on lipid membranes. Surface plasmon resonance experiments show that the protein binds to the nickel lipid in a specific manner and in an oriented fashion, which allows T cell receptor binding. Similar lipid surfaces have been used to grow two-dimensional crystals and to determine the structure of a membrane-anchored murine H-2Kb MHC class I molecule. The docking of the crystallographic structure into the three-dimensional reconstructed structure derived from the two-dimensional crystals allows us to determine that the histidine tag is near the membrane surface and that the MHC molecule is in an upright position, exposing the peptide/α1-α2 domains toward the T cell.
The relevance of crystallographic studies for the interpretation of biological studies is often discussed. The use of soluble extracellular domains of transmembrane proteins for such studies is a controversial issue. Indeed, it is conceivable that some proteins might need to interact with the surface of the membrane and with accessory surface components to achieve their functional conformation. In this respect, the orientation of the extracellular domain of transmembrane molecules relative to membrane surfaces is critical to study. Very recently, techniques of spin relaxation magnetic resonance have been used to determine the positioning of interfacial molecules, such as phospholipase A2, relative to the surface of lipid vesicles (1). We are exploiting the two-dimensional (2D) crystallization and structure determination approach to try to address these questions. Two-dimensional crystallization of soluble protein on lipid monolayers first was introduced by Uzgiris and Kornberg (2) (for a recent review see ref. 3). The technique relies on the interaction between the protein in solution with the polar head group of a lipid spread as a monolayer at the air/water interface. The protein can interact with a natural specific lipid ligand, such as the cholera toxin with GM1 monoganglioside (4) or with a soluble ligand chemically linked to the hydrophilic head group of a lipid, like in the case of the gyrase B subunit with a novobiocin-linked phospholipid (5). In the absence of such lipidic ligand, two-dimensional crystallization also can occur through electrostatic interactions between the protein and a charged lipid (6). However, this latter approach does not rely on a physiological interaction.
We had two goals in our study: to try to reorient the extracellular domain of a transmembrane molecule relative to a lipid membrane and to determine its structure and relationship to the membrane. Achieving these goals would be critical for interpreting high resolution three-dimensional (3D) structures of extracellular domains of integral membrane proteins. As a test case, we chose a system in which both the receptor and the ligand are transmembrane proteins displayed at the surface of two separate cells that will interact very specifically to trigger activation: T cell receptor (TCR) and peptide/MHC antigen complex. We have expressed, purified, crystallized, and determined the 3D x-ray structure of both molecules separately and as a complex (7, 8). Because TCR binds to the most membrane-distant part of peptide MHC, it is critical to determine whether this interactive surface is readily available for binding when the peptide MHC is displayed at the surface of an antigen-presenting cell.
We have engineered most of our soluble molecules by deleting their C-terminal cytoplasmic and/or transmembrane domains and substituting it by a polyhistidine tag to facilitate purification (9–11). For both TCR and MHC, this tag extends the natural connecting peptide of these molecules that links their transmembrane domains to their first membrane-proximal Ig-like domains. As a consequence of this topology, the capture of the histidine tag should orient the molecule correctly in respect to the membrane. To do so, a metal-chelating lipid (12), similar to some previously published ones (13–17), was used to capture MHC-tagged molecules at the surface of liposomes or lipid monolayers.
The results that we obtained with a soluble His-tagged H-2Kb MHC class I molecule are presented here. This protein forms 2D crystals on lipid monolayers only when nickel-chelating lipids are present in the monolayer. The specificity of interaction between His-tagged protein and nickel lipid orientation and function of the immobilized molecule was assessed by surface plasmon resonance (SPR) experiments. A 3D reconstruction model of the 2D crystals was calculated at 14-Å resolution. The docking of the high resolution x-ray crystallographic structure of H-2Kb into this model shows that the His-tag is indeed close to the lipid surface. Finally, electron diffraction data recorded on the same crystals preserved in amorphous ice show that these crystals are ordered to >3.5-Å resolution.
MATERIALS AND METHODS
Materials.
The purification of the soluble form of H-2Kb was performed as described (18). The 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)imidodiacetic acid)succinyl] (DOGS-NTA) (see Scheme S1, top), DOGS-NTA(nickel salt) (DOGS-NTA-Ni) (Scheme S1, bottom), phosphatidylcholine, and dioleoyl phosphatidylcholine (DOPC) lipids were purchased from Avanti Polar Lipids. The fluorescent lipid probe N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-snglycero-3-phosphoethanolamine was purchased from Molecular Probes.
Scheme 1.

Preparation of Liposomes.
