

Abstract
Inactivation of the retinoblastoma tumor suppressor protein (pRb) has been implicated in melanoma cells, but the molecular basis for this phenotype has not yet been elucidated, and the status of additional family members (p107 and p130, together termed pocket proteins) or the consequences on downstream targets such as E2F transcription factors are not known. Because cell cycle progression is dependent on the transcriptional activity of E2F family members (E2F1–E2F6), most of them regulated by suppressive association with pocket proteins, we characterized E2F–pocket protein DNA binding activity in normal versus malignant human melanocytes. By gel shift analysis, we show that in mitogen-dependent normal melanocytes, external growth factors tightly controlled the levels of growth-promoting free E2F DNA binding activity, composed largely of E2F2 and E2F4, and the growth-suppressive E2F4–p130 complexes. In contrast, in melanoma cells, free E2F DNA binding activity (E2F2 and E2F4, to a lesser extent E2F1, E2F3, and occasionally E2F5), was constitutively maintained at high levels independently of external melanocyte mitogens. E2F1 was the only family member more abundant in the melanoma cells compared with normal melanocytes, and the approximately fivefold increase in DNA binding activity could be accounted for mostly by a similar increase in the levels of the dimerization partner DP1. The continuous high expression of cyclin D1, A2, and E, the persistent cyclin-dependent kinase 4 (CDK4) and CDK2 activities, and the presence of hyperphosphorylated forms of pRb, p107, and p130, suggest that melanoma cells acquired the capacity for autonomous growth through inactivation of all three pocket proteins and release of E2F activity, otherwise tightly regulated in normal melanocytes by external growth factors.
Keywords: melanocytes, cyclins, pRb, p107, p130
Introduction
In vitro, malignant melanocytes from advanced primary or metastatic lesions can be defined as cells that have escaped dependency on external growth factors acutely required by normal melanocytes, such as fibroblast growth factors (FGFs) (in particular basic FGF, also known as FGF2), hepatocyte growth factor/scatter factor (HGF/SF), mast/stem cell growth factor (M/SCF), and endothelins (ET-1–ET-3; for reviews, see references 1 2 3 4). Although the molecular basis for this aberrant melanoma cell phenotype is poorly characterized, inactivation of retinoblastoma tumor suppressor protein (pRb) has been implicated, because pRb is constitutively hyperphosphorylated or expressed at extremely low levels in melanoma cells 5 6. The pRb family of proteins (pRb, p107, and p130, termed pocket proteins) are regarded as master regulators of the cell cycle governing the restriction (R) point, i.e., the point of commitment from quiescence (G0/G1) to DNA synthesis (S) 7 8 9 10. In large part, under normal conditions, growth factors mediate transit through the G1 phase of the cell cycle by activating a group of cyclin-dependent kinases (CDKs), mainly CDK2, CDK4, and CDK6, responsible for phosphorylating and inactivating the pocket proteins 11 12 13 14. The CDK holoenzymes, composed of the kinase subunit in association with the positive partner cyclins (cyclin A, cyclin D, and cyclin E), are negatively regulated by cyclin-dependent kinase inhibitors (CKIs) from the inhibitor of CDK (INK) and Kip/Cip family, and by site-specific phosphorylation or dephosphorylation carried out by the CDK activating kinase (CAK) and Cdc25 phosphatase, respectively (for reviews, see references 15 16).
Hyperphosphorylation of pocket proteins releases their suppressive association with five transcription factors from the E2F family (E2F1–E2F5), leading to the accumulation of E2F transcriptional activity and upregulation of genes responsible for cell cycle progression (cyclin A, cyclin D1, cyclin E, cdc2, p107, and p21CIP), DNA synthesis (dihydrofolate reductase and DNA polymerase α), and transcription factors c-MYC, c-MYB, B-MYB, as well as E2F1 and E2F2, that participate in the induction of early and late response genes 10 17 18 19 20. The sixth E2F member, E2F6, does not associate with pocket proteins, but by itself can act as a transcriptional repressor of E2F responsive genes, and can arrest entry into the S phase of the cell cycle when overexpressed 21 22 23.
Therefore, we set out to determine E2F activity in normal versus malignant melanocytes and its regulation by melanocyte growth factors. Using gel shift analyses, we demonstrate that E2F2 and E2F4 were the predominant transcription factors from the E2F family active in proliferating normal melanocytes, melanoma cells, and freshly isolated melanoma tumors. E2F4, in association with p130 and to a lesser extent pRb, were the predominant growth inhibitory complexes in growth-arrested normal melanocytes. There was upregulation of E2F activity in melanoma cells that included, in addition to E2F2 and E2F4, E2F1 and E2F3 and occasionally E2F5. E2F6 was not detected in either cell type. The release of E2F activity in the melanoma cells was dependent on persistent CDK activity that maintained all three pocket proteins in the inactive forms. Inhibition of this activity by flavopiridol also inhibited melanoma cell proliferation. The unregulated E2F activity in melanoma cells is probably sufficient to impart continuous proliferation even in the absence of external growth factors required by normal melanocytes. The absence of E2F6 and the low levels or absence of E2F1 activity are advantageous to melanoma cells, as these two transcription factors can cause growth arrest or apoptosis when overexpressed 21 22 23 24 25 26 27 28.
Materials and Methods
Cell Culture and Proliferation Assay.
Normal human melanocytes were dissociated from newborn foreskins and maintained in Ham's F-10 medium supplemented with glutamine (2 mM), penicillin-streptomycin (100 U/ml), and 7% fetal bovine serum (FBS; all from GIBCO BRL), termed basal medium, that was further enriched with several ingredients required for optimal proliferation. They included 12-O-tetradecanoyl phorbol-13-acetate (TPA, 85 nM), 3-isobutyl-1-methyl xanthine (IBMX, 0.1 mM), cholera toxin (2.5 nM), Na3VO4 (1 μM), and N6,2′-O-dibutyryladenosine 3:5-cyclic monophosphate (dbcAMP, 0.1 mM; Sigma-Aldrich Chemical Co.), termed for short TICVA 2 29 30. Unless otherwise stated, all tests were performed with first passage melanocytes pooled from 6–12 donors (∼4 wk in culture). To assay the effect of specific peptide growth factors, normal melanocytes were seeded in 21-cm2 or 55-cm2 Petri dishes in OptiMEM medium (GIBCO BRL) supplemented with FBS (2%) without or with bFGF (10 ng/ml), HGF/SF (100 ng/ml), ET-1 (10 nM), or combinations thereof as indicated. The pigment cells were highly differentiated as judged by the production of melanin, expression of melanocyte-specific proteins (tyrosinase, gp75/tyrosinase-related protein 1), and dendritic morphology.
DNA synthesis was evaluated by the standard [3H]thymidine assay. Melanocytes were seeded in 24- or 48-well plates, treated with experimental media for the duration indicated, and then exposed to MEMS medium containing 5 μCi/ml [3H]thymidine (New England Nuclear) for 1 or 4 h 31. Each data point is an average of triplicate wells; the SD did not exceed 15% of total counts.
Human metastatic melanoma cells 501 mel, 586 mel, YUSAC2, YUGEN8, YUSIT1, YUZAZ6, and YUBSM were maintained in the basal medium consisting of Ham's F-10 supplemented with glutamine, penicillin-streptomycin, and 7% FBS (GIBCO BRL) as described 6 32 33 34. The cultured melanoma cells were from early (first or second) or late passage (such as passage 24 and after). The YU-designated melanoma cells and the two freshly isolated metastatic melanoma tumors YUBSM and YUHAWK were provided by Yale University Department of Surgical Pathology.
To evaluate the growth-inhibitory effect of flavopiridol, a known CDK inhibitor 35 36 37, melanoma cells were seeded in 6-well plates, treated with flavopiridol (0, 20, 50, or 200 nM) and counted with a Coulter counter over a period of 8 d. These data show averages of duplicate wells.
Electrophoretic Mobility Shift Assays.
Normal or malignant melanocytes were seeded in 55-cm2 Petri dishes at ∼80% confluency, and were exposed to the experimental medium the next day. For deprivation studies, normal melanocytes were washed twice with PBS and then seeded in the experimental medium. Electrophoretic mobility shift assays (EMSAs) were performed under standard conditions 38 39 using nuclear extracts, unless otherwise stated. Extracts (prepared as described 40) were subjected to two successive 20-min centrifugations at 14,000 rpm in a microfuge centrifuge at 4°C to remove melanin, as particulate melanin binds nonspecifically to DNA and interferes with the binding assay (our unpublished observation). Protein concentrations were determined by the Bio-Rad protein assay, and DNA binding assays 31 38 were performed with 4 μg of nuclear proteins (unless otherwise stated) and end-radiolabeled double-stranded DNA fragment (1.5–2 × 105 cpm/assay) containing a single E2F consensus binding site derived from the dihydrofolate reductase (DHFR) promoter (sc-2507; Santa Cruz Biotechnology), termed E2FRE.
