

Abstract
Helicobacter pylori infection induces various gastroduodenal diseases. We examined the role of two genes, vacA and cagE, in the gastric pathogenesis induced by H. pylori using a long-term (62 wk) animal model. Reportedly, both genes are associated with the virulence of H. pylori: vacA encodes vacuolating cytotoxin, and cagE, with other genes in the cag pathogenicity islands, encodes a type IV secretion system. Mongolian gerbils were challenged in this study by a wild-type TN2 strain and its isogenic mutants of cagE or vacA. The wild-type and vacA mutants induced severe gastritis, whereas cagE mutants induced far milder changes. Gastric ulcer was induced at the highest rate (22/23) by the wild-type TN2, followed by the vacA mutant (19/28). No ulcer was found in the gerbils infected with the cagE mutant (0/27) or in controls (0/27). Intestinal metaplasia was also found in the gerbils infected with the wild-type (14/23) or vacA mutant (15/28). Gastric cancer developed in one gerbil with wild-type infection and in one with vacA mutant infection. In conclusion, the knocking out of the cagE gene deprived wild-type H. pylori of the pathogenicity for gastritis and gastric ulcer, suggesting that the secretion system encoded by cag pathogenicity island genes plays an essential role.
Keywords: gastritis, inflammation, mutagenesis, pathogenicity, animal experiments
Introduction
Helicobacter pylori is a gram-negative bacterium infecting human gastric mucosa with worldwide prevalence. This bacterium plays an important role in the pathogenesis of chronic gastritis, peptic ulcers, gastric adenocarcinomas, and gastric mucosa–associated lymphoid tissue lymphomas 1 2 3 4.
Several factors have been proposed as possible virulence determinants. Among them, the CagA protein, encoded by the cagA gene, is found in ∼50–60% of isolates in Western countries and is reportedly associated with advanced gastroduodenal diseases 5 6 7 8. CagA-positive strains reportedly induce IL-8 secretion in gastric epithelial cells 9. Moreover, recent studies showed that the cagA gene is a member of the cag pathogenicity island (PAI), a 40-kb region of supposedly extraneous origin 10. Isogenic mutant studies demonstrated that some cag PAI genes, including cagE, which was initially reported as picB, are essential for IL-8 induction and nuclear factor (NF)-κB activation 10 11 12 13 14. Recent studies indicated that cagE and several other genes of cag PAI code a type IV secretion system, which translocates CagA protein inside the gastric epithelial cells to be tyrosine phosphorylated by a yet unknown mechanism 15 16 17. Indeed, deletion of cagE was found to impair the translocation of CagA.
VacA is a polymorphic gene encoding vacuolating cytotoxin, and the strains producing active cytotoxin are usually cagA positive and toxigenic 18 19 20 21. The VacA protein causes vacuolation in cultured cells and is considered a cytotoxin 19. The strains that possess vacuolating activity are isolated from chronic gastritis and peptic ulcer patients at high frequency 22 23. A recent study showed that VacA alters the intracellular trafficking of proteins, increases the permeability of polarized epithelial cells, inhibits the process of antigen presentation, forms anion-selective channels in lipid bilayers, and interferes with cytoskeleton-dependent cell functions 24. However, in spite of various studies on VacA in vitro and in vivo, the exact role of VacA in gastric pathogenesis in vivo remains to be understood.
Animals used for studying experimental H. pylori infection include monkeys, dogs, piglets, domestic cats, and rodents 25 26 27 28 29 30 31. Several of the described models are not optimal because the animals can not be handled with ease or in large numbers, germ-free conditions are necessary, and infection rates are low. Infection of conventional mice with H. pylori overcomes many of problems, although infection is not easily achieved, and the gastritis in mice induced by H. pylori is less intense than in humans 32. The Mongolian gerbil is an animal in which various gastrointestinal diseases such as gastritis, ulcers, intestinal metaplasia, and gastric cancer can be appropriately developed 33 34 35. We previously reported that TN2GF4, which shares an ancestral strain with TN2, the wild-type (WT) strain used in this study, induced gastric cancer in gerbils without using adjuvant chemical carcinogens 34. It was also reported that isogenic mutants of vacA and cagA colonized well, allowing for examination of the role of virulence markers on gastric pathogenesis in Mongolian gerbils 36 37. We conducted this study to examine the pathological consequences of long-term infection with WT and mutant (ΔcagE, ΔvacA) H. pylori strains using specific pathogen–free Mongolian gerbils to evaluate the role played by those genes in gastric pathogenesis.
Materials and Methods
Bacterial Strains and Culture Conditions.
An H. pylori clinical isolate (TN2) and its two isogenic mutants (TN2ΔcagE and TN2ΔvacA) were used in this study. TN2, a strain positive for the vacuolating cytotoxin and cag PAI, shares an ancestral strain with TN2GF4, which we previously reported to induce gastric cancer in Mongolian gerbils 34. H. pylori cells were cultured in Brucella broth culture medium (Becton Dickinson) containing 2.5% FBS (Cansera International, Inc.) and Glaxo selective supplement A (10 mg/liter vancomycin, 3.3 mg/liter polymyxin B, 20 mg/liter bacitracin, 10.7 mg/liter nalidixic acid, and 5 mg/liter amphotericin B) in a microaerophilic atmosphere generated by CampyPak-Plus (Becton Dickinson) at 37°C for 24 h. Cultures were supplemented with sterilized glycerol at a final concentration of 15% and were stored at −80°C until use.
Construction of Isogenic cagE-negative Mutant (TN2ΔcagE) and vacA-negative Mutant (TN2ΔvacA).
The cagE mutant strain of TN2 was constructed as described previously 13. In brief, cagE gene was amplified using the long PCR method with primers HPPB-1F and HPPB-1R and then cloned into the plasmid vector pCRII (Invitrogen), named pCR/CE. The cloned cagE gene was disrupted by insertion of a kanamycin resistance gene (kmr) cassette at nucleotide position 212, which resulted in pCR/CE::km. DNA from plasmid pCR/CE::km was transferred into the TN2 strain by electroporation. The bacteria were recovered and plated onto nonselective plates and incubated for 24 h. Then, the bacteria were transferred onto agar plates with kanamycin. Kanamycin-resistant H. pylori colonies were screened for the allelic exchange by PCR, and the disruption in cagE gene was confirmed by Southern hybridization.