The lipids DOPC and DOGS-NTA-Ni diluted in chloroform were mixed at a 5:1 or 9:1 (wt/wt) ratio, were dried under argon, and were left under vacuum overnight to remove traces of remaining organic solvent. The dried lipids were hydrated in PBS buffer at a final concentration of 5 mg/ml. The aqueous suspension then was vortexed and freeze-thawed 10 times on dry ice. The liposomes were extruded 5–10 times through 0.1-μm polycarbonate filters by using a mini-extruder (19).
Fluorescence Recovery After Photobleaching Experiments.
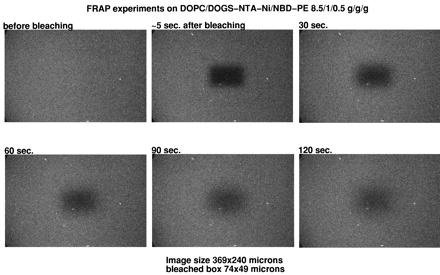
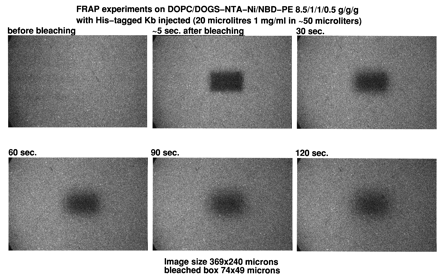
Fluorescence recovery experiments were performed on phospholipid monolayers adsorbed on alkylated glass coverslips prepared according to Tscharner and McConnell (20). In brief, glass coverslips were cleaned with sulfochromic acid, rinsed with distilled water, and dried. The clean coverslips were transferred into a 80% hexadecane, 12% carbon tetrachloride, 8% chloroform, and 0.5% octadecyltrichlorosilane solution and were agitated for 15 minutes. The coverslips were washed three to five times in chloroform, air dried, and attached to clean microscope glass slides with double stick tape, leaving a volume of ≈70 mm3 between the slide and the coverslip. Planar supported monolayers were prepared by injecting 70 μl of a 1 mg/ml liposome suspension containing 5% N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine into the chamber formed between coverslip and glass slide. After a 10-minute incubation, the excess of liposome suspension was removed by injecting 2–3 ml of buffer, followed by immersion overnight of the glass slide and coverslip into the buffer solution under agitation. Care was taken after this stage not to dry out the supported membrane. When desired, a protein solution was injected onto the supported monolayer, and the unbound protein was removed by rinsing with the buffer solution. Before observation, the sides of the chamber were sealed with vaseline. Fluorescence of supported monolayer was observed on a Bio-Rad MRC600 laser scanning confocal microscope attached to a Zeiss IM35M microscope. Photobleaching experiments were made by illuminating at slow speed a rectangular 74- × 49-μm window. Recovery was recorded at different times at normal speed on an enlarged area of 369 × 240 μm centered on the bleached window.
Surface Plasmon Resonance Measurements.
Affinity binding experiments of His-tagged H-2Kb were done by using a BIAcore 2000 (Biacore, Uppsala). Planar supported membranes were made by four successive 100-μl injections of 0.25 mg/ml liposome suspension onto HPA sensor chips at a 5 μl/min flow rate. Before these injections, the surface was primed by a 50-μl injection of 1% N-octyl glucoside. Fusion of liposomes with the hydrophobic surface of the HPA chip was monitored directly by following the change of resonance units. Potential multilamellar structures were rinsed off overnight by a continuous 20 μl/min flow of PBS. For typical binding experiments, histidine-tagged H-2Kb molecules were captured by a single 100-μl injection of a 50–100 μg/ml protein solution at 10 μl/min. The surface then was rinsed at 20 μl/min until the resonance signal reached a stable baseline. All experiments were carried out in PBS at a flow rate of 10 or 20 μl/min. Analytes, such as the TCRs used here, were produced as soluble molecules in fly cells with a polyhistidine tag. The histidines were removed by overnight carboxypeptidase A digestion (Boehringer Mannheim) at a weight-to-weight ratio of 1:100 protease/protein before the last step of purification. Sensorgrams were analyzed by using the global fitting program biaevaluation 3.0 (Biacore). Ligands were injected at 3, 1.5, 0.75, 0.375, and 0.1875 μM.
Electron Microscopy of Proteins Bound to Liposomes.
The DOPC/DOGS-NTA-Ni liposomes and H-2Kb were mixed at a ratio of 1.3 mg/ml protein and 0.7 mg/ml lipid and were incubated overnight at 4°C. Three to five microliters of the protein and liposomes solution were deposited on a glow discharged holey carbon grid; the excess solution was removed with a filter paper, and the grid was flash-frozen in a bath of liquid ethane cooled to −180°C with liquid nitrogen (21).
