

Abstract
Members of the suppressor of cytokine signaling (SOCS) family were discovered as negative regulators of cytokine signaling by inhibition of the Janus kinase–signal transducer and activator of transcription (Jak-STAT) pathway. Among them, cytokine-induced Src homology 2 (SH2) protein (CIS) was found to inhibit the interleukin 3– and erythropietin-mediated STAT5 signaling pathway. However, involvement of SOCS proteins in other signaling pathways is still unknown. This study shows that the expression of CIS is selectively induced in T cells after T cell receptor (TCR) stimulation. In transgenic mice, with selective expression of CIS in CD4 T cells, elevated CIS strongly promotes TCR-mediated proliferation and cytokine production in vitro, and superantigen-induced T cell activation in vivo. Forced expression of CIS also prolongs survival of CD4 T cells after TCR activation. Molecular events immediately downstream from the TCR are not changed in CIS-expressing CD4 T cells, but activation of mitogen-activated protein (MAP) kinase pathways by TCR stimulation is significantly enhanced. Together with the increased MAP kinase activation, a direct interaction of CIS and protein kinase Cθ was also demonstrated. These results suggest that CIS is one of the important regulators of TCR-mediated T cell activation. The functions of CIS, enhancing TCR signaling and inhibiting cytokine signaling, may be important in the regulation of immune response and homeostasis.
Keywords: cytokine-induced SH2 protein, T cell receptor, signal transduction, mitogen-activated protein kinases, T cell activation
Introduction
The antigen engagement of TCRs initiates broad responses required for proper immune function, including cell proliferation, cytokine production, and apoptosis. TCR signaling is initiated by the activation of cytosolic protein tyrosine kinases such as Lck, ZAP-70, and SYK, which phosphorylate conserved motifs, immune receptor tyrosine-based activation motifs, within the cytoplasmic chains of the invariant polypeptide complex of TCR 1 2. The phosphorylation of immune receptor tyrosine-based activation motifs recruits several signaling molecules and activates various signal transduction pathways. Apart from the activation of cytoplasmic kinases, several adaptor proteins have been shown to be involved in TCR signaling. These adaptor proteins lack enzymatic and transcriptional domains but contain multiple motifs and domains that allow binding to other proteins. Therefore, they are capable of integrating the TCR signal into multiple downstream pathways 3 4. After the activation by TCR engagement, the ZAP-70/SYK kinases phophorylate adaptor proteins such as linker for activation of T cells (LAT) and Src homology 2 (SH2) domain–containing leukocyte protein of 76 kD (SLP-76). Phosphorylated LAT and SLP-76 directly bind to key proteins such as phospholipase Cγ, protein kinase C (PKC), and Grb2-Sos. The resulting Ras activation leads to activation of mitogen-activated protein (MAP) kinase pathways and subsequently the activation of transcription factors, including activating protein 1 (AP-1), nuclear factor κB, and nuclear factor of activated T cells 3 4.
Recently, a new adaptor protein family has been identified based on their structural similarities exhibiting an NH2-terminal region of variable length, a highly variable amino acid sequence, a central SH2 domain, and a striking region of COOH-terminal homology, designated as the suppressors of cytokine signaling (SOCS) box 5 6 7 8. Four members (cytokine-induced SH2 protein [CIS], Jak binding protein [JAB]/SOCS1/STAT-induced STAT inhibitor [SSI], SOCS2, and SOCS3) of this family were found to negatively regulate cytokine signal transduction via the interaction with phosphorylated cytoplasmic domains of cytokine receptors 5, with activated Jak kinases 7, or with signal transducer and activator of transcription (STAT) proteins 9. Forced expression of CIS in a myloid cell line, M1, could partially suppress IL-3–induced proliferation 5, and negatively modulated the activation of STAT5 9. More recently, suppression of STAT5 functions in liver, mammary glands, and T cells in CIS transgenic mice, was also discovered 10. However, the involvement of SOCS family proteins in other signaling pathways is unknown. We report in this study that CIS expression is induced in T cells by TCR stimulation. CD4 T cells from CIS transgenic mice, which express the transgene in the CD4 T cell lineage, exhibited enhanced proliferative responses upon TCR stimulation, and prolonged survival of activated T cells. The increased responses in CIS-expressing CD4 T cells are correlated with a significantly increased activation of MAP kinases after TCR stimulation, suggesting that CIS regulates the MAP kinase activation after TCR activation.