The vacA mutant of TN2 was also constructed as described 14, and disruption of the vacA gene was confirmed by means of Southern hybridization and PCR using primers vacA-C1 and vacA-C2 21. A loss of vacuolation activity was checked by use of a vacuolating assay.
Animals.
5-wk-old male specific pathogen–free Mongolian gerbils (MON/Jms/Gbs Slc) were purchased from Japan SLC, Inc. and maintained under standard laboratory conditions (room temperature, 23 ± 2°C; relative humidity 55 ± 5%; 12/12-h light/dark cycle) with free access to a commercial rodent diet (CE-2; Clea Japan) and tap water. All experimental protocols described were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals in the Pharmaceutical Research Division of Takeda Chemical Industries, Ltd. and approved by the Ethical Committee specializing in animal experiments for our division.
Adaptation for Infection.
Six animals were inoculated with broth culture containing 5.4 ± 0.7 × 107 CFUs of TN2 (WT), TN2ΔcagE, or TN2ΔvacA via intragastric gavage after fasting for 24 h. Animals were killed 2–3 wk after inoculation, and stomachs were used for quantitative culture. New gerbils were inoculated with recovered bacteria, and infecting bacteria were recovered 3 wk after inoculation. These steps were repeated three times, and the efficiency of infection was assessed at each stage.
Long-Term Infection Model.
125 animals were divided into four groups. WT groups consisted of 31 animals that were inoculated with 1 ml of H. pylori TN2 broth culture via intragastric gavage after fasting for 24 h. Both the ΔcagE and ΔvacA groups, each consisting of 31 animals, were challenged with TN2ΔcagE or TN2ΔvacA, respectively. Each inoculated broth culture contained 5.5 ± 0.5 × 107 CFUs of bacteria. 32 animals served as intact controls without the challenge of bacterium. The body weight of animals was checked every 3 mo. Animals were killed 62 wk after inoculation, and stomachs were divided into halves. One half was fixed in Carnoy's fixative for histological examination, and the other half was used for quantitative determination of the bacterium by culture.
Culture Study.
The number of the infecting bacteria in the stomach was measured as previously described 34. In brief, after the stomach was opened along the greater curvature, the longitudinal half was homogenized with physiological saline. Modified Skirrow's agar was inoculated with a diluted aliquot and incubated for 4 d at 37°C under microaerobic conditions. Colonies were identified as H. pylori by morphology and urease activity. The number of colonies per plate was counted and calculated as the log CFU per gastric wall. Genotyping by PCR and testing for kanamycin resistance of the recovered bacteria from three animals of each group were performed to confirm the stability of the mutation during infection.
Histological Examination.
The half of the stomach to be used for histopathological examination was stapled onto paper and fixed in Carnoy's fixative. Fixed gastric tissue was cut into longitudinal strips for histopathological examination. After processing for histology by routine methods, paraffin-embedded sections were cut and stained with hematoxylin and eosin (H&E) or with alcian blue, pH 2.5, periodic acid-Schiff (AB-PAS). Histopathological lesions of the glandular stomach were categorized as follows: 1 mild gastritis (gastritis with mild mononuclear cell infiltration and lymphoid follicle formation in the pyloric region); 2 severe gastritis (gastritis with dense neutrophil and mononuclear cell infiltrations, multiple lymphoid follicle formations, and hyperplastic epithelium throughout the pyloric region and a part of the fundic region); 3 ulcer; 4 intestinal metaplasia (diagnosed by the presence of goblet cells positive for AB-PAS staining); 5 adenocarcinoma (diagnosed by the presence of atypical glands locally invading the muscle layer and destroying the original architecture); and 6 carcinoid (diagnosed by neuroendocrine nests greater than three glands in diameter).
DNA Preparation and PCR.
Chromosomal DNA was isolated using SepaGene (Sanko Junyaku) from cultures of TN2, TN2ΔcagE, and TN2ΔvacA before and after three passages through gerbils and cultures isolated from each three animals after long-term infection with TN2, TN2ΔcagE, or TN2ΔvacA. PCR was carried out in 50-μl volumes containing 100 ng of genomic DNA, 0.5 U of AmpliTaq DNA polymerase (Applied Biosystems) or LA Taq (Takara Shuzo), 25 pmol of each primer, and 10 nmol of each deoxynucleoside triphosphate in a standard buffer. Cycling conditions were 30 cycles at 94°C for 15 s, 50°C for 15 s, and 72°C for a time dependent on the expected product size (1 min per kilobase). The PCR primers used in this study are shown in Table 38.
Table 1.
Primers Used in This Study
| Region | Primer | Nucleotide sequence |
|---|---|---|
| cagA | cagA-F | 5′-AACAGGACAAGTAGCTAGCC |
| cagA-R | 5′-GATAACAGGCAAGCTTTTGA | |
| cagE | HPPB-1F | 5′-TCTATAAAGAGAGGGGTGTT |
| HPPB-1R | 5′-GGCTAATCTTTGGTAATCAG | |
| cagF | cagF-F | 5′- ATGAAACAAATTTGCGTGA |
| cagF-R | 5′-TCAATCGTTATTTTTGTTTT | |
| cagH | cagH-F | 5′-ATGGCAGGTACACAAGCTAT |
| cagH-R | 5′-TCACTTCACGATTATTTTAG | |
| cagM | cagM-F | 5′-ATGCTTGCAAAAATTGTTTT |
| cagM-R | 5′-CTATTCAAAGGGATTATTCT | |
| cagT | cagT-F | 5′-CCATGTTTATACGCCTGTGT |
| cagT-R | 5′-CATCACCACACCCTTTTGAT | |
| left end of cag | cagLEC-F | 5′-ACATTTTGGCTAAATAAACGCTG |
| cagLEC-R1 | 5′-TCTCCATGTTGCCATTATGCT | |
| vacA | vacA-C1 | 5′-ATGGAAATACAACAAACACACCGCA |
| vacA-C2 | 5′-TTAGAAACTATACCTCATTCCTAAA | |
| HP0887-0888 | HP887F | 5′-TCATAGCGATTGGCGTGGAA |
| HP888R | 5′-GCTTGGAATTAGAGTCCTTA | |
| HP0888 | HP888F | 5′-ATGAACGCTGGAGTTTTACA |
| HP888R | 5′-GCTTGGAATTAGAGTCCTTA |
Reverse Transcription–PCR.