Two-Dimensional Crystallization.
Two-dimensional crystallization experiments were performed in 4-mm diameter and 1-mm deep Teflon wells. Sixteen microliters of purified His-tagged H-2Kb [0.2 mg/ml in 20 mM Hepes (pH 7.2) and 100 mM NaCl] were placed in a well, and the surface of the solution was coated with 0.5–1 μl of a 0.5 mg/ml 1:5 (wt/wt) mixture of DOGS-NTA-Ni and DOPC diluted in chloroform. The device was incubated overnight at 4°C in a humid chamber. The lipid–protein complexes were transferred onto holey carbon coated grids by depositing the grid directly on the surface of the drop with the carbon facing the surface. The grid was removed and either negatively stained with 2% uranyl acetate or flash frozen in a bath of liquid ethane cooled to −180°C with liquid nitrogen (21).
Electron Microscopy of Two-Dimensional Crystals.
Low dose electron micrographs of negatively stained 2D crystals of H-2Kb were recorded at 52,000× on a Philips (Eindhoven, The Netherlands) CM100 operating at 100 kV on Kodak SO163 film. Images of frozen hydrated liposomes and electron diffractograms were recorded on a Philips CM120 operating at 100 kV and using a Gatan cryoholder (Gatan, Pleasanton, CA). Electron diffraction patterns of 2D crystals were recorded at a nominal camera length of 1.6 meter; no objective or selection area apertures were used.
Image Processing.
Micrographs of tilted and untilted negatively stained 2D crystals were selected by optical diffractometry for quality of the diffraction data, absence of astigmatism, and specimen drift. Selected micrographs were digitized by using a Perkin–Elmer 1010-G microdensitometer with 25-μm spot and step sizes (4.81 Å at the specimen). Numerical arrays were Fourier transformed, and reciprocal lattices were indexed by using the graphical interface spectra (22). Subsequent analysis, including correction for lattice distortions, merging of untilted and tilted data, and 3D reconstruction, was performed by using the mrc suite of computer programs (23, 24).
RESULTS
Binding of the His-Tagged Protein to the Nickel- Lipid.
The nickel-chelating lipid used in this study was DOGS-NTA-Ni. The ability of DOGS-NTA-Ni to mix with other lipids and form liposomes was tested with phosphatidyl choline and/or DOPC by using the sonication and extrusion technique (19). Size homogeneity and the absence of multilamellar “onion-like” structure was examined directly by electron microscopy. Liposomes of DOPC/DOGS-NTA-Ni, 100-nm average diameter, were used for most of the present experiments. The mixing of these liposomes with soluble histidine tagged molecules resulted in the decoration of the liposomes as shown in Fig. 1 for soluble recombinant histidine-tagged H-2Kb molecules. The protein distribution at the liposome surface appears homogeneous, indicating that there was no phase separation of the two lipids during liposome formation.
Figure 1.

Electron micrograph of frozen hydrated DOGS-NTA-Ni/DOPC (1:9 wt/wt) liposomes without protein (Left) and decorated with histidine-tagged recombinant soluble H-2Kb molecules (Right). (Bar = 200 nm.)
For surface plasmon resonance experiments, the same liposomes were fused to hydrophobic HPA sensor chips to form supported monolayers. Histidine-tagged molecules were captured specifically on this surface. Carboxypeptidase A removal of the tag (11) resulted in a complete loss of binding (Fig. 2), and surfaces made of DOPC only exhibited no significant binding (data not shown). The specificity of binding was confirmed by the fact that the His-tagged immobilized molecule could be released from the surface by 500 mM imidazole and that immobilized molecules lost most of their reactivity to antihistidine antibodies while keeping their reactivity with other antibodies (Fig. 2). After extensive washes, the decay of these surfaces appeared to be extremely slow and allowed standard SPR experiments to be carried out. Furthermore the surfaces were found to be stable between 4 and 37°C (data not shown).
Figure 2.

SPR monitoring of DOGS-NTA-Ni/DOPC surfaces and function of the immobilized ligand. (A) The same amount of H-2Kb without (carboxypeptidase A-treated) and with histidine tag were injected successively on the supported layer. (B) Immobilized H-2Kb binds the antihistidine antibody marginally whereas the anti-α1-α2 domain antibody Y3 binds very well. (C) The immobilized H-2Kb is functional and binds the H-2Kb ova8-specific TCR 149–42 whereas the anti-IAk TCR D10 does not.
