

Abstract
β-Catenin is a protein that plays a role in intercellular adhesion as well as in the regulation of gene expression. The latter role of β-catenin is associated with its oncogenic properties due to the loss of expression or inactivation of the tumor suppressor adenomatous polyposis coli (APC) or mutations in β-catenin itself. We now demonstrate that another tumor suppressor, PTEN, is also involved in the regulation of nuclear β-catenin accumulation and T cell factor (TCF) transcriptional activation in an APC-independent manner. We show that nuclear β-catenin expression is constitutively elevated in PTEN null cells and this elevated expression is reduced upon reexpression of PTEN. TCF promoter/luciferase reporter assays and gel mobility shift analysis demonstrate that PTEN also suppresses TCF transcriptional activity. Furthermore, the constitutively elevated expression of cyclin D1, a β-catenin/TCF–regulated gene, is also suppressed upon reexpression of PTEN. Mechanistically, PTEN increases the phosphorylation of β-catenin and enhances its rate of degradation. We define a pathway that involves mainly integrin-linked kinase and glycogen synthase kinase 3 in the PTEN-dependent regulation of β-catenin stability, nuclear β-catenin expression, and transcriptional activity. Our data indicate that β-catenin/TCF–mediated gene transcription is regulated by PTEN, and this may represent a key mechanism by which PTEN suppresses tumor progression.
Keywords: integrin-linked kinase, glycogen synthase kinase 3, cyclin D1, prostate cancer, protein kinase B
Introduction
β-Catenin was originally identified as a cytoplasmic component of cell–cell adherens junctions, where it interacts with the cytoplasmic domains of cadherin molecules and links them via α-catenin to the actin cytoskeleton (Kemler 1993; Geiger et al. 1995). β-Catenin is also part of the wnt-signaling pathway (Peifer and Wieschaus 1990; Peifer et al. 1993; Wodarz and Nusse 1998). Upon stimulation of this pathway, β-catenin accumulates in the cytoplasm and subsequently translocates to the nucleus in association with members of the T cell factor (TCF)/lymphoid enhancer factor (LEF) DNA-binding transcription factor family (Miller and Moon 1996; Willert and Nusse 1998). In the nucleus, this bipartite complex binds to DNA at the TCF/LEF binding sites and induces the transcription and expression of specific genes (Riese et al. 1997; van de Wetering et al. 1997). The cyclin D1 gene has been identified as a key transcriptional target of β-catenin/TCF through a TCF-4/LEF-1 binding site in the cyclin D1 promoter (Shtutman et al. 1999; Tetsu and McCormick 1999; Behrens 2000; Lin et al. 2000). In addition to cyclin D1, two other recently defined targets, c-myc and matrilysin, may also be responsible for the oncogenic potential of β-catenin (He et al. 1998; Brabletz et al. 2000). In the absence of growth and differentiation signals, cytoplasmic β-catenin is efficiently degraded by the ubiquitin-proteosome system (Aberle et al. 1997; Orford et al. 1997; Salomon et al. 1997). Protein degradation by the proteosome is under the control of the adenomatous polyposis coli (APC), glycogen synthase kinase 3 proteins (GSK-3; Munemitsu et al. 1995; Rubinfeld et al. 1996), and adapter proteins axin and conductin (Behrens 2000; Polakis 2000). The process involves targeted phosphorylation of highly conserved serine and threonine residues (Yost et al. 1996) and ubiquitination of the NH2 terminus of β-catenin (Aberle et al. 1997; Orford et al. 1997). Mutation of these NH2 terminus phosphorylation sites of β-catenin stabilize the protein, promote its nuclear accumulation, and hence its availability to serve as a transcription factor (Yost et al. 1996; Korinek et al. 1997; Morin et al. 1997; Barker et al. 2000). Loss of functional APC protein also results in stabilization and accumulation of β-catenin, leading to subsequent dysregulation of the signaling pathway (Munemitsu et al. 1995; Tetsu and McCormick 1999). Dysregulation of β-catenin signaling due to loss of APC or mutations in β-catenin itself is an important event in the progression of colorectal cancer (Voeller et al. 1998; Morin 1999). Although increased nuclear β-catenin accumulation and TCF activity are frequently found in other malignancies, for example prostate cancers, β-catenin mutations (Voeller et al. 1998) or APC mutations (Suzuki et al. 1994; Watanabe et al. 1996) do not appear to be involved in this activation.
A very frequent and prominent feature of prostate cancer (as well as a wide variety of other human cancers) is the mutational inactivation of the tumor suppressor PTEN (Stambolic et al. 1998; Sun et al. 1999). PTEN is a lipid and protein phosphatase that is a negative regulator of phosphatidylinositol 3 (PI-3) kinase–dependent signaling pathway. The latter regulates cell survival and growth via integrated signaling from growth factor receptors and components of the extracellular matrix (Stambolic et al. 1998; Sun et al. 1999). Loss of expression or mutational inactivation of PTEN results in the suppression of apoptosis (Stambolic et al. 1998) and accelerated cell cycle progression (Ramaswamy et al. 1999; Sun et al. 1999). Reexpression of PTEN in PTEN null cells has been shown to induce growth suppression (Ramaswamy et al. 1999; Sun et al. 1999) and this is thought to be mediated by the inhibition of signaling through the PI-3 kinase pathway (Ramaswamy et al. 1999; Sun et al. 1999). We have also recently demonstrated that reexpression of PTEN induced apoptosis and G1 cell cycle arrest in PTEN null prostate cancer cells (Persad et al. 2000). Although the exact biochemical mechanisms by which PTEN exerts its growth inhibitory effects are still largely unknown, it has been shown that PI-3 kinase and PTEN regulate the phosphorylation, and hence activation, of the antiapoptotic protein, protein kinase B (PKB; Ramaswamy et al. 1999; Sun et al. 1999). We have recently shown that the integrin-linked kinase (ILK) is an intermediate between PTEN and PKB, such that inhibition of ILK activity also results in the inhibition of activation of PKB and in an increase in apoptosis and G1 phase cell cycle arrest (Persad et al. 2000).
In this study, we demonstrate that in the absence of any overt APC (Suzuki et al. 1994; Watanabe et al. 1996) or β-catenin (Voeller et al. 1998) mutations, the PTEN null prostate cancer cell lines PC3 and LNCaP exhibit constitutively high levels of nuclear β-catenin, as well as cyclin D1. Upon reexpression of wild-type PTEN (PTEN-WT), GSK-3 activity is increased, leading to an increase in β-catenin phosphorylation, increased β-catenin degradation, and subsequent decrease in the expression of the protein in the nucleus. Transfection of PC3 cells with the dominant negative form of ILK (kinase-deficient ILK [ILK-KD]) or GSK-3-WT also suppressed the elevated expression of nuclear β-catenin. Furthermore, PTEN, ILK-KD, and GSK-3-WT induced comparable suppression of TCF/β-catenin transcriptional activity in PC3 cells. In correlation with this, we noted that cyclin D1 expression and promoter activity, which are constitutively elevated in PC3 cells, are suppressed upon reexpression of PTEN or expression of ILK-KD and GSK-3-WT. These results delineate a novel role for PTEN in the regulation of cell growth and proliferation via its regulation of nuclear β-catenin and subsequently cyclin D1.
Materials and Methods
Cell Culture and Transfections
The PTEN null prostate cancer cell lines PC3 and LNCaP were used in this study. PC3 cells were cultured in DME containing 10% FBS, and LNCaP cells were cultured in RPMI-1640 media supplemented with 10% FBS. All cells were passaged in 5% CO2 at 37°C. The cells were transiently transfected with His-V5–tagged ILK-WT cDNA, His-V5–tagged ILK-KD cDNA, green fluorescent protein (GFP)-tagged PTEN-WT cDNA (Persad et al. 2000), GFP-tagged pEGFP cDNA, hemagglutinin (HA)-tagged dominant negative PKB (PKB-AAA) cDNA (Wang et al. 1999) or HA-tagged GSK-3-WT cDNA. Transfection of PC3 and LNCaP cells was carried out by using lipofectin reagent (GIBCO BRL) according to the manufacturers guidelines, and using 4 μl of lipofectin reagent and 2–8 μg cDNA. Control cells were transfected with an empty vector. The ILK cDNAs, PKB-AAA cDNA, and GSK-3 cDNA were in the pc-DNA-3 plasmid, whereas the PTEN cDNA was in the p-EGFP plasmid. The efficiency of the transfections were evaluated by flow cytometric analysis of cells cotransfected with pEGFP cDNA and the various plasmids used in the study. The PTEN plasmid is tagged with GFP and therefore was not cotransfected with pEGFP. The percentage of cells expressing GFP was then evaluated using the WinMDI analysis program. All transfections were carried out overnight and the cells were processed for immunofluorescence or harvested for lysis 24–48 h later.
Western Blot
Equivalent protein concentrations were resolved by SDS-PAGE electrophoresis and proteins were transferred to polyvinylidene difluoride membrane. Western blot analysis using the enhanced chemiluminescence detection system (Amersham Pharmacia Biotech) was carried out as described previously (Delcommenne et al. 1998). The following antibodies were used in this study: anti–β-catenin antibody (mouse monoclonal; Transduction Laboratories), anti–β-catenin antibody (rabbit polyclonal; Santa Cruz Biotechnology, Inc.), N-cadherin (mouse monoclonal), anti–TCF-4 antibody (mouse monoclonal; Upstate Biotechnology), anti–Lef-1 antibody (mouse monoclonal; Oncogene Research Products); anti-Histone H1 antibody (mouse monoclonal; Chemicon International, Inc.); anti-GFP antibody (mouse monoclonal; Boehringer); anti-Cyclin D1 (mouse monoclonal; Upstate Biotechnology), anti-p27Kip (rabbit polyclonal; Upstate Biotechnology), anti-p21Cip (rabbit polyclonal; Upstate Biotechnology), anti-ILK antibody (rabbit polyclonal; StressGen Biotechnologies), anti-PTEN antibody (mouse monoclonal; Oncogene Research Products), anti-PKB antibody (rabbit polyclonal; New England Biolabs, Inc.), anti–GSK-3 antibody (mouse monoclonal; Transduction Laboratories); antiphospho–β-catenin (Ser33/37/Thr41) antibody (rabbit polyclonal; Cell Signaling Technologies).
Cells and Fluorescence Microscopy
PC3 cells were plated onto Lab-Tek® chamber slides (Nunc) 24 h before transfection. Cells were transfected overnight with PTEN-GFP (3 μg) using Lipofectin reagent (4 μl) according to the manufacturers guidelines. Cells were fixed and permeabilized 48 h posttransfection with methanol at −20°C for 20 min. Immunofluorescence analysis was done using mouse monoclonal β-catenin antibody followed by rhodamine (TRITC)-conjugated anti–mouse IgG (Jackson ImmunoResearch Laboratories). Images were obtained by fluorescent microscopy (ZEISS). The percentage of cells with nuclear β-catenin was determined by evaluating the number of cells expressing high amounts of nuclear β-catenin in a field with reference to the total number of cells in that field.