RNA was prepared from 24-h broth cultures of WT or mutant H. pylori strains using ISOGEN (NipponGene). A 10-μg sample of each RNA was treated with RNase-free DNase I (Roche Diagnostics) to remove contaminating DNA. We next prepared cDNA from a 2-μg aliquot of the DNase-treated RNA samples by use of Super Script II RNase H− Reverse Transcriptase (GIBCO BRL) and random hexamer oligonucleotide primers. The reaction mixtures were diluted to 20 μl with H2O, and 1 μl of each was used as a template for PCR with oligonucleotide primers specific for cagA, cagF, cagH, or HP0888 (Table ). DNase-treated RNA samples were also diluted at the same concentration and used as a template for PCR to confirm that DNA was removed from the RNA samples.
Statistical Methods.
The body weights of gerbils were compared by one-factor ANOVA (analysis of variance). The incidence of gastric lesion formation was compared by Fisher's exact probability test followed by the Holm's correction for multiple comparison. The differences were considered significant when the P value was <0.05.
Results
Colonization Efficiency of WT and Isogenic Mutant H. pylori Strains.
To assess colonization ability in the gerbil's stomach, gerbils were inoculated with WT or mutants of H. pylori and killed 2–3 wk after inoculation, and the stomachs were then cultured for H. pylori (Table ). Recovered bacteria were cultured in vitro and then inoculated in other animals up to the third stage of in vivo passage. The strain TN2ΔcagE did not achieve an efficient rate of infection during the first stage, whereas WT and TN2ΔvacA infected the gerbils without failure. However, the rate of successful infection improved after in vivo passage. Those strains recovered after three passages were used for the 62-wk long-term infection study.
Table 2.
Adaptation for Infection after Mutagenesis
| 1st stage | 2nd stage | 3rd stage | |
|---|---|---|---|
| TN2 (WT) | 6/6 (100) | 5/5 (100) | 5/5 (100) |
| TN2ΔcagE | 2/6 (33) | 5/5 (100) | 5/5 (100) |
| TN2ΔvacA | 6/6 (100) | 5/5 (100) | 5/5 (100) |
Number of infected animals/total number of challenged animals (percentage with infection) are shown.
Long-Term Infection of WT and Mutant Strains.
31 animals in each group were inoculated with TN2, TN2ΔvacA, or TN2ΔcagE, and 32 animals served as the intact control group. During the 62 wk after inoculation, eight, four, three, and five animals died in the WT, ΔcagE, ΔvacA, and control groups, respectively. The mean body weight of surviving animals at 62 wk was 66 g in the WT, 77 g in the ΔcagE, 76 g in the ΔvacA, and 90 g in the control group (Fig. 1). Concerning mortality, there were no significant differences between groups other than the body weight curves, which were significantly lower in the WT than in the mutant and control group. Culture and histological examination were performed in 23, 27, 28, and 27 animals of each group (WT, ΔcagE, ΔvacA, and control, respectively; Table ). H. pylori was recovered from all animals in the challenged groups except for one animal in the WT group, and H. pylori was not cultured from animals in the control group. Bacteria density (log CFU per gastric wall) was (mean ± SE) 5.48 ± 0.28, 5.53 ± 0.15, 5.53 ± 0.20, and zero in the WT, ΔcagE, ΔvacA, and control groups, respectively (Table ).
Figure 1.

Changes in body weight of Mongolian gerbils inoculated orally with H. pylori TN2 (○) and its isogenic vacA (▴) or cagE (□) mutants and intact control gerbils (♦). The data and error bars represent means and the standard error of body weights.
Table 3.
Bacterial Counts within the Gastric Wall of Mongolian Gerbils 62 wk after Oral Challenge of H. pylori
| Strain | Infection rate | Bacterial recovery | |
|---|---|---|---|
| n | Positive (%) | Log CFU per stomach | |
| TN2 (WT) | 23 | 22 (96) | 5.48 ± 0.28 |
| TN2ΔcagE | 27 | 27 (100) | 5.53 ± 0.15 |
| TN2ΔvacA | 28 | 28 (100) | 5.53 ± 0.20 |
| Intact control | 27 | 0 (0) | Not detected |
Histological Examination.
The histopathologic changes in the gastric mucosa of gerbils infected with H. pylori for 62 wk are shown in Fig. 2 Fig. 3 Fig. 4 and summarized in Table . Inflammation was significantly reduced in the gerbils infected with TN2ΔcagE compared with those infected with TN2 or TN2ΔvacA. However, no significant differences were found in histopathological changes induced by TN2 or TN2ΔvacA. Severe gastritis was observed in all animals (100%) infected with TN2 and in 26 out of 28 animals (93%) infected with TN2ΔvacA. In these animals, dense infiltration of neutrophils and mononuclear cells and multiple lymphoid follicle formation were observed in the lamina propria and submucosa throughout the pyloric region as well as the adjacent part of the fundic region (Fig. 2A, Fig. C, Fig. E, and Fig. G). The normal mucosal architecture was almost completely lost in these areas and replaced with hyperplastic epithelium. The mucosal layer was thickened remarkably, and pseudopyloric gland formation, cystic glandular dilatation, and invagination into the submucosa of hyperplastic glands were frequently noted. Animals infected with TN2ΔcagE, however, showed much milder gastritis (Fig. 2B and Fig. F) restricted to the pyloric region and characterized by mild mononuclear cell infiltration with lymphoid follicle formation. Neutrophil activity and epithelial changes were rarely seen. No evidence of gastritis was found in any of the control animals (Fig. 2D and Fig. H).
Figure 2.

Histopathological findings in the gastric mucosa of gerbils 62 wk after H. pylori inoculation. Gastritis (A–D, pyloric region; and E–H, fundic region): severe gastritis characterized by dense neutrophil and mononuclear cell infiltrations and epithelial hyperplasia can be seen in the animals infected with TN2 (A and E) and TN2ΔvacA (C and G). The gastritis extends throughout the pyloric region (A and C) and into a part of the fundic region (E and G). Mucosal thickness increases remarkably as compared with uninfected animals (D and H). Only mononuclear cell infiltration and lymphoid follicle formation are noted in the pyloric region of animals infected with TN2ΔcagE, and neutrophil infiltration and epithelial alterations are not evident (B). There was no inflammation in the fundic regions of animals infected with TN2ΔcagE (F). H&E stain; original magnifications 35×.
Figure 3.

Ulcers, extending to the muscular layer, developed in the region of the fundus–pylorus border of animals infected with TN2 (A) or TN2ΔvacA (C). No gastric ulcers are seen in animals infected with TN2ΔcagE (B) or uninfected animals (D). H&E stain; original magnifications 10×.