Lateral Diffusion in Supported Monolayers.
Free lateral diffusion of membrane protein is an important property of natural membranes. It has been shown that the formation of planar supported lipid bilayers from cell membranes resulted in abolishing the free lateral diffusion of integral membrane proteins (25). We wanted to show that the lipids into a supported monolayer were free to diffuse laterally, either interacting or not with His-tagged molecules. Because SPR experiments were carried out by using HPA sensor chips, we used alkylated glass coverslips as hydrophobic planar supports to test for lateral motions in our supported monolayers. Liposomes were made with the addition of 1% of the fluorescent lipid N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine and were fused onto an alkylated coverslip identical to HPA chip surfaces. Fluorescence was visualized by laser scanning confocal microscopy. Fluorescence recovery after photobleaching experiments were performed by bleaching a large, 3,600 μm2 area and recording images of an enlarged area centered on the photobleached region (data not shown). Independently of the presence of DOGS-NTA-Ni in the lipid layer, >50% of the fluorescence was recovered after 2–3 minutes despite the large size of the bleached box. The fluorescence recovery was slowed 20–30% when His-tagged molecules were bound to the DOGS-NTA-Ni inserted into the monolayer. However, very significant recovery always was noted for the different molecules that were tested, suggesting strongly that the lipids are free to diffuse laterally on the hydrophobic support, even when a protein is bound to the hydrophilic head group. (See Figs. 5–11, which are published as supplemental data on the PNAS web site, www.pnas.org.)
Orientation of H-2Kb Within the Supported Monolayer and Function of the Immobilized Molecule.
After capture of His-tagged H-2Kb molecules, the accessibility of the surface to antibodies was tested. First, an anti-6 histidines monoclonal antibody that reacts very strongly with randomly immobilized histidine-tagged molecules on CM5 chips (2.5 nM affinity) (H.C., unpublished results) showed very limited binding to lipid-captured H-2Kb (Fig. 2), indicating that the C-terminal tag was not accessible to antibody binding. On the contrary, the anti-α1α2 domain antibody Y3 that binds specifically to the peptide-binding groove of H-2Kb (26) reacted normally (Fig. 2). Thus, these data suggested that the molecule was anchored into the membrane through the histidine tag and that its binding site was exposed to the solvent. The orientation and functionality of the immobilized molecule was confirmed by binding experiments with soluble T cell receptor. H-2Kb molecules were loaded with the high affinity peptide ova-8 (SIINFEKL) and were captured on a DOGS-NTA-Ni surface. Binding was tested with the 149–42 TCR (27), which recognizes specifically H-2Kb ova-8 complexes. D10, a TCR specific for I-Ak conalbumin complex (28), was used as a negative control. Both TCRs were digested with carboxypeptidase A to remove their histidine tag before the last step of purification. Only 149–42 TCR showed binding to the H-2Kb ova-8 surface (Fig. 2). Five successive 2-fold dilutions of these TCRs were injected over the surface to analyze binding. The data were fitted satisfactorily (Chi2:0.49) on a 1:1 Langmuir model by using a global fit program (biaevaluation 3.0). The association constant (Kass) was evaluated at 3.79⋅104 M−1⋅s−1, and the dissociation constant (Kdiss) was 7.97 × 10−3⋅s−1 for a calculated Kd of 2.46 × 10−7 M, which is relatively low for a TCR/MHC interaction. That same interaction measured by using random coupling on CM5 chips was a log higher (L.T., unpublished results; ref. 2). These results indicate that correct orientation of the ligand is indeed critical for SPR measurements (30), especially for low affinities such as seen for TCR/MHC pairs (31). Of interest, the Kd on nickel-lipids is very close to the one measured with soluble MHC peptide complexes on living cells (32).
Two-Dimensional Crystallization of H-2Kb.
The His-tagged H-2Kb forms 2D crystals on lipid monolayer containing DOGS-NTA-Ni. Crystals were not observed on monolayer lacking the DOGS-NTA lipid or if the crystallization buffer contained 100 mM imidazole or more. Electron micrographs of negatively stained crystals diffracted to 14-Å resolution. The average lattice parameters of the 2D crystals, computed from six untilted negatively stained crystals, were a = 92.1 ± 1.3 Å, b = 69.5 ± 0.5 Å, and γ = 109.7 ± 1.3°. These images were analyzed and combined together to generate a data set of 40 averaged reflections up to 14-Å resolution. After refinement to 0 or 180°, the mean phase deviation was 8.5°, indicating that the protein crystallized in P2 space group. The unit cell in the fully symmetrized and averaged projection map contains two asymmetric units related to each other by a two-fold axis (Fig. 3a). Each asymmetric unit is composed of a 40- × 30-Å elongated main density (domain 1), connected to two circular densities 20 Å in diameter (domains 2 and 3). Examination of the maps computed from single images showed that the connectivity between the three domains is not clearly defined.