For competition studies, the DNA binding assays also included 10 ng of unlabeled E2FRE, mutant E2FRE double-stranded oligonucleotide with a CG to AT substitution at the E2F binding motif (E2FREmut, sc-2508), or double-stranded oligonucleotide containing an MYB response element (MRE) as a nonspecific competitor 38. To identify the proteins in complex with the E2F consensus site, extracts were preincubated with rabbit polyclonal antibodies (0.5–2 μg each) to E2F1 (sc-193 X), E2F2 (sc-633 X), E2F3 (sc-878 X or sc-879 X), E2F4 (sc-866 X), E2F5 (sc-1083 X or sc-999 X), E2F6 (sc-8175), DP1 (sc-610 X), DP2 (sc-829 X), pRb (sc-050 X), p107 (sc-318 X), p130 (sc-317 X), or CDK2 (sc-163), mAb to cyclin A2 (sc-BF683 X), cyclin E (sc-248 X), or Sp1 (sc-420 X), all from Santa Cruz Biotechnology. Antibodies to the microphthalmia-associated transcription factor (MITF) were from David Fisher (Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA). Reaction mixtures were loaded onto precast 6% polyacrylamide DNA retardation gels (NOVEX), and dried gels were exposed to reflection film (BioMax MR; Eastman Kodak Co.) at room temperature for 1–2 d.
To estimate fold difference, x-ray films were scanned with a Computing Densitometer (Molecular Dynamics), and the optical density of each band was determined after subtracting adjacent, cold competition background.
Immunoblot Analysis.
For pocket proteins and cyclins, normal melanocytes and melanoma cells were lyzed in RIPA buffer containing a cocktail of protease inhibitors (Complete EDTA-free; Boehringer Mannheim) and phosphatase inhibitors (100 mM NaF, 10 mM Na4P2O7, 1 mM Na3VO4, 0.2 mM ZnCl2 and 20 mM β-glycerol phosphate). Proteins (30–40 μg/lane, as measured by the Bio-Rad protein assay reagent) were fractionated in 6, 8, or 10% polyacrylamide precast gel (NOVEX) as indicated and Western blotted under standard conditions 34. Specific antibodies recognizing pRb (IF8), p130, p107, cyclin A (described above), cyclin E (sc-248; Santa Cruz Biotechnology), or actin (A2066; Sigma Immunochemicals) were used at 1 μg/ml.
To assess the levels of E2F and DP1 transcription factors, nuclear extracts were prepared as described for gel shift analysis and subjected to Western blotting (20 μg/lane) using E2F1 mAb KH95 (Santa Cruz Biotechnology), and the antibodies to E2F2–E2F6, DP1, and DP2 as described above. Fold differences in protein expression were estimated by the optical density of the respective bands as mentioned above.
Immune Complex Kinase Assays.
Kinase assays were performed as described 41 with slight modifications. Cells were collected by scraping, washed in cold PBS supplemented with 50 μM Na3VO4, lyzed in buffer (50 mM Hepes, 200 mM NaCl, pH 7.5, supplemented with the protease and the phosphatase inhibitors mentioned above, and 2% 3-[(3-chlamidopropyl)-dimethylammonio]-1-propanesulfonate [CHAPS] or 0.1% Tween 20) and slightly sonicated. After 10 min on ice, the cell lysates were centrifuged for 10 min at 12,000 rpm, and aliquots of the supernatants (180–250 μg protein/assay for CDK2 and 500 μg/assay for CDK4) were incubated with antibodies (2 μg/each) against CDK2 (M2, sc-163-G; Santa Cruz Biotechnology), CDK4 (H-22-G, sc-601-G; Santa Cruz Biotechnology), or p130, using goat or rabbit IgG as a control, for 2 h on ice. Antibody–antigen complexes were captured on 50 μl slurry of protein G–Sepharose beads (GammaBind G; Amersham Pharmacia Biotech) by rotating for 30 min in the cold. The beads were then washed successively three times with lysis buffer, and once with kinase buffer (30 mM Hepes at pH 7.5, 10 mM MgCl2, 1.0 mM dithiothreitol, 5 mM benzamidine), and the immune complex–bound beads were incubated with 20 μl kinase buffer supplemented with 25 μM ATP and 2 μCi of [γ-32P]ATP (3,000 Ci/mmol; NEN Research Products), plus 2.5 μg histone (for CDK2) or 2.5 μg glutathione S-transferase (GST)-pRb(773–928) (for CDK4 activity), at 37°C for 30 min. The reactions were terminated by the addition of 10 μl 3× SDS sample buffer and 5 min heating at 95°C. After centrifugation, supernatants were fractionated by SDS-PAGE electrophoresis, and dried gels were autoradiographed. Histone 1 or GST-pRb(773–928) protein bands were excised from the gels and radioactivity was measured in a scintillation counter.
Results
E2F Binding Activity, Expression Pattern of Pocket Proteins, and Cyclins in Normal Melanocyte as a Function of External Stimuli.
We first characterized the pattern of E2F activity and pocket protein phosphorylation in normal melanocytes in response to external stimuli, as this has not yet been reported. As shown previously (for a review, see reference 2), normal melanocytes, unlike melanoma cells from advanced lesions, rapidly exit the cell cycle when shifted to serum-supplemented, but growth factor–deprived medium (Fig. 1 A). Under these conditions, exposure to individual growth factors, such as bFGF or HGF/SF, but not ET-1, promoted low levels of DNA synthesis that were further enhanced by costimulation with ET-1 and bFGF, ET-1 and HGF/SF, bFGF and HGF/SF, or all three factors combined (Fig. 1 A, b; as indicated).
Figure 1.

Growth factor regulation of E2F DNA binding activity and expression of growth-regulatory proteins in normal human melanocytes. Melanocytes (first passage) were used in parallel to assay for proliferative responses (A); E2F DNA binding activity (B); and pocket protein and cyclin expression (C). (A, a) Melanocytes grown in TICVA/supplemented basal medium were seeded in 24-well plates (1.4 × 105 melanocytes/well) in either TICVA-supplemented (0) or deprived medium for the indicated period of time. [3H]Thymidine incorporation was for 1 h at different time points as indicated. Data are averages of triplicate wells. SE did not exceed 10%. (A, b) Melanocytes (0.7 × 105) were seeded into 48-well plates and exposed to OptiMEM medium with or without peptide growth factors. [3H]Thymidine was added 24 h after the addition of experimental medium for 4 h. Data are averages of triplicate wells normalized to 1 h/1.4 × 105 melanocytes, as in b. Growth factors included: bFGF/FGF2 (H, 10 ng/ml), HGF/SF (F, 100 ng/ml), ET-1 (E, 0.1 μM), or combinations thereof. NA, no additions. SD did not exceed 13% of total counts. (B) EMSA of whole cell lysates prepared from melanocytes grown in TICVA/serum-supplemented medium and harvested at the exponential phase of growth (lanes 1–3), or after TICVA deprivation for the time indicated (lanes 4–8), as in A, a. Alternatively, melanocytes were incubated with peptide growth factors as in A, b, and harvested 24 h later (lanes 9 and 10). E2F EMSA reactions contained 4 μg of total cellular proteins and an end-labeled DNA fragment derived from the DHFR promoter. Excess (10 ng) unlabeled double-stranded oligonucleotide containing wild-type E2F responsive element site (E2FRE) or mutant E2FRE (E2FREmut) were added to gel shift assays where indicated. GF, growth factor; additional abbreviations as indicated in the legend to A, b. (C) Western blot analyses of pocket proteins and cyclins of melanocytes treated as in A, a (lanes 1–6) and A, b (lanes 7–14) with antibodies as indicated. Equal loading of the 6% gel used to fractionate pocket proteins is indicated by the spurious band (control), whereas equal loading of the 10% used to fractionate the cyclins is represented by actin staining (actin).
Growth arrest was associated with decreased total E2F binding activity and a shift in the pattern of E2F complex formation as revealed by gel retardation assays (Fig. 1 B). A major, fast migrating E2F binding activity (Fig. 1 B, a, at bracket) and two additional minor complexes migrating at intermediate and slower rates (Fig. 1 B, b, solid arrow, and c, open arrow) were apparent in proliferating melanocytes. The binding activity in all complexes was due to E2F, as it was competed out by excess unlabeled E2FRE, but not by E2FREmut (Fig. 1 B; compare lane 2 to 1 and 3, and lane 10 to 9 and 11).
