

Abstract
Bax is a member of the Bcl-2 family of proteins known to regulate mitochondria-dependent programmed cell death. Early in apoptosis, Bax translocates from the cytosol to the mitochondrial membrane. We have identified by confocal and electron microscopy a novel step in the Bax proapoptotic mechanism immediately subsequent to mitochondrial translocation. Bax leaves the mitochondrial membranes and coalesces into large clusters containing thousands of Bax molecules that remain adjacent to mitochondria. Bak, a close homologue of Bax, colocalizes in these apoptotic clusters in contrast to other family members, Bid and Bad, which circumscribe the outer mitochondrial membrane throughout cell death progression. We found the formation of Bax and Bak apoptotic clusters to be caspase independent and inhibited completely and specifically by Bcl-XL, correlating cluster formation with cytotoxic activity. Our results reveal the importance of a novel structure formed by certain Bcl-2 family members during the process of cell death.
Keywords: apoptosis, Bcl-2, green fluorescent protein, mitochondria, clusters
Introduction
Apoptosis is a regulated mechanism to selectively eliminate unwanted cells, keeping the delicate balance between cell proliferation and death. Cell death stimuli induce the activation of cell executioners, a family of cysteine proteases called caspases. Mitochondria-dependent and -independent pathways activate these caspases that subsequently lead to cell death. The mitochondria-dependent pathway is regulated by the Bcl-2 family of proteins. Bcl-2 was discovered to be a protooncogene that inhibited cell death induced by interleukin 3 deprivation (Vaux et al. 1988) and was shown subsequently to inhibit death induced by many other stimuli. The Bcl-2 family consists of two opposing clans: one subfamily including Bcl-2, Bcl-XL, Ced-9, Bcl-w, and Mcl-1 that promotes cell survival and another that promotes cell death. The proapoptotic subfamily can be further divided into those proteins consisting of Bcl-2 homology (BH) 1–3, like Bax, Bak, and Bok, and those consisting of only the BH3 region, like Bad, Bid, Bik, Bim, and Hrk (Adams and Cory 1998).
One of the intriguing aspects of apoptosis regulation by members of the Bcl-2 family is their subcellular localization and translocation. Certain pro- and antiapoptotic members such as Bcl-2, Bcl-XL, and Bak reside predominantly in the mitochondria, whereas other members such as Bax, Bid, and Bad reside in the cytosol of healthy cells. Early during the initiation of apoptosis, these cytosolic Bcl-2 family members translocate to the outer mitochondrial membrane (Gross et al. 1999a). After translocation of Bax and Bid to mitochondria, mitochondria lose their membrane potential (ΔΨm) and release intermembrane space proteins. However, the mechanisms linking proapoptotic Bcl-2 family members to these subsequent events in apoptosis are unclear.
Translocation of these proapoptotic proteins involves distinct mechanisms. Phosphorylation and 14-3-3 binding (Zha et al. 1996b) regulate Bad translocation, whereas Bid is regulated by caspase 8 cleavage as part of the Fas signaling pathway (Li et al. 1998; Luo et al. 1998). The regulation of Bax translocation is different from that of Bad and Bid but little understood. Bax translocation involves a conformational change that exposes the NH2 terminus and a hydrophobic COOH terminus that targets mitochondria (Gross et al. 1998; Nechushtan et al. 1999). It has also been suggested that Bid binds to cytosolic Bax to initiate this change in conformation and targeting of mitochondria (Desagher et al. 1999).
Translocation of Bax to mitochondria has been linked to the release of the mitochondrial intermembrane protein, cytochrome c, to the cytosol (Eskes et al. 1998; Jurgensmeier et al. 1998; Pastorino et al. 1998), and the question of how Bax might participate in this process is of current interest. Bax was shown to act as a pore-forming protein (Antonsson et al. 1997; Schlesinger et al. 1997; Eskes et al. 1998) and has also been suggested to interact with the voltage-dependent anion channel (VDAC) (Narita et al. 1998) or adenine nucleotide transporter (ANT) (Marzo et al. 1998). VDAC and ANT, the two most abundant proteins of the outer and inner mitochondrial membranes (respectively) are components of the permeability transition pore complex thought to be involved in cytochrome c release. On the other hand, other studies have indicated that the cytochrome c release pathway can be uncoupled from the mitochondrial permeability transition pore and Bax (Eskes et al. 1998; Jurgensmeier et al. 1998; Kim et al. 2000). Current hypotheses about the function of Bax assume that the critical site of Bax activity is the mitochondria. In this study, we show that Bax resides in the mitochondria only during a brief period in the process of apoptosis. After this period, Bax leaves the mitochondria and coalesces into clusters that are adjacent to the mitochondria. In addition, our observations indicate that cluster formation may be essential for Bax and Bak promotion of cell death. These findings provide a new framework for understanding the roles of Bax and Bak in the cell death program.
Materials and Methods
Restriction and modifying enzymes and oligonucleotide primers were obtained from GIBCO BRL. DNA sequencing was performed with a Sequenase II kit (United States Biochemical Corp.). All of the media and antibiotics were obtained from Biofluid, Inc. Except as noted, reagents were purchased from Sigma-Aldrich.
Generation of Expression Constructs
Wild-type full-length Bax, Bcl-XL, full-length and truncated Bid, and full-length Bad were synthesized by a standard PCR using as templates the cDNA for human Bax and mouse Bid (a gift from S.J. Korsmeyer, Harvard Medical School, Boston, MA), the cDNA for human Bcl-XL (a gift from C. Thompson, University of Pennsylvania, Philadelphia, PA), and the cDNA for mouse Bad (a gift from M.E. Greenberg, Harvard Medical School, Boston, MA). The primers used in the amplification reactions introduced an XhoI site for the NH2 terminus and an EcoRI site for the COOH terminus of the Bax and Bad. The EcoRI site for the NH2 terminus and a BamHI site for the COOH terminus were introduced in the amplifications of full-length and truncated Bid. All PCR reactions were done by 30 cycles of 1 min at 94°C, 2 min at 60°C, and 3 min at 72°C using Pfu polymerase (Stratagene). The resulting PCR products were digested with the appropriate restriction enzymes and cloned into the mammalian expression vectors, ECFP-C1 for Bax, EYFP-N1 for Bid, and EYFP-C1 for Bcl-XL and Bad. All vectors were purchased from CLONTECH Laboratories, Inc. The yellow fluorescent protein (YFP) fusion to the mutated COOH terminus of Bax, lacking serine 184 (YFP-20) in EYFP-C1, and the full-length point mutant Bax-S184V in ECFP-C1 (CLONTECH Laboratories, Inc.) were cloned as described previously (Nechushtan et al. 1999). Human VDAC-1 (a gift from Michael Forte, Vollum Institute, Oregon Health Sciences University, Portland, OR) was cloned into the DsRed vector (CLONTECH Laboratories, Inc.). Cytochrome c–green fluorescent protein (GFP) was a kind gift from Anna-Liisa Nieminen (Case Western Reserve University, Cleveland, OH). All constructs were confirmed by restriction endonuclease digestion and DNA sequence analysis. Escherichia coli strain DH5α (Stratagene) was used for all plasmid transformations and propagations.