Figure 4.
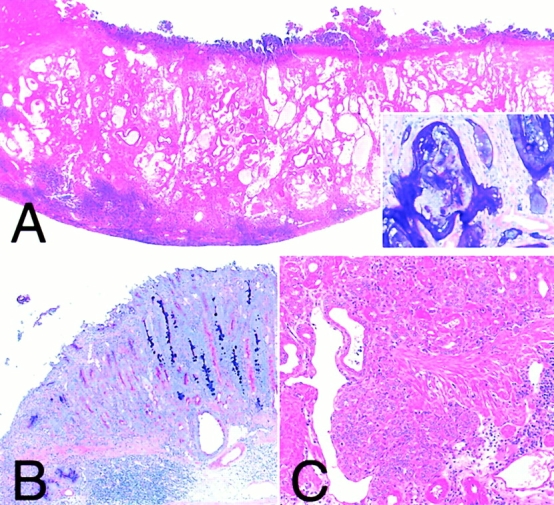
Other histopathological findings. Adenocarcinoma (A): an adenocarcinoma observed in animals infected with TN2. Neoplastic glands consisted of well-differentiated intestinal-type epithelium (inset) and penetrated into the muscle layer. Intestinal metaplasia (B): intestinal metaplasia observed in animals infected with TN2. Metaplastic epithelium containing AB-PAS–positive goblet cells frequently formed near the ulcer. Carcinoids (C): carcinoids detected in the animal infected with TN2ΔvacA. A and C, H&E stain; original magnifications 15 (A) and 80× (C). A, inset, and B, AB-PAS: original magnifications 80 (A, inset) and 30× (B).
Table 4.
Histological Findings in the Stomachs of Gerbils 62 wk after Oral Challenge of H. pylori
| Strain: | TN2 (WT) | TN2ΔcagE | TN2ΔvacA | Intact control |
|---|---|---|---|---|
| No. of animals: | 23 | 27 | 28 | 27 |
| Gastritis | 23 (100) | 27 (100) | 28 (100) | 0 (0)* |
| (Severe) | 23 (100) | 0 (0)* | 26 (93) | 0 (0)* |
| Gastric ulcer | 22 (96) | 0 (0)* | 19 (68)** | 0 (0)* |
| Intestinal metaplasia | 14 (61) | 0 (0)* | 15 (54) | 0 (0)* |
| Adenocarcinoma | 1 (4) | 0 (0) | 1 (4) | 0 (0) |
| Carcinoid | 1 (4) | 0 (0) | 5 (18) | 0 (0) |
Rates are shown as percentages in parentheses. Significant differences against WT infection by means of Fisher's exact probability are indicated as the following: *P < 0.0001 and **P = 0.013.
Gastric ulcers were found in 22 out of 23 animals (96%) with TN2 and in 19 out of 28 animals (68%) with TN2ΔvacA. Ulcers were localized in the region of the fundus–pylorus border, and all ulcer lesions reached the submucosa or the proper muscular layer (Fig. 3A and Fig. C). No gastric ulcers were found in animals with TN2ΔcagE or animals of the control group (Fig. 3B and Fig. D). The prevalence of gastric ulcers was significantly higher in animals challenged with TN2 or TN2ΔvacA than in animals with TN2ΔcagE or in controls (P < 0.0001 for all pairs). In the ΔvacA group, the number of animals suffering from gastric ulcers was reduced significantly compared with the WT group (P = 0.013). Intestinal metaplasia, formed mainly near the ulcer (Fig. 4 B), was found in 14 out of 23 (61%), zero out of 27 (0%), 15 out of 28 (54%), and zero out of 27 (0%) animals in the WT, ΔcagE, ΔvacA, and control group, respectively.
An adenocarcinoma developed in two animals, one in the WT group and another in the ΔvacA group. Both tumors were located in the pyloric region, grew downward, and penetrated into the muscle layer (Fig. 4 A). The tumors retained a well-preserved glandular structure and consisted of well-differentiated columnar intestinal-type epithelium. In addition, carcinoids derived from the fundic mucosa were detected in one animal (4%) in the WT group and five animals (18%) in the ΔvacA group. These carcinoids were arranged in solid sheets or small nests in the deep lamina propria and occasionally spread into the submucosa (Fig. 4 C). Neither adenocarcinomas nor carcinoids were found in the gerbils in the ΔcagE group and control group. The incidence of gastric cancer and carcinoid did not differ significantly among the groups.
Genomic Stability of H. pylori through Infection Study.
The stability of the H. pylori genome through in vivo passages was examined for the intended mutation and for the cag PAI. The disruption of the cagE and vacA genes in recovered bacteria from three animals of each infected group was confirmed as stable based on the size of the amplicons by PCR as compared with the original mutants. All recovered mutants were resistant to kanamycin, indicating that the kanamycin cassette still worked after 62 wk of infection (data not shown). The presence of five regions (cagA, cagF, cagM, cagT, and cagLEC) in cag PAI was examined by PCR. All cultures examined revealed positive amplification for the five regions (data not shown).
Detection of Gene Expression by Reverse Transcription–PCR.
To investigate whether insertion of an antibiotic resistance cassette into cagE or vacA had polar effects on expression of the flanking genes located downstream of insertion, we performed reverse transcription (RT)-PCR on RNA samples prepared from WT strain (TN2), cagE mutant, and vacA mutant. Control PCR experiments verified that DNA was efficiently removed from RNA samples by DNase treatment. We then applied cDNA prepared from these samples to PCR experiments with oligonucleotide primers specific for the cagA, cagF, cagH, and HP0888 genes (Table ). A primer pair for the cagA gene, used as positive control of RT-PCR, produced amplicons of expected size in these strains to similar extent, indicating that cDNA was properly prepared from each sample. The RT-PCR product of the cagF or cagH gene was also amplified in TN2ΔcagE as well as in TN2, indicating that the kmr cassette insertion into cagE had no polar effect on transcription of cagF or cagH. This finding agrees with the previous report with Northern blot showing that cagE (picB) was cotranscribed with only picA, which is located upstream of cagE 12. Transcription of the HP0888 gene, which is located downstream of vacA (HP0887) in strain 26695, was also analyzed by RT-PCR 39. The primer pair encompassing the vacA gene and HP0888 amplified the expected product in the TN2 genome, indicating that the HP0888 gene is located downstream of vacA in TN2 genome as in that of the 26695 genome. RT-PCR analysis showed no difference in the transcription of HP0888 between TN2 and TN2ΔvacA, suggesting that the insertion of the kmr cassette into vacA gene did not affect the transcription of HP0888. This result is consistent with the previous report that vacA gene is most likely transcribed alone 40.