Luciferase Assay for TCF and Cyclin D1 Promoter Activities
Luciferase assays were done according to the manufacturer's instructions (Promega). A dual luciferase reporter assay was performed whereby the experimental TCF promoter/luciferase reporter gene (TOPFLASH; Korinek et al. 1997) or the cyclin D1 promoter/luciferase reporter (Tetsu and McCormick 1999) and a control reporter (pRenilla) were cotransfected with ILK-WT, ILK-KD, PTEN-WT, PKB-AAA, or GSK-3-WT. The effect of ILK-WT, ILK-KD, PTEN-WT, PKB-AAA, or GSK-3-WT upon the TCF reporter gene activity and the effect of ILK-WT, ILK-KD, PTEN-WT, or GSK-3-WT on cyclin D1 reporter gene activity were evaluated. The promoter activity was normalized to the activity of the control (pRenilla). Two different TCF-luciferase reporter genes were used in this study: an intact wild-type TCF-luciferase reporter construct (TOPFLASH) and a mutated TCF-luciferase reporter construct (FOPFLASH), which served as a negative control for TOPFLASH activity. Protein concentrations were determined by Bradford assay and the results were expressed as relative light units per microgram of protein.
Nuclear Extracts and Electrophoretic Mobility Shift Assays
Nuclear extracts were prepared by the miniextraction method as described previously (Andrews and Faller 1991). PC3 cells transfected with empty vector (control), ILK-KD, or PTEN-WT were washed with ice cold PBS and harvested by being scraped in 1.5 ml PBS. Cells were then pelleted and resuspended in 400 μl of 10 mM Hepes–potassium hydroxide, pH 7.9, 1.5 mM magnesium chloride, 10 mM potassium chloride, 0.5 mM dithiothreitol, 0.2 mM PMSF. After 10 min of incubation on ice, nuclei were pelleted by being spun for 10 s and resuspended in 50 μl of 20 mM Hepes–potassium hydroxide, pH 7.9, 25% glycerol, 420 mM sodium chloride, 1.5 mM magnesium chloride, 0.2 mM EDTA, 0.5 mM dithiothreitol, 0.2 mM PMSF. Tubes were incubated for 20 min on ice and then centrifuged to clear the cellular debris. Nuclear extracts were stored at −70°C. Electrophoretic mobility shift assays were performed by using 4 μg of the nuclear extracts for 20 min at room temperature with a 32P end-labeled DNA fragment containing the putative protein binding site (for TCF binding site from CD1 promoter, 5′-TGC CGG GCT TTG ATC TTT GCT-3′; [γ-32P]ATP was from Amersham Pharmacia Biotech). Reaction products were analyzed on a nondenaturing 5% polyacrylamide gel (0.5% Tris-borate-EDTA, 3.5% glycerol). The specificity of the DNA–protein interaction was established by competition experiments using 10× cold TCF oligonucleotide as the competitor. For the supershift assays, 10 μg of mouse anti–β-catenin antibody or nonspecific IgG was added to the reaction mixture, subsequent to the addition of the 32P-labeled oligonucleotide probe, and the mixture was incubated for 45 min at room temperature. Complexes were resolved by electrophoresis as described for the mobility shift assays.
Nuclear Extracts and Coimmunoprecipitation Assay
PC3 cells were transfected with an empty vector (control), ILK-KD, or PTEN-WT cDNA and harvested at 48 h posttransfection. The cells were washed in cold PBS and nuclear extracts were prepared by the miniextraction method as described previously (Andrews and Faller 1991). Immunoprecipitation of β-catenin was performed using 400 μg of nuclear lysate and polyclonal anti–β-catenin antibody. Immunocomplexes were isolated with protein A/G-PLUS agarose (Santa Cruz Biotechnology, Inc.) and separated on 7.5% SDS-PAGE gels. The gels were then Western blotted with either monoclonal anti–TCF-4 or monoclonal anti–Lef-1 antibodies to detect presence of these transcription factors in the immunocomplexes.
Pulse–Chase Analysis
PC3 cells were cultured to 70–80% confluence and transfected with empty vector (control) or PTEN-WT. For each chase time point the cells were washed once with DME without cysteine/methionine (starve media; Sigma-Aldrich) and incubated in the starve media for 1 h at 37°C. Cells were pulsed for 1 h at 37°C using [35S]Promix (Amersham Pharmacia Biotech), 100 μCi/ml. Cells were then chased with DME/10% FBS for the indicated time points. The cells were harvested at the various time points and lysed in RIPA buffer. Immunocomplexes were isolated with protein G Sepharose (Santa Cruz Biotechnology, Inc.) and separated on 7.5% SDS-PAGE gels, stained with Coomassie blue, destained, and incubated with Amplify (Amersham Pharmacia Biotech) fluorographic reagent, dried, and exposed to film.
Northern Blot Analysis of Cyclin D1
Northern blot analysis of cyclin D1 was carried out on total RNA from serum-starved PC3 cells. Total RNA was isolated from in vitro cultured PC3 cells using TRIZOL reagent (Life Technologies). The PC3 cells were transfected with empty vector (control), ILK-WT, ILK-KD, PTEN-WT, or GSK-3-WT. 15 μg of total RNA from each sample was subjected to electrophoresis on 1.2% agarose-formaldehyde gels and transferred to nylon membranes (Gelman Sciences) overnight. The RNA blots were hybridized with a cyclin D1 cDNA probe labeled with [32P]dCTP by random primer labeling.
GSK-3 Kinase Assay
GSK-3 kinase activity was determined in cell extracts from PC3 cells transiently transfected with either empty vector (control), ILK-WT, ILK-KD, PTEN-WT or dominant negative PKB (PKB-AAA). Cells were lysed in 50 mM Hepes buffer, pH 7.5, containing NaCl (150 mM), NP-40 (1%), sodium deoxycholate (0.5%), leupeptin (10 μg/ml), PMSF (1 mM), aprotinin (2.5 μl/ml), sodium fluoride (5 mM) and sodium orthovanadate (1 mM). Equivalent protein concentrations of cell lysates (determined by Bradford Assay) were precleared with nonspecific IgG and protein A Sepharose. After centrifugation the supernatants were immunoprecipitated with anti–GSK-3α antibody bound to protein A Sepharose beads. Kinase assay was carried out as described previously (Delcommenne et al. 1998) using GS-1 peptide as a substrate. Phosphorylated GS-1 peptide was electrophoresed on a tricine gel, then visualized and quantified by autoradiography or phosphoimage analysis.
ILK Kinase Assay
Equivalent protein concentrations of cell lysates were precleared with nonspecific IgG and protein A Sepharose. After centrifugation, the supernatants were immunoprecipitated with anti-ILK antibody (Upstate Biotechnology) bound to protein A Sepharose. Kinase assays were carried out as described previously (Delcommenne et al. 1998) using purified GSK-3-KD as substrate and [γ-32P]ATP as the phosphate donor. The ILK kinase assays were carried out in the presence or absence of either DMSO or a highly selective small molecule inhibitor of ILK, KP-SD-1 (Persad et al. 2001). Phosphorylated GSK-3 was electrophoresed on a 7.5% SDS-PAGE gel, then visualized and quantified by autoradiography and phosphoimage analysis.
In addition, in vitro ILK kinase assays were carried out in which purified recombinant ILK prepared in insect cells and GSK-3-KD were coincubated in the presence of kinase reaction buffer and ATP as described previously (Delcommenne et al. 1998). The kinase assays were carried out in the presence or absence of the ILK inhibitor, KP-SD-1. Phosphorylated GSK-3 was resolved by SDS-PAGE and detected by Western blot analysis using the anti–GSK-3-Ser-9-P antibody (rabbit polyclonal; New England Biolabs, Inc.).
Results
Inhibition of Nuclear β-Catenin Expression and Induction of E-Cadherin Expression in PTEN-negative Cells by Reexpression of PTEN
Upon examination of β-catenin expression in prostate cancer cell lines, we noted that a significant population of the PTEN-negative prostate cancer cells (PC3; ∼70%) express high levels of β-catenin in the nucleus (Fig. 1 A). Constitutively high levels of β-catenin are also observed in nuclear extracts from PC3 cells as well as another PTEN null prostate cancer cell line, LNCaP (Fig. 1b and Fig. c). Transient transfection of increasing amounts of PTEN-WT into these PTEN null cells (LNCaP and PC3) leads to inhibition of nuclear β-catenin expression in these cells (Fig. 1b and Fig. c). The transfection efficiency of the PTEN-WT plasmid is ∼80% in PC3 and LNCaP prostate cancer cells. There was no change in nuclear β-catenin expression when PC3 cells were transfected with increasing amounts of pEGFP cDNA (Fig. 1 C). Total cellular β-catenin expression was unaffected by the PTEN transfections. Immunofluorescence microscopy of PC3 cells transiently transfected with PTEN-GFP also showed that β-catenin is mostly present outside the nucleus in cells expressing PTEN-GFP, and a much smaller number of cells (∼16%) express nuclear β-catenin (Fig. 1 D). Surprisingly, reexpression of PTEN also induced the expression of E-cadherin in PC3 cells, which do not normally express E-cadherin (Fig. 1 C). However, PC3 cells do express N-cadherin and the expression of this cadherin remains unchanged by the reexpression of PTEN (Fig. 1 C).
Figure 1.