Cloning of Human Bak cDNA
RNA was extracted and isolated from hepatocellular carcinoma Hep G2 cells using RNA Iso-lator (Genosys Biotechnology). RNA (2 mg) was reverse transcribed to a single strand of cDNA using reverse primers and Moloney murine leukemia virus reverse transcriptase. Bak cDNA was amplified using the forward primer 5′-TCAGATCTCGAGCTATGGCTTCGGGGCAAGGCCC-3′ and the reverse primer 5′-ACTGCAGAATTCTCATGATTTGAAGAATCTTCGTACC-3′, introducing XhoI and EcoRI sites, respectively. PCR was performed using a PerkinElmer Cetus thermal cycler in a volume of 100 μl by 30 cycles of 1 min at 94°C, 2 min at 60°C, and 2 min at 72°C using Pfu polymerase (Stratagene). The resulting PCR product was digested with XhoI and EcoRI and cloned into the mammalian expression vector EYFP-C1 (CLONTECH Laboratories, Inc.). The resulting YFP-Bak construct was confirmed by restriction endonuclease digestion and DNA sequence analysis.
Cell Culture and Measurement of Cell Viability
Cos-7 green monkey renal epithelial, HeLa human cervical carcinoma, and Hep G2 hepatocellular carcinoma cell lines (American Type Culture Collection) were grown in DME (Cos-7) and Earle's minimum essential medium (HeLa and Hep G2), each supplemented with 10% heat-treated FCS, 2 mM glutamine, nonessential amino acids, 2.5 mM sodium pyruvate, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were cultured at 37°C in 5% CO2.
To evaluate the ability of Bcl-XL to counter Bak and Bax mutant induction of apoptosis of cells in culture, we used luciferase assay as described previously (Nechushtan et al. 1999). In short, the mammalian expression vector pGL3 (Promega) carrying the firefly luciferase (Luc) structural gene was cotransfected along with either Bak or Bax mutant S184V (Nechushtan et al. 1999) with or without the antiapoptosis protein Bcl-XL. A 4:16:1 molar ratio of the tested gene to Bcl-XL or control DNA plasmid and Luc were used to cotransfect Cos-7 cells in 6-well tissue culture. 24 h after transfection, the cells were washed with PBS and harvested using the luciferase assay system (Promega). Luciferase activity was measured by liquid scintillation counting using 20 μl of the cellular extract. In every experiment, each sample was repeated six times, and cell viability is shown as the relative luc activity of the tested construct.
Transfection of Cells in Culture
For confocal microscopy, 104 cells were plated on a 2-well Lab-Tek chambered coverglass (Nunc). For the EM, 5 × 103 cells were plated on a 4-well Lab-Tek permanox chamber slide (Nunc), and both were transfected 48 h later using 1 μg plasmid DNA and 3 μl of FuGENE 6 (Roche) per chamber for the confocal microscopy and half of the amount for the EM.
Confocal Microscopy
Confocal microscope images of live Cos-7 and HeLa cells were captured on an LSM 510 microscope with a 60 × 1.3 NA Apochromat objective (ZEISS). Cells were grown and transfected as described above on 1 thickness 2-well Lab-Tek chambered coverglass (Nunc). After 24–48 h, confocal microscopy of live cells was performed after incubation with 20 ng/ml of a mitochondrion-specific dye (Mitotracker red CMXRos; Molecular Probes, Inc.) as indicated in individual experiments. The excitation wavelength for cyan fluorescent protein (CFP), GFP, YFP, and Mitotracker red CMXRos were 413, 488, 514, and 543 nm, respectively. The pH was maintained by addition of 25 mM Hepes, pH 7.4, to the culture medium.
Quantitation of Bax Molecules in the Apoptotic Clusters
Cos-7 cells were transfected with enhanced GFP (EGFP)-Bax as described above. Confocal microscope images were captured on an LSM 510 (ZEISS) microscope using 488 nm laser excitation for EGFP with low intensity illumination (2% laser power). Images for quantitation of Bax cluster fluorescence were captured with a 60× 1.4 NA Apochromat objective (ZEISS) and an open pinhole to collect fluorescence from the entire depth of the cell. Estimation of the number of Bax molecules in the apoptotic clusters was made by comparing the total fluorescence in a cluster with a standard curve calculated from a serial dilution of recombinant EGFP protein (CLONTECH Laboratories, Inc.). 10–15 clusters were measured in each cell, and 10 different cells were analyzed including healthy ones. Background fluorescence was measured in four different regions in each tested cell in areas of the cytosol free of clusters, and the average background value was subtracted from the fluorescence values measured from the apoptotic clusters. The analysis of the confocal images was done using MetaMorph (Universal Imaging Corp.).
Thin Section Immunoelectron Microscopy
Preembedding immunocytochemistry was performed as described previously (Tanner et al. 1996). In brief, samples were fixed with 2% paraformaldehyde and 0.1% glutaraldehyde in 0.1 M phosphate buffer at pH 7.4 for 30 min, washed, permeabilized, and incubated with one of the following antibodies: antiuniversal Bax 6A7 and the anti–human Bax 1F6 monoclonal antibodies, both recognize monkey Bax (Hsu and Youle 1997); and for all GFP variants, 3E6 anti-AFP was used (Quantum). Treatment of samples with the secondary antibody conjugated to Nanogold (Nanoprobes) was followed by washing and silver enhancement (HQ kit; Nanoprobes). Samples were treated with 0.2% OsO4, dehydrated, and embedded in Epoxy resin.
Results
Bax Leaves the Mitochondria and Clusters during Apoptosis
The translocation of Bax to the outer mitochondrial membrane is an early step in apoptosis. Translocation is associated with a conformational change that exposes the NH2 terminus and a hydrophobic portion of the COOH terminus. This 21–amino acid portion of the COOH terminus alone, minus serine 184, targets the mitochondria in both healthy and dying cells (Nechushtan et al. 1999). When we compared the localization of wild-type Bax fused to CFP-Bax and the COOH-terminal fragment fused to YFP-20, we were surprised to find that the two proteins colocalize on the outer mitochondrial membrane only during a brief period after induction of apoptosis. As apoptosis proceeds, YFP-20 remains, circumscribing the mitochondrial membrane (Fig. 1 and Fig. 2 A), whereas CFP-Bax coalesces into clusters (Fig. 1). The clusters are small initially (<0.5 μm) but progressively enlarge and remain closely associated with mitochondria. Each mitochondrion appears to be associated with one to three of the clusters, and some clusters appear to be shared between two to three mitochondria (Fig. 1, enlargement).
