

Abstract
Epidemiologic investigations have shown that exercise reduces morbidity and mortality from coronary artery disease. In this study, using a rat model, we attempted to determine whether exercise can reduce ischemic injury to the heart and elucidate a mechanism for the cardioprotective effect of exercise. Results showed that exercise significantly reduced the magnitude of a myocardial infarction in biphasic manner. The time course for cardioprotection resembled that of the change in manganese superoxide dismutase (Mn-SOD) activity. The administration of the antisense oligodeoxyribonucleotide to Mn-SOD abolished the expected decrease in infarct size. We showed that the level of tumor necrosis factor α (TNF-α) and interleukin 1β (IL-1β) increased after exercise. The simultaneous administration of the neutralizing antibodies to the cytokines abolished the exercise-induced cardioprotection and the activation of Mn-SOD. Furthermore, TNF-α can mimic the biphasic pattern of cardioprotection and activation of Mn-SOD. An antioxidant completely abolished cardioprotection and the activation of Mn-SOD by exercise or the injection of TNF-α as well as exercise-induced increase in TNF-α and IL-1β. The production of reactive oxygen species and endogenous TNF-α and IL-1β induced by exercise leads to the activation of Mn-SOD, which plays major roles in the acquisition of biphasic cardioprotection against ischemia/reperfusion injury in rats.
Keywords: free radical, tumor necrosis factor, interleukin 1, infarct size, N-2-mercaptopropionyl glycine
Epidemiologic evidence demonstrates that regular physical exercise is associated with a reduced incidence of coronary artery disease (CAD).1 Exercise can reduce such risk factors for CAD as hyperlipidemia, diabetes, obesity, and, perhaps, hypertension. Well-designed clinical investigations have demonstrated that exercise is an independent cardioprotective factor, even in the presence of other risk factors (1, 2). The mechanisms of this protective effect of exercise are supposed to include anatomic and physiologic changes in the coronary arteries, alterations in autonomic tone, and other effects, which lead to an increase in coronary vascular transport activity (3). However, the direct proof that exercise has a beneficial effect on myocardial ischemia/ reperfusion injury has not yet been presented.
The preconditioning phenomenon, in which exposure to brief sublethal ischemia increases the tolerance of the heart to subsequent lethal ischemia, is acquired both soon after and 24 h after preconditioning in a biphasic manner, as reported by our group and other authors (4–7). The preconditioning phenomenon reportedly can be related to the production of reactive oxygen species (8–11). The induction of an intrinsic radical scavenger, manganese-superoxide dismutase (Mn-SOD), by a brief period of ischemia has been suggested to cause the heart to become tolerant to ischemia/reperfusion injury (12–14). TNF and IL-1 are both reported to be involved in cardioprotection against ischemia/ reperfusion injury (15–17).
Exercise has been reported to result in increased free radical production in the heart and other tissues (18, 19). Cytokines such as TNF and IL-1 are produced after exercise (20). These cytokines induce Mn-SOD in myocardium (16, 17). Therefore, we can hypothesize that the acquisition of cardioprotective effect by exercise and brief ischemia may involve a common mechanism that functions through an increase in Mn-SOD activity via the production of reactive oxygen species and cytokines.
In this study, we examined (a) whether a single session of exercise reduces the size of the myocardial infarct after reperfusion; (b) whether cardioprotection induced by exercise may be related to the activation of an intrinsic radical scavenger, Mn-SOD; (c) whether proinflammatory cytokines TNF-α and IL-1β are involved in the activation of Mn-SOD after exercise; and (d) whether a reactive oxygen species–mediated mechanism is involved in the acquisition of tolerance after exercise.
Materials and Methods
Exercise Protocol.
Male Wistar rats (weighing 240–300 g) were initially screened by determining whether they could run on a motor-driven treadmill at 20–25 m/min, 0% grade, for 20 min (exercise began and ended with a 5-min “warm-up” and “cool-down” at 15 m/min and 0% grade; the duration of exercise included the warm-up and cool-down periods). At least 2 wk later, only rats that completed the exercise screening test were used for all experiments described in this study. For exercise experiments, rats received one session of exercise, which consisted of running on a treadmill at 27–30 m/min, 0% grade, for 25–30 min, including the warm-up and cool-down periods as described for the screening test. Rats in the sham-treated control group were placed on the treadmill apparatus without exercise for 30 min and allowed to recover at room temperature for a defined interval, as indicated in the figure legends. Some rats were not placed on the treadmill apparatus (untreated controls). After the exercise session, rats were anesthetized with sodium pentobarbital, and the chest of each rat was opened via a right parasternal sternomectomy. The heart was exposed, and 7-0–type silk thread was then passed around the left coronary artery (LCA) 3–4 mm distal to the origin of LCA. After a 10-min period of stabilization, the LCA was ligated. The snare was released after 20 min of coronary occlusion. Reperfusion was indicated by a change in the color of the ventricular surface. Arrhythmias were monitored by electrocardiogram. Ventricular fibrillation (VF) was defined according to the criteria of the Lambeth Conventions (21). If VF occurred during ischemia and did not resolve spontaneously within 3 s, manual cardioversion was attempted by gentle palpation of the nonischemic region of the heart. We excluded from infarct size analysis rats in which VF persisted for more than 6 s or in which cardioversion had to be performed >3 times.
Measurement of Myocardial Infarct Size.
48 h after reperfusion, the rats were killed by an overdose of sodium pentobarbital (100 mg/kg). The heart was excised and the size of the infarct was evaluated by a method of double staining using Evans blue dye and triphenyltetrazolium chloride (TTC) as previously described (22, 23). The size of the myocardial infarct, i.e., the area without TTC staining, was expressed as a percentage of the ischemic area at risk, i.e., the area free of Evans blue dye.
The rate–pressure product showed no significant difference among the groups before ischemia, at the end of the ischemic period, or 30 min after reperfusion (data not shown).
SOD Activity and Content in Myocardium.