As starved melanocytes exit the cell cycle, there was a sharp reduction in complex a after 20 h (Fig. 1 B, lanes 4–8, at bracket). Complex b persisted for 16 h and was drastically reduced at 20 h, with little remaining at 24 h of deprivation (Fig. 1 B, compare lane 7 to 8 and 6, solid arrow), time points corresponding to complete inhibition of DNA synthesis (Fig. 1 A, a). On the other hand, complex c disappeared within 5 h of deprivation (Fig. 1 B, compare lane 4 to 1 and 3, open arrow). A new complex that migrated to an intermediate position between complex b and c became visible 10 h after deprivation and persisted thereafter (Fig. 1 B, lanes 5–8, 9, and 11, at d, arrowhead). The timing of complex d coincided with a precipitous drop in DNA synthesis (Fig. 1 A, a).
The levels of complex a also correlated with the extent of DNA synthesis in medium supplemented with specific peptide growth factors (compare relevant bands marked with bracket in Fig. 1 B, lanes 9–18, with the histogram in Fig. 1 A, b). Interestingly, complex a was more prominent, and complex c was hardly detected in peptide growth factor–supplemented medium, compared with TICVA-supplemented medium (compare lane 1 to 12, 13, and 15–18, open arrow), conditions also associated with higher levels of DNA synthesis (compare [3H]thymidine incorporation in Fig. 1 A, a to Fig. 1 A, b).
The migration pattern in Western blots analyses revealed that, as expected, all three pocket proteins became dephosphorylated in starved melanocytes, observed as a shift from slow (hyper-phosphorylated) to fast (hypo- or unphosphorylated) migrating forms (Fig. 1 C, as indicated). Loss of slow migrating forms was clearly visible within 10 h of deprivation, especially with p130, (Fig. 1 C, compare lane 1 to 3 in panels marked p130 and p107), coinciding with the appearance of complex d described above (Fig. 1 B, compare lane 1 to 5, arrowhead). Noticeable changes in pRb gel migration patterns were detected only 20 h after deprivation and included the loss of the slow and fast migrating forms and reduction in the total level of the protein, changes particularly accentuated in peptide growth factor–supplemented medium (Fig. 1 C, compare lanes 1–4 to 6 and 7, and 11–14 to 7 and 10; panel marked pRb). A sharp decline in p107 to almost undetectable levels was observed 20 and 24 h after deprivation, time points characterized by complete growth arrest, loss of the fast, and accumulation of the slow migrating form of E2F DNA binding activity, consistent with p107 regulation by E2F1 and E2F2 42 43.
The phosphorylation of pocket proteins is coordinated by the levels of cyclins that bind and activate CDKs, such as cyclin D1 in complex with CDK4/CDK6, and cyclin A and E in complex with CDK2 (for reviews, see references 9, 15, 44). However, normal melanocytes are different from other cell types in that the presence of serum was not sufficient to maintain high cyclins levels. Cyclin D1 and cyclin A2 were maintained only in medium supplemented with serum and TICVA or specific growth factors (Fig. 1 C). Cyclin D1 and A2 levels abruptly disappeared 20 h after deprivation of TICVA. The gradual decline in cyclin D1 levels during the initial 16 h of starvation coincided with the gradual loss of p107 high molecular weight species, i.e., highly phosphorylated forms, consistent with loss of CDK4/6 activity. Synergistic growth factors also synergized in promoting cyclin D1 levels. All together, these results reflect the accepted paradigm in which E2F activity is tightly regulated in response to external stimuli required for melanocyte proliferation, positively correlating with the intensity of DNA synthesis and the phosphorylation status of pocket proteins, driven by cyclins and/or CDK holoenzymes.
Identification of the Components in the E2F DNA Binding Activity.
The addition of specific antibodies to the EMSA reaction mixtures established that the predominant species bound to the E2F site in exponentially proliferating melanocytes were E2F2 and E2F4, as detected by suppressed or supershifted complexes (Fig. 2, compare lanes 1 and 3 to 5 and 7, at bracket; supershift marked by bar). However, low levels of E2F1, E2F3, and possibly E2F5, were also present (Fig. 2, compare lanes 1 and 3 to 4, 6, and 8, at bracket). In medium supplemented with peptide growth factors (bFGF/HGF/ET-1), E2F4 was still the most prominent binding activity (Fig. 2, compare lanes 9 and 11 to 15), whereas E2F2, E2F3, and E2F5 were present to a lesser extent (Fig. 2, lanes 13, 14, and 16), with E2F1 not detectable (Fig. 2, lane 12). The reduced suppression of DNA binding activity by antibodies to E2F1, E2F3, or E2F5 was not due to lower binding efficiency, as these antibodies were effective in EMSA studies with HeLa (data not shown), and as can be seen below with melanoma cell nuclear extracts.
Figure 2.

Identification of the components in the E2F DNA binding activity in nuclear extracts. Gel shift analyses of melanocytes grown in TICVA-supplemented basal medium (lanes 1–8), or in OptiMEM supplemented with 2% FBS and peptide growth factors bFGF/FGF2, HGF/SF, and ET-1, for 24 h. E2F EMSA reactions contained 4 μg of nuclear proteins and an end-labeled DNA fragment as described in the legend to Fig. 1. Antibodies specific for each of the E2F family members, E2F1–E2F5, or each of the pocket proteins, pRb, p107, or p130, or the E2F dimerization partner, DP1, were added to the binding reactions as indicated to identify proteins in specific complexes. Bracket marks free E2F species, solid and open arrows (marked b and c, respectively) point at complex E2F binding activity, and bars on the left and right of the figure mark the positions of the supershifts.
The antibodies also detected E2F4 in the slow migrating complex c (Fig. 2, lane 7, open arrow), and E2F1 in complex b (Fig. 2, lane 4, solid arrow). In both culture conditions, the E2Fs were in complex with DP1 (Fig. 2, lane 20; and Fig. 3 A, lane 17). Because antibodies to pRb, p107, or p130 had no effect on the fast migrating DNA binding activity (Fig. 2, lanes 17–19; and Fig. 3 A, lanes 5–8, 14–16, at bracket), we concluded that complex a contained unbound E2Fs, termed free E2F or E2F.
Figure 3.
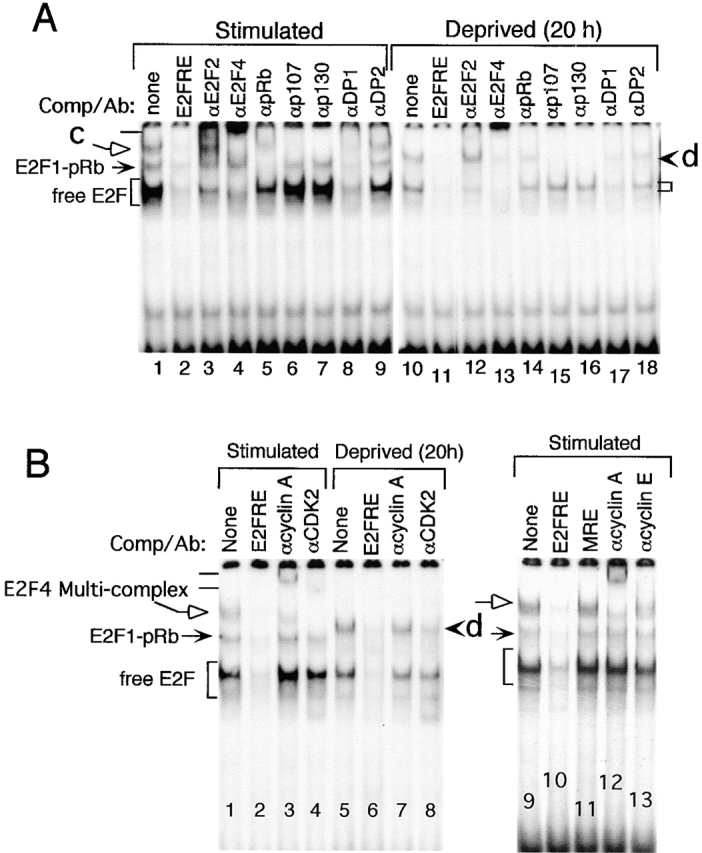
Gel shift analyses of nuclear extracts from stimulated versus growth-arrested melanocytes. (A and B) Melanocytes grown in TICVA plus serum-supplemented medium were harvested at the exponential phase of growth (Stimulated), or after transfer to basal medium without TICVA, for 20 h (Deprived [20 h]). E2F EMSA reactions contained end-labeled DNA fragment derived from the DHFR promoter, 4 μg of nuclear proteins, cold competitors, or antibodies, where indicated and as described above. Free E2F binding activity is marked by brackets (free E2F), and complex activities by solid (E2F1-pRb) and open arrows (c, or E2F4 Multi-complex), and arrowhead (d). Antibodies to specific proteins were added to the reaction mixtures as indicated. Bars mark the supershifts.