Discussion
In this study, the role of virulence factors in the pathogenesis of H. pylori infection was investigated using Mongolian gerbils as an in vivo animal model. For this purpose, targeted genes were disrupted by homologous recombination, and the pathogenicity was compared in WT strains and isogenic mutants. All gerbils except one were infected with WT and mutant strains throughout this long-term study. Disruption of the genes did not affect the ability of H. pylori to colonize and replicate in the gerbil's stomach. Animals in every group survived for 62 wk, with only a few deaths, and the survival rate of each group did not differ significantly. However, gains in body weight differed significantly.
In humans, the effect of H. pylori infection on nutritional status of the host remains controversial. Reportedly, sustained H. pylori infection is associated with growth failure in Gambian infants 41. Fall et al. 42 also reported that in England, low weight at 1 y of age was associated with increased H. pylori seropositivity rates in males, although they did not decide whether H. pylori infection was the cause of growth failure or whether small infants were more susceptible to infection. On the other hand, there were reports from Bangladesh, Guatemala, and Nicaragua that found no association between H. pylori infection and malnutrition in children or infants 43 44 45. Quinonez et al. 44 concluded that the small differences in the nutritional status between the infected and uninfected children may be due to sociodemographic factors. The results of this study show that experimental infection with H. pylori, especially the WT, has led to a significant reduction in body weight 3 mo after inoculations. This infection model is purely experimental and, in design, is not affected by factors other than infection. Therefore, our observations indicate that infection with H. pylori may interfere with normal growth in Mongolian gerbils and further suggests that the same can occur in humans.
The colonization ability of H. pylori strains, especially TN2ΔcagE, was improved after the three passages through gerbils. It is known that the cag PAI often is lost during human infection, and it is discussed that strains that lost cag PAI may grow better than the cag PAI–positive parental strains in a subset of humans 46. To characterize the isolates obtained after the three passages through gerbils and the isolates after long-term infection in comparison with the pre-challenge strains, we examined the existence of genes in cag PAI by PCR. No major deletion of cag PAI was found, and it is possible that cag PAI does not affect adaptational capacity for infection in gerbils.
Deletions of the cag PAI and several cag insertion mutations reportedly block the induction of proinflammatory cytokine IL-8 in gastric epithelial cells in vitro 10 13 47. Recently, reports have noted the presence of cag PAI in association with H. pylori–mediated NF-κB activation in gastric epithelial cells, which is a critical regulator of the genes involved in inflammation, proliferation, and apoptosis 14 48. The proteins encoded by genes located in cag PAI are suggested to function as secretion machinery exporting molecules possibly involved in H. pylori–host cell interaction 10 49. In addition, CagA protein is noted to be translocated into gastric cells via cag PAI system and to be phosphorylated 15 16 17. The cagE gene is a homologue of the transporter component in Agrobacterium tumefaciens and Bordetella pertussis that engages in the transcellular transport of molecules 12. In this study, the role of the cagE gene product was assessed using gene disruption in an animal model suitable for the assessment of the pathogenesis of H. pylori infection. CagE knockout mutants, which possibly have a defect in the secretion apparatus, could not cause severe gastritis or induce the metaplastic changes often followed by the development of cancer 50. The WT and vacA-disrupted strains, however, induced severe gastritis and metaplasia in the gastric mucosa of gerbils, indicating the role of CagE protein in the pathogenesis of inflamed gastric mucosa. Moreover, it is tempting to speculate that the product of the cagE gene, one of the functional genes in cag PAI, may play a crucial role in the carcinogenesis that accompanies H. pylori infection. Reportedly, other genes in cag PAI are also required in host–bacterial interactions in vitro; however, their in vivo role was not assessed in this study. Further investigations are necessary to elucidate the roles of other cag PAI genes in vivo.
The VacA protein has been regarded as a potential candidate responsible for the virulence of H. pylori as well as the cause of inflammation and ulcer formation in the stomach and duodenum 22 23 24 51. However, as the VacA-positive strains often have intact cag PAI 52 53, the responsibility of VacA in inducing gastritis and ulcer formation is still controversial. Oral administration of the purified cytotoxin caused ulceration in mice 51, and recent investigations revealed the pathogenic effect of VacA in epithelial cells. VacA inhibited the conversion of procathepsin D and the intracellular degeneration of epidermal growth factor 54 and also interfered with cytoskeleton-dependent cell functions and the transmission of signals related to cell spreading and growth 24. On the other hand, Ghiara et al. 55 suggest that VacA does not play an important role in eliciting inflammation using a mouse model with sonic extracts of H. pylori. Weel et al. 7 also reported that their analysis of clinical isolates showed an association of peptic ulcer disease with only CagA, not VacA.
In our study, Mongolian gerbils were challenged with the vacA knockout strain to assess the role played by vacuolating activity in pathogenesis in vivo. The vacA-disrupted strain caused inflammatory changes in the stomachs of gerbils to a degree similar to that in the WT, indicating that VacA or the vacuolating activity of bacteria is not essential for the inflammatory response of gastric mucosa. Gastric ulcer was induced in 96% of the animals infected with the WT strain, 68% of those infected with TN2ΔvacA, and in none of those infected with TN2ΔcagE. This result also indicates the important role, direct or indirect, of the cagE gene product in the pathogenesis of gastric ulcers. On the other hand, the disruption of vacA gene reduced the incidence of gastric ulcers significantly, although gastric ulcers were observed nevertheless in more than half of the animals in the ΔvacA group. These results suggest that vacA is not involved in the inflammation of gastric mucosa but may be partially responsible for gastric ulcer formation. However, there is a possibility that VacA is not directly involved in ulcerogenesis but still worsens gastric ulcer or delays its healing by degenerating epidermal growth factor 54 or interfering with cytoskeleton-dependent cell function 24.