Figure 1.

Bax coalesces adjacent to mitochondria during apoptosis. Cos-7 and HeLa cells transiently expressing CFP-Bax together with the outer mitochondrial membrane marker, YFP-20, were treated with 1 μM STS for 5 h to induce apoptosis and then were examined by laser scanning confocal microscopy. CFP-Bax (red) was located in small punctate structures, whereas YFP-20 (green) was located in larger round mitochondrial-shaped structures. In overlay images, the Bax clusters appeared to be located next to or on top of the YFP-20–labeled structures. Bars, 25 μm.
Figure 2.
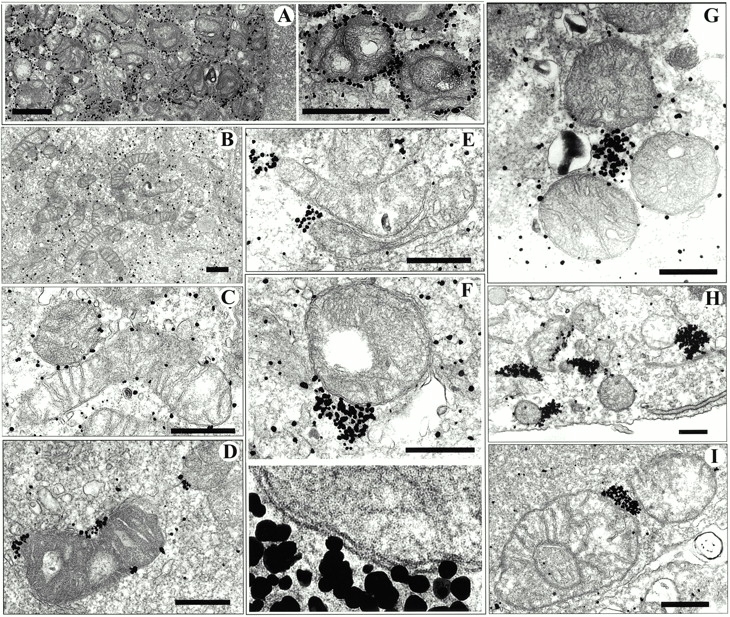
Ultrastructural localization of Bax and YFP-20. (A) Silver-enhanced immunogold labeling of YFP-20 in apoptotic Cos-7 cells showed specific labeling of gold particles around the outer mitochondria membrane. (B) Healthy Cos-7 cells transfected with wild-type Bax show a cytosolic diffuse distribution of Bax. (C) An early stage of apoptosis showing cytosolic and mitochondria-bound wild-type Bax labeling. (D) Apoptotic nontransfected cells showing labeling of endogenous Bax clusters. (E and F) Apoptotic cells transfected with wild-type Bax. The bottom micrograph in F shows a 4× enlargement of the contact site between the Bax cluster and the mitochondria. (G–I) Apoptotic cells transfected with GFP-Bax. In A and H, cells were labeled with 3E6 monoclonal anti-GFP (all variants), B–G were labeled with 1F6 monoclonal anti–human Bax, and I was labeled with 6A7 monoclonal anti-universal Bax. In STS-treated samples (all panels except A and B), the diffuse labeling of Bax in the cytoplasm is greatly diminished, whereas mitochondria-associated clusters of various sizes appear. Bars: (A) 1 μm; (C–J) 0.5 μm.
Previous work suggested that Bax forms oligomers during apoptosis (Oltvai et al. 1993; Sedlak et al. 1995; Tan et al. 1999; Antonsson et al. 2000; Saito et al. 2000), but the sizes of the Bax apoptotic clusters we observed were too large for them to represent individual dimers, tetramers, or even 30-mers as had been suggested. To assess the number of Bax molecules in single clusters, we employed a quantitative fluorescence imaging technique. Confocal microscope images of GFP-Bax–expressing cells were compared with images taken of serial dilutions of commercial recombinant GFP under the same conditions (as described in Materials and Methods). A typical concentration of GFP-Bax molecules measured in the cytosol of healthy cells was 4 μM. Our calculations indicated that the number of Bax molecules in the apoptotic clusters ranged from 1 × 103 to 2 × 104 molecules. These values are likely to underestimate the numbers of molecules because they are based on the assumption that each cluster was contained in a single 0.8-μm optical section, which was true for some small clusters but not for large ones. Thus, the novel structures we have found associated with mitochondria in apoptotic cells contain several thousands of Bax molecules.
To further characterize the large Bax clusters identified by confocal microscopy, we examined healthy and apoptotic cells by preembedding electron microscopic immunocytochemistry. We examined cells transfected with GFP-Bax or Bax alone and nontransfected cells. The cells were treated with staurosporine (STS), a broad specificity kinase inhibitor used extensively to trigger apoptosis (Weil et al. 1996), and were fixed at a time when Bax was clustered in a high percentage of the cells as determined in cells expressing GFP-Bax. Inspection of the cultures after they were embedded in plastic showed that some of the apoptotic cells were lost during processing, reducing the percentage of apoptotic cells relative to healthy cells. In immunolabeled cultures examined by EM, we observed cells in which Bax was cytosolic (Fig. 2 B), Bax was uniformly distributed around the surfaces of mitochondria (Fig. 2 C), and Bax was clustered at sites adjacent to mitochondria (Fig. 2, D–I). We interpret the third group to represent cells in which Bax appeared clustered by light microscopy. The Bax clusters were not enclosed in mitochondrial membranes, which were clearly delineated despite some disruption of inner mitochondrial structure (Fig. 2 F). Although Bax initially coated the mitochondria membranes as seen in Fig. 2 C when Bax coalesced into clusters, most of the outer mitochondrial membranes were cleared of all detectable Bax (Fig. 2, D–I).
Clustering of Bax was detected with two different monoclonal anti-Bax antibodies: 1F6 that recognizes human and monkey Bax and 6A7 that recognizes the Bax protein from mice, rats, monkeys, cows, and humans (Fig. 2, D–G and I) and with a monoclonal anti-GFP antibody, 3E6 anti-AFP, that recognizes all GFP variants (Fig. 2 H). All three antibodies showed the same pattern of Bax clustering in mitochondria-associated structures in apoptotic cells. One of the two antibodies we used to detect Bax, 6A7, only binds to Bax that has undergone a conformational change associated with translocation from the cytosol to the mitochondria (Hsu and Youle 1998; Nechushtan et al. 1999). The binding of this antibody to the clusters (Fig. 2 I) indicates that the clusters contain an activated form of Bax.