Figure 3.

(a) Two-dimensional projection map calculated out of the averaged structure factors from six crystalline arrays corrected for lattice distortions. The P2 symmetry has been imposed. The solid black contour lines outlines stain excluding regions where biological material is located; the dotted black lines are negative values. The unit cell (a = 92.1 Å, b = 69.5 Å, γ = 109.7°) is shown with the solid blue lines. The three different domains of one asymmetric unit described in the text are shown in colored dotted lines: domain 1, red; domain 2, blue; domain 3, green. (b) Electron diffractogram of a frozen hydrated 2D crystal of H-2Kb. The background has been normalized to show both high and low diffraction spots. The dashed circle corresponds to 3.5-Å resolution. The directions of the two crystallographic vectors a* and b* are indicated.
The resolution achievable with negatively stained specimens is 10–15 Å. It is noteworthy that the 2D crystals are actually highly ordered. Electron diffraction patterns recorded from frozen hydrated crystals suspended into holes showed diffraction spots beyond 3.5-Å resolution (Fig. 3b). The high resolution peaks are directly visible on electron micrographs, without any further processing. This result demonstrates that these crystals are suitable for high resolution electron crystallography.
Three-Dimensional Reconstruction.
Nine tilt series of negatively stained 2D crystals were selected to calculate a 3D structure of H-2Kb. Fifty-seven images of crystals tilted 10–60° to the incident electron beam were analyzed. The generated data sets (h, k, amplitude, phase) were combined together, taking the average 0° tilt data set as reference and assuming P2 symmetry. Only reflections showing an intensity quality of 4 or better were used in the analysis. The distribution of amplitudes and phases along different lattice lines was plotted to a resolution cutoff of 14 Å, and smooth curves were fitted to the data with the program latline (24). Sampling of the smooth curves at regular intervals provided the 3D data set (h, k, l, amplitude, phase) from which the 3D map was calculated (see the supplemental data on the PNAS web site, www.pnas.org). Viewed from the top with the plane of the lipid behind, the main domain 1 and the two smaller 2 and 3 identified in the 2D projection map are evident (Fig. 4a). A side view shows that domains 2 and 3 are close to the lipids (Fig. 4b). Domain 1 has a pyramidal shape 50 Å high, with the apex toward the lipid surface. Domain 2 is an elongated cylinder 30 Å long, with its main axis making an angle of ≈50° with the lipid film; one extremity of the cylinder is in close proximity with the lipid film whereas the other connects domain 1. The domain 3 is roughly triangular, ≈20 Å in diameter, and connects both domains 1 contained in one unit cell, as well as domain 2 of the neighboring unit cell.
Figure 4.

Views of the three-dimensional reconstruction of H-2Kb [volume visualization program volvis (38)]. The threshold value used to determine the outer surface of the model has been chosen to minimize the contacts between the adjacent unit cells in the crystal. The different domains are shown for one asymmetric unit on a. (a) Top view. (b) Ninety-degree rotation from an horizontal axis. The drawing in b is a representation of the lipid film to show the position of the lipids compared with the protein (not to scale). (c and d) Docking of the crystallographic structure of H-2Kb into the 3D model. c and d correspond to a and b, respectively. The atomic structure of H-2Kb is a ribbon representation. The heavy chain is colored in blue, the β2m in green, and the peptide in red. The 6-histidine tag is not represented but is directly adjacent to the C-terminal residue shown in yellow.
Docking of H-2Kb in the 3D Model.
The docking of the x-ray atomic structure of H-2Kb ova-8 (33) into the 3D reconstructed model was done interactively on a graphics workstation and as a rigid body. The quality of the docking was estimated visually and was judged satisfactory if the different domains of the atomic structure filled the volume of the 3D model. In the best fit, domain 2 corresponds to the α3 domain of H-2Kb whereas domain 1 comprises α1, α2, and β2 microglobulin (β2m) (Fig. 4 c and d). The small domain 3 density is unaccounted for in any of the fits that were explored. The elongated part of domain 1, which comes to contact with the lipid layer, corresponds to β2m, although the bulk of it is filled with the α1 and α2 domains, which are oriented in such a way that the peptide-binding groove and peptide face away from the lipid surface and are exposed to the aqueous phase. The physical connection between the α2 and α3 domains in the atomic structure is located in the 3D model at the interface between domains 1 and 2. The polyhistidine tag of H-2Kb, which is at the C terminus of the molecule, appears in domain 2 and is in close proximity to the lipid layer.