Similar analysis showed that pocket proteins were present in complex b, c, and d. In growth-stimulated melanocytes, the E2F1 containing complex b was inhibited by anti-pRb but not by anti-p107 or anti-p130 antibodies (Fig. 3 A, compare lane 1 to 5, solid arrow) and was thus designated E2F1-pRb. In contrast, complex c was completely abolished by antibodies to p107 or p130 (Fig. 3 A, compare lane 1 to 6 and 7, open arrow). In growth-arrested melanocytes, complex d was reduced by the anti-pRb antibodies, and was completely abolished by anti-p107 or anti-p130 antibodies (Fig. 3 A, compare lane 10 to 14–16, arrowhead).
The apparent interference by anti-p107 antibodies with E2F DNA binding activity in 20 h–deprived melanocytes (Fig. 3 A, lane 15), coinciding with drastic reduction in p107 (Fig. 1 C, lane 5), raised the possibility that the antibodies were not specific to p107. Indeed, a reciprocal immunoprecipitation and immunoblotting experiment performed with cell extracts prepared from 24–h starved melanocytes in which p107 was almost undetectable (Fig. 1 C, lane 6) contained ample p130 (data not shown). On the other hand, antibodies to p130 did not detect p107. Therefore, we concluded that although complex c may contain p107 and p130, complex d contained, in addition to pRb, only p130, very likely both in association with E2F4 (Fig. 3 A, lanes 4 and 13; and Fig. 2, lane 7). This EMSA also confirmed that among the DPs, DP1 was the predominant E2F dimerization partner (Fig. 3 A, lanes 8, 9, 17, 18).
Previous reports showed the presence of multimeric E2F DNA binding activities in other cell types, such as CDK2–cyclin A and CDK2–cyclin E in association with p107–E2F or with p130–E2F (reference 10 [for a review], 18, 19, 45, 46), as well as in the melanoma cell line HO-1 47. Similar analysis revealed that indeed CDK2 and cyclin A were in the E2F4–p130 (or p107) complex c, as visualized by supershifts with the respective antibodies (Fig. 3 B, compare lane 1 to 3 and 4, supershifts shown by bars). Complex d was also suppressed by antibodies to CDK2, but there was no supershift by antibodies to cyclin A (Fig. 3 B, compare lane 8 to 5 and 7, at arrowhead). Cyclin A was the most abundant cyclin in the complex, as only low levels of cyclin E could be detected (Fig. 3 B, compare lane 12 to 13, open arrow). Multimeric complexes composed of CDK2–cyclin A with E2F and p107 or p130 are characteristic of cells entering the S phase of the cell cycle 46 48, consistent with the proliferative state of the melanocytes. In contrast, E2F4 in complex with pRb or p107 was shown to suppress E2F-dependent transcription and to mediate growth arrest 49, and E2F in association with p130 was also detected in HO-1 melanoma cells undergoing growth arrest and differentiation in response to interferon (IFN-β) plus mezerein treatment 47. All together, these results demonstrate that growth factors tightly control E2F DNA activity in normal melanocytes, with E2F2 and E2F4 being the major species. Growth arrest was associated with low levels of free E2F and the accumulation of pRb and p130 suppressive complexes, possibly in association with E2F4.
Aging-dependent Changes in E2F Binding Activity in Normal Melanocytes.
Normal human melanocytes, unlike melanoma cells, have a definite life span in culture and become progressively unresponsive to external growth stimuli (50, 51, and our unpublished results). Since all of the experiments described above have employed highly proliferating first passage melanocytes, we extended the gel shifts analyses to fourth passage melanocytes. Indeed, late passage melanocytes grown continuously in the presence of TICVA (∼12 population doubling times representing ∼3 mo in culture) displayed a pattern of E2F DNA binding activity similar to that of growth-arrested first passage melanocytes (Fig. 4, compare first to late passage cells at bracket). Here again, free E2F was composed mostly of E2F4, and to lesser extent E2F2, both present at lower levels than in first passage melanocytes (Fig. 4). pRb-associated complexes were not detected (Fig. 4, lane 17), but E2F4, in association with p130, was present (Fig. 4, lanes 11, 18, 19, and 29). The anti-p107 suppressive effect is likely due to cross-reactive activity with p130, as p107 was hardly detected in fourth passage melanocytes (see Fig. 6 C). There was no change in DP1 activity (Fig. 4, lane 16). These results suggest a diminishing ability to respond to external growth factors as the cells age in culture.
Figure 4.

A shift to growth-arrest pattern of E2F DNA binding activity in senescing normal melanocytes. Nuclear extracts were prepared from fourth (lanes 2–19, 25–29) or first passage melanocyte cultures grown in TICVA-supplemented basal medium and harvested at the exponential growth phase. DNA binding reactions contained 0–8 μg of nuclear proteins (as indicated) and an end-labeled DNA fragment derived from the DHFR promoter as described above. The reaction mixtures contained excess (10 ng) double-stranded oligonucleotide encoding wild-type E2F site (E2FRE), the unrelated MRE site, or specific antibodies as indicated. Brackets and arrowheads point at the positions of the free E2F and E2F4-p130 suppressive complexes, respectively. The position of E2F1–pRb and the multimeric E2F4–p130 (p107)/cyclin A–CDK2 are indicated by solid and open arrows, respectively.
Figure 6.
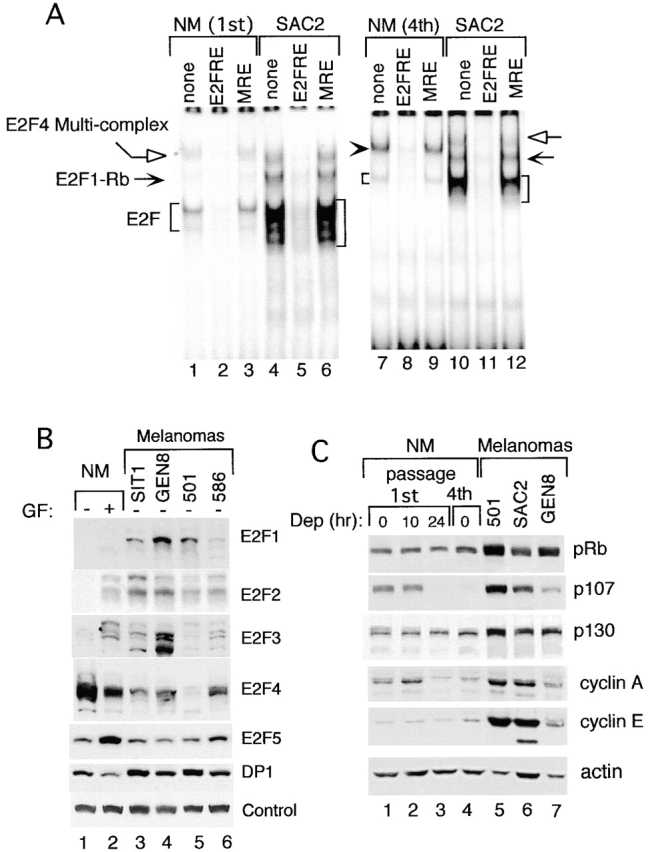
Upregulation of E2F DNA binding activity in proliferating melanoma cells compared with normal melanocytes as a function of pocket protein and E2F expression. (A) EMSA showing the relative amounts of E2F DNA binding activity in normal melanocytes (NM, first and fourth passage) compared with melanoma cells YUSAC2 (SAC2), all harvested at the exponential growth phase. Each reaction mixture contained 4 μg nuclear proteins, radiolabeled probe, and/or competitors (where indicated) as described above. Normal melanocytes were grown in TICVA-supplemented basal medium, whereas melanoma cells YUSAC2 were grown in basal medium without TICVA. (B) Expression of E2F1–E2F5 and DP1 in normal melanocytes compared with melanoma cells. Shown are Western blots with the respective antibodies (as marked) using nuclear extracts (prepared as described for gel shift analysis) harvested from exponentially growing (+) or 24 h TICVA–deprived normal melanocytes (NM), or from proliferating melanoma cells YUSIT1 (SIT1), YUGEN8 (GEN8), 501 mel (501), or 586 mel (586). Proteins were fractionated in 8% polyacrylamide precast gels. Equal protein loading is indicated by staining of a spurious band (Control). (C) Western blots of pocket proteins and cyclins using whole cell extracts from normal melanocytes (NM, first or fourth passage), harvested at the exponential growth phase (0), or 10 or 24 h after exposure to TICVA-deprived basal medium (Dep 10, 24), also used to grow the melanoma cells 501 mel (501), YUSAC2 (SAC2), and YUGEN8 (GEN8). Proteins were fractionated in 8% polyacrylamide precast gels. Actin staining was used to indicate the loading in each well.
Free E2F Binding Activity Is Abundant in Melanoma Cells and Tumors.