We measured the activity and content of SODs, intrinsic radical scavengers that are supposed to protect the heart against ischemia/reperfusion injury. At the time after exercise indicated in the figures, rats in the exercise and control groups were killed by administering an overdose of sodium pentobarbital. The blood was washed out of the left and right coronary arteries with PBS from the ascending aorta in a retrograde manner. The left ventricular myocardium was rapidly frozen in liquid nitrogen and stored at −80°C. SOD activity of the myocardial samples was determined by the nitroblue tetrazolium method (24) with a modification for sample preparation (23). Mn-SOD content was measured by means of an ELISA, as we previously reported (12). The measurements of Mn-SOD activity and content were performed at least in triplicate. The activity and content of Mn-SOD was corrected for the protein concentration in the supernatant determined according to the Lowry method (24a).
TNF-α and IL-1β Levels in Myocardium.
To examine whether TNF-α and IL-1β are involved in the activation of Mn-SOD after exercise, the levels of TNF-α and IL-1β in myocardium were measured. Myocardium was homogenized in 20 mM PBS containing 1 mM EDTA and centrifuged at 900 g for 15 min. The levels of TNF-α and IL-1β in the supernatant were measured by means of ELISA kits (TNF-α, Genzyme Co.; IL-1β, Immuno-Biological Labs. Co.). The measurements of cytokines were performed in triplicate. The levels of TNF-α and IL-1β were corrected for the protein concentration in the supernatant determined by the Lowry method (24a).
Administration of Reagents.
To manipulate the level of expression of Mn-SOD, 22-mer phosphorothioate derivative of the antisense oligodeoxyribonucleotide ([ASODN]; CACGCCGCCCGACACAACATTG), sense oligodeoxyribonucleotide ([SODN]; CAATGTTGTGTCGGGCGGCGTG), or scrambled ODN (TCTCAGTGAGCCCTCATTCTGT) was injected intraperitoneally just after exercise at a dose of 10 mg/kg. To optimize the experimental conditions for the in vivo delivery of systemically injected ODNs, we evaluated the time course of their accumulation in the heart. In experiments with 5′ FITC–labeled ASODN to Mn-SOD, we found that significant labeling of these tissues occurred at the following times after the intraperitoneal injection: at 2–4 h in endothelial cells, at 4 h in vascular smooth muscle, and at 8 h in cardiac myocytes (unpublished observation). We could not determine the subcellular distribution of ASODN in the cardiac myocytes due to technical limitations.
To neutralize the increases of TNF-α and IL-1β after exercise, anti-murine TNF-α antibody (0.5 ml) and/or anti-murine IL-1β antibody (0.5 mg), obtained from Genzyme, were infused intraperitoneally 30 min before exercise. Both antibodies cross-react with rat cytokines.
Recombinant murine TNF-α and recombinant murine IL-1β were obtained from Genzyme. The recombinant cytokines were diluted in pathogen-free saline on the day of injection. Rats received an intravenous injection of cytokine via the right femoral vein for 2 min at a dose of 1.5 μg/kg body weight to examine the involvement of TNF-α in exercise-induced cardioprotection. N-2-mercaptopropionyl glycine (MPG) is a low molecular weight, synthetic analogue of glutathione and a diffusible and membrane-permeable antioxidant. This agent especially scavenges hydrogen peroxide and hydroxyl radicals. To determine the involvement of reactive oxygen species during exercise in the acquisition of tolerance against ischemia/reperfusion injury, MPG (100 mg/kg) was infused intraperitoneally 10 min before the 20-min exercise session or 30 min before the injection of TNF-α. As the half-time for elimination of MPG is ∼7 min in vivo (25), MPG should not be effective during myocardial ischemia/reperfusion period. Treatment with MPG also did not alter the area at risk (data not shown), indicating that its effect on infarct size may not be mediated via changes in collateral flow.
Statistical Methods.
Figure data are expressed as mean ± SEM. Intergroup comparisons were assessed for significance by using one-way analysis of variance (ANOVA) with Bonferroni's post hoc test for multiple comparisons. A level of P < 0.05 was accepted as statistically significant.
Results
Exclusion due to VF and Death.
A total of 28 rats developed serious VF during occlusion (10 in sham-treated control group, 4 in exercise group, 3 in AODN-treated group, 2 in exercise group treated with AODN, 2 in exercise group pretreated with anti-murine TNF-α and anti-murine IL-1β antibodies, 4 in TNF-α–treated group, 2 in exercise group pretreated with MPG, and 1 in sham-treated control group pretreated with MPG) and were excluded from the evaluation of myocardial infarct size. 29 rats died prematurely (probably because of arrhythmia or heart failure) during the 48-h reperfusion period (7 in sham-treated control group, 4 in exercise group, 3 in AODN-treated group, 1 in SODN-treated group, 1 in exercise group treated with AODN, 1 in exercise group treated with scrambled ODN, 1 in exercise group pretreated with anti-murine TNF-α antibody, 2 in exercise group pretreated with anti-murine IL-1β antibody, 2 in exercise group pretreated with anti-murine TNF-α and anti-murine IL-1β antibodies, 5 in TNF-α–treated group, 1 in exercise group pretreated with MPG, and 1 in sham-treated control group pretreated with MPG).
Size of Myocardial Infarct after Exercise.
We examined the size of myocardial infarct after occlusion (20 min)/reperfusion (48 h) in the LCA in rats, which received one session of exercise. There were no significant differences in the size of the myocardial infarct for recovery intervals of 0.5–72 h among the sham-treated control groups (data not shown). As shown in Fig. 1 (top), the size of the myocardial infarct after reperfusion in the sham-treated control rats did not differ significantly from the size of the infarct observed in rats at 3, 24, and 72 h after exercise. However, rats subjected to ischemia at 0.5, 36, 48, and 60 h after the exercise session exhibited a marked decrease relative to the control rats in the size of the myocardial infarct after reperfusion. The infarct-limiting effect of exercise was slightly less at 36 and 60 h than at 0.5 and 48 h after exercise. The area at risk did not differ significantly among the groups (data not shown).
Figure 1.