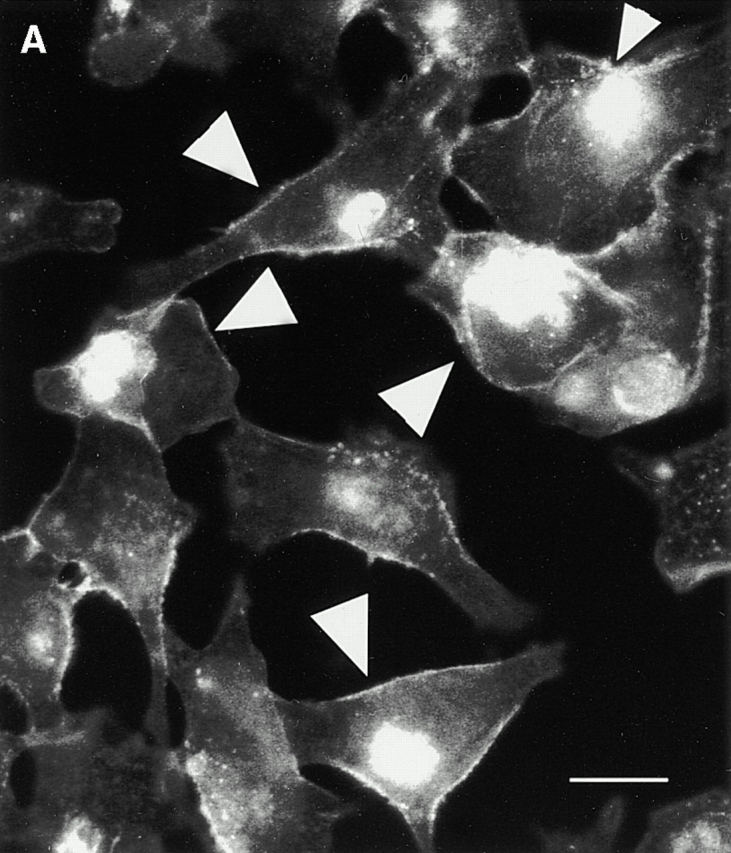

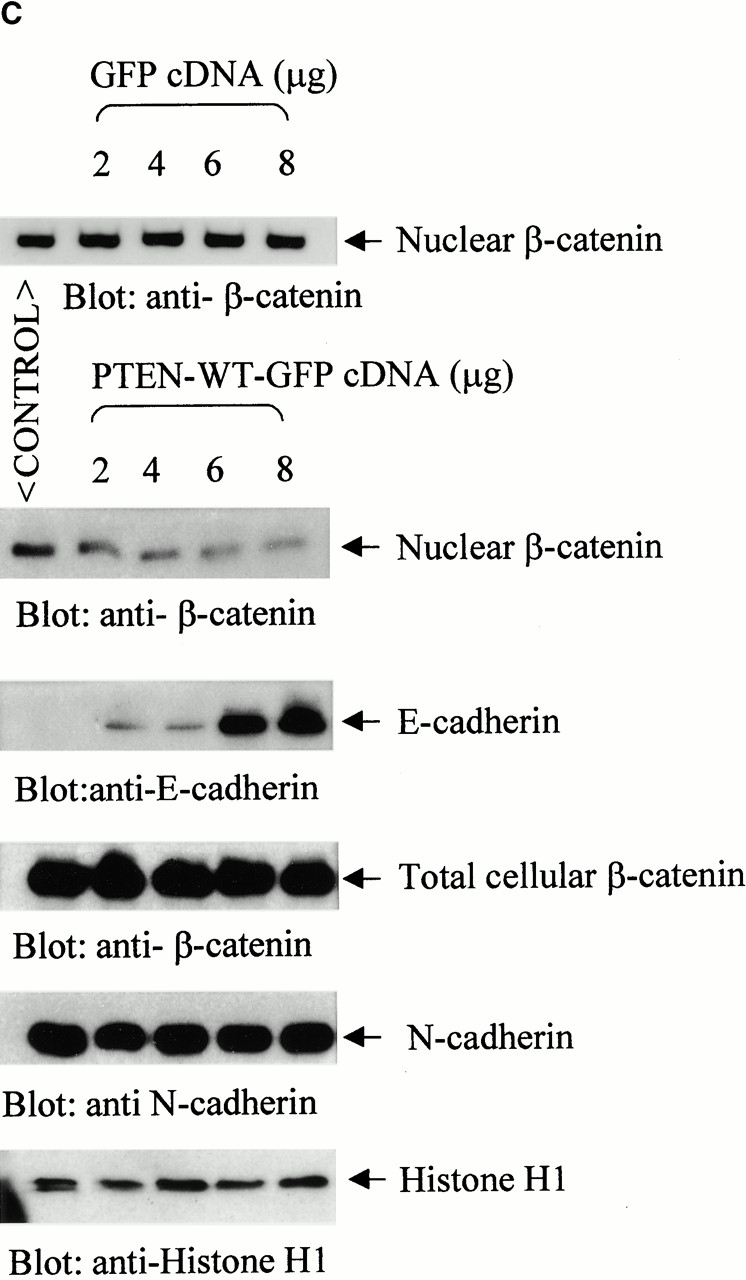

Expression of nuclear β-catenin is reduced by reexpression of PTEN in PTEN null prostate cancer cells. (A) Immunofluorescence analysis of PTEN null prostate cancer cells, PC3, shows that a significant population (70%) of these cells express high levels of β-catenin in their nucleus (arrowheads). Reexpression of PTEN in PTEN null prostate cancer cells LNCaP (B) and PC3 (C) results in a dose-dependent reduction in the expression of nuclear β-catenin. (D) Immunofluorescence analysis of β-catenin confirms that unlike nontransfected PC3 cells (arrowheads), PC3 cells transfected with PTEN (arrows) exhibit anuclear localization β-catenin. A significantly lower proportion of cells in this case express high levels of nuclear β-catenin (16%). Bar, 5 μm.
Constitutive Expression of Nuclear β-Catenin in PC3 Cells Is Reduced by Dominant Negative ILK and GSK-3-WT
To identify a pathway via which PTEN may be regulating β-catenin, we investigated the effect of the various members of the pathway downstream of PI-3 kinase/PTEN, namely, ILK, PKB, and GSK-3, on nuclear β-catenin expression (Fig. 2). GSK-3 is known to be a member of the complex that regulates cellular levels of β-catenin in a phosphorylation-dependent manner (Morin 1999; Behrens 2000).
Figure 2.
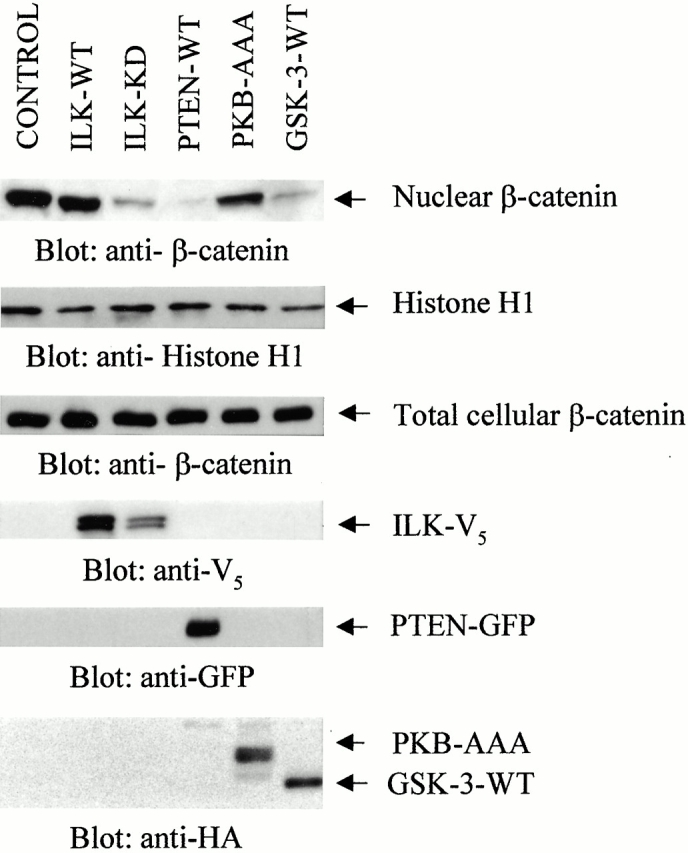
Nuclear β-catenin is mainly regulated by PTEN and ILK via GSK-3. The expression of nuclear β-catenin is dramatically and comparably abrogated by expression of PTEN (82%), ILK-KD (79%), and GSK-3-WT (78%) into PC3 cells. Nuclear β-catenin expression is altered to a lesser extent by the dominant negative PKB-AAA (79%) and completely unaffected by expression of ILK-WT (78%).Values in brackets indicate the transfection efficiency of the various plasmids. The absence of any alterations in the total cellular β-catenin expression demonstrates the specific effect of the various components upon nuclear β-catenin.
We have recently demonstrated a critical role for the ILK in PTEN-dependent cell cycle regulation (Persad et al. 2000). Also, ILK has been shown to inhibit GSK-3 activity upon cell–extracellular matrix interaction (Troussard et al. 1999). In this study we show that although transient transfection of ILK-WT has no effect on nuclear β-catenin expression, expression of the dominant negative ILK (Persad et al. 2000) or GSK-3-WT resulted in significant reductions in nuclear β-catenin expression (Fig. 2). The dramatic inhibitory effects of the plasmids are a reflection of their high transfection efficiency (ILK-KD, 79%; GSK-3, 78%) into PC3 cells. Since dominant negative ILK has been shown to inhibit PKB serine 473 phosphorylation and its activity in PC3 cells, we wanted to determine whether the ILK-KD– and PTEN-induced inhibition of nuclear β-catenin involved PKB activity. Surprisingly, even though the transfection efficiency of PKB-AAA is quite comparable (79%) to the other plasmids, we found that its effect upon nuclear β-catenin expression was lower compared with dominant negative ILK, PTEN-WT, or GSK-3-WT. Total cellular levels of β-catenin remained unchanged by all the plasmid transfections.
The results in Fig. 2 suggest that PTEN suppresses nuclear accumulation of β-catenin and this involves the downstream effectors, ILK and GSK-3, and to a lesser extent, PKB.
Expression of PTEN or Inhibition of ILK Inhibits Complex Formation between β-Catenin and TCF and between β-Catenin/TCF and TCF Consensus Oligonucleotide
It is known that β-catenin interacts with transcription factors of the TCF/LEF family and subsequently activates genes that are responsive to TCF/LEF family members (Giese et al. 1995; Behrens et al. 1996; Clevers and Grosschedl 1996; Molenaar et al. 1996; van de Wetering et al. 1997). Therefore, we analyzed the ability of β-catenin to form complexes with a TCF-4 consensus oligonucleotide in the presence of either PTEN-WT or dominant negative ILK. PC3 cells were transfected with empty vector, PTEN-WT, or ILK-KD, and nuclear extracts for gel mobility shift assays were prepared. The gel mobility shift assay demonstrated the presence of significant levels of protein–DNA complex in PC3 cells transfected with empty vector (Fig. 3 A, panel 1). However, the level of the protein–DNA complex was significantly reduced in PC3 cells transfected with either PTEN-WT or ILK-KD (lanes 2 and 3) compared with cells transfected with empty vector (lane 1).
Figure 3.