The persistent phosphorylation of pRb commonly observed in melanoma cells 5 6 is expected to relieve E2F–pRb transcriptional repression and to generate free E2F activity. Indeed, the gel retardation assays revealed uniformly abundant unbound E2F in nuclear extracts from two uncultured melanoma tumors and seven cultured melanoma cell strains, represented by selected examples in Fig. 5 (at brackets). As judged by the level of displacement of the E2F complex with specific antibodies, E2F2 and E2F4 were again, invariably, the predominant DNA binding proteins (Fig. 5 A, lanes 9, 11, 17, and 19; B, lanes 6 and 8; and C, lanes 6 and 8). However, the other E2F family members were also present; E2F1, E2F3, and E2F5 were detected in melanoma 501 mel (Fig. 5 A, lanes 8, 10, and 12), E2F1 and E2F3 in YUGEN8 (Fig. 5 A, lanes 16 and 18), E2F3 in YUSAC2 (Fig. 5 B, lane 7), and E2F5 was detected at high levels in the melanoma tumor YUHAWK (Fig. 5 C, lane 9). These binding activities represented free E2F because antibodies to pocket proteins or to CDK2/cyclins had no effect (Fig. 5 A–C, antibodies as marked). There were no differences in the pattern of E2F binding activity between cultured cells and freshly isolated tumor derived from the same patient (data not shown).
Figure 5.
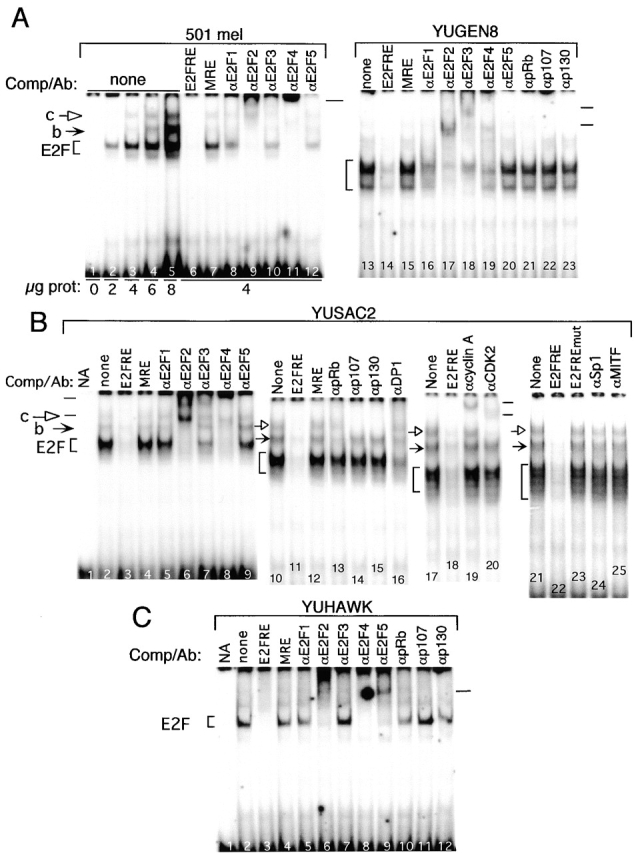
Free E2F DNA binding activity is abundant in melanoma cells and tumors. Nuclear extracts (A, 0–8 μg/assay; B and C, 4 μg/assay) from metastatic melanoma cells (501 mel, YUGEN8, and YUSAC2) or a freshly isolated melanoma tumor (YUHAWK) were used in EMSAs as described above. All cells in culture were from the exponential growth phase. Excess unlabeled competitors or specific antibody were added to the reaction mixtures as described above. Complexes are marked as above. NA, reaction contained radioactive E2FRE without cell extracts or any other additions.
Nuclear extracts from melanoma cells also contained low levels of the b and c E2F complexes described in proliferating normal melanocytes. Displacement of the complexes by antibodies showed that, as before, complex b represented E2F bound to pRb (as in Fig. 5 B, lane 13), and complex c contained E2F in association with p130 (and possibly p107), CDK2, and cyclin A (Fig. 5 B, lanes 14, 15, 19, and 20). The E2F4-p130/pRb complex d, characteristic of growth-arrested melanocytes, was not detected. The effects of the antibodies were specific, as no suppression or supershift was observed by antibodies to the transcription factors Sp1 and MITF (Fig. 5 B, lanes 24 and 25), reported to associate with pRb 52 53.
Persistent Hyperactivity of E2F DNA Binding in Melanoma Cells: The Consequence of Continuous Pocket Protein Hyperphosphorylation and DP1 Overexpression.
Parallel EMSAs showed that the persistent E2F DNA binding activity in melanoma cells, free as well as in multimeric complexes, was ∼5–10-fold higher compared with the first passage proliferating normal melanocytes (Fig. 6 A, compare lane 1 and 3 to 4 and 6) and with fourth passage melanocytes (Fig. 6 A, compare lane 7 and 9 to 10 and 12), as estimated by a scanning densitometer.
To determine the cause of this persistently high activity, we assessed the levels of E2Fs, DPs, pocket proteins, cyclins, and CDK activity. Western blots of the six E2Fs and two DPs family members revealed that E2F1, E2F2, and E2F3 were present only in growth factor–stimulated normal melanocytes, but were constitutively expressed in melanomas cells exposed to the same medium as “deprived” melanocytes (Fig. 6 B, compare lane 1 to 2–6 of the respective blots). In normal human melanocytes, E2F4 was expressed regardless of culture conditions at even slightly higher levels compared with melanoma cells (Fig. 6 B, compare lane 1 and 2 to 3–6, blot marked E2F4). E2F5 was slightly induced by growth factors in normal human melanocytes, but it was not overproduced in melanoma cells, and E2F6 was not detected in either cell type (data not shown). Most importantly, the immunoblots clearly showed an approximately fourfold increase in the abundance of the dimerization partner DP1 in melanoma cells over proliferating normal melanocytes (Fig. 5 B, compare lane 2 to 3–6). The higher levels of this dimerization partner, absolutely required for E2F DNA binding activity, are probably the major reason for the approximately fivefold increase in E2F2/4 DNA binding activity in the melanoma cells, as the levels of the respective proteins are similar in proliferative normal and malignant melanocytes (Fig. 5 B, compare lane 2 to 3–6). As with the gel shift, DP2 was not detected by this method as well (data not shown).
The phosphorylation state of the three pocket proteins assessed by their pattern of migration in Western blots confirmed previous observations 5 6, that all forms (unhypo- and hyperphosphorylated) of pRb were more abundant in melanoma cells compared with normal melanocytes (Fig. 6 C). Likewise, the three melanoma cell lines also expressed higher levels of all forms of p107 and p130, as well as cyclin A and cyclin E (Fig. 6 C). Interestingly, growth-stimulated fourth passage melanocytes displayed pocket protein and cyclin expression patterns similar to that of growth-arrested first passage melanocytes (Fig. 6 C, compare lane 4 to 1 and 3), in agreement with the pattern and low levels of free E2F activity (Fig. 6 A, lanes 7–9, and Fig. 4).
Since cyclin D1 was shown previously to be persistently present in melanoma cells 6, we presumed that the release of E2F activity in these cells was due to persistent activity of cyclin D1–CDK4/CDK6, cyclin A–CDK2, and cyclin E–CDK2, known to coordinate the continuous phosphorylation and/or inactivation of the three pocket proteins (for reviews, see references 9, 15, 44). This assumption was confirmed, as shown by the immune complex kinase assays (Fig. 7). The activity of CDK4, CDK2, and p130-associated kinase was not dependent on growth factors in the three melanoma cell lines tested, but was drastically reduced (CDK2 and p130-associated kinase) or undetected (CDK4) in normal melanocytes deprived of growth factors for 24 h. Furthermore, CDK4 activity was 10–15-fold higher in the melanoma cell lines compared with proliferating melanocytes.
Figure 7.

CDK activity in normal melanocytes versus melanoma cells. Immune complex kinase assays were performed with cell extracts from first passage normal melanocytes (NM) harvested 24 h after TICVA starvation (GF−, lanes marked 1) or at the exponential growth phase (GF+, lanes marked 2), and from melanoma cells YUGEN8 (GEN8, lanes marked 3), 586 mel (586, lanes marked 4), or YUZAZ6 (ZAZ6, lanes marked 5), grown in TICVA-deprived basal medium (GF−). Antibodies to CDK4, CDK2, p130, control goat (for CDK4 and CDK2), or rabbit IgG (for p130), are as indicated. Autoradiograms show radioactive substrates representing GST-pRb(773–928) in the case of CDK4, or histone 1 in the case of CDK2 and p130 immune complex kinase assays. The histograms represent the radioactivity in the excised substrate bands of anti-CDK4, CDK2, or p130 immune complex minus the respective control antibody.