Effect of exercise on the size of the myocardial infarct (top) and the activity (center) and content (bottom) of Mn-SOD. Rats were subjected to occlusion of the LCA followed by reperfusion at 0.5, 3, 24, 36, 48, 60, and 72 h after exercise. C, sham-treated control 0.5 h after sham treatment. Top, the size of the infarct was evaluated 48 h after reperfusion. The size of the area of myocardial infarct (the area without TTC staining) was expressed as a percentage of the ischemic area at risk (the area without Evans blue dye); n = 9–10. Center, Mn-SOD activity in the myocardial tissue was examined between 0.5 and 72 h after one session of exercise. Mn-SOD activity divided by total protein content is shown; n = 3–4. Bottom, Mn-SOD content in myocardial tissue was examined between 0.5 and 72 h after one trial of exercise. Mn-SOD content divided by total protein content is shown; n = 3–4. Data are expressed as mean ± SEM; *P < 0.05 vs. sham-treated control by one-way ANOVA with Bonferroni's post hoc test for multiple comparisons.
Mn-SOD Activity after Exercise.
Reactive oxygen species produced during ischemia/reperfusion in the heart have been believed to induce myocardial cell damage (26– 28). An intrinsic radical scavenger, Mn-SOD protects hearts against oxygen free radicals. To examine the involvement of Mn-SOD in the cardioprotective effect of exercise, we measured Mn-SOD activity after treadmill exercise. There were no significant differences in the activity and content of Mn-SOD for recovery intervals of 0.5–72 h among the sham-treated control groups (data not shown). Mn-SOD activity was unchanged at 3, 24, and 72 h after exercise but was significantly increased at 0.5 and 48 h after exercise, as compared with the sham-treated control (Fig. 1, center). The biphasic time course of the change in Mn-SOD activity in the myocardium of rats exposed to exercise resembled the time course for the protection against ischemia/ reperfusion injury. The myocardial content of Mn-SOD did not change relative to the sham-treated control group within 24 h after exercise. Thereafter, however, the Mn-SOD level increased significantly, reaching 137% of the control value 48 h after exercise (Fig. 1, bottom). The Mn-SOD level returned to control values within 72 h after exercise. The activity of the cytosolic isoform of SOD (Cu, Zn-SOD) was unaffected by exercise (data not shown).
We examined the relationship between the acquisition of tolerance to ischemia/reperfusion and the induction of Mn-SOD in the myocardium 48 h after exercise. We manipulated the level of expression of Mn-SOD using ASODN. The administration of ASODN completely inhibited the increases in Mn-SOD activity and content 48 h after exercise (Fig. 2, center and bottom). However, SODN did not attenuate the increases in Mn-SOD activity and content induced by exercise. As shown in Fig. 2 (top), the expected decrease in infarct size induced by exercise was also abolished in rats treated with ASODN to Mn-SOD, in which the induction of Mn-SOD was specifically inhibited. SODN, which did not attenuate the induction of Mn-SOD in the myocardium after exercise, did not abolish the protective effect of exercise. Administration of the scrambled oligonucleotide had no effect on infarct size or on Mn-SOD activity as seen with SODN. Administration of ASODN decreased the activity and content of Mn-SOD and increased the infarct size after reperfusion in sham-treated control rats.
Figure 2.
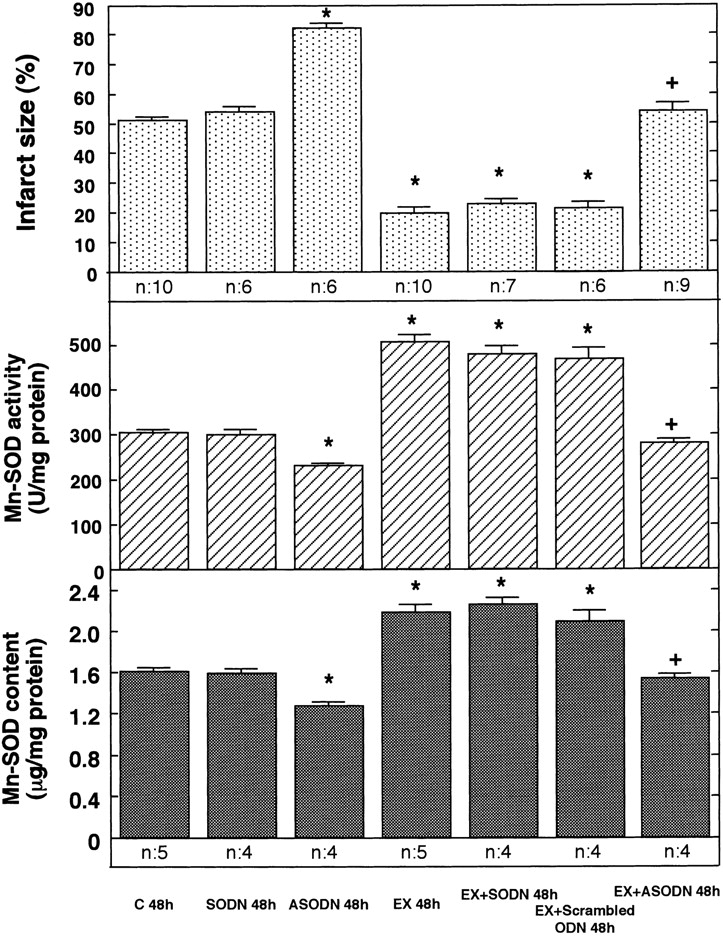
Effects of ASODN to Mn-SOD on the size of the myocardial infarct (top) and the activity (center) and content (bottom) of MnSOD. SODN, ASODN, or scrambled ODN to Mn-SOD were injected intraperitoneally into rats just after exercise (EX). In 48 h, rats were subjected to occlusion of the LCA for 20 min, followed by reperfusion for 48 h. The size of the myocardial infarct was measured after reperfusion (n = 6–10). The activity and content of Mn-SOD (n = 4–5) was evaluated 48 h after exercise. C 48h, sham-treated control 48 h after sham treatment. Data are expressed as mean ± SEM; *P < 0.05 vs. sham-treated control; + P < 0.05 vs. without ASODN by one-way ANOVA with the Bonferroni's post hoc test for multiple comparisons.
Induction of Cardioprotection by Cytokines.