Clustering of Bax was not a consequence of overexpression because clusters were observed in nontransfected apoptotic cells (Fig. 2 D); nor was it GFP-related aggregation since cells overexpressing wild-type Bax showed the same clustering (Fig. 2E and Fig. F) as GFP-Bax (Fig. 2, G–I). The number of silver particles per cluster was smaller in nontransfected cells than in cells overexpressing Bax, indicating that there were fewer Bax molecules per cluster.
We reported previously that the Bax protein, even when overexpressed well beyond the endogenous level, would translocate completely from the cytosol to the mitochondria (Nechushtan et al. 1999). We concluded at that time that there must be unlimited binding sites for Bax on the mitochondria. The present finding of large apoptotic clusters of Bax provides an alternative explanation for this complete translocation. These clusters serve as the final target for the translocating Bax, making the number of mitochondria binding sites irrelevant. It is of note that both Bax clusters and monomers were never found to be associated with any other membranes or organelles, such as the nucleus or ER, by either confocal microscopy or EM.
Bak Colocalizes with Bax in the Apoptotic Clusters whereas Bid and Bad Do Not
Among the proapoptotic members of the Bcl-2 family, Bak is closest to Bax in terms of sequence homology, size (Adams and Cory 1998), and biological activity. Studying mice lacking both Bax and Bak genes (bax−/− bak−/−) showed overlapping roles of these proteins in the regulation of apoptosis (Lindsten et al. 2000). Like Bax, Bak has been shown to induce the loss of mitochondrial membrane potential and lead to cytochrome c release, whereas Bcl-2 and Bcl-XL prevent these events (Eskes et al. 1998; Shimizu et al. 1999; Chatterjee et al. 2000; Wei et al. 2000). However, Bax exists in the cytosol of healthy cells and translocates to the mitochondria during apoptosis, whereas Bak constitutively resides on mitochondrial membranes in healthy cells. To determine whether Bak forms similar clusters during apoptosis, cells were cotransfected with CFP-Bak and either YFP-20 as a marker for mitochondria or YFP-Bax to visualize Bax apoptotic clusters. Bak is located on mitochondria in healthy cells and fully colocalizes with YFP-20 (Fig. 3 A, top row) in contrast to Bax, which resides in the cytosol (Fig. 3 B, top row, second panel). However, upon induction of apoptosis by STS, Bak segregates from YFP-20 and the mitochondria and coalesces into mitochondria-associated clusters (Fig. 3 A, bottom row). These clusters have the same characteristics as those formed by Bax in terms of size, location, and number. Bak and Bax colocalize in the same clusters adjacent to mitochondria during apoptosis (Fig. 3 B, bottom row, overlay). The localization of Bak and Bax is significantly different from that of the mitochondria (Fig. 3 B, bottom row, overlay, blue staining). To further investigate these apoptotic clusters of Bak revealed by the fluorescence images of living cells, we examined Cos-7 cells expressing CFP-Bak using EM (Fig. 3). Bak circumscribes the outer mitochondrial membrane in healthy cells (Fig. 3 C, control) and coalesces in large multimeric clusters in apoptotic ones (Fig. 3 C, STS). The number and distribution of the Bak clusters are similar to those found for Bax (Fig. 2).
Figure 3.



Bak clusters and colocalizes with Bax in dying cells. Healthy and apoptotic Cos-7 cells transiently coexpressing CFP-Bak with either YFP-20 (A) or YFP-Bax (B) were examined by laser scanning confocal microscopy. Bak (red) was associated with mitochondria in healthy cells (control) and formed clusters adjacent to mitochondria labeled with YFP-20 (green). Bak had a distinctive distribution from Bax in healthy cells (B, control) but colocalized with Bax in dying cells (B, STS). Mitochondria were labeled with Mitotracker red CMXRos in B (blue). (C) Silver-enhanced immunogold labeling of CFP-Bak in Cos-7 cells showing specific labeling of gold particles around the mitochondria outer membrane in healthy cells (control) and in large clusters associated with the mitochondria in apoptotic cells (STS). Apoptosis was induced by addition of 1 μM STS for 5 h. Bars: (A and B) 25 μm; (C) 0.5 μm.
Bid and Bad structurally resemble Bax and Bak only in their BH3 domain (Gross et al. 1999a), and like Bax they are cytosolic in healthy cells and translocate to mitochondria during apoptosis (Zha et al. 1996b; Li et al. 1998). These resemblances led us to ask whether Bid and Bad coalesce in clusters along with Bax and Bak. We cotransfected Cos-7 cells with Bid-YFP or YFP-Bad and CFP-Bax. In apoptotic cells, Bid-YFP and YFP-Bad did not colocalize with Bax clusters but instead remained circumscribing the mitochondria (Fig. 4). Thus, two BH3-only members of the Bcl-2 family, Bid and Bad, do not coalesce during apoptosis in contrast to Bax and Bak. Interestingly, over-expressed Bid and Bad, unlike Bax, do not translocate completely to the mitochondria in apoptotic cells. Instead, a significant portion of the cellular Bid and Bad remains in the cytosol. The Bcl-2 family member Bid is cytosolic in healthy cells and translocates to mitochondria during an early stage of apoptosis (Li et al. 1998; Luo et al. 1998). Several groups reported that after cleavage by caspase 8, Bid triggers Bax redistribution from cytosol to membrane (Eskes et al. 1998; Jurgensmeier et al. 1998; Desagher et al. 1999; Gross et al. 1999b). Interestingly, when we compared the time course of Bid and Bax translocation from cytosol to mitochondria during STS-induced apoptosis in cells coexpressing both proteins we found that often Bax translocation differed from that of Bid. In some cells, Bax translocated to the mitochondria before Bid. In other cells, Bax and Bid translocation was simultaneous or Bid translocation preceded that of Bax (data not shown), indicating that although Bax and Bid migrate to mitochondria in approximately the same time frame, it is unlikely that Bid serves as a direct activator of Bax translocation.
Figure 4.

Bid and Bad do not localize with Bax apoptotic clusters. Cos-7 cells transiently coexpressing CFP-Bax (red) with Bid-YFP or YFP-Bad (green) were treated with 1 μM STS for 5 h to induce apoptosis and visualized by laser fluorescence confocal microscopy. Bid and Bad were localized in structures with shapes typical of mitochondria in dying cells at stages when Bax was localized in clusters. The magnified image (×5) shows parts of the same fields. Apoptosis was induced by addition of 1 μM STS for 5 h. Bar, 25 μm.
To determine whether other proteins involved in mitochondria-dependent apoptosis might coalesce into clusters with Bax and Bak, we examined by confocal microscopy Cos-7 cells transfected with cytochrome c–GFP or DsRed-VDAC. In apoptotic cells (STS treatment), VDAC remained in the mitochondria, whereas cytochrome c became cytosolic (data not shown) as shown previously by others (Liu et al. 1996; Kluck et al. 1997). Therefore, it is most likely that VDAC and cytochrome c are not part of the Bax and Bak apoptotic clusters.