Materials and Methods
Differential Display.
Purified CD4 spleen T cells (5 × 106) were stimulated for 6 h with or without plate-bound anti-CD3 antibody (2C11, 10 μg/ml; PharMingen). After stimulation, poly-A RNA was purified and submitted to differential display analysis as described 11 using the Delta Differential Display kit (Clontech). Putative differentially expressed cDNA fragments were excised from dried sequencing gels, eluted in Tris-EDTA (TE) buffer, and reamplified using the same primer pair originally used in differential display. The differential expression of isolated fragments was confirmed by Northern blot analysis. PCR products from confirmed fragments were cloned into a TA cloning vector (Invitrogen), and sequenced.
Reverse Transcriptase PCR.
CD4 T cells from mouse spleen were stimulated with anti-CD3. Total RNA was extracted with Trizol solution (GIBCO BRL), reverse transcribed with a first strand cDNA synthesis kit (Amersham Pharmacia Biotech) using oligo-T primer, and amplified for expression of CIS, SOCS1, SOCS2, SOCS3, SOCS5, and IL-2. Primer pairs for amplification were: 5′-ACATGGTCCTCTGCGTACA-3′ and 5′-CAGCTGTCACATGCATGC-3′ for CIS; 5′-CGCCAACGGAACTGCTTCTTC-3′ and 5′-TCAGGTAGTCACGGAGTACC-3′ for SOCS1; 5′-TATATCCGTTAAGACGTCAGC-3′ and 5′-CTTGTTGGTAAAGGCAGTCC-3′ for SOCS2; 5′-TGTGTACTCAAGCTGGTGCAC-3′ and 5′-CATACTGATCCAGGAACTCC-3′ for SOCS3; and 5′-ATAAGTGGAGATGGTTCTGC-3′ and 5′-TCCTCCTGTGCAGAGTCC-3′ for SOCS5. The primers specific for glyceraldehyde 3-phosphate dehydrogenase (G3PDH) confirmed that equal amounts of RNA were present in each sample.
Transgenic Mice.
A murine full-length CIS cDNA was isolated from a mouse liver cDNA library (Stratagene), as described 12. The CIS cDNA in pBlueScript was excised and cloned into the SalI site of plasmid CD4 promoter vector p428 (a gift from N. Killeen, University of California, San Francisco). The vector-free CD4-CIS fragment was excised by NotI from the sense-oriented clone and used for construction of transgenic mice by injection of the linearized promoter transgene cassette (free of plasmid DNA) into the pronuclei of fertilized oocytes. Since the silencer of CD4 regulatory elements has been deleted, the CIS transgene is expressed in all T cells 13. All animals used in this study were 4–6 wk old. Normal and CIS transgenic mice of the same age were used in each experiment. T cell activation with SEA was by intraperitoneal injection of 10 μg of staphylococcal enterotoxin A (SEA) dissolved in saline as described 14.
Cell Proliferation Assay.
CD4 T cells were isolated from spleen cells by anti-CD4–coated magnetic beads (Dynabeads L3T4; Dynal). After each separation, the efficiency of CD4 cell purification was analyzed by flow cytometry and was routinely >95%. Cells at 105/well were cultured in 2 μg/ml anti-CD3, or as indicated, 2C11 (PharMingen), precoated plates, or anti-CD3 plus 4 μg/ml anti-CD28 (PharMingen) or 2 μg/ml Con A (PharMingen). The same amount of CD4 T cells were also cultured with PMA and ionomycin (40 ng/ml each; Sigma Chemical Co.). The proliferative response to IL-2 was analyzed under the condition that CD4 T cells were first stimulated with 5 μg/ml anti-CD3 for 48 h and then washed and recounted before addition of 100 U/ml IL-2 (R&D Systems). The cells were used for analysis at day 3 or as indicated in the figures after an 8-h pulse with 1 μCi/well [3H]thymidine. The incorporation of [3H]thymidine was measured with a β-counter.
ELISA and Flow Cytometry.
T cell culture supernatants were assayed in triplicate for IL-2 and IFN-γ by ELISA (PharMingen). The FITC-labeled mAbs anti-CD3, anti-CD4, as well as the PE-labeled anti-CD8 were purchased from PharMingen. Unseparated spleen or thymus cells were stained for 30 min at 4°C, washed, and analyzed on a Becton Dickinson FACScan™ flow cytometer.