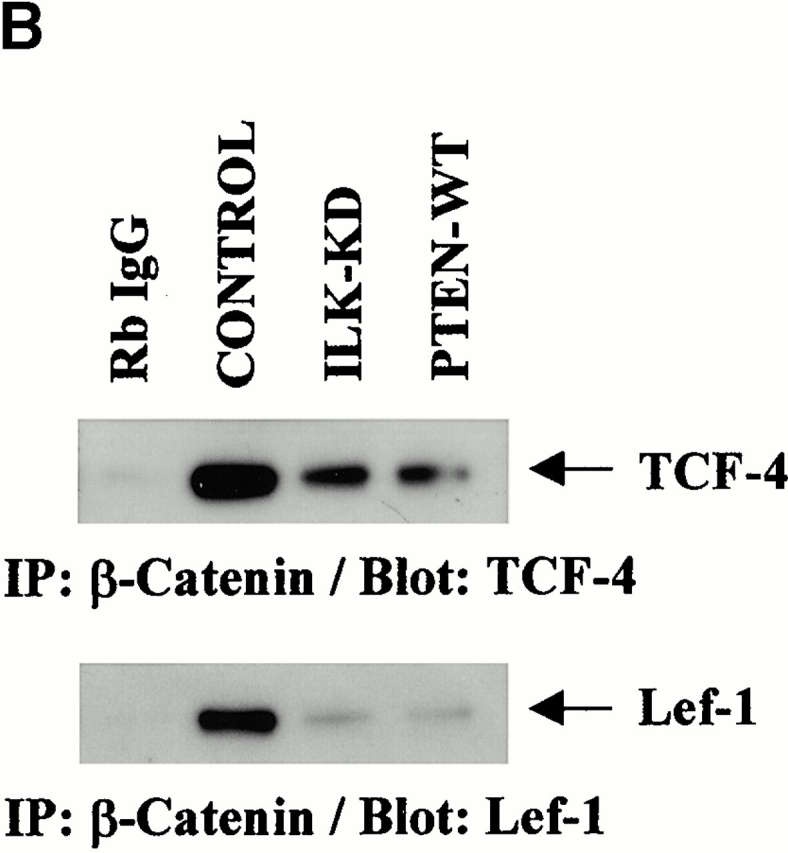

PTEN and ILK-KD inhibit DNA binding activities of β-catenin–TCF complex. (A) Electrophoretic mobility shift assays, using an oligonucleotide containing a potential binding site for TCF, demonstrated an abundance of DNA–protein complex in empty vector–transfected PC3 cells (panel 1, lane 1). The quantities of the protein–DNA complex are reduced significantly in PC3 cells transfected with ILK-KD or PTEN (panel 1, lanes 2 and 3). Electrophoretic mobility shift assays performed in the presence of a supershifting antibody (anti–β-catenin antibody) confirmed that the transcription factor complex binding to the oligonucleotide includes β-catenin (panel 2). (B) Coimmunoprecipitation of TCF-4 or Lef-1 with β-catenin using nuclear lysates from empty vector (control), ILK-KD–, or PTEN-WT–transfected PC3 cells show reduced complex formation in ILK-KD– and PTEN-WT–transfected cells compared with control cells. (C) The relative β-catenin/TCF activities (TOPFLASH/FOPFLASH luciferase reporter) in response to the various components of the PI-3 kinase/PTEN pathway. PC3 cells were transiently transfected with 0.5 μg of TOPFLASH reporter together with 2.5 μg of empty vector, ILK-WT, ILK-KD, PTEN-WT, PKB-AAA, or GSK-3-WT. β-Catenin/TCF activity was dramatically reduced due to PTEN, ILK-KD, and GSK-3-WT. The effect of PKB-AAA upon TOPFLASH activity was more modest in comparison. Parallel cotransfections with the various plasmids and FOPFLASH served as a negative control for TOPFLASH activity.
To determine if the protein–DNA complex in the PC3 cells contained β-catenin, we carried out a supershift assay using anti–β-catenin antibody to detect the presence of β-catenin in the complex. As seen in Fig. 3 A, panel 2, anti–β-catenin antibody induced a protein–DNA mobility shift of the complex, highlighting the presence of β-catenin in the complex. In agreement with the reduced levels of protein–DNA complex in PC3 cells transfected with PTEN-WT or ILK-KD, the supershift bands, while quite evident in the empty vector–transfected cells (panel 2, lane 1), were negligible to absent in PTEN or ILK-KD–transfected cells (panel 2, lanes 2 and 3). Furthermore, Western blot analysis of nuclear β-catenin immunoprecipitates with anti–TCF-4 or anti–Lef-1 antibodies identified substantially reduced β-catenin–TCF-4 and β-catenin–Lef-1 complexes in ILK-KD– and PTEN-WT–transfected PC3 cells compared with controls (Fig. 3 B).
In view of the fact that the nuclear β-catenin is complexed to TCF and likely induces the activity of genes containing a TCF DNA binding site in their promoter region, our next aim was to determine the effect of the various members of the PI-3 kinase/PTEN pathway (PTEN, ILK, PKB, and GSK-3) upon this β-catenin–mediated transactivation. A multimeric synthetic TCF-4 binding site upstream of a luciferase reporter gene (TOPFLASH; van de Wetering et al. 1997) was cotransfected into PC3 cells together with either ILK-WT, ILK-KD, PTEN-WT, PKB-AAA, or GSK-3-WT cDNAs. Parallel experiments, where each of the experimental cDNAs were cotransfected into PC3 cells with a construct containing a mutated TCF-4 binding site upstream of a luciferase reporter gene (FOPFLASH) served as a negative control. The results presented in Fig. 3 C show a dramatic decrease (75–80%) in luciferase activity due to transfection and expression of PTEN-WT, ILK-KD, or GSK-3-WT into PC3 cells. Interestingly, the inhibition of TOPFLASH activity due to transfection of PKB-AAA was much lower (25%).
Collectively, the results in Fig. 3 show that in PTEN null cells β-catenin likely accumulates in the nucleus in complex with TCF/LEF. The β-catenin–TCF–LEF bipartite complex binds to TCF DNA response element and induces genes that possess the TCF DNA binding site in their promoter region. In the presence of dominant negative ILK or upon reexpression of PTEN, β-catenin accumulation in the nucleus is inhibited, resulting in a decrease in transcription of target genes. The results also suggest that PTEN regulates β-catenin–TCF–LEF-mediated transcriptional activity via ILK and GSK-3, with a smaller contribution from PKB.
GSK-3 Activity Is Stimulated by PTEN-WT and Dominant Negative ILK in PTEN Null Cells
To further understand the relative roles of ILK and PTEN in the GSK-3–mediated regulation of β-catenin, we evaluated GSK-3 activity in PC3 cells transiently transfected with ILK-WT, ILK-KD, PKB-AAA, or PTEN-WT. As shown in Fig. 4 A, although both PTEN-WT and dominant negative ILK-KD induced a significant increase in GSK-3 activity (∼3–4-fold) the effect of dominant negative PKB-AAA was lower (∼1.6-fold). This difference is unlikely to be due to differences in transfection efficiencies, since all three plasmids had comparable transfection efficiencies (see legend to Fig. 4). Transfection of ILK-WT cDNA did not alter the activity of GSK-3 compared with the control. These data confirm that GSK-3 activity, which is known to be involved in controlling β-catenin levels in cells (Morin 1999; Behrens 2000), is regulated to various extents by PTEN, ILK, and PKB. Although both ILK (Delcommenne et al. 1998; Troussard et al. 1999) and PKB (Cross et al. 1995) are known to phosphorylate GSK-3, the greater stimulatory effect of PTEN and ILK-KD upon GSK-3 activity compared with PKB-AAA indicate that in these cells, GSK-3 is largely regulated by PTEN in an ILK-dependent manner. The critical protein in the regulation of β-catenin phosphorylation and stability is GSK-3, whose activity is also regulated by phosphorylation at serine 9. Although PKB can phosphorylate GSK-3 at this site and inhibit its activity, our results also implicate a direct role for ILK in the regulation of GSK-3 activity. To determine if ILK can directly phosphorylate GSK-3, we coincubated ILK purified from serum-starved PC3 cells by immunoprecipitation with anti-ILK antibody with purified GSK-3-KD in the presence of [γ-32P]ATP and kinase reaction buffer. As shown in Fig. 4 B, top, GSK-3 was successfully phosphorylated by the immunoprecipitated ILK and this phosphorylation was significantly inhibited in the presence of the ILK inhibitor KP-SD-1 (Persad et al. 2001), demonstrating the specificity of the phosphorylation of GSK-3 by ILK. Although it is conceivable that PKB, which may have coimmunoprecipitated along with ILK, is responsible for the GSK-3 phosphorylation, the inhibitory effect of the ILK inhibitor, KP-SD-1 argues against this. KP-SD-1 is ineffective in inhibiting the activity of purified PKB in vitro (Persad et al. 2001). This is further supported by an in vitro kinase assay that shows that ILK can directly phosphorylate GSK-3 at serine 9 in the absence of PKB. This was done by using recombinant ILK protein prepared in insect cells, purified GSK-3-KD as the substrate, and ATP as the phosphate group donor, followed by Western blotting with anti–GSK-3-Ser-9-P antibody. As shown in Fig. 4 B, bottom, ILK can indeed promote the direct phosphorylation of GSK-3 on serine 9 and this phosphorylation is inhibited in the presence of the ILK inhibitor, KP-SD-1.
Figure 4.