Inhibition of CDK Activity by Flavopiridol Arrests Melanoma Cell Growth Mediated by Downregulated E2F Activity.
We reasoned that if melanoma cell proliferation is driven by the persistent activity of CDKs, the cells should be efficiently arrested by agents that inhibit CDK activity, such as flavopiridol 35 36 37. Indeed, exposure of melanoma cells YUSIT1 and YUGEN8 (as well as others not shown) to 200 nM flavopiridol, levels known to completely inhibit CDK activity 36, induced growth arrest (Fig. 8 A). Western blot analysis revealed progressive loss of phosphorylated forms of pRb, p107, and p130, as well as reduction in pRb and p107 protein levels in response to flavopiridol (Fig. 8 B), reminiscent of growth factor–deprived normal melanocytes (Fig. 1 C) and confirming results obtained by others for pRb and p107 35. However, for the first time we show here the strong impact of flavopiridol on E2F activity. The gel shift analysis demonstrated a sharp decrease in free E2F binding activity that affected the four E2Fs' binding activity in the melanoma cells, i.e., E2F1, E2F2, E2F3, and E2F4 (Fig. 8 C, compare lanes 1–8 to 9–16, at bracket). Furthermore, the complex containing E2F1 in association with pRb (formerly designated complex b) accumulated in response to flavopiridol (Fig. 8 C).
Figure 8.
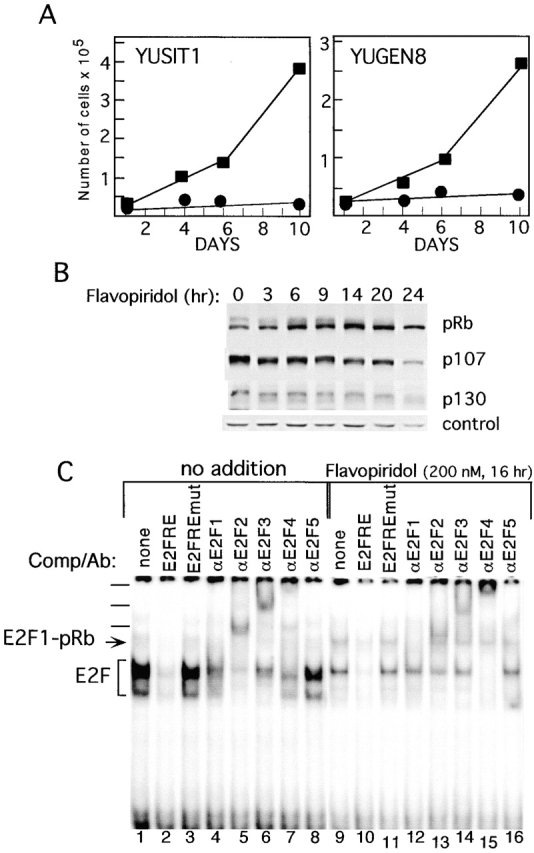
Flavopiridol suppresses melanoma cell growth, pocket protein phosphorylation, and E2F DNA binding activity. (A) Growth–response to flavopiridol. Metastatic melanoma cells YUSIT1 and YUGEN8 were seeded in basal medium without (▪) or with (•) 200 nM flavopiridol in 6-well plates. Cell were harvested at the indicated times and counted with the Coulter counter. Each data point is an average of two wells. (B) Progressive dephosphorylation of pocket proteins in response to flavopiridol. Melanoma cells YUGEN8 were exposed to 200 nM flavopiridol and harvested at the indicated time. Proteins were fractionated in 6% polyacrylamide precast gels and Western blotted with the indicated antibodies. A spurious band is presented to indicate protein loading in each well. (C) Reduction in total E2F activity in response to flavopiridol. Melanoma cells YUGEN8 were exposed to 200 nM flavopiridol and harvested 16 h later. Whole cell lysates (4 μg/assay) were subjected to EMSA in the absence or presence of competitors or antibodies as described above. The positions of complexes containing free E2F (E2F) or E2F1–pRb are indicated, and supershifts are marked with bars.
Discussion
Here, for the first time, we characterized the regulation of E2F DNA binding activity in normal human melanocytes in response to growth factors compared with melanoma cells and tumors. We show that in highly proliferating normal melanocytes, binding activity to an oligonucleotide probe comprising a single E2F consensus site is due mostly to E2F2 and E2F4, and to a lesser extent, to E2F1 and E2F3. In growth-arrested melanocytes, total levels of E2F DNA binding activity declined, and E2F4 in complex with p130, and to a lesser extent with pRb, accumulated. Unexpectedly, the presumably transcriptionally suppressive E2F1–pRb complexes were more abundant in proliferating compared with growth-arrested melanocytes. Furthermore, we show that tumor-dependent changes in melanoma cells included upregulation of E2F2 and E2F4 DNA binding activity, and to various extents, also that of E2F1, E2F3, and E2F5. Except for E2F1, the increase in E2F DNA binding was not due to increased expression of the respective E2F members, but rather to a proportional increase in the expression of the dimerization partner DP1.
All three pRb family members were highly abundant in their hypo- and hyperphosphorylated forms in the melanoma cells tested. In melanoma cells, the tumor-suppressor pocket proteins were inactivated by continuous CDK activity driven by the persistent presence of high levels of at least three cyclins, cyclin D1, cyclin A2, and cyclin E. Inhibition of CDK activity in melanoma cells by flavopiridol-induced growth arrest was accompanied by reduction in free E2F activity and an increase in E2F1–pRb transcriptional suppressive complexes. Therefore, we suggest that melanoma cell autonomy is supported by dysregulated cyclins that lead to upregulation of E2F transcriptional activity.
Low levels of E2F1–pRb complexes, potentially with transcriptional suppressive function, were observed in proliferating normal and malignant melanocytes. Although in contradiction to present models of cell cycle control, such complexes were observed already in proliferating mouse embryo fibroblasts 54, hyperproliferative primary keratinocytes derived from transgenic K5 E2F1 mice expressing up to 80-fold more E2F1 than nontransgenic keratinocytes 55, and in the cytokine-dependent 6-1 pro-B lymphoid cell line 56. Interestingly, the lymphoid cell line also shares other characteristics with the melanocyte–melanoma system, in that growth inhibition was mediated by p130-containing complexes, and acquisition of cytokine independence by E2F4-driven transcriptional stimulation 56.
Constitutive CDK4 and CDK2 kinase activities are the likely cause for the highly phosphorylated forms of pocket proteins in melanoma cells, as shown here by the immune kinase assays of the respective complexes. It is possible that a combination of genetic alterations frequently encountered in melanomas, i.e., loss of the CKI p16INK4A (for reviews, see references 6, 57, 58) and aberrant expression of the protooncogene bFGF/FGF2 (for a review, see reference 2) are the cause for persistent CDK activity. Ectopic expression of bFGF activates the FGFR1, which in turn can induce high levels of the positive CDK regulators cyclin D1, cyclin A, and cyclin E, frequently observed in melanoma cells (6, and results shown here). Constitutive expression of cyclins can be the major culprit for melanoma cell–dysregulated proliferation, since loss of p16INK4A by itself is not sufficient to alter the cell dependency on growth factors 31 or to induce melanomas in humans 58. In contrast, cyclin D1 is capable by itself of inducing constitutive CDK activity 59 60, and overexpression of cyclin E induced S phase and cell division, independently of E2F activation 61. In breast cancer cells, overexpressed cyclin E, in complex with CDK2, can replace the loss of cyclin D–dependent kinase complexes in maintaining persistent pRb phosphorylation 62. In melanomas, continuous expression of cyclin D1 is not solely due to genetic alteration, as amplification of chromosomal region 11q13, the site of cyclin D1 (previously PRAD1), occurs in only 1 out of 10 melanomas 63 64. Thus, high levels of cyclin D1, cyclin A, and cyclin E in melanoma cells must be the result of increased synthesis and/or decreased degradation.
The persistently high free E2F activity and lack of E2F-p130 inhibitory complexes in melanoma cells, even in the absence of external growth factors, most likely contribute to their ability to proliferate by affecting the expression of several target genes known to be required for cell cycle progression. One of these genes, B-MYB, known to be mostly affected by deletion of p107/p130, may have a particular significance. B-MYB expression was 25-fold higher in quiescent p107/p130 double null mouse embryo fibroblasts compared with controls 54. We found that c-MYB and B-MYB mRNA are overexpressed in melanoma cell lines compared with normal melanocytes, and that ectopic expression of murine c-MYB in SK-MEL-2 human melanoma cells resulted in increased expression of FGF2 mRNA and FGF2 protein 38. Therefore, it is possible that E2F transcriptional activity and elimination of E2F4–p107/p130 transcriptional repression set in motion a loop of autoregulatory gene expression that further enhances the malignant phenotype.