To determine whether cluster formation is caspase dependent, we induced apoptosis while inhibiting caspase activation with benzyloxycarbonyl-Val-Ala-Asp-(OMe)-fluoromethylketone (zVAD-fmk), a tetrapeptide inhibitor of caspases that prolongs the induction of apoptosis induced by various agents (Dolle et al. 1994). We cotransfected CFP-Bax or CFP-Bak with the mitochondria marker YFP-20 in the presence of 50 μM zVAD-fmk and treated those cells with STS. As can be seen in Fig. 5, both CFP-Bax and CFP-Bak formed mitochondria-associated clusters similar to those formed without the treatment of caspase inhibitor (Fig. 1 Fig. 2 Fig. 3). These results place Bax and Bak translocation and cluster formation during apoptosis upstream of the caspase activation or caspase independent.
Figure 5.

Bax and Bak clustering is caspase independent. Transient coexpression of CFP-Bax or CFP-Bak and YFP-20 in apoptotic Cos-7 cells shows that mitochondria-associated clusters of Bax and Bak form in the presence of 50 μM zVAD-fmk. The transfected cells were treated with 1 μM STS for 5 h and visualized by laser scanning confocal microscopy. Both Bax and Bak coalesce into mitochondria-associated clusters (red) different from YFP-20 (green) that remains on the outer mitochondrial membrane. Bars, 25 μm.
Bcl-XL Blocks Bax and Bak Clustering and Toxicity but Does Not Prevent Constitutive Outer Mitochondrial Membrane Localization
Bcl-XL has been shown to inhibit apoptosis induced by different stimuli in various cell types. Bcl-XL prevents the cell death induced by overexpression of Bax or Bak and reduces the rate of cell loss during STS treatment (Chittenden et al. 1995; Vander Heiden et al. 1997; Wolter et al. 1997; Griffiths et al. 1999). Furthermore, Bcl-XL does not require a direct interaction with Bax to inhibit its promotion of cell death (Cheng et al. 1996). To analyze the biological significance of cluster formation, we asked what effect Bcl-XL might have on this process. Bcl-XL is known to inhibit Bax translocation to mitochondria during apoptosis (Wolter et al. 1997). Thus, to specifically test the role of Bcl-XL on cluster formation we focused on Bax-S184V because this protein constitutively associates with mitochondria. The S184V mutant form of Bax is constitutively targeted to the mitochondria in healthy cells and has a proapoptotic activity similar to wild-type Bax (Nechushtan et al. 1999). We have found that, like Bax, Bax-S184V also forms mitochondrial-associated clusters in apoptotic cells (Fig. 6 A). When we coexpressed Bax-S184V and Bcl-XL in Cos-7 cells, we found that Bcl-XL completely prevented the cluster formation of the Bax mutant with or without treatment of STS (Fig. 6 B). Cells cotransfected with Bcl-XL and Bax-S184V showed a marked reduction in apoptosis compared with cells singly transfected with Bax-S184V (Fig. 7 A). CFP-Bax-S184V targeted the mitochondria regardless of the absence or presence of overexpressed YFP-Bcl-XL, showing that Bcl-XL does not inhibit the mitochondria-binding step (Fig. 7 C). However, the presence of Bcl-XL completely inhibited coalescence into clusters as is observed in cells singly transfected with Bax (Fig. 1) or Bax-S184V (Fig. 6 A). Analysis of CFP-Bcl-XL showed that during apoptosis, Bcl-XL always circumscribes the mitochondria (data not shown). Bak, like Bax-S184V, is bound to mitochondria in healthy cells. Cotransfection of Bcl-XL and Bak caused no change in Bak localization (Fig. 7 D). However, when apoptosis was induced by incubation with STS, no Bak cluster formation occurred (data not shown) as occurs in the absence of Bcl-XL (Fig. 3). Cotransfection with Bcl-XL also markedly inhibited Bak-stimulated apoptosis (Fig. 7 B). Furthermore, analysis of CFP-Bcl-XL showed that Bcl-XL always circumscribes the mitochondria in the presence of either Bax-S184V or Bak (Fig. 7C and Fig. D) and remains on the mitochondria during apoptosis (data not shown). The observation that Bcl-XL specifically blocks cluster formation and the bioactivity of Bax-S184V and Bak indicates that the clusters are the biologically active proapoptotic structures.
Figure 6.


Bcl-XL inhibits the formation of apoptotic clusters of the constitutive mitochondria-docking Bax mutant, S184V. (A) Transient expression of CFP–Bax-S184V and YFP-20 in apoptotic Cos-7 cells shows the formation of mitochondria-associated clusters by the S184V Bax mutant. (B) Quantitation of Bax-S184V cluster formation. Cos-7 cells were transfected with Bax-S184V, with or without Bcl-XL, and two of the groups were treated with 1 μM STS for 6 h. 150 cells in each treatment group were examined for the presence of clusters under the confocal microscope. Results are displayed as the percentage of cells with clusters of the total cells counted. This experiment was repeated four times.
Figure 7.

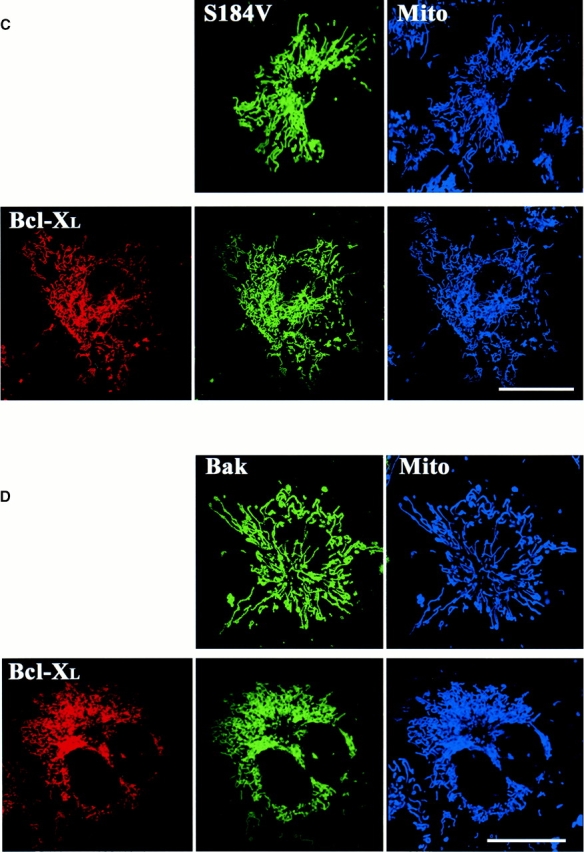
Bcl-XL does not prevent mitochondrial targeting of Bak or Bax-S184V but inhibits their proapoptotic activity. Cos-7 cells were transiently cotransfected with the pGL3 luciferase reporter construct, Bcl-XL, and CFP-Bax-S184V (A) or CFP-Bak (B) in 1:16:4 ratios. The ECFP-C1 expression vector was used as a control. After 24 h of incubation, cells were harvested and processed for luciferase activity. Results were quantitated with a scintillation counter and displayed as 106 counts of light intensity. The error bars show a mean standard error determined from six independent measurements. (C and D) Healthy Cos-7 cells transiently expressing CFP-S184V and CFP-Bak with and without YFP-Bcl-XL were incubated with Mitotracker red CMXRos (Mito) to reveal mitochondria and visualized by laser scanning confocal microscopy. If Bcl-XL was to inhibit binding of Bax-S184V or Bak to mitochondria as a protection mechanism, both Bak and Bax-S184V proteins (green) would show a higher degree of cytosolic distribution. Both Bax-S184V (C, green) and Bak (D, green) localized to mitochondria (blue) similarly in the presence or absence of Bcl-XL. Bars, 25 μm.