To examine the contribution of the cytokines to exercise-induced cardioprotection, we investigated the time course of the induction of TNF-α and IL-1β in the myocardium after exercise using the ELISA method. There were no significant differences in TNF-α and IL-1β contents for recovery intervals of 0–30 min among the sham-treated control groups (data not shown). As illustrated in Fig. 3 (top), an increase in TNF-α content was evident immediately after exercise. The level of TNF-α peaked 10 min after exercise and then decreased rapidly toward the baseline within 20 min. Immediately after exercise, the myocardial level of IL-1β also increased significantly, as compared with the sham-treated control group, and declined to the control level within 10 min (Fig. 3, bottom).
Figure 3.

Time course of myocardial levels of TNF-α (top) and IL-1β (bottom) after exercise. After exercise, the myocardial levels of TNF-α and IL-1β were measured with an ELISA (○). Increases in TNF-α and IL-1β were neutralized by their respective antibodies (•). MPG abolished the increases in TNF-α and IL-1β related to exercise (▪). C, sham-treated control just after sham treatment. Data are expressed as mean ± SEM; n = 3–4. *P < 0.05 vs. sham-treated control (C); + P < 0.05 vs. without antibody or MPG by one-way ANOVA with Bonferroni's post hoc test for multiple comparisons.
To determine whether the production of these cytokines after exercise may be involved in the acquisition of tolerance to ischemia/reperfusion, we administered the antibody to these cytokines intraperitoneally 30 min before exercise. The intraperitoneal administration of anti-murine TNF-α and anti-murine IL-1β antibody 30 min before exercise inhibited the observed increase in levels of TNF-α and IL-1β, respectively, after exercise (Fig. 3). As shown in Fig. 4 (top), the administration of an antibody to TNF-α did not influence the size of infarct at 0.5 or 48 h after exercise. The administration of an antibody to IL-1β also did not alter the size of the myocardial infarct at 0.5 or 48 h after exercise. However, the simultaneous administration of the antibodies to TNF-α and IL-1β abolished the protection against ischemic damage at 0.5 and 48 h after exercise. Antibody to TNF-α or IL-1β had no effect on the increase in Mn-SOD activity induced by exercise (Fig. 4, bottom). The simultaneous administration of the antibodies to these cytokines eliminated the activation of Mn-SOD at 0.5 and 48 h after exercise.
Figure 4.
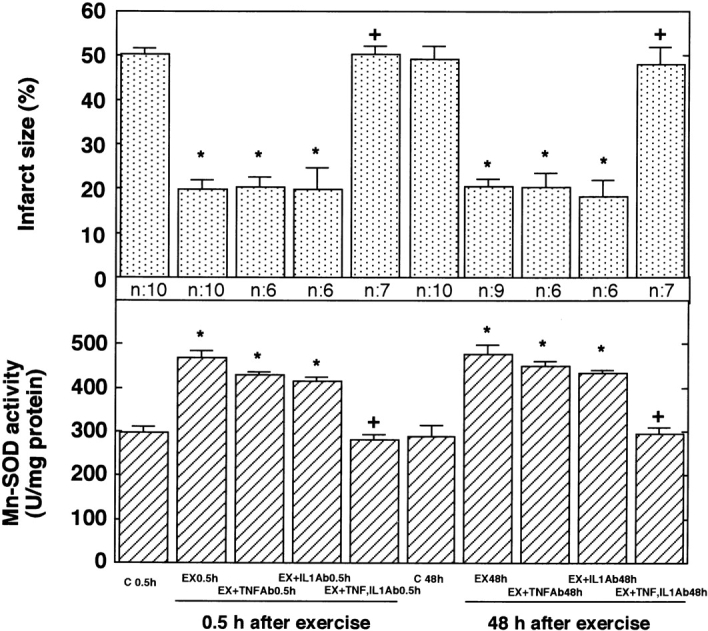
Effects of anti–TNF-α and anti–IL-1β antibodies on infarct size (top) and Mn-SOD activity (bottom). Anti-murine TNF-α and/or anti-murine IL-1β antibodies were infused intraperitoneally 30 min before exercise. 0.5 or 48 h after exercise (EX), rats were subjected to occlusion of the LCA followed by reperfusion. C 0.5h and C 48h indicate sham-treated control 0.5 and 48 h after sham treatment, respectively. The size of the myocardial infarct was measured 48 h after reperfusion (n = 6–10). The activity of Mn-SOD (n = 4–5) was evaluated 0.5 or 48 h after exercise. Data are expressed as mean ± SEM; *P < 0.05 vs. corresponding sham-treated control; + P < 0.05 vs. without the antibodies by one-way ANOVA with Bonferroni's post hoc test for multiple comparisons.
We next investigated whether TNF-α could mimic biphasic cardioprotection. In the untreated control groups, the size of myocardial infarct and Mn-SOD activity remained constant after vehicle (saline) injection in the time course of the experiment (data not shown). As shown in Fig. 5 (top), the infusion of murine rTNF-α reduced the size of the infarct in a biphasic pattern. The first phase of protection occurred 0.5 h after the administration of TNF-α. When such tolerance had disappeared, the second phase of protection was observed. Peak protection during the second phase was achieved 48 h after administration of a bolus injection of TNF-α. The time course was identical to that for the cardioprotection induced by exercise (Fig. 1, top). IL-1β also mimicked this biphasic cardioprotective effect (data not shown).
Figure 5.