DISCUSSION
The goal of structural biology is to support and help the interpretation of biological experiments. Most structural information is provided by x-ray crystallography, which, like any technique, has its limitations: (i) crystals represents a one-conformational state, and (ii) crystals are grown in a nonphysiological environment. Therefore, experiments have to be designed to support crystallographic studies. In immunology, many receptors and ligands are transmembrane molecules. The engineering of soluble secreted portions of these molecules has allowed structural investigation of their extracellular domains. The relationships between these domains and the cell membrane have yet to be studied. To investigate these relationships, we substituted a polyhistidine tag for the transmembrane segment and captured the molecules on the surface of a lipid film via a nickel-chelating lipid. This approach should mimic the protein–lipid membrane relationships as in vivo on the cell membrane.
Results are presented for the murine MHC class I molecule H-2Kb, a transmembrane molecule that is a ligand for the T cell receptor. The high resolution x-ray structure of the soluble extracellular domain of H-2Kb is known (33), but the relationship of this molecule with the membrane has never been studied directly. MHC accessibility to TCR is a critical feature of T cell recognition. The presence of hydrophobic patches at its surface, such as seen on β2m, could bend the molecule toward the surface and could limit the exposure of the α1α2 domains/peptide surface.
Here, we have shown the specificity of the interaction between the nickel-lipid and the histidine-tagged MHC by SPR and 3D electron microscopy analysis. SPR experiments were carried out on phospholipid monolayers supported on alkanethiol treated sensor chips (34, 35). This format offers a number of advantages: (i) absence of diffusion limitations through a dextran layer such as seen on CM5 and NTA chips (36, 37); (ii) easy regeneration of the surface by imidazole injection; (iii) usage of the same protein source for immobilized ligand and analyte by using carboxypeptidase A digestion of the C-terminal histidine tag; (iv) stability of the surface between 4°C and 37°C; and (v) lateral diffusion of the lipid within the supported layer. This later property should allow the immobilization of multiple histidine-tagged molecules on the same surface to study lateral interactions and oligomerization. Limitations of the NTA capture of tagged molecules include a narrow pH range of binding (7.0 to 8.0) and the impossibility of using metal chelators in the buffer.
On nickel-chelating lipid monolayers, the crystallization of H-2Kb was spontaneous and abolished by the addition of imidazole. This crystallization process is likely to occur because of a unique orientation of the protein on the lipid film resulting from the interaction of the histidine tag with the NTA-Ni group. The same crystal form was observed independently of peptide content and for molecules closely related to H-2Kb such as H-2Kbm3 (data not shown). A 14-Å resolution cut off was chosen even though some negative crystals showed optical diffraction beyond that limit. It is important to emphasize that electron diffractograms of frozen hydrated samples of the same crystals showed visible spots beyond 3.5-Å resolution.
To understand how the x-ray structure of the molecule is oriented on the lipids, we calculated a 3D map by tilt series reconstruction of negatively stained 2D crystals. The 3D volume of one asymmetric unit showed three main densities corresponding to the stain excluded regions of the protein. Domains 1 and 2 were connected whereas domains 1 and 3 had no clear continuity. Docking of the atomic structure of H-2Kb as a rigid body into the electron microscopy map shows that domain 2 is filled with the α3 domain of H-2Kb as domain 1 accounts for the α1 and α2 domains and the β2m. The connection between domain 1 and 2 corresponds to the loop connecting α2 and α3. However, this model did not account for domain 3, which cannot be attributed to a staining artifact because it also was found on projections calculated from frozen hydrated crystals (data not shown). A possible explanation for it would be the staining of some of the N-linked carbohydrates of the α1-α2 domains. This hypothesis is being directly tested by attempting the crystallization of nonglycosylated H-2Kb molecules. An alternative explanation that cannot be excluded is that lipid-bound H-2Kb molecules have a different structure compared with the x-ray structure. This possibility will be answered by the high resolution structure from frozen hydrated crystals. In any instance, it is likely that the orientation of H-2Kb on the lipid is indeed correct, with the peptide-binding groove being oriented toward the aqueous phase and accessible to TCR binding as seen in SPR experiments with the H-2Kb ova8-specific TCR.