Discussion
Bax redistributes from the cytosol to the mitochondria (Hsu et al. 1997; Wolter et al. 1997) and undergoes conformational changes during the early stages of apoptosis (Hsu and Youle 1998; Desagher et al. 1999; Nechushtan et al. 1999). Recent studies have suggested three main models for the Bax mechanism of activating apoptosis after mitochondrial insertion: Bax may neutralize antiapoptotic members of the Bcl-2 family (Yin et al. 1994; Han et al. 1996; Zha et al. 1996a), bind and regulate mitochondrial proteins like VDAC (Narita et al. 1998) and ANT (Marzo et al. 1998), or act as a pore-forming protein by homooligomerization to release apoptosis activating factors (Antonsson et al. 1997, Antonsson et al. 2001; Schlesinger et al. 1997; Eskes et al. 1998; Mikhailov et al. 2001). All of these models place Bax across the mitochondria membranes at the end of apoptotic activation. Our present results indicates that targeting the mitochondrial membranes is only the first step (Fig. 8) in the Bax mechanism of action and that it is essential that Bax complete a second step, coalescing into apoptotic clusters, during cell death. Bak, which resides in the mitochondrial membranes of healthy cells, shares this second step as it coalesces into the same apoptotic clusters upon cell death.
Figure 8.

Two steps in the Bax apoptotic pathway. A schematic representation of Bax (red) localization in cells undergoing apoptosis. Step 1 shows cytosolic Bax targeting the mitochondria and coating the outer membrane, as do Bid and Bad. Step 2 shows Bax leaving the mitochondrial membranes and coalescing in apoptotic clusters as does Bak. The colored boxes indicate the changes in conformation and localization of the four proapoptotic proteins. Although Bax and Bak change conformation and complete step 2, Bid and Bad after cleavage and dephosphorylation, respectively, complete only step 1.
Overexpression of Bcl-XL specifically blocks Bax and Bak cluster formation and prevents cell death, indicating that the clusters are required for death to occur. When GFP–wild-type Bax was coexpressed with Bcl-XL and apoptosis was induced, neither docking of GFP-Bax to the mitochondria nor clustering could be detected (Wolter et al. 1997). The model that Bcl-XL inhibits cluster formation merely by blocking Bax docking to mitochondria cannot explain the Bcl-XL inhibition of Bax-S184V and Bak clustering, since they reside in the mitochondrial membrane before apoptosis. Overall, our data suggest that the inhibition of Bax clustering (step 2) by Bcl-XL somehow inhibits the docking of Bax on the outer mitochondrial membrane (step 1). Previously, we and others have assumed that Bax insertion to the mitochondria was a critical event in the progress of apoptosis (Goping et al. 1998; Murphy et al. 2000). However, there are reports that Bax inserts into mitochondria membrane in cells that remain healthy (Desagher et al. 1999). Bax was also reported to undergo a rapid translocation to mitochondria as cells were detached from the extracellular matrix and returned to the cytosol upon reattachment (Gilmore et al. 2000). This short time redistribution to the mitochondria did not result in commitment to apoptosis and cell death, possibly due to the Bcl-XL antiapoptotic effect of inhibiting cluster formation (Fig. 8, block of step 2). We predict that the Bax that can bind mitochondria in healthy cells coats the outer mitochondrial membrane, awaiting an apoptotic triggering signal, and does not form the apoptotic clusters.
The ability of Bcl-XL to block Bax and Bak apoptosis promotion through inhibition of cluster formation addresses one of the fundamental questions involving the opposing biological activities of members of the Bcl-2 family. It is still not established if Bax activity promotes cell death and Bcl-XL inhibits Bax or if Bcl-XL actively promotes cell viability and Bax inhibits Bcl-XL. Aside from their opposing apoptotic activity, Bax, Bak, and Bcl-XL are very similar. Bax and Bcl-XL have similar three-dimensional structures (Suzuki et al. 2000), and both form channels in lipid bilayers (Minn et al. 1997; Schlesinger et al. 1997). The formation of the large mitochondria-associated clusters is one of the few distinguishing molecular differences between Bax and Bcl-XL. Our data indicate that Bcl-XL inhibits Bax cluster formation and apoptosis induction. These results are consistent with the model that Bax, through cluster formation, is actively inducing cell death, and Bcl-XL is the inhibitor.
Since initially described, the role of Bax dimerization has been subject to debate. The rheostat model in which the ratio of Bax homodimers to the heterodimers with Bcl-2 determines survival or death was proposed originally (Oltvai et al. 1993). However, other studies showed that cytosolic Bax is a monomer (Hsu and Youle 1997, Hsu and Youle 1998). Since then, various studies have proposed different numerical associations among Bax molecules during apoptosis. Antonsson et al. 2001, for example, suggested that the channel-forming Bax oligomer is composed of six to eight Bax molecules. Another study showed that the number of Bax molecules that participate in the active membrane pore increases in a concentration-dependent manner up to a maximum of four molecules (Saito et al. 2000). In the present study, the EM and fluorescent microscopy were unable to detect small complexes like dimers or tetramers; however, complexes of thousands of GFP-Bax molecules were found in cells undergoing apoptosis.