Acknowledgments
We thank Hoechst Marion Roussel, Inc. (Bridgewater, NJ) for the gift of flavopiridol, and Dr. David Fisher (Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA) for the MITF antibodies.
This work was supported by National Institutes of Health grant 2R01-CA44542 to R. Halaban.
Footnotes
Abbreviations used in this paper: CDK, cyclin-dependent kinase; CKI, cyclin-dependent kinase inhibitor; DHFR, dihydrofolate reductase; EMSA, electrophoretic mobility shift assay; ET, endothelin; FBS, fetal bovine serum; FGF, fibroblast growth factor; GST, glutathione S-transferase; HGF, hepatocyte growth factor; INK, inhibitor of CDK; MITF, microphthalmia-associated transcription factor; MRE, Myb response element; pRb, retinoblastoma tumor suppressor protein; RE, response element; SF, scatter factor.
References
- Halaban R. Melanoma cell autonomous growththe Rb/E2F pathway. Cancer Metastasis Rev. 1999;8:333–343. doi: 10.1023/a:1006396104073. [DOI] [PubMed] [Google Scholar]
- Halaban R. Growth factors and melanomas. Semin. Oncol. 1996;23:673–681. [PubMed] [Google Scholar]
- Rodeck U., Melber K., Kath R., Menssen H.D., Varello M., Atkinson B., Herlyn M. Constitutive expression of multiple growth factor genes by melanoma cells but not normal melanocytes. J. Investig. Dermatol. 1991;97:20–26. doi: 10.1111/1523-1747.ep12477822. [DOI] [PubMed] [Google Scholar]
- Rodeck U. Growth factor independence and growth regulatory pathways in human melanoma development. Cancer Metastasis Rev. 1993;12:219–226. doi: 10.1007/BF00665954. [DOI] [PubMed] [Google Scholar]
- Bartkova J., Lukas J., Guldberg P., Alsner J., Kirkin A.F., Zeuthen J., Bartek J. The p16-cyclin D/Cdk4-pRb pathway as a functional unit frequently altered in melanoma pathogenesis. Cancer Res. 1996;56:5475–5483. [PubMed] [Google Scholar]
- Halaban R., Miglarese M.R., Smicun Y., Puig S. Melanomas, from the cell cycle point of view. Int. J. Mol. Med. 1998;1:419–425. doi: 10.3892/ijmm.1.2.419. [DOI] [PubMed] [Google Scholar]
- Pardee A. G1 events and regulation of cell proliferation. Science. 1989;246:603–608. doi: 10.1126/science.2683075. [DOI] [PubMed] [Google Scholar]
- Bartek J., Bartkova J., Lukas J. The retinoblastoma protein pathway and the restriction point. Curr. Opin. Cell Biol. 1996;8:805–814. doi: 10.1016/s0955-0674(96)80081-0. [DOI] [PubMed] [Google Scholar]
- Planas-Silva M.D., Weinberg R.A. The restriction point and control of cell proliferation. Curr. Opin. Cell Biol. 1997;9:768–772. doi: 10.1016/s0955-0674(97)80076-2. [DOI] [PubMed] [Google Scholar]
- Nevins J.R. Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ. 1998;9:585–593. [PubMed] [Google Scholar]
- Adams P.D., Kaelin W.G., Jr. Transcriptional control by E2F. Semin. Cancer Biol. 1995;6:99–108. doi: 10.1006/scbi.1995.0013. [DOI] [PubMed] [Google Scholar]
- Adams P.D., Kaelin W.G., Jr. The cellular effects of E2F overexpression. Curr. Top. Microbiol. Immunol. 1996;208:79–93. doi: 10.1007/978-3-642-79910-5_4. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. Growth factor-regulated G1 cyclins. Stem Cells. 1994;1:47–55. [PubMed] [Google Scholar]
- Sherr C.J. G1 phase progressioncycling on cue. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. D-type cyclins. Trends Biochem. Sci. 1995;20:187–190. doi: 10.1016/s0968-0004(00)89005-2. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. Mammalian G1 cyclins and cell cycle progression. Proc. Assoc. Am. Physicians. 1995;107:181–186. [PubMed] [Google Scholar]
- Jacks T., Weinberg R.A. Cell-cycle control and its watchman. Nature. 1996;381:643–644. doi: 10.1038/381643a0. [DOI] [PubMed] [Google Scholar]
- Grana X., Garriga J., Mayol X. Role of the retinoblastoma protein family, pRB, p107 and p130 in the negative control of cell growth. Oncogene. 1998;17:3365–3383. doi: 10.1038/sj.onc.1202575. [DOI] [PubMed] [Google Scholar]
- Johnson D.G., Schneider-Broussard R. Role of E2F in cell cycle control and cancer. Front. Biosci. 1998;3:d447–d448. doi: 10.2741/a291. [DOI] [PubMed] [Google Scholar]
- Hiyama H., Iavarone A., Reeves S.A. Regulation of the CDK inhibitor p21 gene during cell cycle progression is under the control of the transcription factor E2F. Oncogene. 1998;16:1513–1523. doi: 10.1038/sj.onc.1201667. [DOI] [PubMed] [Google Scholar]
- Trimarchi J.M., Fairchild B., Verona R., Moberg K., Andon N., Lees J.A. E2F-6, a member of the E2F family that can behave as a transcriptional repressor. Proc. Natl. Acad. Sci. USA. 1998;95:2850–2855. doi: 10.1073/pnas.95.6.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright P., Muller H., Wagener C., Holm K., Helin K. E2F-6a novel member of the E2F family is an inhibitor of E2F-dependent transcription. Oncogene. 1998;17:611–623. doi: 10.1038/sj.onc.1201975. [DOI] [PubMed] [Google Scholar]
- Gaubatz S., Wood J.G., Livingston D.M. Unusual proliferation arrest and transcriptional control properties of a newly discovered E2F family member, E2F-6. Proc. Natl. Acad. Sci. USA. 1998;95:9190–9195. doi: 10.1073/pnas.95.16.9190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalik T.F., DeGregori J., Schwarz J.K., Nevins J.R. E2F1 overexpression in quiescent fibroblasts leads to induction of cellular DNA synthesis and apoptosis. J. Virol. 1995;69:2491–2500. doi: 10.1128/jvi.69.4.2491-2500.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips A.C., Bates S., Ryan K.M., Helin K., Vousden K.H. Induction of DNA synthesis and apoptosis are separable functions of E2F-1. Genes Dev. 1997;11:1853–1863. doi: 10.1101/gad.11.14.1853. [DOI] [PubMed] [Google Scholar]
- Holmberg C., Helin K., Sehested M., Karlstrom O. E2F-1-induced p53-independent apoptosis in transgenic mice. Oncogene. 1998;17:143–155. doi: 10.1038/sj.onc.1201915. [DOI] [PubMed] [Google Scholar]
- DeGregori J., Leone G., Miron A., Jakoi L., Nevins J.R. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc. Natl. Acad. Sci. USA. 1997;94:7245–7250. doi: 10.1073/pnas.94.14.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan B., Farmer A.A., Lee W.H. The molecular basis of E2F-1/DP-1-induced S-phase entry and apoptosis. Cell Growth Differ. 1996;7:689–697. [PubMed] [Google Scholar]
- Halaban R., Ghosh S., Duray P., Kirkwood J.M., Lerner A.B. Human melanocytes cultured from nevi and melanomas. J. Invest. Dermatol. 1986;87:95–101. doi: 10.1111/1523-1747.ep12523594. [DOI] [PubMed] [Google Scholar]
- Halaban R. Growth regulation in normal and malignant melanocytes. In: Ferrone S., editor. Human MelanomaImmunology, Diagnostics and Therapy. Springer-Verlag; New York: 1989. pp. 3–14. [Google Scholar]
- Halaban R., Cheng E., Zhang Y., Mandigo C.E., Miglarese M.R. Release of cell cycle constraints in mouse melanocytes by overexpressed mutant E2F1E132, but not by deletion of p16INK4A or p21WAF1/CIP1. Oncogene. 1998;16:2489–2501. doi: 10.1038/sj.onc.1201773. [DOI] [PubMed] [Google Scholar]
- Zakut R., Perlis R., Eliyahu S., Yarden Y., Givol D., Lyman S.D., Halaban R. KIT ligand (mast cell growth factor) inhibits the growth of KIT-expressing melanoma cells. Oncogene. 1993;8:2221–2229. [PubMed] [Google Scholar]
- Halaban R., Rubin J.S., Funasaka Y., Cobb M., Boulton T., Faletto D., Rosen E., Chan A., Yoko K., White W. Met and hepatocyte growth factor/scatter factor signal transduction in normal melanocytes and melanoma cells. Oncogene. 1992;7:2195–2206. [PubMed] [Google Scholar]