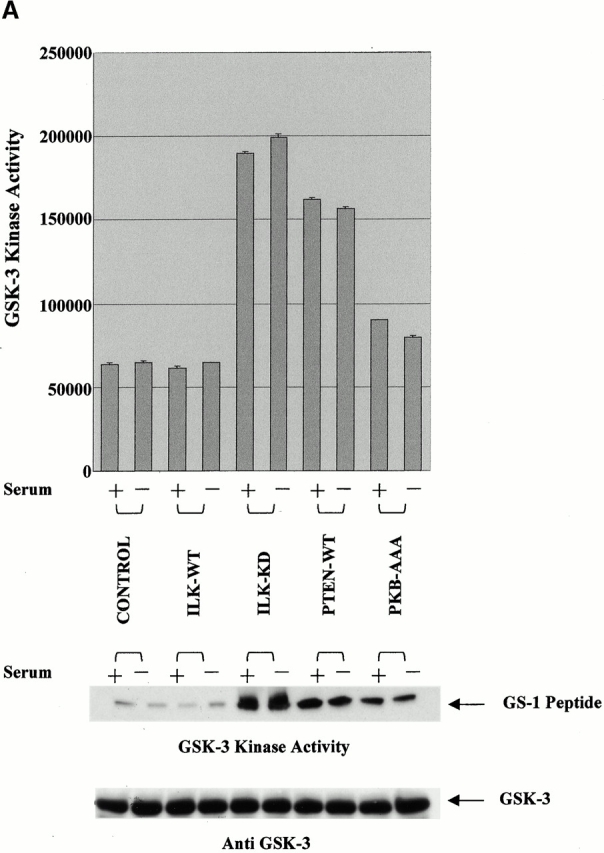
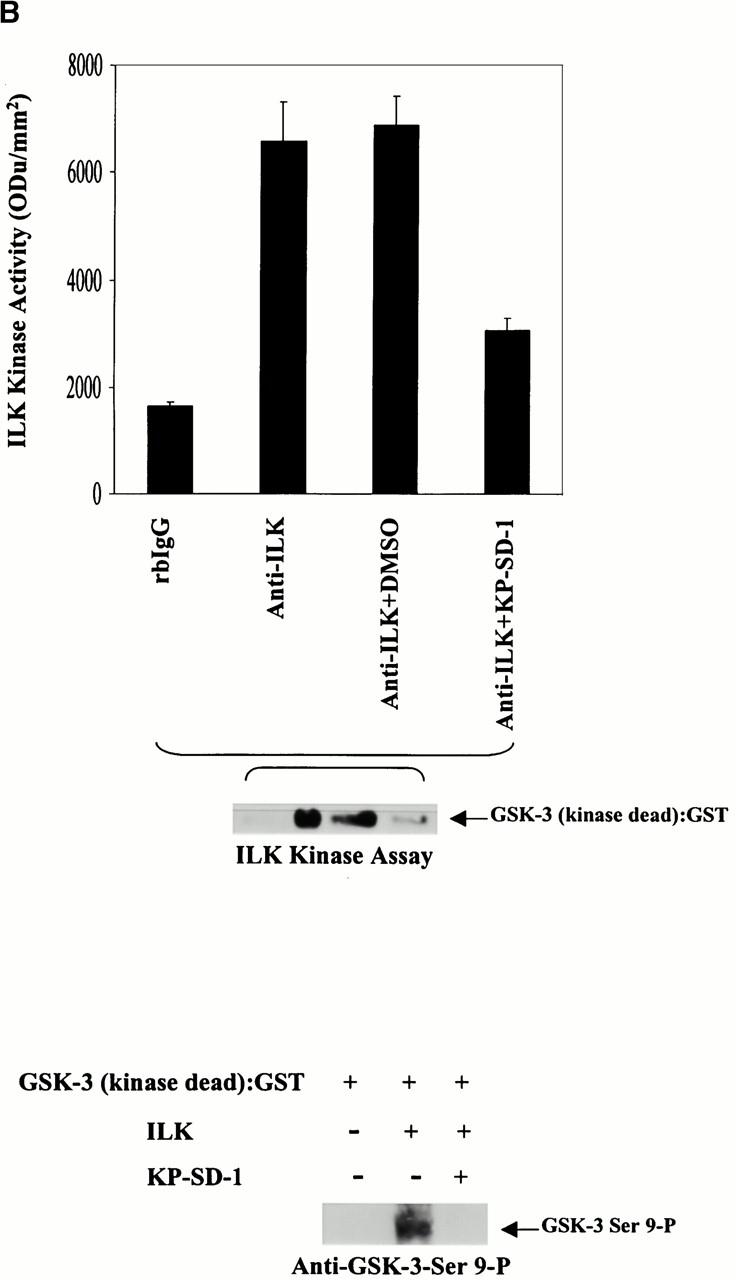
(A) Bar graph represents quantification of GSK-3 kinase activities by densitometric analysis (Odu/mm2) in PC3 cells transiently transfected with empty vector (control), ILK-WT (78%), ILK-KD (79%), PKB-AAA (79%), or PTEN-WT (82%). Values in brackets indicate the transfection efficiency of the various plasmids. Top panel is a representative autoradiograph of GSK-3 kinase activities in the various transfectants. To evaluate stimulation of GSK-3 activity, transfected cells were serum starved for 18 h, refed with serum for 1 h, and then analyzed for GSK-3 kinase activity by using GS-1 peptide as a substrate. Although PTEN and ILK-KD induced a dramatic increase in GSK-3 kinase activity (∼3–4-fold) the effect of PKB-AAA was more modest (∼1.6-fold). GSK-3 kinase activity was determined as described in Materials and Methods. Immunoblot with anti–GSK-3 antibody shows equivalent amounts of GSK-3 in each extract (bottom). (B) Bar graph represents quantification of ILK kinase activity by densitometric analysis (Odu/mm2) in serum-starved (18 h) PC3 cells. ILK, purified by immunoprecipitation with anti-ILK antibody, was coincubated with purified GSK-3-KD and [γ-32P]ATP in the presence or absence of the ILK inhibitor KP-SD-1. Bottom panel represents an in vitro ILK kinase assay, where recombinant ILK prepared in insect cells was coincubated with GSK-3-KD and ATP in the presence or absence of an ILK inhibitor, KP-SD-1. Phosphorylated GSK-3 was detected by Western blot analysis using anti–GSK-3-Ser-9-P antibody. Odu, optical density units.
PTEN Stimulates Phosphorylation of β-Catenin and Accelerates Its Degradation
We examined the effect of PTEN on the stability of β-catenin by pulse–chase analysis of PC3 cells transfected with PTEN. We observed that the rate of degradation of β-catenin was dramatically increased upon reexpression of PTEN in these PTEN null cells (Fig. 5 A).
Figure 5.
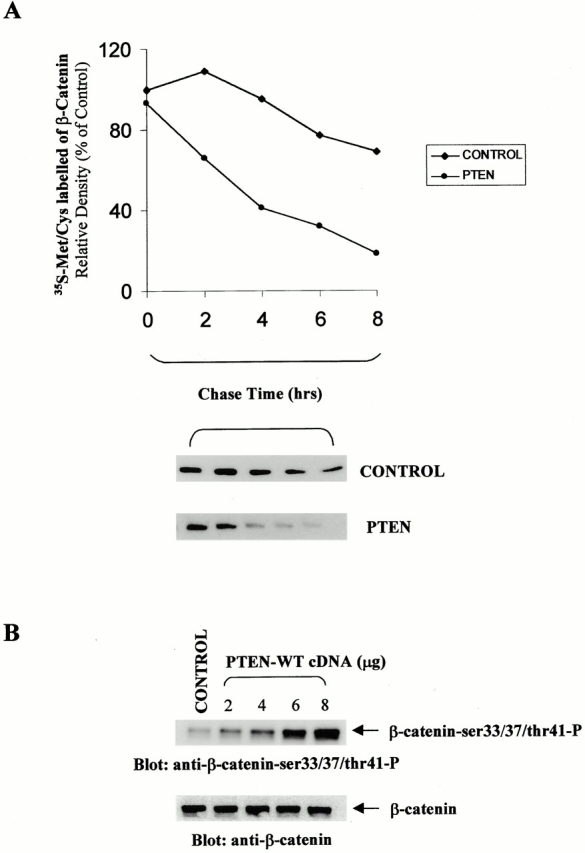
PTEN stimulates phosphorylation of β-catenin and enhances its rate of degradation. (A) PC3 cells transfected with empty vector (control) or PTEN were starved with DME without cysteine and methionine, pulsed with [35S]promix for 1 h, and then chased for the indicated time-points with cold DME containing cysteine/methionine and FBS. Cells were then harvested, lysed, and immunoprecipitated for β-catenin. Results were analyzed by densitometry and expressed as a percentage of the value at 0 h. (B) In correlation with the enhanced degradation of β-catenin on reexpression of PTEN in PC3 cells, the phosphorylation at ser33/37 and thr41 of β-catenin also increases with transfection of increasing amounts of PTEN into PC3 cells.
Since degradation of β-catenin is initiated by the phosphorylation of this protein at serine 33/37 and threonine 41, we evaluated phosphorylation of β-catenin in PC3 cells transfected with empty vector (control) or increasing amounts of PTEN using antiphospho-β-catenin-Ser 33/37/Thr41 antibody (Fig. 5 B). Our results show that the phosphorylation of β-catenin at Ser 33/37 and Thr 41 is stimulated with increasing quantities of PTEN transfected into the cells.
Inhibition of Serum-independent Cyclin D1 Expression in PTEN-negative Cells by PTEN-WT, Dominant Negative ILK, and GSK-3-WT
We have shown previously that reexpression of PTEN-WT in PTEN null cells induces G1/S cell cycle arrest (Persad et al. 2000). Since cyclin D1 has been established as a target for β-catenin, our next aim was to examine the status of this cyclin in PTEN null cells. As shown in Fig. 6, the expression level of cyclin D1 is high in PC3 cells, and is serum-independent. When these cells are transfected with PTEN-WT, the expression of cyclin D1 is significantly inhibited. Consistent with our earlier finding that ILK is inhibited by PTEN (Morimoto et al. 2000; Persad et al. 2000), transfection of dominant negative ILK also resulted in profound inhibition of the expression of cyclin D1 (Fig. 6). Transfection of ILK-WT did not alter the expression of this cyclin, demonstrating the specificity of the dominant negative effect of ILK-KD. In agreement with the fact that cellular β-catenin levels are regulated by GSK-3, the expression of cyclin D1 is also decreased dramatically by transfection of GSK-3-WT (Fig. 6). A surprising observation in this study was the fact that transfection with PTEN-WT, ILK-KD, or GSK-3-WT in PTEN null PC3 cells (Fig. 6) did not alter the expression level of the cyclin-dependent kinase (CDK) inhibitors p27Kip and p21Cip. This is in contrast to the findings of Sun et al. 1999, who reported that PTEN appears to regulate the levels of CDK inhibitors in PTEN−/− ES cells.
Figure 6.

Alteration in the expression level of cyclin D1 and the cyclin-CDK inhibitory proteins p27Kip and p21Cip, in serum-starved PC3 cells transiently transfected with empty vector (control), ILK-WT (78%), ILK-KD (79%), PTEN-WT (82%), or GSK-3-WT (78%). Values in brackets indicate the transfection efficiency of each plasmid measured as described in Materials and Methods. Cells transfected with the various plasmids were deprived of serum for 18 h, commencing 48 h posttransfection. Equal amounts of protein from the various treatments were then analyzed for cyclin D1, p27Kip, and p21Cip expression by Western blot using the respective antibodies. Although cyclin D1 expression is constitutively elevated in a serum-independent manner in PC3 cells transfected with either empty vector or ILK-WT, it was decreased in cells transfected with ILK-KD, PTEN-WT, or GSK-3-WT. The p27Kip and p21Cip content was unaffected by all the plasmid transfections. ILK-WT-V5, ILK-KD-V5, PTEN-WT-GFP, and GSK-3-WT-HA transgene expressions were determined in the transfectants by Western blot analysis with anti-V5, anti-GFP, and anti-HA antibodies, respectively.
Collectively, the results shown in Fig. 6 demonstrate that PTEN and ILK regulate cyclin D1 expression via GSK-3. Similar findings have recently been reported for mammary carcinoma cells (D'Amico et al. 2000). Dysregulation of cyclin D1 expression in PTEN mutant cells can be restored by either inhibiting ILK, replacing PTEN, or augmenting GSK-3 expression. Furthermore, the data indicate that PTEN and ILK regulate the expression of cyclin D1 in a parallel manner to their regulation of β-catenin.