Both electron microscope data and fluorescent intensity show a variation in the sizes of the Bax apoptotic clusters. Our analysis of fluorescent images of GFP-Bax compared with GFP recombinant protein in solution indicates that the clusters contain on the order of several thousands to several tens of thousands of GFP-Bax molecules. Since the clusters grow as cell death progresses, small clusters may exist at early stages in apoptosis, but these would be difficult to discern because of the presence of cytosolic GFP-Bax at this stage. Considering the size of the apoptotic clusters and their localization outside the mitochondria with only a small proportion of the total molecules in contact with the outer membrane (Fig. 2, D–I), it may be possible to purify the Bax clusters. However, different centrifugation techniques, including sucrose gradients, were unable to separate the clusters from the mitochondria without dispersing Bax into soluble small molecular weight forms (our unpublished data).
Bax undergoes conformational changes during apoptosis (Hsu and Youle 1998; Desagher et al. 1999; Nechushtan et al. 1999). The S184V Bax mutant when already docked to mitochondria does not expose the 6A7 epitope, and like the wild-type Bax it exposes that epitope during apoptosis (Nechushtan et al. 1999). Our EM studies show that the 6A7 epitope is exposed in the Bax clusters (Fig. 2 I). Similarly, Bak exposes its NH2 terminus during apoptosis (Griffiths et al. 1999). Thus, it appears that the conformational change in Bax and Bak is an obligatory step in the mitochondria-associated clusters formation and apoptosis induction. Our data showing that Bak takes part in the same apoptotic clusters as Bax may indicate a cooperative action with Bax. On the other hand, Bax and Bak may represent either a back up and/or parallel system in which one protein can substitute for the other, or one in which the proteins are similar tools triggered by different stimuli/pathways in the execution of the cell death program.
Surprisingly, proapoptotic Bid and Bad do not form apoptotic clusters and continue to coat the mitochondria of dying cells. This suggests that Bid and Bad have a fundamentally different mechanism of cell death promotion. At this stage, we can only speculate on the mechanism of Bax and Bak mode of action. Whether it is the autonomous function of the apoptotic clusters that contributes to the mitochondrial dysfunction or the actual clustering and departure of proapoptotic proteins from the outer mitochondrial membrane, the significance of the clusters to the apoptotic process has been introduced.
Acknowledgments
We thank J. Barrick, P. Johnson, J-H. Tao-Cheng, and the National Institute of Neurological Disorders and Stroke Electron Microscopy facility staff for their technical assistance.
Footnotes
Abbreviations used in this paper: ANT, adenine nucleotide transporter; BH, Bcl-2 homology; CFP, cyan fluorescent protein; EGFP, enhanced GFP; GFP, green fluorescent protein; STS, staurosporine; VDAC, voltage-dependent anion channel; YFP, yellow fluorescent protein.
References
- Adams J.M., Cory S. The Bcl-2 protein familyarbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- Antonsson B., Conti F., Ciavatta A., Montessuit S., Lewis S., Martinou I., Bernasconi L., Bernard A., Mermod J.J., Mazzei G. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997;277:370–372. doi: 10.1126/science.277.5324.370. [DOI] [PubMed] [Google Scholar]
- Antonsson B., Montessuit S., Lauper S., Eskes R., Martinou J.C. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem. J. 2000;345:271–278. [PMC free article] [PubMed] [Google Scholar]
- Antonsson B., Montessuit S., Sanchez B., Martinou J.C. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J. Biol. Chem. 2001;276:11615–11623. doi: 10.1074/jbc.M010810200. [DOI] [PubMed] [Google Scholar]
- Chatterjee D., Pantazis P., Li G., Bremner T.A., Hendrickson E.A., Wyche J.H. Susceptibility to apoptosis is restored in human leukemia HCW-2 cells following induction and stabilization of the apoptotic effector Bak. Oncogene. 2000;19:4108–4116. doi: 10.1038/sj.onc.1203757. [DOI] [PubMed] [Google Scholar]
- Cheng E.H., Levine B., Boise L.H., Thompson C.B., Hardwick J.M. Bax-independent inhibition of apoptosis by Bcl-XL. Nature. 1996;379:554–556. doi: 10.1038/379554a0. [DOI] [PubMed] [Google Scholar]
- Chittenden T., Flemington C., Houghton A.B., Ebb R.G., Gallo G.J., Elangovan B., Chinnadurai G., Lutz R.J. A conserved domain in Bak, distinct from BH1 and BH2, mediates cell death and protein binding functions. EMBO J. 1995;14:5589–5596. doi: 10.1002/j.1460-2075.1995.tb00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desagher S., Osen-Sand A., Nichols A., Eskes R., Montessuit S., Lauper S., Maundrell K., Antonsson B., Martinou J.C. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 1999;144:891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolle R.E., Hoyer D., Prasad C.V., Schmidt S.J., Helaszek C.T., Miller R.E., Ator M.A. P1 aspartate-based peptide alpha-([2,6-dichlorobenzoyl]oxy)methyl ketones as potent time-dependent inhibitors of interleukin-1 beta-converting enzyme. J. Med. Chem. 1994;37:563–564. doi: 10.1021/jm00031a003. [DOI] [PubMed] [Google Scholar]
- Eskes R., Antonsson B., Osen-Sand A., Montessuit S., Richter C., Sadoul R., Mazzei G., Nichols A., Martinou J.C. Bax-induced cytochrome C release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J. Cell Biol. 1998;143:217–224. doi: 10.1083/jcb.143.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore A.P., Metcalfe A.D., Romer L.H., Streuli C.H. Integrin-mediated survival signals regulate the apoptotic function of Bax through its conformation and subcellular localization. J. Cell Biol. 2000;149:431–446. doi: 10.1083/jcb.149.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goping I.S., Gross A., Lavoie J.N., Nguyen M., Jemmerson R., Roth K., Korsmeyer S.J., Shore G.C. Regulated targeting of BAX to mitochondria. J. Cell Biol. 1998;143:207–215. doi: 10.1083/jcb.143.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths G.J., Dubrez L., Morgan C.P., Jones N.A., Whitehouse J., Corfe B.M., Dive C., Hickman J.A. Cell damage–induced conformational changes of the proapoptotic protein Bak in vivo precede the onset of apoptosis. J. Cell Biol. 1999;144:903–914. doi: 10.1083/jcb.144.5.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A., Jockel J., Wei M.C., Korsmeyer S.J. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 1998;17:3878–3885. doi: 10.1093/emboj/17.14.3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A., McDonnell J.M., Korsmeyer S.J. BCL-2 family members and the mitochondria in apoptosis Genes Dev 13 1999. 1899 1911a [DOI] [PubMed] [Google Scholar]