In our previous study, 37% of gerbils contracted gastric cancer 62 wk after inoculation with the same WT H. pylori strain, TN2. In this study, two gerbils (one from the WT group and another from the ΔvacA group) developed gastric cancer, and none from the ΔcagE or control group did. Thus, the incidence of cancer development (4%) in this study was smaller than that reported in our previous study 34. Possibly, employment of a different breeder (Japan SLC, Inc.) of Mongolian gerbils (MON/Jms/Gbs Slc) in this study caused the difference in the incidence of cancer. Intestinal metaplasia also developed in fewer animals with WT TN2 (61%) than in our previous study (85%) when using the same time interval of 62 wk after inoculation. Therefore, a possible reason for the low incidence of cancer in this study may be the slow progression of chronic changes in the gastric mucosa as metaplasia due to genetic diversity of the host or unexpected differences in environmental factors. In fact, another long-term (∼64 wk) experiment using 28 gerbils with TN2 infection confirmed the incidence of gastric adenocarcinoma as 21% (data not shown), with gerbils (MGS/Sea) purchased from the same breeder (Seiwa Experimental Animals) that was used in the previous study. Both breeders provided us with the descendants of 20 pairs of Mongolian gerbils captured at the Amur Basin in 1935. However, the ancestries of MGS/Sea diverged in 1973 and were separately inbred since that time, and those of MON/Jms/Gbs Slc diverged in 1979 and were inbred. Therefore, it is possible that there is a genetic diversity between the MGS/Sea and MON/Jms/Gbs Slc gerbils that may affect the inflammatory response as well as the process of carcinogenesis. Thus, it will be extremely interesting to further study the relation of gastric pathogenesis with genetic diversity of the host (Mongolian gerbil) and that of the infecting agent (H. pylori) and may eventually reveal the underlying mechanism of gastric carcinogenesis.
In conclusion, we showed in this study that cagE is responsible for various gastric lesions induced in the gerbils model, and in the future, this experimental animal model may help to elucidate the virulence factors responsible for gastric carcinogenesis. It has also been suggested that cag PAI may play an important role in the pathogenesis of gastric diseases related to H. pylori infection also in humans.
Acknowledgments
This study was supported in part by a Grant-in-aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan (11557040).
Footnotes
Abbreviations used in this paper: NF, nuclear factor; PAI, pathogenicity island; RT, reverse transcription; WT, wild-type.
References
- Graham D.Y., Lew G.M., Klein P.D., Evans D.G., Evans D.J.J., Saeed Z.A., Malaty H.M. Effect of treatment of Helicobacter pylori infection on the long-term recurrence of gastric or duodenal ulcer. A randomized, controlled study. Ann. Intern. Med. 1992;116:705–708. doi: 10.7326/0003-4819-116-9-705. [DOI] [PubMed] [Google Scholar]
- Parsonnet J., Friedman G.D., Vandersteen D.P., Chang Y., Vogelman J.H., Orentreich N., Sibley R.K. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 1991;325:1127–1131. doi: 10.1056/NEJM199110173251603. [DOI] [PubMed] [Google Scholar]
- Nomura A., Stemmermann G.N., Chyou P.H., Kato I., Perez P.G., Blaser M.J. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N. Engl. J. Med. 1991;325:1132–1136. doi: 10.1056/NEJM199110173251604. [DOI] [PubMed] [Google Scholar]
- Forman D., Newell D.G., Fullerton F., Yarnell J.W., Stacey A.R., Wald N., Sitas F. Association between infection with Helicobacter pylori and risk of gastric cancerevidence from a prospective investigation. Br. Med. J. 1991;302:1302–1305. doi: 10.1136/bmj.302.6788.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covacci A., Censini S., Bugnoli M., Petracca R., Burroni D., Macchia G., Massone A., Papini E., Xiang Z., Figura N. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. USA. 1993;90:5791–5795. doi: 10.1073/pnas.90.12.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummuru M.K., Cover T.L., Blaser M.J. Cloning and expression of a high-molecular-mass major antigen of Helicobacter pylorievidence of linkage to cytotoxin production. Infect. Immun. 1993;61:1799–1809. doi: 10.1128/iai.61.5.1799-1809.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weel J.F., van der Hulst R.W., Gerrits Y., Roorda P., Feller M., Dankert J., Tytgat G.N., van der Ende A. The interrelationship between cytotoxin-associated gene A, vacuolating cytotoxin, and Helicobacter pylori-related diseases. J. Infect. Dis. 1996;173:1171–1175. doi: 10.1093/infdis/173.5.1171. [DOI] [PubMed] [Google Scholar]
- Blaser M.J., Perez-Perez G.I., Kleanthous H., Cover T.L., Peek R.M., Chyou P.H., Stemmermann G.N., Nomura A. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer. Res. 1995;55:2111–2115. [PubMed] [Google Scholar]
- Crabtree J.E., Farmery S.M. Helicobacter pylori and gastric mucosal cytokinesevidence that CagA-positive strains are more virulent. Lab. Invest. 1995;73:742–745. [PubMed] [Google Scholar]
- Censini S., Lange C., Xiang Z., Crabtree J.E., Ghiara P., Borodovsky M., Rappuoli R., Covacci A. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. USA. 1996;93:14648–14653. doi: 10.1073/pnas.93.25.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.D., Kersulyte D., Lindley I.J., Neelam B., Berg D.E., Crabtree J.E. Multiple genes in the left half of the cag pathogenicity island of Helicobacter pylori are required for tyrosine kinase-dependent transcription of interleukin-8 in gastric epithelial cells. Infect. Immun. 1999;67:3893–3899. doi: 10.1128/iai.67.8.3893-3899.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummuru M., Sharma S., Blaser M. Helicobacter pylori picB, a homologue of the Bordetella pertussis toxin secretion protein, is required for induction of IL-8 in gastric epithelial cells. Mol. Microbiol. 1995;18:867–876. doi: 10.1111/j.1365-2958.1995.18050867.x. [DOI] [PubMed] [Google Scholar]