Transcriptional Regulation of Cyclin D1 Expression by PTEN, ILK, and GSK-3
Since β-catenin regulates cyclin D1 expression at the transcriptional level and PTEN, ILK, and GSK-3 regulate β-catenin, we measured cyclin D1 promoter activity by transiently cotransfecting PC3 cells with a plasmid containing the cyclin D1 promoter and a luciferase reporter gene with these elements of the PTEN/PI-3-kinase pathway. The cyclin D1 reporter gene was cotransfected with empty vector (control), ILK-WT, ILK-KD, PTEN-WT, or GSK-3-WT and the cyclin D1 promoter activity was evaluated. As shown in Fig. 7 A, the cyclin D1 reporter gene activity is constitutively elevated in the control PC3 cells in the absence and presence of serum. Cotransfection of ILK-WT does not further enhance the cyclin D1 promoter gene activity in these cells. However, PTEN or dominant negative ILK imposes a substantial inhibition of the cyclin D1 promoter activity. Interestingly, cotransfection of GSK-3-WT resulted in an inhibition of the cyclin D1 promoter activity by a similar magnitude to that observed due to PTEN-WT or ILK-KD. It should be pointed out that the transfection efficiency of all the plasmids was very similar and the enhancement of the endogenous levels of proteins due to transfection of the respective plasmids is quite comparable (Fig. 7 A).
Figure 7.
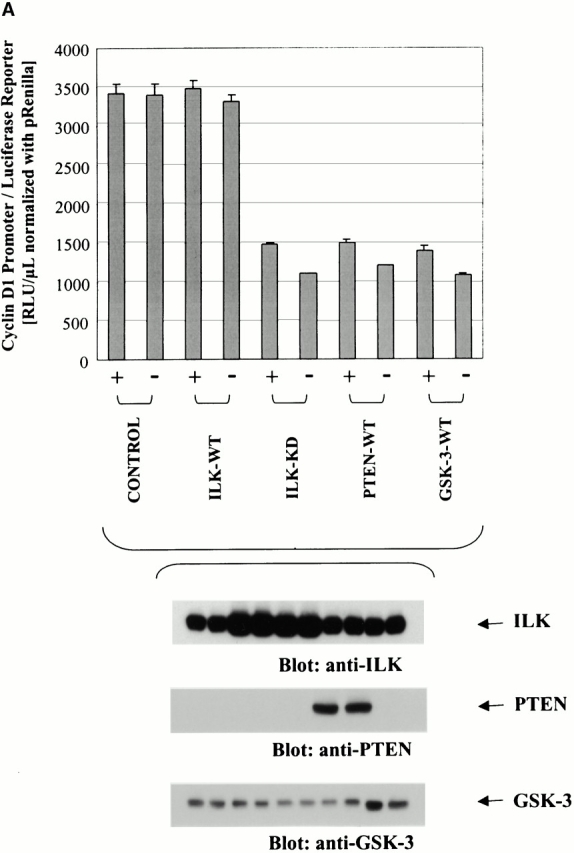

(A) Transcription of cyclin D1 is inhibited by PTEN-WT, ILK-KD, and GSK-3. Effect of PTEN, ILK, and GSK-3 on cyclin D1 promoter activity. PC3 cells were cotransfected with pGL3-cyclin D1 and empty vector, ILK-WT, ILK-KD, PTEN-WT, or GSK-3-WT. Cells were serum starved for 18 h commencing 48 h post transfection, refed with serum for 1 h, lysed, and assessed for cyclin D1 activity by a luciferase reporter gene assay. Bar graph demonstrates that cyclin D1 promoter activity is significantly reduced by comparable magnitude by the reexpression of PTEN or expression of ILK-KD and GSK-3-WT. Bottom panels show the comparable enhancement of the expression of ILK, PTEN, and GSK-3 upon transfection of ILK-WT or ILK-KD, PTEN-WT, and GSK-3-WT, respectively. (B) Northern blot of cyclin D1 in serum-starved PC3 cells. Total RNA was prepared by the TRIZOL method from serum-starved PC3 cells transfected with empty vector (control), ILK-KD, PTEN-WT, and GSK-3-WT. Transcriptional expression determined by probing the blot with a cyclin D1 probe randomly labeled with [32P]dCTP demonstrated highly comparable reductions due to transfection of PTEN, ILK-KD, or GSK-3-WT.
Northern blot analysis of total RNA from serum-starved PC3 cells was used to determine the effects of dominant negative ILK, PTEN-WT, and GSK-3-WT on cyclin D1 transcriptional expression. As shown in Fig. 7 B, transfection of PC3 cells with ILK-KD, PTEN-WT, or GSK-3-WT results in a dramatic decrease in transcriptional expression of cyclin D1.
These results indicate that PTEN and ILK are linked in a common pathway that regulates cyclin D1 gene transcriptional activity. The fact that GSK-3 inhibits the cyclin D1 promoter activity by a similar degree to that observed due to PTEN-WT and dominant negative ILK indicates that GSK-3 is very likely the downstream effector of ILK via which this regulation of the cyclins is achieved.
Discussion
Dysregulated β-catenin expression and signaling is now recognized as an important event in the genesis of several malignancies, especially in colorectal cancer (Voeller et al. 1998; Morin 1999). β-Catenin mutations (Morin 1999) or loss-of-function mutations (Brabletz et al. 2000) of the APC tumor suppressor gene appear to be crucial steps in the progression of this disease. In this study, we have demonstrated elevated levels of nuclear β-catenin in prostate cancer cells PC3 and LNCaP, where mutations in β-catenin (Voeller et al. 1998) or APC (Suzuki et al. 1994; Watanabe et al. 1996) are not known to be present. Instead, these cells do not express the tumor suppressor PTEN (Stambolic et al. 1998; Sun et al. 1999). In this study we demonstrate that the reexpression of PTEN in these cells leads to a reduction of nuclear β-catenin, indicating the regulation of the protein by the PTEN/PI-3 kinase pathway. The fact that dominant negative ILK-KD also has a similar effect on nuclear β-catenin expression and its function, as does PTEN, indicates that ILK is an important intermediate in this pathway. Since the dominant negative ILK-KD can inhibit PKB phosphorylation and activity in PC3 cells (Persad et al. 2000), and since PKB can phosphorylate and inactivate GSK-3 (Cross et al. 1995), it was necessary to evaluate the PKB dependence of PTEN- and ILK-mediated regulation of GSK-3 and subsequently β-catenin. Our data, using a dominant negative form of PKB, PKB-AAA (Wang et al. 1999), suggest that the effect of PKB upon GSK-3 activity, nuclear β-catenin expression, and TCF promoter activity are minor compared with the effects of PTEN-WT, ILK-KD, or GSK-3-WT. These results suggest that although PTEN and ILK-KD inhibit PKB phosphorylation and activation in PC3 cells (Persad et al. 2000), this inhibition of PKB activity contributes less to GSK-3 phosphorylation and activity in these cells than does inhibition of ILK. This would imply that ILK can directly regulate GSK-3 phosphorylation and activity. Indeed, it has been demonstrated previously that ILK can phosphorylate GSK-3 in vitro (Delcommenne et al. 1998) and that it can regulate GSK-3 activity in a PKB-independent manner (Troussard et al. 1999); here we have demonstrated that ILK can phosphorylate GSK-3 on serine 9 in the absence of PKB.
Regulation of β-catenin by GSK-3 (Morin 1999; Behrens 2000), as well as regulation of GSK-3 by ILK in a PI-3 kinase–dependent manner, has been described (Troussard et al. 1999; Tan et al. 2001). However, the regulation of β-catenin by the PTEN/PI-3 kinase pathway is a novel finding in this study that has not been described before. Hypothetically, the reduction in nuclear β-catenin can lead to reduced complex formation with TCF, resulting in decreased transactivation of genes containing the TCF-4/LEF-1 binding sequence near their promoter, such as cyclin D1 (Morin 1999; Shtutman et al. 1999; Behrens 2000; Lin et al. 2000). This theory is supported by our finding that reexpression of PTEN in PTEN null PC3 cells resulted in reduced activation of the TCF promoter–reporter construct, TOPFLASH. In addition, the fact that binding of nuclear proteins to a TCF-4 oligonucleotide was reduced upon reexpression of PTEN in PC3 cells supports this finding. The fact that both PTEN and ILK-KD inhibited β-catenin containing nuclear complex formation with TCF-oligonucleotide lends additional support to this hypothesis.
Mechanistically, our results indicate that PTEN induces an increase in the phosphorylation of β-catenin, thereby increasing its relative rate of degradation in PTEN-transfected PC3 cells compared with the control cells. It is likely that this increased phosphorylation is a direct result of the observed increase in GSK-3 activity induced by PTEN. It is well known that β-catenin stability is regulated by phosphorylation of the protein at Ser 33/37/45 and Thr 41 by GSK-3 at its NH2 terminus (Yost et al. 1996), followed by ubiquitination proteasome-mediated degradation. This increased degradation of β-catenin may effectively lower cellular concentration of the protein and prevent its further accumulation in the nucleus, leading to decreased nuclear β-catenin. It is also possible that PTEN may regulate the translocation of β-catenin from the nucleus and subsequently induce its degradation in the cytosol. However, further studies are required to explore this latter hypothesis.
PTEN-transfected PC3 cells also appear to exhibit a more prominent membranal localization of β-catenin. This may be related to the fact that PTEN appears to induce the transcription of E-cadherin in PC3 cells which do not normally express E-cadherin (Davies et al. 2000). Also, it is possible that the reexpression of E-cadherin in PC3 cells may contribute to the observed decrease in nuclear β-catenin. β-Catenin is known to interact with the cytoplasmic domains of E-cadherin, linking it to the actin cytoskeleton (Sadot et al. 1998; Shtutman et al. 1999). Thus, the more prominent localization of β-catenin to the cell surface may be related to the reappearance of its cell membrane anchor, E-cadherin, in the PTEN-reexpressing cells. However, we were unable to detect any E-cadherin–β-catenin complexes in PTEN-transfected PC3 cells (data not shown). Also, the lack of interaction between β-catenin and E-cadherin demonstrates that stimulation of E-cadherin expression is unlikely to play a significant role in the observed decreased expression of nuclear β-catenin. However, it should be noted that PC3 cells do express N-cadherin (Tran et al. 1999) and this expression is unaffected by reexpression of PTEN. Loss of E-cadherin expression has been linked to the acquisition of an invasive and/or metastatic phenotype (Bussemakers et al. 2000; Mialhe et al. 2000) and it is this antiinvasive and/or antimetastatic property of E-cadherin that may be of significance in relation to the tumor suppressor PTEN. In support of this, reexpression of E-cadherin by transfection has been shown to suppress the invasive phenotype in E-cadherin–negative prostate tumor cell clones (Luo et al. 1999). Overexpression or constitutive activation of ILK has been shown to result in an invasive phenotype concomitant with downregulation of E-cadherin expression, translocation of β-catenin to the nucleus, and formation of the LEF-1–β-catenin bipartite complex (Novak et al. 1998; Tan et al. 2001). Also, ILK has been shown to be regulated by the tumor suppressor PTEN (Morimoto et al. 2000; Persad et al. 2000).