- Gross A., Yin X.M., Wang K., Wei M.C., Jockel J., Milliman C., Erdjument-Bromage H., Tempst P., Korsmeyer S.J. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death J. Biol. Chem 274 1999. 1156 1163b [DOI] [PubMed] [Google Scholar]
- Han J., Sabbatini P., Perez D., Rao L., Modha D., White E. The E1B 19K protein blocks apoptosis by interacting with and inhibiting the p53-inducible and death-promoting Bax protein. Genes Dev. 1996;10:461–477. doi: 10.1101/gad.10.4.461. [DOI] [PubMed] [Google Scholar]
- Hsu Y.T., Youle R.J. Nonionic detergents induce dimerization among members of the Bcl-2 family. J. Biol. Chem. 1997;272:13829–13834. doi: 10.1074/jbc.272.21.13829. [DOI] [PubMed] [Google Scholar]
- Hsu Y.T., Youle R.J. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J. Biol. Chem. 1998;273:10777–10783. doi: 10.1074/jbc.273.17.10777. [DOI] [PubMed] [Google Scholar]
- Hsu Y.T., Wolter K.G., Youle R.J. Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc. Natl. Acad. Sci. USA. 1997;94:3668–3672. doi: 10.1073/pnas.94.8.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgensmeier J.M., Xie Z., Deveraux Q., Ellerby L., Bredesen D., Reed J.C. Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.H., Zhao Y., Barber M.J., Kuharsky D.K., Yin X.M. Bid-induced cytochrome c release is mediated by a pathway independent of mitochondrial permeability transition pore and Bax. J. Biol. Chem. 2000;275:39474–39481. doi: 10.1074/jbc.M003370200. [DOI] [PubMed] [Google Scholar]
- Kluck R.M., Bossy-Wetzel E., Green D.R., Newmeyer D.D. The release of cytochrome c from mitochondriaa primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Li H., Zhu H., Xu C.J., Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Lindsten T., Ross A.J., King A., Zong W.-X., Rathmell J.C., Shiels H.A., Ulrich E., Waymire K.G., Mahar P., Frauwirth K. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Kim C.N., Yang J., Jemmerson R., Wang X. Induction of apoptotic program in cell-free extractsrequirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Luo X., Budihardjo I., Zou H., Slaughter C., Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- Marzo I., Brenner C., Zamzami N., Jurgensmeier J.M., Susin S.A., Vieira H.L., Prevost M.C., Xie Z., Matsuyama S., Reed J.C. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- Mikhailov V., Mikhailova M., Pulkrabek D.J., Dong Z., Venkatachalam M.A., Saikumar P. Bcl-2 prevents bax oligomerization in the mitochondrial outer membrane. J. Biol. Chem. 2001;276:18361–18374. doi: 10.1074/jbc.M100655200. [DOI] [PubMed] [Google Scholar]
- Minn A.J., Velez P., Schendel S.L., Liang H., Muchmore S.W., Fesik S.W., Fill M., Thompson C.B. Bcl-x(L) forms an ion channel in synthetic lipid membranes. Nature. 1997;385:353–357. doi: 10.1038/385353a0. [DOI] [PubMed] [Google Scholar]
- Murphy K.M., Streips U.N., Lock R.B. Bcl-2 inhibits a Fas-induced conformational change in the Bax N terminus and Bax mitochondrial translocation. J. Biol. Chem. 2000;275:17225–17228. doi: 10.1074/jbc.C900590199. [DOI] [PubMed] [Google Scholar]
- Narita M., Shimizu S., Ito T., Chittenden T., Lutz R.J., Matsuda H., Tsujimoto Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. USA. 1998;95:14681–14686. doi: 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechushtan A., Smith C.L., Hsu Y.T., Youle R.J. Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J. 1999;18:2330–2341. doi: 10.1093/emboj/18.9.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltvai Z.N., Milliman C.L., Korsmeyer S.J. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Pastorino J.G., Chen S.T., Tafani M., Snyder J.W., Farber J.L. The overexpression of Bax produces cell death upon induction of the mitochondrial permeability transition. J. Biol. Chem. 1998;273:7770–7775. doi: 10.1074/jbc.273.13.7770. [DOI] [PubMed] [Google Scholar]
- Saito M., Korsmeyer S.J., Schlesinger P.H. BAX-dependent transport of cytochrome c reconstituted in pure liposomes. Nat. Cell Biol. 2000;2:553–555. doi: 10.1038/35019596. [DOI] [PubMed] [Google Scholar]
- Schlesinger P.H., Gross A., Yin X.M., Yamamoto K., Saito M., Waksman G., Korsmeyer S.J. Comparison of the ion channel characteristics of proapoptotic BAX and antiapoptotic BCL-2. Proc. Natl. Acad. Sci. USA. 1997;94:11357–11362. doi: 10.1073/pnas.94.21.11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlak T.W., Oltvai Z.N., Yang E., Wang K., Boise L.H., Thompson C.B., Korsmeyer S.J. Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proc. Natl. Acad. Sci. USA. 1995;92:7834–7838. doi: 10.1073/pnas.92.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S., Narita M., Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- Suzuki M., Youle R.J., Tjandra N. Structure of Bax. Coregulation of dimer formation and intracellular localization. Cell. 2000;103:645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- Tan Y.J., Beerheide W., Ting A.E. Biophysical characterization of the oligomeric state of Bax and its complex formation with Bcl-XL. Biochem. Biophys. Res. Commun. 1999;255:334–339. doi: 10.1006/bbrc.1999.0222. [DOI] [PubMed] [Google Scholar]
- Tanner V.A., Ploug T., Tao-Cheng J.H. Subcellular localization of SV2 and other secretory vesicle components in PC12 cells by an efficient method of preembedding EM immunocytochemistry for cell cultures. J. Histochem. Cytochem. 1996;44:1481–1488. doi: 10.1177/44.12.8985140. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G., Chandel N.S., Williamson E.K., Schumacker P.T., Thompson C.B. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- Vaux D.L., Cory S., Adams J.M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- Wei M.C., Lindsten T., Mootha V.K., Weiler S., Gross A., Ashiya M., Thompson C.B., Korsmeyer S.J. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- Weil M., Jacobson M.D., Coles H.S., Davies T.J., Gardner R.L., Raff K.D., Raff M.C. Constitutive expression of the machinery for programmed cell death. J. Cell Biol. 1996;133:1053–1059. doi: 10.1083/jcb.133.5.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolter K.G., Hsu Y.T., Smith C.L., Nechushtan A., Xi X.G., Youle R.J. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X.M., Oltvai Z.N., Korsmeyer S.J. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature. 1994;369:321–323. doi: 10.1038/369321a0. [DOI] [PubMed] [Google Scholar]
- Zha H., Aime-Sempe C., Sato T., Reed J.C. Proapoptotic protein Bax heterodimerizes with Bcl-2 and homodimerizes with Bax via a novel domain (BH3) distinct from BH1 and BH2 J. Biol. Chem 271 1996. 7440 7444a [DOI] [PubMed] [Google Scholar]
- Zha J., Harada H., Yang E., Jockel J., Korsmeyer S.J. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell 87 1996. 619 628b [DOI] [PubMed] [Google Scholar]