- Halaban R., Cheng E., Zhang Y., Moellmann G., Hanlon D., Michalak M., Setaluri V., Hebert D.N. Aberrant retention of tyrosinase in the endoplasmic reticulum mediates accelerated degradation of the enzyme and contributes to the dedifferentiated phenotype of amelanotic melanoma cells. Proc. Natl. Acad. Sci. USA. 1997;94:6210–6215. doi: 10.1073/pnas.94.12.6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrump D.S., Matthews W., Chen G.A., Mixon A., Altorki N.K. Flavopiridol mediates cell cycle arrest and apoptosis in esophageal cancer cells. Clin. Cancer Res. 1998;4:2885–2890. [PubMed] [Google Scholar]
- Carlson B.A., Dubay M.M., Sausville E.A., Brizuela L., Worland P.J. Flavopiridol induces G1 arrest with inhibition of cyclin-dependent kinase (CDK) 2 and CDK4 in human breast carcinoma cells. Cancer Res. 1996;56:2973–2978. [PubMed] [Google Scholar]
- Patel V., Senderowicz A.M., Pinto D., Jr., Igishi T., Raffeld M., Quintanilla-Martinez L., Ensley J.F., Sausville E.A., Gutkind J.S. Flavopiridol, a novel cyclin-dependent kinase inhibitor, suppresses the growth of head and neck squamous cell carcinomas by inducing apoptosis. J. Clin. Invest. 1998;102:1674–1681. doi: 10.1172/JCI3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miglarese M.R., Halaban R., Gibson N.W. Regulation of FGF-2 expression in melanoma cells by the c-Myb proto-oncoprotein. Cell Growth Differ. 1997;8:1199–1210. [PubMed] [Google Scholar]
- Nevins J.R., DeGregori J., Jakoi L., Leone G. Functional analysis of E2F transcription factor. Methods Enzymol. 1997;283:205–219. doi: 10.1016/s0076-6879(97)83017-0. [DOI] [PubMed] [Google Scholar]
- Dignam J.D., Lebovitz R.M., Roeder R.G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushime H., Quelle D.E., Shurtleff S.A., Shibuya M., Sherr C.J., Kato J.Y. D-type cyclin-dependent kinase activity in mammalian cells. Mol. Cell. Biol. 1994;14:2066–2076. doi: 10.1128/mcb.14.3.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E.J., Leone G., Nevins J.R. Distinct mechanisms control the accumulation of the Rb-related p107 and p130 proteins during cell growth. Cell Growth Differ. 1998;9:297–303. [PubMed] [Google Scholar]
- Garriga J., Limon A., Mayol X., Rane S.G., Albrecht J.H., Reddy E.P., Andres V., Grana X. Differential regulation of the retinoblastoma family of proteins during cell proliferation and differentiation. Biochem. J. 1998;1:645–654. doi: 10.1042/bj3330645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg R.A. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Claudio P.P., De Luca A., Howard C.M., Baldi A., Firpo E.J., Koff A., Paggi M.G., Giordano A. Functional analysis of pRb2/p130 interaction with cyclins. Cancer Res. 1996;56:2003–2008. [PubMed] [Google Scholar]
- Cobrinik D., Whyte P., Peeper D.S., Jacks T., Weinberg R.A. Cell cycle-specific association of E2F with the p130 E1A-binding protein. Genes Dev. 1993;7:2392–2404. doi: 10.1101/gad.7.12a.2392. [DOI] [PubMed] [Google Scholar]
- Jiang H.P., Lin J., Young S.M., Goldstein N.I., Waxman S., Davila V., Chellappan S.P., Fisher P.B. Cell cycle gene expression and E2F transcription factor complexes in human melanoma cells induced to terminally differentiate. Oncogene. 1995;11:1179–1189. [PubMed] [Google Scholar]
- Lees E., Faha B., Dulic V., Reed S.I., Harlow E. Cyclin E/cdk2 and cyclin A/cdk2 kinases associate with p107 and E2F in a temporally distinct manner. Genes Dev. 1992;6:1874–1885. doi: 10.1101/gad.6.10.1874. [DOI] [PubMed] [Google Scholar]
- Furukawa Y., Iwase S., Kikuchi J., Nakamura M., Yamada H., Matsuda M. Transcriptional repression of the E2F-1 gene by interferon-alpha is mediated through induction of E2F-4/pRB and E2F-4/p130 complexes. Oncogene. 1999;18:2003–2014. doi: 10.1038/sj.onc.1202500. [DOI] [PubMed] [Google Scholar]
- Medrano E.E., Yang F., Boissy R., Farooqui J., Shah V., Matsumoto K., Nordlund J.J., Park H.Y. Terminal differentiation and senescence in the human melanocyterepression of tyrosine-phosphorylation of the extracellular signal-regulated kinase 2 selectively defines the two phenotypes. Mol. Biol. Cell. 1994;5:497–509. doi: 10.1091/mbc.5.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad M.M., Xu W., Medrano E.E. Aging in epidermal melanocytescell cycle genes and melanins. J. Investig. Dermatol. Symp. Proc. 1998;3:36–40. [PubMed] [Google Scholar]
- Noe V., Alemany C., Chasin L.A., Ciudad C.J. Retinoblastoma protein associates with SP1 and activates the hamster dihydrofolate reductase promoter. Oncogene. 1998;16:1931–1938. doi: 10.1038/sj.onc.1201718. [DOI] [PubMed] [Google Scholar]
- Yavuzer U., Keenan E., Lowings P., Vachtenheim J., Currie G., Goding C.R. The microphthalmia gene product interacts with the retinoblastoma protein in vitro and is a target for deregulation of melanocyte-specific transcription. Oncogene. 1995;10:123–134. [PubMed] [Google Scholar]
- Hurford R.K., Jr., Cobrinik D., Lee M.H., Dyson N. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev. 1997;11:1447–1463. doi: 10.1101/gad.11.11.1447. [DOI] [PubMed] [Google Scholar]
- Pierce A.M., Gimenez-Conti I.B., Schneider-Broussard R., Martinez L.A., Conti C.J., Johnson D.G. Increased E2F1 activity induces skin tumors in mice heterozygous and nullizygous for p53. Proc. Natl. Acad. Sci. USA. 1998;95:8858–8863. doi: 10.1073/pnas.95.15.8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshikawa Y., Mori A., Amimoto K., Iwabe K., Hatakeyama M. Control of retinoblastoma protein-independent hematopoietic cell cycle by the pRB-related p130. Proc. Natl. Acad. Sci. USA. 1998;95:8574–8579. doi: 10.1073/pnas.95.15.8574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamb A. Human melanoma genetics. J. Investig. Dermatol. Symp. Proc. 1996;1:177–182. [PubMed] [Google Scholar]
- Kamb A. Cyclin-dependent kinase inhibitors and human cancer. Curr. Top. Microbiol. Immunol. 1998;227:139–148. doi: 10.1007/978-3-642-71941-7_7. [DOI] [PubMed] [Google Scholar]
- Bartkova J., Lukas J., Strauss M., Bartek J. Cyclin D1 oncoprotein aberrantly accumulates in malignancies of diverse histogenesis. Oncogene. 1995;10:775–778. [PubMed] [Google Scholar]
- Hall M., Peters G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv. Cancer Res. 1996;68:67–108. doi: 10.1016/s0065-230x(08)60352-8. [DOI] [PubMed] [Google Scholar]
- Lukas J., Herzinger T., Hansen K., Moroni M.C., Resnitzky D., Helin K., Reed S.I., Bartek J. Cyclin E-induced S phase without activation of the pRb/E2F pathway. Genes Dev. 1997;11:1479–1492. doi: 10.1101/gad.11.11.1479. [DOI] [PubMed] [Google Scholar]
- Gray-Bablin J., Zalvide J., Fox M.P., Knickerbocker C.J., DeCaprio J.A., Keyomarsi K. Cyclin E, a redundant cyclin in breast cancer. Proc. Natl. Acad. Sci. USA. 1996;93:15215–15220. doi: 10.1073/pnas.93.26.15215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaban R., Funasaka Y., Lee P., Rubin J., Ron D., Birnbaum D. Fibroblast growth factors in normal and malignant melanocytes. Ann. NY Acad. Sci. 1991;638:232–243. doi: 10.1111/j.1749-6632.1991.tb49034.x. [DOI] [PubMed] [Google Scholar]
- Gaudray P., Szepetowski P., Escot C., Birnbaum D., Theillet C. DNA amplification at 11q13 in human cancerfrom complexity to perplexity. Mutat. Res. 1992;276:317–328. doi: 10.1016/0165-1110(92)90018-5. [DOI] [PubMed] [Google Scholar]