- Ogura K., Takahashi M., Maeda S., Ikenoue T., Kanai F., Yoshida H., Shiratori Y., Mori K., Mafune K.I., Omata M. Interleukin-8 production in primary cultures of human gastric epithelial cells induced by Helicobacter pylori . Dis. Dis. Sci. 1998;43:2738–2743. doi: 10.1023/a:1026671815512. [DOI] [PubMed] [Google Scholar]
- Maeda S., Yoshida H., Ogura K., Mitsuno Y., Hirata Y., Yamaji Y., Akanuma M., Shiratori Y., Omata M. Helicobacter pylori activates NF-κB through a signaling pathway involving IκB kinases, NF-κB-inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology. 2000;119:97–108. doi: 10.1053/gast.2000.8540. [DOI] [PubMed] [Google Scholar]
- Asahi M., Azuma T., Ito S., Ito Y., Suto H., Nagai Y., Tsubokawa M., Tohyama Y., Maeda S., Omata M. Helicobacter pylori CagA protein can be tyrosine phosphorylated in gastric epithelial cells. J. Exp. Med. 2000;191:593–602. doi: 10.1084/jem.191.4.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M., Rappuoli R., Covacci C. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc. Natl. Acad. Sci. USA. 2000;97:1263–1268. doi: 10.1073/pnas.97.3.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odenbreit S., Puls J., Sedlmaier B., Gerland E., Fischer W., Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- Leunk R.D., Johnson P.T., David B.C., Kraft W.G., Morgan D.R. Cytotoxic activity in broth-culture filtrates of Campylobacter pylori . J. Med. Microbiol. 1988;26:93–99. doi: 10.1099/00222615-26-2-93. [DOI] [PubMed] [Google Scholar]
- Cover T.L., Tummuru M.K., Cao P., Thompson S.A., Blaser M.J. Divergence of genetic sequences for the vacuolating cytotoxin among Helicobacter pylori strains. J. Biol. Chem. 1994;269:10566–10573. [PubMed] [Google Scholar]
- Atherton J.C., Cao P., Peek R.M.J., Tummuru M.K., Blaser M.J., Cover T.L. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J. Biol. Chem. 1995;270:17771–17777. doi: 10.1074/jbc.270.30.17771. [DOI] [PubMed] [Google Scholar]
- Ogura K., Kanai F., Maeda S., Yoshida H., Ogura M., Lan K., Hirota K., Kawabe T., Shiratori Y., Omata M. High prevalence of cytotoxin positive Helicobacter pylori in patients unrelated to the presence of peptic ulcers in Japan. Gut. 1997;41:463–468. doi: 10.1136/gut.41.4.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox J.G., Correa P., Taylor N.S., Thompson N., Fontham E., Janney F., Sobhan M., Ruiz B., Hunter F. High prevalence and persistence of cytotoxin-positive Helicobacter pylori strains in a population with high prevalence of atrophic gastritis. Am. J. Gastroenterol. 1992;87:1554–1560. [PubMed] [Google Scholar]
- Tee W., Lambert J.R., Dwyer B. Cytotoxin production by Helicobacter pylori from patients with upper gastrointestinal tract diseases. J. Clin. Microbiol. 1995;33:1203–1205. doi: 10.1128/jcm.33.5.1203-1205.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai R., Cover T.L., Tarnawski A.S. Helicobacter pylori vacuolating cytotoxin (VacA) disorganizes the cytoskeletal architecture of gastric epithelial cells. Biochem. Biophys. Res. Commun. 1999;262:245–250. doi: 10.1006/bbrc.1999.1194. [DOI] [PubMed] [Google Scholar]
- Dubois A., Berg D.E., Incecik E.T., Fiala N., Heman-Ackah L.M., Perez-Perez G.I., Blaser M.J. Transient and persistent experimental infection of nonhuman primates with Helicobacter pyloriimplications for human disease. Infect. Immun. 1996;64:2885–2891. doi: 10.1128/iai.64.8.2885-2891.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois A., Fiala N., Heman-Ackah L.M., Drazek E.S., Tarnawski A., Fishbein W.N., Perez-Perez G.I., Blaser M.J. Natural gastric infection with Helicobacter pylori in monkeysa model for spiral bacteria infection in humans. Gastroenterology. 1994;106:1405–1417. doi: 10.1016/0016-5085(94)90392-1. [DOI] [PubMed] [Google Scholar]
- Fox J.G., Batchelder M., Marini R., Yan L., Handt L., Li X., Shames B., Hayward A., Campbell J., Murphy J.C. Helicobacter pylori-induced gastritis in the domestic cat. Infect. Immun. 1995;63:2674–2681. doi: 10.1128/iai.63.7.2674-2681.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karita M., Li Q., Cantero D., Okita K. Establishment of a small animal model for human Helicobacter pylori infection using germ-free mouse. Am. J. Gastroenterol. 1994;89:208–213. [PubMed] [Google Scholar]
- Lambert J.R., Borromeo M., Pinkard K.J., Turner H., Chapman C.B., Smith M.L. Colonization of gnotobiotic piglets with Campylobacter Pyloridis—an animal model? J. Infect. Dis. 1987;155:1344. doi: 10.1093/infdis/155.6.1344. [DOI] [PubMed] [Google Scholar]
- Radin M.J., Eaton K.A., Krakowka S., Morgan D.R., Lee A., Otto G., Fox J. Helicobacter pylori gastric infection in gnotobiotic beagle dogs. Infect. Immun. 1990;58:2606–2612. doi: 10.1128/iai.58.8.2606-2612.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuto R., Fujioka T., Kubota T., Nasu M. Experimental gastritis induced by Helicobacter pylori in Japanese monkeys. Infect. Immun. 1993;61:933–939. doi: 10.1128/iai.61.3.933-939.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti M., Arico B., Burroni D., Figura N., Rappuoli R., Ghiara P. Development of a mouse model of Helicobacter pylori infection that mimics human disease. Science. 1995;267:1655–1658. doi: 10.1126/science.7886456. [DOI] [PubMed] [Google Scholar]
- Hirayama F., Takagi S., Kusuhara H., Iwao E., Yokoyama Y., Ikeda Y. Induction of gastric ulcer and intestinal metaplasia in Mongolian gerbils infected with Helicobacter pylori . J. Gastroenterol. 1996;31:755–757. doi: 10.1007/BF02347631. [DOI] [PubMed] [Google Scholar]
- Watanabe T., Tada M., Nagai H., Sasaki S., Nakao M. Helicobacter pylori infection induces gastric cancer in Mongolian gerbils. Gastroenterology. 1998;115:642–648. doi: 10.1016/s0016-5085(98)70143-x. [DOI] [PubMed] [Google Scholar]