In conclusion, this report describes a combination of approaches to study the relationships between extracellular domains of transmembrane proteins and cell membranes. The approach allows SPR studies as well as structural studies at low and high resolution. Lateral motions within the supported layer also should allow the study of lateral interactions and oligomerization within membranes. In this respect, these artificial membranes will be an essential complement to x-ray structure determination for the study of cell surface receptors.
Supplementary Material
Acknowledgments
We thank R. Diaz-Avalos and M. Whittaker for help with computing, R. Stefanko and M. Bondad for technical assistance, and K. C. Garcia and H. Sosa for helpful discussions. This work was supported by National Institute of Health Grant AI42267 (to L.T.). H.C. is a recipient of a National Cancer Institute Fellowship. This is the Scripps Research Institute manuscript 11722-IMM.
ABBREVIATIONS
- TCR
T cell receptor
- 2D
two-dimensional
- 3D
three-dimensional
- SPR
surface plasmon resonance
- DOGS-NTA-Ni
1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)imidodiacetic acid)succinyl] (nickel salt)
- DOPC
dioleoyl phosphatidylcholine
References
- 1.Lin Y, Nielsen R, Murray D, Hubbell W L, Mailer C, Robinson B H, Gelb M H. Science. 1998;279:1925–1929. doi: 10.1126/science.279.5358.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uzgiris E E, Kornberg R D. Nature (London) 1983;301:134–136. doi: 10.1038/301125a0. [DOI] [PubMed] [Google Scholar]
- 3.Chiu W, Avila-Sakar A J, Schmid M F. Adv Biophys. 1997;34:161–172. doi: 10.1016/s0065-227x(97)89638-4. [DOI] [PubMed] [Google Scholar]
- 4.Ludwig D S, Ribi H O, Schoolnik G K, Kornberg R D. Proc Natl Acad Sci USA. 1986;83:8585–8588. doi: 10.1073/pnas.83.22.8585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lebeau L, Regnier E, Schultz P, Wang J C, Mioskowski C, Oudet P. FEBS Lett. 1990;267:38–42. doi: 10.1016/0014-5793(90)80282-n. [DOI] [PubMed] [Google Scholar]
- 6.Schultz P, Celia H, Riva M, Darst S A, Colin P, Kornberg R D, Sentenac A, Oudet P. J Mol Biol. 1990;216:353–362. doi: 10.1016/S0022-2836(05)80326-2. [DOI] [PubMed] [Google Scholar]
- 7.Garcia K C, Degano M, Stanfield R L, Brunmark A, Jackson M R, Peterson P A, Teyton L, Wilson I A. Science. 1996;274:209–219. [PubMed] [Google Scholar]
- 8.Garcia K C, Degano M, Pease L R, Huang M, Peterson P A, Teyton L, Wilson I A. Science. 1998;279:1166–1172. doi: 10.1126/science.279.5354.1166. [DOI] [PubMed] [Google Scholar]
- 9.Janknecht R, DeMartynoff G, Lou J, Hipskind R A, Nordheim A, Stunnenberg H G. Proc Natl Acad Sci USA. 1991;88:8972–8976. doi: 10.1073/pnas.88.20.8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scott C A, Garcia K C, Carbone F R, Wilson I A, Teyton L. J Exp Med. 1996;183:2087–2095. doi: 10.1084/jem.183.5.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia K C, Tallquist M D, Pease L R, Brunmark A, Scott C A, Degano M, Stura E A, Peterson P A, Wilson I A, Teyton L. Proc Natl Acad Sci USA. 1997;94:13838–13843. doi: 10.1073/pnas.94.25.13838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson-Kubalek E M, Brown R E, Celia H, Milligan R A. Proc Natl Acad Sci USA. 1998;95:8040–8045. doi: 10.1073/pnas.95.14.8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kubalek E M, LeGrice S F J, Brown P O. J Struct Biol. 1994;113:117–123. doi: 10.1006/jsbi.1994.1039. [DOI] [PubMed] [Google Scholar]