Cyclin D1 is known to be one of the oncogenic targets of β-catenin (Morin 1999; Shtutman et al. 1999; Behrens 2000; Brabletz et al. 2000; Lin et al. 2000). We have demonstrated that in PC3 cells, the expression level of cyclin D1 is upregulated in a constitutive, serum-independent manner. More importantly, replacement of PTEN or inhibition of ILK results in dramatic suppression of the expression levels of cyclin D1. Furthermore, transient overexpression of GSK-3-WT also suppresses cyclin D1 expression. These results are supported by our previous observation that overexpression of ILK stimulates cyclin D1 expression (Radeva et al. 1997). Also, ILK has been shown recently to regulate cyclin D1 transcription via a pathway involving GSK-3 and the cyclic AMP response element binding protein transcription factor (D'Amico et al. 2000). We note that cyclin D1 expression in PC3 cells changes in a parallel manner to β-catenin expression in response to reexpression of PTEN or expression of ILK-KD and GSK-3. Therefore, we propose that the alterations in cyclin D1 expression very likely represent the physiological end result of the regulation of β-catenin by PTEN and ILK via GSK-3. It should be pointed out that although nuclear β-catenin and cyclin D1 expressions undergo parallel alterations due to reexpression of PTEN or inhibition of ILK, the expression of the CDK inhibitors p27Kip and p21Cip remain unchanged. This illustrates the specificity of the alterations induced upon β-catenin and cyclin D1. Our hypothesis is confirmed by cotransfection experiments with either TOPFLASH or the cyclin D1 promoter luciferase reporter gene. The results clearly show that PTEN, ILK-KD, and GSK-3-WT all dramatically reduce both TOPFLASH and the cyclin D1 promoter activity by a comparable degree. Furthermore, Northern blot analysis demonstrates that ILK-KD, PTEN-WT, and GSK-3 induce dramatic inhibitory effects upon cyclin D1 transcriptional expression. This supports our working hypothesis that PTEN and ILK can regulate nuclear β-catenin through GSK-3. This is in agreement with the fact that β-catenin is known to be regulated by GSK-3 (Morin 1999; Behrens 2000) and recent studies have identified GSK-3 as a critical regulatory component for the transcriptional activity and binding of the TCF–LEF-1–β-catenin complex (Eastman and Grosschedl 1999) transcription factors.
In conclusion, we have demonstrated a novel pathway involving PI-3 kinase/PTEN, ILK, and GSK-3 that maintains tight control over the levels and localization of β-catenin (Fig. 8). In prostate cancer cells, as well in other malignancies where PTEN is either lost or inactive, this control may be eliminated, resulting in elevated β-catenin levels, its accumulation in the nucleus, and increased transcription of its oncogenic targets. Cyclin D1 is a known target of β-catenin and it is also the first participant of the chain of cyclins and CDKs that control progression through the G1 and S phase of the cell cycle. Therefore, it is conceivable that PTEN and ILK, by virtue of their capacity to regulate β-catenin and subsequently cyclin D1, may ultimately regulate the progression of cells through the cell cycle. By virtue of their ability to regulate the expression of E-cadherin, PTEN/PI-3 kinase and ILK may also control the metastatic potential of cancer cells. Therefore, the inhibition of a potent regulator such as ILK may present a feasible alternative means of treating the numerous forms of tumors where the PI-3 kinase–dependent signal transduction pathway is dysregulated due to mutations of the tumor suppressor PTEN.
Figure 8.
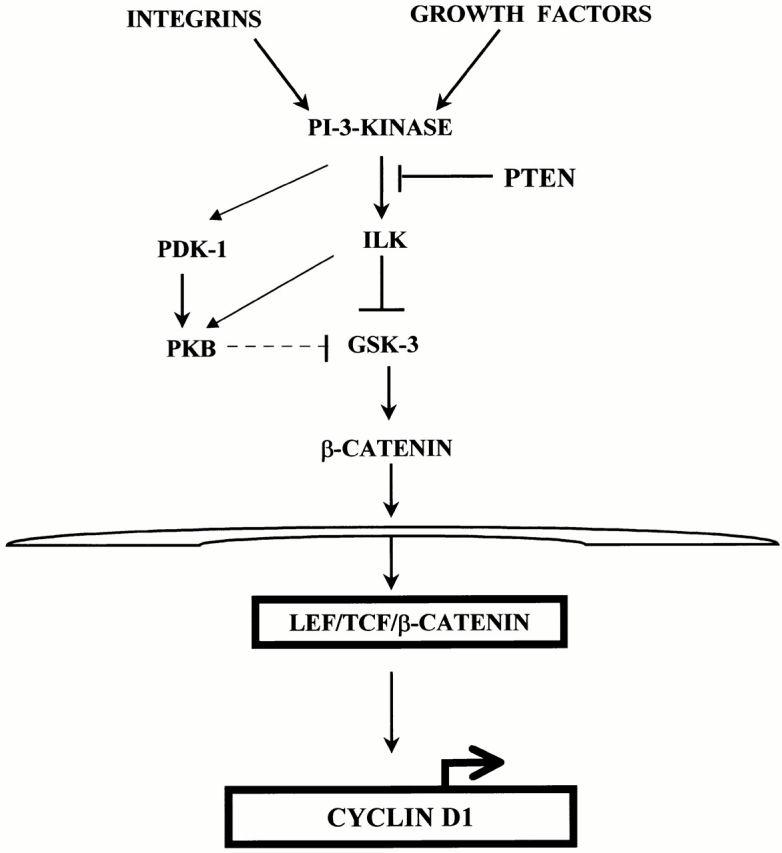
Schematic representation of the regulation of cyclin D1 from integrins and growth factor receptors via ILK and GSK-3. ILK activity is regulated in a PI-3 kinase–dependent manner and activated ILK regulates β-catenin and subsequently cyclin D1, and therefore cell proliferation, mainly via direct phosphorylation of GSK-3 and to a lesser extent via PKB. Stimulation of ILK results in a phosphorylation-mediated inhibition of GSK-3 activity. Inhibition of GSK-3 can lead to the accumulation of β-catenin, which activates the cyclin D1 gene by complexing with transcription factors of the TCF/LEF-1 family and binding to the TCF/LEF-1 binding site in the cyclin D1 promoter. PTEN suppresses the expression of cyclin D1 by inhibiting PI-3 kinase–dependent activation of ILK.
Acknowledgments
We thank Dr. J. Woodgett and Dr. N. Auersperg for providing the GSK-3-WT cDNA and N-cadherin antibody, respectively. The TOPFLASH and FOPFLASH constructs were provided by Dr. H. Clevers (University Hospital, Utrect, Netherlands). The cyclin D1 promoter construct was provided by Dr. Tetsu (University of California at San Francisco, San Francisco, CA).
S. Persad is a research fellow of the National Cancer Institute of Canada and was supported with funds provided by the Terry Fox Run. This work was supported by grants to S. Dedhar from the National Cancer Institute of Canada and by the Terry Fox Program project grant on prostate cancer progression.
Footnotes
Abbreviations used in this paper: APC, adenomatous polyposis coli; CDK, cyclin-dependent kinase; GFP, green fluorescent protein; GSK-3, glycogen synthase kinase 3; HA, hemagglutinin; ILK, integrin-linked kinase; KD, kinase deficient; LEF, lymphoid enhancer factor; PI-3, phosphatidylinositol 3; PKB, protein kinase B; TCF, T cell factor; WT, wild-type.