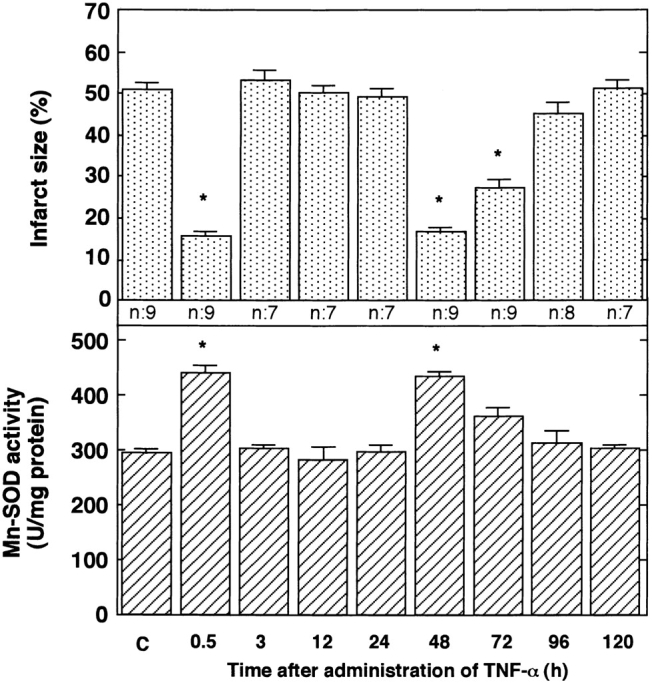
Effects of TNF-α on size of myocardial infarct (top) and activity of Mn-SOD (bottom). After the intravenous injection of rTNF-α, rats were subjected to LCA occlusion followed by reperfusion at the times indicated. Infarct size was evaluated 48 h after reperfusion (n = 7–9). The activity of Mn-SOD was measured at the indicated times after TNF-α administration (n = 4). C, untreated control 0.5 h after vehicle (saline) injection. Data are expressed as mean ± SEM; *P < 0.05 vs. control (C) by one-way ANOVA with Bonferroni's post hoc test for multiple comparisons.
The activity of Mn-SOD increased significantly in a biphasic manner 48 as well as 0.5 h after the bolus injection of TNF-α (Fig. 5, bottom). No change in Mn-SOD activity was observed 3, 24, and 72 h after TNF-α administration. IL-1β also activated Mn-SOD in a biphasic manner (data not shown). The time course of cardioprotection coincided with that for the increase of Mn-SOD activity after the administration of TNF-α.
Involvement of Reactive Oxygen Species.
To determine whether the generation of reactive oxygen species during exercise may be involved in the acquisition of tolerance to ischemia/reperfusion, we administered an antioxidant, MPG, 10 min before exercise. Although this agent did not alter the size of the myocardial infarct in the sham-treated control rats, MPG completely abolished the protection against ischemia/reperfusion injury at 0.5 and 48 h after exercise (Fig. 6, top).
Figure 6.
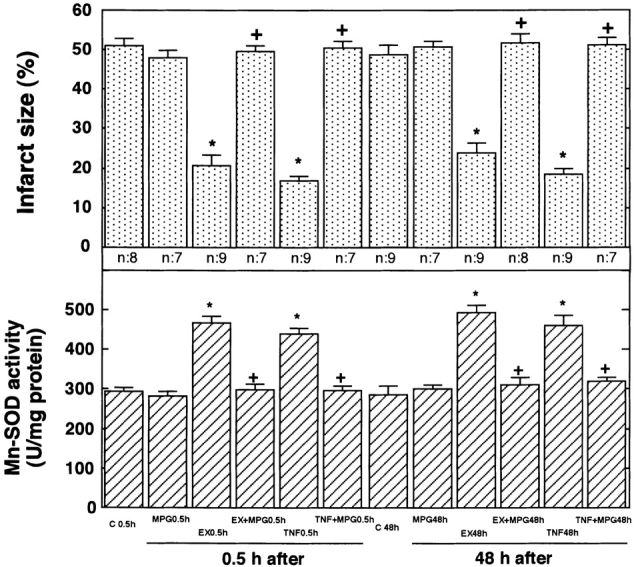
Effects of MPG on size of infarct (top) and activity of Mn-SOD (bottom). MPG was infused intraperitoneally 10 min before a 20-min exercise period or 30 min before the injection of TNF-α. At 30 min and 48 h after exercise (EX) or the injection of TNF-α, rats were subjected to occlusion of the LCA for 20 min, followed by reperfusion for 48 h. The size of the infarct was then evaluated (n = 7–9). The activity of Mn-SOD was measured 0.5 and 48 h after exercise or the injection of TNF-α (n = 4). C 0.5h and C 48h indicate sham-treated control 0.5 and 48 h after sham treatment, respectively. Both controls received vehicle (saline) injection. There were no differences in the size of myocardial infarct or Mn-SOD activity between the untreated and sham-treated control rats. Data are expressed as mean ± SEM; *P < 0.05 vs. corresponding sham-treated control; + P < 0.05. vs. without MPG by one-way ANOVA with Bonferroni's post hoc test for multiple comparisons.
MPG did not alter Mn-SOD activity in sham-treated control rats, whereas it completely abolished the early (0.5-h) and late (48-h) peaks in Mn-SOD activity that followed either exercise or the injection of TNF-α (Fig. 6, bottom). MPG completely abolished the exercise-induced increase in TNF-α and IL-1β in myocardial tissue (Fig. 3).
Discussion
Exercise Induces Cardioprotection.
A substantial body of epidemiologic evidence demonstrates that exercise reduces cardiovascular mortality. Although exercise can improve other known risk factors for CAD, such as elevated plasma lipid level, obesity, and glucose intolerance, exercise appears to exert an independent cardioprotective effect (1, 2). The present results provide the first demonstration that the physiological stimulus of exercise has a directly beneficial effect on myocardial ischemia/reperfusion injury in a biphasic manner in an animal model. Exercise appears to induce structural and/or functional adaptation, leading to an increase in coronary vascular transport capacity (3). Bloor et al. reported that exercise training could contribute to the development of the collateral circulation and the tissue salvage in the myocardial ischemia seen in pigs (29). However, we observed no difference in the area at risk among the experimental groups, indicating that the beneficial effects of exercise are unlikely to be related to the development of collateral circulation.
Involvement of Mn-SOD in Cardioprotection.
The mechanism that underlies the cardioprotection observed at both the early phase and the late phase after exercise appeared to be related to an increase in Mn-SOD activity. A mechanism remains to be elucidated for the activation of Mn-SOD at the early phase (0.5 h) after exercise, where there was no difference in Mn-SOD at the protein level between the exercise and sham-treated control groups. These results indicated that a precursor or inactive form of Mn-SOD that possesses antigenicity but lacks enzyme activity may be modulated by exercise. The increase of Mn-SOD activity disappeared by 3 h after exercise, suggesting that a rapid inactivation should follow the activation of Mn-SOD. It is possible that a reversible phenomenon, such as a phosphorylation–dephosphorylation mechanism, might be involved in this step.