- 14.Dietrich C, Schmitt L, Tampé R. Biochemistry. 1995;92:9014–9018. doi: 10.1073/pnas.92.20.9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frey W, Schief W R, Pack D W, Chen C T, Chilkoti A, Stayton P, Vogel V, Arnold F H. Proc Natl Acad Sci USA. 1996;93:4937–4941. doi: 10.1073/pnas.93.10.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barklis E, McDermott J, Wilkens S, Schabtach E, Schmid M F, Fuller S, Karanjia S, Love Z, Jones R, Rui Y, et al. EMBO J. 1997;16:1199–1213. doi: 10.1093/emboj/16.6.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vénien-Bryan C, Balavoine F, Toussaint B, Mioskowski C, Hewat E A, Helme B, Vignais P M. J Mol Biol. 1997;274:687–692. doi: 10.1006/jmbi.1997.1431. [DOI] [PubMed] [Google Scholar]
- 18.Matsumura M, Saito Y, Jackson M R, Song E S, Peterson P A. J Biol Chem. 1992;267:23589–23595. [PubMed] [Google Scholar]
- 19.MacDonald R C, MacDonald R I, Menco B P M, Takeshita K, Subbarao N K, Hu L. Biochim Biophys Acta. 1991;1061:297–303. doi: 10.1016/0005-2736(91)90295-j. [DOI] [PubMed] [Google Scholar]
- 20.Von Tscharner V, McConnell H M. Biophys J. 1981;36:421–427. doi: 10.1016/S0006-3495(81)84741-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dubochet J, Adrian M, Chang J J, Homo J C, Lepault J, McDowell A W, Schultz P. Q Rev Biophys. 1988;21:129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- 22.Schmid M F, Dargahi R, Tam M W. Ultramicroscopy. 1993;48:251–264. doi: 10.1016/0304-3991(93)90099-j. [DOI] [PubMed] [Google Scholar]
- 23.Amos L A, Henderson R, Unwin P N T. Prog Biophys Mol Biol. 1982;39:183–231. doi: 10.1016/0079-6107(83)90017-2. [DOI] [PubMed] [Google Scholar]
- 24.Henderson R, Baldwin J M, Downing K H, Lepault J, Zemlin F. Ultramicroscopy. 1986;19:147–178. [Google Scholar]
- 25.Brian A A, McConnell H M. Proc Natl Acad Sci USA. 1984;81:6159–6163. doi: 10.1073/pnas.81.19.6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ajitkumar P, Geier S S, Kesari K V, Borriello F, Nakagawa M, Bluestone J A, Saper M A, Wiley D C, Nathenson S G. Cell. 1988;54:47–56. doi: 10.1016/0092-8674(88)90178-x. [DOI] [PubMed] [Google Scholar]
- 27.Hogquist K A, Jameson S C, Heath W R, Howard J L, Bevan M J, Carbone F R. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 28.Sant’Angelo D B, Waterbury G, Preston-Hurlburt P, Tim Yoon S, Medzhitov R, Hong S, Janeway C A. Immunity. 1996;4:367–376. doi: 10.1016/s1074-7613(00)80250-2. [DOI] [PubMed] [Google Scholar]
- 29.Alam S M, Travers P J, Wung J L, Nasholds W, Redpath S, Jameson S C, Gascoigne N R. Nature (London) 1996;381:616–620. doi: 10.1038/381616a0. [DOI] [PubMed] [Google Scholar]
- 30.Lyons D S, Lieberman S A, Hampl J, Boniface J J, Chien Y, Berg L J, Davis M M. Immunity. 1996;5:53–61. doi: 10.1016/s1074-7613(00)80309-x. [DOI] [PubMed] [Google Scholar]
- 31.Davis M M, Boniface J J, Reich Z, Lyons D, Hampl J, Arden B, Chien Y. Annu Rev Immunol. 1998;16:523–544. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- 32.Sykulev Y, Brunmark A, Tsomides T J, Kageyama S, Jackson M, Peterson P A, Eisen H N. Proc Natl Acad Sci USA. 1994;91:11487–11491. doi: 10.1073/pnas.91.24.11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fremont D H, Stura E A, Matsumara M, Peterson P A, Wilson I A. Proc Natl Acad Sci USA. 1995;92:2479–2483. doi: 10.1073/pnas.92.7.2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terrettaz S, Stora T, Duschl C, Vogel H. Langmuir. 1993;9:1361–1369. [Google Scholar]
- 35.Plant A L, Brigham-Burke M, Petrella E C, O’Shanessy D J. Anal Biochem. 1995;226:342–348. doi: 10.1006/abio.1995.1234. [DOI] [PubMed] [Google Scholar]
- 36.Gershon P D, Khilko S. J Immunol Methods. 1995;183:65–76. doi: 10.1016/0022-1759(95)00032-6. [DOI] [PubMed] [Google Scholar]
- 37.Sigal G B, Bamdad C, Barberis A, Strominger J, Whitesides G M. Anal Chem. 1996;68:490–497. doi: 10.1021/ac9504023. [DOI] [PubMed] [Google Scholar]
- 38.Avila R, Hong H L, Kaufman A, Pfister H, Silva C, Sobierajski L, Wang S. IEEE Visualization Proceedings. Washington, DC: IEEE Computer Society Press; 1994. pp. 31–38. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}