- Ikeno T., Ota H., Sugiyama A., Ishida K., Katsuyama T., Genta R.M., Kawasaki S. Helicobacter pylori-induced chronic active gastritis, intestinal metaplasia, and gastric ulcer in Mongolian gerbils. Am. J. Pathol. 1999;154:951–960. doi: 10.1016/S0002-9440(10)65343-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth H.P., Beins M.H., Yang M., Tham K.T., Blaser M.J. Experimental infection of Mongolian gerbils with wild-type and mutant Helicobacter pylori strains. Infect. Immun. 1998;66:4856–4866. doi: 10.1128/iai.66.10.4856-4866.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek R.M., Jr., Wirth H.P., Moss S.F., Yang M., Abdalla A.M., Tham K.T., Zhang T., Tang L.H., Modlin I.M., Blaser M.J. Helicobacter pylori alters gastric epithelial cell cycle events and gastrin secretion in Mongolian gerbils. Gastroenterology. 2000;118:48–59. doi: 10.1016/s0016-5085(00)70413-6. [DOI] [PubMed] [Google Scholar]
- Maeda S., Yoshida H., Ikenoue T., Ogura K., Kanai F., Kato N., Shiratori Y., Omata M. Structure of cag pathogenicity island in Japanese Helicobacter pylori isolates. Gut. 1999;44:336–341. doi: 10.1136/gut.44.3.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomb J.F., White O., Kerlavage A.R., Clayton R.A., Sutton G.G., Fleischmann R.D., Ketchum K.A., Klenk H.P., Gill S., Dougherty B.A. The complete genome sequence of the gastric pathogen Helicobacter pylori . Nature. 1997;338:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- Phadnis S.H., Ilver D., Janzon L., Normark S., Westblom T.U. Pathological significance and molecular characterization of the vacuolating cytotoxin gene of Helicobacter pylori . Infect. Immun. 1994;62:1557–1565. doi: 10.1128/iai.62.5.1557-1565.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale A., Thomas J.E., Darboe M.K., Coward W.A., Harding M., Weaver L.T. Helicobacter pylori infection, gastric acid secretion, and infant growth. J. Pediatr. Gastroenterol. Nutr. 1998;26:393–397. doi: 10.1097/00005176-199804000-00006. [DOI] [PubMed] [Google Scholar]
- Fall C.H., Goggin P.M., Hawtin P., Fine D., Duggleby S. Growth in infancy, infant feeding, childhood living conditions, and Helicobacter pylori infection at age 70. Arch. Dis. Child. 1997;77:310–314. doi: 10.1136/adc.77.4.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens J., Albert M.J., Rao M., Huda S., Qadri F., Van Loon F.P., Pradhan B., Naficy A., Banik A. Sociodemographic, hygienic and nutritional correlates of Helicobacter pylori infection of young Bangladeshi children. Pediatr. Infect. Dis. J. 1996;15:1113–1118. doi: 10.1097/00006454-199612000-00012. [DOI] [PubMed] [Google Scholar]
- Quinonez J.M., Chew F., Torres O., Begue R.E. Nutritional status of Helicobacter pylori-infected children in Guatemala as compared with uninfected peers. Am. J. Trop. Med. Hyg. 1999;61:395–398. doi: 10.4269/ajtmh.1999.61.395. [DOI] [PubMed] [Google Scholar]
- Kehrt R., Becker M., Brosicke H., Kruger N., Helge H. Prevalence of Helicobacter pylori infection in Nicaraguan children with persistent diarrhea, diagnosed by the 13C-urea breath test. J. Pediatr. Gastroenterol. Nutr. 1997;25:84–88. doi: 10.1097/00005176-199707000-00014. [DOI] [PubMed] [Google Scholar]
- Kersulyte D., Chalkauskas H., Berg D.E. Emergence of recombinant strains of Helicobacter pylori during human infection. Mol. Microbiol. 1999;31:31–43. doi: 10.1046/j.1365-2958.1999.01140.x. [DOI] [PubMed] [Google Scholar]
- Akopyants N.S., Clifton S.W., Kersulyte D., Crabtree J.E., Youree B.E., Reece C.A., Bukanov N.O., Drazek E.S., Roe B.A., Berg D.E. Analyses of the cag pathogenicity island of Helicobacter pylori . Mol. Microbiol. 1998;28:37–53. doi: 10.1046/j.1365-2958.1998.00770.x. [DOI] [PubMed] [Google Scholar]
- Glocker E., Lange C., Covacci A., Bereswill S., Kist M., Pahl H.L. Proteins encoded by the cag pathogenicity island of Helicobacter pylori are required for NF-kappaB activation. Infect. Immun. 1998;66:2346–2348. doi: 10.1128/iai.66.5.2346-2348.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covacci A., Telfold J.L., Giudice G.D., Parsonnet J., Rappuoli R. Helicobacter pylori virulence and genetic geography. Science. 1999;284:1328–1333. doi: 10.1126/science.284.5418.1328. [DOI] [PubMed] [Google Scholar]
- Kuipers E.J. Review articleexploring the link between Helicobacter pylori and gastric cancer Aliment. Pharmacol. Ther. 13Suppl. 11999. 3 11 [DOI] [PubMed] [Google Scholar]
- Telford J.L., Ghiara P., Dell'Orco M., Comanducci M., Burroni D., Bugnoli M., Tecce M.F., Censini S., Covacci A., Xiang Z. Gene structure of the Helicobacter pylori cytotoxin and evidence of its key role in gastric disease. J. Exp. Med. 1994;179:1653–1658. doi: 10.1084/jem.179.5.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z., Censini S., Bayeli P.F., Telford J.L., Figura N., Rappuoli R., Covacci A. Analysis of expression of CagA and VacA virulence factors in 43 strains of Helicobacter pylori reveals that clinical isolates can be divided into two major types and that CagA is not necessary for expression of the vacuolating cytotoxin. Infect. Immun. 1995;63:94–98. doi: 10.1128/iai.63.1.94-98.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S., Ogura K., Yoshida H., Kanai F., Ikenoue T., Kato N., Shiratori Y., Omata M. Major virulence factors, VacA and CagA, are commonly positive in Helicobacter pylori isolates in Japan. Gut. 1998;42:338–343. doi: 10.1136/gut.42.3.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satin B., Norais N., Telford J., Rappuoli R., Murgia M., Montecucco C., Papini E. Effect of Helicobacter pylori vacuolating toxin on maturation and extracellular release of procathepsin D and on epidermal growth factor degradation. J. Biol. Chem. 1997;272:25022–25028. doi: 10.1074/jbc.272.40.25022. [DOI] [PubMed] [Google Scholar]
- Ghiara P., Marchetti M., Blaser M.J., Tummuru M.K., Cover T.L., Segal E.D., Tompkins L.S., Rappuoli R. Role of the Helicobacter pylori virulence factors vacuolating cytotoxin, CagA, and urease in a mouse model of disease. Infect. Immun. 1995;63:4154–4160. doi: 10.1128/iai.63.10.4154-4160.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]