Inhibition of Mn-SOD induction at the late phase by the administration of ASODN to Mn-SOD abolished the protection against ischemia/reperfusion injury induced by exercise. Therefore, at the late phase, the induction of Mn-SOD leads to an increase in its enzyme activity, resulting in the acquisition of cardioprotection against ischemia/reperfusion injury.
Signal Transduction for Cardioprotection.
Previous reports (20) and this study showed that TNF and IL-1 are produced after exercise. Our results clearly showed that TNF-α and IL-1β are involved in exercise-induced cardioprotection via activation of Mn-SOD. Because the effects of TNF-α and IL-1β show some redundancy (30), either of these cytokines is sufficient for both biphasic Mn-SOD activation and cardioprotection, and the blockade of both cytokines is necessary for the inhibition of Mn-SOD activation and cardioprotection induced by exercise. Although the mechanism for the activation of Mn-SOD appears to be different between early and late phases after exercise, these cytokines can activate Mn-SOD at both phases.
It has been known that exercise produces reactive oxygen species in the heart (18, 19). Our results indicated that (i) reactive oxygen species induce TNF-α and IL-1β, (ii) the activation of Mn-SOD by these cytokines is mediated by the reactive oxygen species, and (iii) the cardioprotective effect of TNF-α administration is mediated through the production of reactive oxygen species. Based on these observations, we could elucidate the mechanism for the exercise-induced cardioprotection as follows. Reactive oxygen species, produced during exercise, increase the levels of TNF-α and IL-1β in myocardium. Then, the cytokines activate Mn-SOD during the early phase and induce Mn-SOD during the late phase of cardioprotection, possibly via the production of reactive oxygen species. Reactive oxygen species are present upstream as well as downstream of the cytokines in the exercise-activated signaling pathway. It is unclear whether the loop of these cytokines and reactive oxygen species is necessary for the activation of Mn-SOD. The production of reactive oxygen species by the cytokines may take part in a positive feedback mechanism in this signal transduction system. The role of the reactive oxygen species might be dose dependent. A high dose of reactive oxygen species causes damage to cardiac myocytes, whereas a low dose of such species acts as a signal transduction messenger in cells.
The transcriptional factors activator protein 1 and nuclear factor κB are subjected to redox regulation (31–36). The activation of nuclear factor κB induces such cytokines as TNF-α and IL-1β (37–39). TNF-α and IL-1β also stimulate the production of reactive oxygen species in cells (40– 44). The induction of Mn-SOD is regulated under a redox state (45) and is mediated by cytokines (46). Exercise may activate these transcriptional factors via the production of reactive oxygen species and lead to cytokine production. However, the relationship between the activation of these transcriptional factors and Mn-SOD as observed in the early phase of cardioprotection remains to be established.
Cardioprotection by Sublethal Stress.
The time course of the cardioprotection induced by exercise resembles that produced by a brief period of cardiac ischemia (ischemic preconditioning) (4–7). This tolerance seems to be related to the production of reactive oxygen species (8–11). The induction of Mn-SOD by a brief period of ischemia is responsible for the preconditioning phenomenon (12–14). TNF and IL-1 are both reported to be involved in cardioprotection against ischemia/reperfusion injury (15–17) as well as the radioresistance induced by sublethal ionizing radiation or LPS (47). The sequence and nature of the signal transduction steps in the preconditioning phenomenon and radioresistance remain to be elucidated. The acquisition of cardioprotection following sublethal stress such as brief ischemia, exercise, ionizing radiation, and LPS may involve a common mechanism that functions through an induction and activation of Mn-SOD via the production of reactive oxygen species and cytokines.
Acknowledgments
We thank Drs. Naoyuki Taniguchi and Keiichiro Suzuki of Osaka University for the gift of polyclonal antibody to rat Mn-SOD.
This work was supported in part by a grant-in-aid from the Meiji Life Foundation of Health and Welfare (to N. Yamashita) and a research grant from the Ministries of Education, Science, and Culture of Japan (to T. Kuzuya).
Abbreviations used in this paper
- ANOVA
analysis of variance
- ASODN
antisense oligodeoxyribonucleotide
- CAD
coronary artery disease
- LCA
left coronary artery
- MPG
N-2-mercaptopropionyl glycine
- TTC
triphenyltetrazolium chloride
- Mn-SOD
manganese superoxide dismutase
- SODN
sense oligodeoxyribonucleotide
References
- 1.Bijnen FC, Caspersen CJ, Mosterd WL. Physical inactivity as a risk factor for coronary heart disease: a WHO and International Society and Federation of Cardiology position statement. Bull World Health Organ. 1994;72:1–4. [PMC free article] [PubMed] [Google Scholar]
- 2.Fletcher GF, Blair SN, Blumenthal J, Caspersen C, Chaitman B, Epstein S, Falls H, Froelicher ES, Froelicher VF, Pina IL. Statement on exercise. Benefits and recommendations for physical activity programs for all Americans. A statement for health professionals by the Committee on Exercise and Cardiac Rehabilitation of the Council on Clinical Cardiology, American Heart Association. Circulation. 1992;86:340–344. doi: 10.1161/01.cir.86.1.340. [DOI] [PubMed] [Google Scholar]