References
- Aberle H., Bauer A., Stappert J., Kispert A., Kemler R. β-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews N.C., Faller D.V. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19:2499. doi: 10.1093/nar/19.9.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N., Morin P.J., Clevers H. The yin-yang of TCF/beta-catenin signalling. Adv. Cancer Res. 2000;77:1–24. doi: 10.1016/s0065-230x(08)60783-6. [DOI] [PubMed] [Google Scholar]
- Behrens J. Control of beta-catenin signalling in tumor development. Ann. NY Acad. Sci. 2000;10:21–33. doi: 10.1111/j.1749-6632.2000.tb06698.x. [DOI] [PubMed] [Google Scholar]
- Behrens J., von Kries J.P., Kuhl M., Bruhn L., Wedlich D., Grosschedl R., Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- Brabletz T., Herrmann K., Jung A., Faller G., Kirchner T. Expression of nuclear beta-catenin and c-myc is correlated with tumor size but not with proliferative activity of colorectal adenomas. Am. J. Pathol. 2000;156:865–870. doi: 10.1016/s0002-9440(10)64955-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussemakers M.J., van Bokhoven A., Tomita K., Jansen C.F., Schalken J.A. Complex cadherin expression in human prostate cancer cells. Int. J. Cancer. 2000;85:446–450. [PubMed] [Google Scholar]
- Clevers H., Grosschedl R. Transcriptional control of lymphoid developmentlessons from gene targeting. Immunol. Today. 1996;17:336–343. doi: 10.1016/0167-5699(96)10019-0. [DOI] [PubMed] [Google Scholar]
- Cross D.A.E., Alessi D.R., Cohen P., Andjelkovic M., Hemmings B.A. Inhibition of glycogen synthase kinase by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- D'Amico M., Hulit J., Amanatullah D.F., Zafonte B.T., Albanese C., Bouzahzah B., Fu M., Augenlicht L.H., Donehower L.A., Takemaru K.I. The integrin-linked kinase regulates the cyclin D1 gene through glycogen synthase kinase 3 beta and cAMP-responsive element-binding protein-dependent pathways. J. Biol. Chem. 2000;275:32649–32657. doi: 10.1074/jbc.M000643200. [DOI] [PubMed] [Google Scholar]
- Davies G., Jingo W.A.G., Mason M.D. Cell-cell adhesion molecules and signalling intermediates and their role in the invasive potential of prostate cancer cells. J. Urol. 2000;163:985–992. [PubMed] [Google Scholar]
- Delcommenne M., Tan C., Gray V., Ruel L., Woodgett J., Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/Akt by integrin-linked kinase. Proc. Natl. Acad. Sci. USA. 1998;95:11211–11216. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman Q., Grosschedl R. Regulation of LEF-1/TCF transcription factors by Wnt and other signals. Curr. Opin. Cell Biol. 1999;11:233–240. doi: 10.1016/s0955-0674(99)80031-3. [DOI] [PubMed] [Google Scholar]
- Geiger B., Yehuda-Levenberg S., Bershadsky A.D. Molecular interactions in the submembrane plaque of cell-cell and cell-matrix adhesions. Acta Anat. 1995;154:46–62. doi: 10.1159/000147751. [DOI] [PubMed] [Google Scholar]
- Giese K., Kingsley C., Kirshner J., Grosschedl R. Assembly and function of a TCF-alpha enhance complex is dependent on LEF-1-induced DNA bending and multiple protein-protein interactions. Genes Dev. 1995;9:995–1008. doi: 10.1101/gad.9.8.995. [DOI] [PubMed] [Google Scholar]
- He T., Sparks A., Rago H., Hermeking L., Zawel L., da Costa L., Morin P., Vogelstein B., Kinzler K. Identification of c-myc as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Kemler R. From cadherins to cateninscytoplasmic protein interactions and regulation of cell adhesion. Trends Genet. 1993;9:317–321. doi: 10.1016/0168-9525(93)90250-l. [DOI] [PubMed] [Google Scholar]
- Korinek V., Barker N., Morin P.J., van Wichen D., de Weger R., Kinzler K.W., Vogelstein B., Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC −/− colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- Lin S.-Y., Xia W., Wang J.C., Kwong K.Y., Spohn B., Wen Y., Pestell R.G., Hung M.-C.H. β-catenin, a novel prognostic marker for breast cancerIts role in cyclin D1 expression and cancer progression. Proc. Natl. Acad. Sci. USA. 2000;97:4262–4266. doi: 10.1073/pnas.060025397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J., Lubaroff D.M., Hendrix M.J. Suppression of prostate cancer invasive potential and matrix metalloproteinase activity by E-cadherin transfection. Cancer Res. 1999;59:3552–3556. [PubMed] [Google Scholar]
- Mialhe A., Levacher G., Champelovier P., Martel V., Serres M., Knudsen K., Seigneurin D. Expression of E-, P-, N-cadherins and catenins in human bladder carcinoma cell lines. J. Urol. 2000;164:826–835. doi: 10.1097/00005392-200009010-00057. [DOI] [PubMed] [Google Scholar]
- Miller J.R., Moon R.T. Signal transduction through β-catenin and specification of cell fate during embryogenesis. Genes Dev. 1996;10:2527–2539. doi: 10.1101/gad.10.20.2527. [DOI] [PubMed] [Google Scholar]
- Molenaar M., van de Wetering M., Oosterwegel M., Peterson-Maduro J., Godsave S., Korinek V., Roose J., Destree O., Clevers H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- Morimoto A.M., Tomlinson M.G., Nakatani K., Bolen J.B., Roth R.A., Herbst R. The MMAC1 tumor suppressor phosphatase inhibits phospholipase C and integrin linked kinase activity. Oncogene. 2000;19:200–209. doi: 10.1038/sj.onc.1203288. [DOI] [PubMed] [Google Scholar]
- Morin P.J. Beta-catenin signalling and cancer. Bioessays. 1999;21:1021–1030. doi: 10.1002/(SICI)1521-1878(199912)22:1<1021::AID-BIES6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Morin P.J., Sparks A.B., Korinek V., Barker N., Clevers H., Vogelstein B., Kinzler K.W. Activation of beta-catenin-Tcf signalling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Munemitsu S., Albert I., Souza B., Rubinfeld B., Polakis P. Regulation of intracellular β-catenin levels by the adenomatous polyposis coli (APC) tumor suppressor protein. Proc. Natl. Acad. Sci. USA. 1995;92:3046–3050. doi: 10.1073/pnas.92.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak A., Hsu S.-C., Leung-Hagesteijn C., Radeva G., Papkoff J., Montesano R., Roskelly C., Grosschedl R., Dedhar S. Cell adhesion and the integrin-linked kinase regulate the LEF-1 and β-catenin signalling pathways. Proc. Natl. Acad. Sci. 1998;95:4374–4379. doi: 10.1073/pnas.95.8.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orford K., Crockett C., Jensen J., Weissman A., Byers S. Serine phosphorylation-regulated ubiquitination and degradation of β-catenin. J. Biol. Chem. 1997;272:24735–24738. doi: 10.1074/jbc.272.40.24735. [DOI] [PubMed] [Google Scholar]
- Peifer M., Wieschaus E. The segment polarity gene armadillo encodes a functionally modular protein that is the Drosophila homolog of human plakoglobin. Cell. 1990;63:1167–1176. doi: 10.1016/0092-8674(90)90413-9. [DOI] [PubMed] [Google Scholar]
- Peifer M., Orsulie S., Pai L., Loureiro J. A model system for cell adhesion and signal transduction in Drosophila . Dev. Suppl. 1993;1993:163–176. [PubMed] [Google Scholar]
- Persad S., Attwell S., Gray V., Delcommenne M., Troussard A., Sanghera J., Dedhar S. Inhibition of integrin-linked kinase (ILK) suppresses activation of protein kinase B/Akt and induces cell cycle arrest and apoptosis in PTEN null prostate cancer cells. Proc. Natl. Acad. Sci. USA. 2000;97:3207–3212. doi: 10.1073/pnas.060579697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persad S., Attwell S., Gray V., Mawji N., Deng J.T., Leung D., Yan J., Sanghera J., Walsh M.P., Dedhar S. Regulation of protein kinase B/Akt-serine-473 phosphorylation by integrin linked kinase (ILK)critical roles for kinase activity and amino acid arginine-211 and serine-343. J. Biol. Chem. 2001;In press doi: 10.1074/jbc.M102940200. [DOI] [PubMed] [Google Scholar]
- Polakis P. Wnt signalling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- Radeva G., Petrocelli T., Behrend E., Leung-Hagesteijn C., Filmus J., Slingerland J., Dedhar S. Overexpression of integrin-linked kinase promotes anchorage-independent cell cycle progression. J. Biol. Chem. 1997;272:13937–13944. doi: 10.1074/jbc.272.21.13937. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S., Nakamura N., Vasquez F., Batt D.B., Perera S., Roberts T.M., Sellers W.R. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol-3 kinase/Akt pathway. Proc. Natl. Acad. Sci. USA. 1999;96:2110–2115. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riese J., Yu X., Munnerlynn A., Eresh S., Hsu S.-C., Grosschedl R., Bienz M. LEF-1, a nuclear factor coordinating signaling inputs from wingless and decapentaplegic. Cell. 1997;88:777–787. doi: 10.1016/s0092-8674(00)81924-8. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B., Albert I., Porfiri E., Fiol C., Munemitsu S., Polakis P. Binding of GSK-3β to the APC-β-catenin complex and regulation of complex assembly. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- Sadot E., Simcha I., Shtutman M., Ben-Ze'ev A., Geiger B. Inhibition of β-catenin-mediated transactivation by cadherin derivatives. Proc. Natl. Acad. Sci. USA. 1998;95:15339–15344. doi: 10.1073/pnas.95.26.15339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon D., Sacco P., Roy S., Simcha I., Johnson K., Wheelock M., Ben-Ze'ev A. Regulation of beta-catenin levels and localization by overexpression of placoglobin and inhibition of the ubiquitin-proteasome pathway. J. Cell Biol. 1997;139:1325–1335. doi: 10.1083/jcb.139.5.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtutman M., Zhurinsky J., Simcha I., Albanese C., D'Amico M., Pestell R., Ben-Ze'ev A. The cyclin D1 gene is a target of the β-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. USA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V., Suzuki A., de la Pompa J.L., Brothers G.M., Mirtsos C., Sasaki T., Ruland J., Penninger J.M., Siderovsky D.P., Mak T.W. Negative regulation of Akt-dependent cell survival by tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Sun H., Lesche R., Li D., Liliental J., Zhang H., Gao J., Gavrilova N., Muller B., Liu X., Wu H. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5-trisphosphate and Akt/protein kinase B signalling pathway. Proc. Natl. Acad. Sci. USA. 1999;96:6199–6204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H., Aida S., Akimoto S., Igarashi T., Yatani R., Shimazaki J. State of adenomatous polyposis coli gene and ras oncogenes in Japanese prostate cancer. Jpn. J. Cancer. Res. 1994;85:847–852. doi: 10.1111/j.1349-7006.1994.tb02957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan C., Costello P., Sanghera J., Dominguez D., Baulida J., Garcia de Herreros A., Dedhar S. Inhibition of integrin linked kinase (ILK) suppresses β-catenin-Lef/Tcf-dependent transcription and expression of the E-cadherin repressor, snail, in APC−/− human colon carcinoma cells. Oncogene. 2001;20:133–140. doi: 10.1038/sj.onc.1204052. [DOI] [PubMed] [Google Scholar]
- Tetsu O., McCormick F. β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- Tran N.L., Nagle R.B., Cress A.E., Heimark R.L. N-cadherin expression in human prostate carcinoma cell lines. An epithelial-mesenchymal transformation mediating adhesion with stromal cells. Am. J. Pathol. 1999;155:787–798. doi: 10.1016/S0002-9440(10)65177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troussard A.A., Tan C., Yoganathan N., Dedhar S. Cell-extracellular matrix interactions stimulate the AP-1 transcription factor in an integrin-linked kinase- and glycogen synthase kinase 3-dependent manner. Mol. Cell. Biol. 1999;19:420–427. doi: 10.1128/mcb.19.11.7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering M., Cavallo R., Dooijes D., van Beest M., van Es J., Loureiro J., Ypma A., Hursh D., Jones T., Bejsovec A. Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell. 1997;88:789–799. doi: 10.1016/s0092-8674(00)81925-x. [DOI] [PubMed] [Google Scholar]
- Voeller H.J., Truica C.I., Gelmann E.P. β-catenin mutations in human prostate cancer. Cancer Res. 1998;58:2520–2523. [PubMed] [Google Scholar]
- Wang Q., Somwar R., Bilan P.J., Liu Z., Jin J., Woodgett J.R., Klip A. Protein kinase B/Akt participates in GLUT4 translocation by insulin in L6 myoblasts. Mol. Cell. Biol. 1999;19:4008–4018. doi: 10.1128/mcb.19.6.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M., Kakiuchi H., Kato H., Shiraishi T., Yatani R., Sugimura T., Nagao M. APC gene mutations in human prostate cancer. Jpn. J. Clin. Oncol. 1996;26:77–81. doi: 10.1093/oxfordjournals.jjco.a023188. [DOI] [PubMed] [Google Scholar]
- Willert K., Nusse R. Beta-catenina key mediator of Wnt signalling. Curr. Opin. Genet. Dev. 1998;8:95–102. doi: 10.1016/s0959-437x(98)80068-3. [DOI] [PubMed] [Google Scholar]
- Wodarz A., Nusse R. Mechanism of wnt signalling in development. Annu. Rev. Cell Dev. Biol. 1998;14:59–88. doi: 10.1146/annurev.cellbio.14.1.59. [DOI] [PubMed] [Google Scholar]
- Yost C., Torres M., Miller J.R., Huang E., Kimelman D., Moon R.T. The axis-inducing activity, stability, and subcellular distribution of β-catenin regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]