- 3.Laughlin MH, McAllister RM. Exercise training-induced coronary vascular adaptation. J Appl Physiol. 1992;73:2209–2225. doi: 10.1152/jappl.1992.73.6.2209. [DOI] [PubMed] [Google Scholar]
- 4.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 5.Murry CE, Jennings RB, Reimer KA. New insights into potential mechanisms of ischemic preconditioning. Circulation. 1991;84:442–445. doi: 10.1161/01.cir.84.1.442. [DOI] [PubMed] [Google Scholar]
- 6.Marber MS, Latchman DS, Walker JM, Yellon DM. Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction. Circulation. 1993;88:1264–1272. doi: 10.1161/01.cir.88.3.1264. [DOI] [PubMed] [Google Scholar]
- 7.Kuzuya T, Hoshida S, Yamashita N, Fuji H, Oe H, Hori M, Kamada T, Tada M. Delayed effects of sublethal ischemia on the acquisition of tolerance to ischemia. Circ Res. 1993;72:1293–1299. doi: 10.1161/01.res.72.6.1293. [DOI] [PubMed] [Google Scholar]
- 8.Hoshida S, Kuzuya T, Yamashita N, Oe H, Fuji H, Hori M, Tada M, Kamada T. Brief myocardial ischemia affects free radical generating and scavenging systems in dogs. Heart Vessels. 1993;8:115–120. doi: 10.1007/BF01744795. [DOI] [PubMed] [Google Scholar]
- 9.Chen W, Gabel S, Steenbergen C, Murphy E. A redox-based mechanism for cardioprotection induced by ischemic preconditioning in perfused rat heart. Circ Res. 1995;77:424–429. doi: 10.1161/01.res.77.2.424. [DOI] [PubMed] [Google Scholar]
- 10.Zhou X, Zhai X, Ashraf M. Direct evidence that initial oxidative stress triggered by preconditioning contributes to second window of protection by endogenous antioxidant enzyme in myocytes. Circulation. 1996;93:1177–1184. doi: 10.1161/01.cir.93.6.1177. [DOI] [PubMed] [Google Scholar]
- 11.Sun J-Z, Tang X-L, Park S-W, Qiu Y, Turrens JF, Bolli R. Evidence for an essential role of reactive oxygen species in the genesis of late preconditioning against myocardial stunning in conscious pigs. J Clin Invest. 1996;97:562–576. doi: 10.1172/JCI118449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamashita N, Nishida M, Hoshida S, Igarashi J, Hori M, Kuzuya T, Tada M. a1-Adrenergic stimulation induces tolerance of cardiac myocytes to hypoxia through induction and activation of Mn-SOD. Am J Physiol. 1996;271:H1356–H1362. doi: 10.1152/ajpheart.1996.271.4.H1356. [DOI] [PubMed] [Google Scholar]
- 13.Yamashita N, Nishida M, Hoshida S, Kuzuya T, Hori M, Taniguchi N, Kamada T, Tada M. Induction of manganese superoxide dismutase in rat cardiac myocytes increases tolerance to hypoxia 24 hours after preconditioning. J Clin Invest. 1994;94:2193–2199. doi: 10.1172/JCI117580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamashita N, Hoshida S, Nishida M, Igarashi J, Taniguchi N, Tada M, Kuzuya T, Hori M. Heat shock-induced manganese superoxide dismutase enhances the tolerance of cardiac myocytes to hypoxia-reoxygenation injury. J Mol Cell Cardiol. 1997;29:1805–1813. doi: 10.1006/jmcc.1997.0415. [DOI] [PubMed] [Google Scholar]
- 15.Brown JM, White CW, Terada LS, Grosso MA, Shanley PF, Mulvin DW, Banerjee A, Whitman GJ, Harken AH, Repine JE. Interleukin 1 pretreatment decreases ischemia/reperfusion injury. Proc Natl Acad Sci USA. 1990;87:5026–5030. doi: 10.1073/pnas.87.13.5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eddy LJ, Goeddel DV, Wong GH. Tumor necrosis factor-alpha pretreatment is protective in a rat model of myocardial ischemia-reperfusion injury. Biochem Biophys Res Commun. 1992;184:1056–1059. doi: 10.1016/0006-291x(92)90698-k. [DOI] [PubMed] [Google Scholar]
- 17.Maulik, N., R.M. Engelman, Z. Wei, D. Lu, J.A. Rousou, and D.K. Das. 1993. Interleukin-1 alpha preconditioning reduces myocardial ischemia reperfusion injury. Circulation. 88: II-387–II-394. [PubMed]
- 18.Salo DC, Donovan CM, Davies KJ. HSP70 and other possible heat shock or oxidative stress proteins are induced in skeletal muscle, heart, and liver during exercise. Free Radic Biol Med. 1991;11:239–246. doi: 10.1016/0891-5849(91)90119-n. [DOI] [PubMed] [Google Scholar]
- 19.Kumar CT, Reddy VK, Prasad M, Thyagaraju K, Reddanna P. Dietary supplementation of vitamin E protects heart tissue from exercise-induced oxidant stress. Mol Cell Biochem. 1992;111:109–115. doi: 10.1007/BF00229581. [DOI] [PubMed] [Google Scholar]
- 20.Shepard RJ, Shek PN. Impact of physical activity and sport on the immune system. Rev Environ Health. 1996;11:133–147. doi: 10.1515/reveh.1996.11.3.133. [DOI] [PubMed] [Google Scholar]
- 21.Walker MJ, Curtis MJ, Hearse DJ, Cambell RW, Janse MJ, Yellon DM, Cobbe SM, Coker SJ, Harness JB, Harron DJ. The Lambeth Conventions: guidelines for the study of arrhythmias in ischaemia infarction, and reperfusion. Cardiovasc Res. 1988;22:447–455. doi: 10.1093/cvr/22.7.447. [DOI] [PubMed] [Google Scholar]
- 22.Yamashita N, Hoshida S, Nishida M, Igarashi J, Aoki K, Hori M, Kuzuya T, Tada M. Time course of tolerance to ischemia-reperfusion injury and induction of heat shock protein 72 by heat stress in the rat heart. J Mol Cell Cardiol. 1997;29:1815–1821. doi: 10.1006/jmcc.1997.0416. [DOI] [PubMed] [Google Scholar]
- 23.Yamashita N, Hoshida S, Taniguchi N, Kuzuya T, Hori M. A “second window of protection” occurs 24 hours after ischemic preconditioning in the rat heart. J Mol Cell Cardiol. 1998;30:1181–1189. doi: 10.1006/jmcc.1998.0682. [DOI] [PubMed] [Google Scholar]
- 24.Hoshida S, Kuzuya T, Fuji H, Yamashita N, Oe H, Hori M, Suzuki K, Taniguchi N, Tada M. Sublethal ischemia alters myocardial antioxidant activity in canine heart. Am J Physiol. 1993;264:H33–H39. doi: 10.1152/ajpheart.1993.264.1.H33. [DOI] [PubMed] [Google Scholar]
- 24a.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 25.Horwitz LD, Fennessey PV, Shikes RH, Kong Y. Marked reduction in myocardial infarct size due to prolonged infusion of an antioxidant during reperfusion. Circulation. 1994;89:1792–1801. doi: 10.1161/01.cir.89.4.1792. [DOI] [PubMed] [Google Scholar]
- 26.Jolly SR, Kane WJ, Bailie MB, Abrams GD, Lucchesi BR. Canine myocardial reperfusion injury. Its reduction by the combined administration of superoxide dismutase and catalase. Circ Res. 1984;54:277–284. doi: 10.1161/01.res.54.3.277. [DOI] [PubMed] [Google Scholar]
- 27.Hammond B, Hess ML. The oxygen free radical system: potential mediator of myocardial injury. J Am Coll Cardiol. 1985;6:215–218. doi: 10.1016/s0735-1097(85)80278-3. [DOI] [PubMed] [Google Scholar]
- 28.Opie LH. Reperfusion injury and its pharmacologic modification. Circulation. 1989;80:1049–1062. doi: 10.1161/01.cir.80.4.1049. [DOI] [PubMed] [Google Scholar]
- 29.Bloor CM, White FC, Sanders TM. Effects of exercise on collateral development in myocardial ischemia in pigs. J Appl Physiol. 1984;56:656–665. doi: 10.1152/jappl.1984.56.3.656. [DOI] [PubMed] [Google Scholar]
- 30.Neta R, Sayers TJ, Oppenheim JJ. Relationship of TNF to interleukins. Immunol Ser. 1992;56:499–566. [PubMed] [Google Scholar]
- 31.Abate C, Patel L, Rauscher J, Curran T. Redox regulation of fos and jun DNA-binding activity in vitro. Science. 1990;249:1157–1161. doi: 10.1126/science.2118682. [DOI] [PubMed] [Google Scholar]
- 32.Staal FJ, Roedere M, Herzenberg LA, Herzenberg LA. Intracellular thiols regulate activation of nuclear factor kB and transcription of human immunodeficiency virus. Proc Natl Acad Sci USA. 1990;87:9943–9947. doi: 10.1073/pnas.87.24.9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Menon SD, Qin S, Gu GR, Tan YH. Differential induction of nuclear NF-kB by protein phosphatase inhibitors in primary and transformed human cells: requirement for both oxidation and phosphorylation in nuclear translocation. J Biol Chem. 1993;268:26805–26812. [PubMed] [Google Scholar]
- 34.Hayashi T, Ueno Y, Okamoto T. Oxidoreductive regulation of nuclear factor kB: involvement of a cellular reducing catalyst thioredoxin. J Biol Chem. 1993;268:11380–11388. [PubMed] [Google Scholar]
- 35.Schenk H, Klein M, Erdbrugger W, Droge W, Schulze-Osthoff K. Distinct effects of thioredoxin and antioxidants on the activation of transcription factor NF-kB and AP-1. Proc Natl Acad Sci USA. 1994;91:1672–1676. doi: 10.1073/pnas.91.5.1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pinkus R, Wiener LM, Daniel V. Role of oxidants and antioxidants in the induction of AP-1, NF-kB, and glutathione S-transferase gene expression. J Biol Chem. 1996;271:13422–13429. doi: 10.1074/jbc.271.23.13422. [DOI] [PubMed] [Google Scholar]
- 37.Greene WC. Regulation of HIV-1 gene expression. Annu Rev Immunol. 1990;8:453–475. doi: 10.1146/annurev.iy.08.040190.002321. [DOI] [PubMed] [Google Scholar]
- 38.Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 39.Verma IM, Stevenson JK, Svhwarz EM, Van-Antwerp D, Miyamoto S. Rel/NF-kappa B/I kappa B family. Gene Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 40.Meier B, Radeke HH, Selle S, Younes M, Sies H, Resch K, Habermehl GG. Human fibroblasts release reactive oxygen species in response to interleukin-1 or tumour necrosis factor-alpha. Biochem J. 1989;263:539–545. doi: 10.1042/bj2630539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Radeke HH, Meier B, Topley N, Floge J, Habermehl GG, Resch K. Interleukin 1-alpha and tumor necrosis factor-alpha induce oxygen radical production in mesangial cells. Kidney Int. 1990;37:767–775. doi: 10.1038/ki.1990.44. [DOI] [PubMed] [Google Scholar]
- 42.Feng L, Xia Y, Garcia GE, Hwang D, Wilson CB. Involvement of reactive oxygen intermediates in cyclooxygenase-2 expression induced by interleukin-1, tumor necrosis factor-alpha, and lipopolysaccharide. J Clin Invest. 1995;95:1669–1675. doi: 10.1172/JCI117842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tao W, Dougherty R, Johnston P, Pickett W. Recombinant bovine GM-CSF primes superoxide production but not degranulation induced by recombinant bovine interleukin-1 beta in bovine neutrophils. J Leukoc Biol. 1993;53:679–684. doi: 10.1002/jlb.53.6.679. [DOI] [PubMed] [Google Scholar]
- 44.Nelson SK, Wong GH, McCord JM. Leukemia inhibitory factor and tumor necrosis factor induce manganese superoxide dismutase and protect rabbit hearts from reperfusion injury. J Mol Cell Cardiol. 1995;27:223–229. doi: 10.1016/s0022-2828(08)80021-1. [DOI] [PubMed] [Google Scholar]
- 45.Warner BB, Stuart L, Gebb S, Wispe JR. Redox regulation of manganese superoxide dismutase. Am J Physiol. 1996;271:L150–L158. doi: 10.1152/ajplung.1996.271.1.L150. [DOI] [PubMed] [Google Scholar]
- 46.Suzuki K, Tatsumi H, Satoh S, Senda T, Nakata T, Fujii J, Taniguchi N. Manganese-superoxide dismutase in endothelial cells: localization and mechanism of induction. Am J Physiol. 1993;265:H1173–H1178. doi: 10.1152/ajpheart.1993.265.4.H1173. [DOI] [PubMed] [Google Scholar]
- 47.Neta R, Oppenheim JJ, Schreiber RD, Chizzonite R, Ledney GD, MacVittie TJ. Role of cytokines (interleukin 1, tumor necrosis factor, and transforming growth factor b) in natural and lipopolysaccharide-enhanced radioresistance. J Exp Med. 1991;173:1177–1182. doi: 10.1084/jem.173.5.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]