Immunoprecipitation and Immunoblot Analysis.
Cell lysates of anti-CD3–stimulated CD4 T cells were prepared in 1% NP-40 lysis buffer with protease inhibitors (Boehringer Mannheim) and sodium vanadate, and immunoprecipitated with anti–ZAP-70 (Santa Cruz Biotechnology) or anti-CIS (Santa Cruz Biotechnology). Immune complexes were resolved by 10% SDS-PAGE and immunoblotted with antiphosphotyrosine (4G10; Upstate Biotechnology, Inc.) for detection of phospho–ZAP-70 or with anti-PKCθ (Santa Cruz Biotechnology) for detection of coprecipitated PKCθ. For confirming the expression of the CIS transgene, CD4 T cells from CIS transgenic mice were lysed and blotted by anti-CIS antiserum.
In Vitro Kinase Assay.
c-Jun NH2-terminal kinase (JNK) and extracellular signal–regulated kinase (ERK) from cell lysates of anti-CD3 (2 μg/ml) stimulated CD4 T cells were purified by immunoprecipitation with anti-JNK (Santa Cruz Biotechnology) or anti-ERK antibody (Santa Cruz Biotechnology). As described previously 15, kinase activity of JNK and ERK was measured by incubation of immunoprecipitated JNK or ERK with glutathione S-transferase (GST)-c-Jun or myelin basic protein (MBP), respectively, in the presence of 10 μCi [γ-32P]ATP in kinase buffer containing 20 mM Hepes (pH 7.0), 5 mM 2-ME, 10 mM MgCl2, 0.1 mg/ml BSA, 10 μM unlabeled ATP at 37°C for 20 min. The phosphorylated GST-c-Jun or MBP was analyzed by SDS-PAGE. The counts from each band were quantitated by PhosphorImager® using the ImageQuant® software (Molecular Dynamics).
Results
CIS Is an Immediate Early Gene Induced by TCR Stimulation in T Cells.
In search for molecules involved in TCR-stimulated T cell activation, poly(A)-selected RNA from CD4 T cells with or without anti-CD3 stimulation for 6 h, was displayed by arbitrarily primed PCR fingerprinting (differential display) as described previously 11. Eight differentially displayed bands were revealed from anti-CD3–stimulated CD4 T cells. PCR products were purified from these bands, reamplified, and sequenced. One of these was CIS (Fig. 1 a, lanes 1 and 2), indicated by its identical nucleotide sequence to the 3′ portion of the cDNA encoding CIS protein 5. To confirm the induction of CIS expression by TCR stimulation, naive T cells were incubated with or without anti-CD3 for different periods of time, and then total RNA was isolated and analyzed for CIS expression by Northern blot. CIS RNA was not expressed in naive CD4 T cells, but was induced after 30 min stimulation with anti-CD3 (Fig. 1 b). Immunohistochemical analysis demonstrated that CIS protein was detected exclusively in activated T blast cells after anti-CD3 stimulation (Fig. 1 c). For investigation of the dependence of CIS expression on TCR signaling in TCR-activated T cells, expression of CIS was examined in the TCR-activated T cells after removal of anti-CD3 for 16 h. The expression of CIS was barely detected either at the protein or RNA levels after withdrawal of anti-CD3 stimulation (Fig. 1b and Fig. c), suggesting that TCR signaling regulates CIS expression transcriptionally.
Figure 1.

CIS was induced by TCR activation. (a) Differentially displayed CIS transcript in anti-CD3–stimulated CD4 T cells. (b) Northern analysis of mRNA from CD4 T cells treated with anti-CD3 for different periods of time showing induced expression of CIS (lanes 1–4), and dependence on TCR activation (lane 5). The effect of cycloheximide or cyclosporin A on anti-CD3–induced CIS expression was analyzed by incubation with cyclosporin A together with anti-CD3 or addition of cycloheximide after anti-CD2 stimulation for 6 h (lanes 6–8). Probe specific for G3PDH confirmed that equal amounts of RNA were present in each sample. (c) The expression of CIS in CD4 T cells was analyzed by immunostaining with anti-CIS antibody (Santa Cruz Biotechnology) as described (reference 41). Induced CIS expression was detected in activated T blasts after 20-h stimulation with anti-CD3. Expressed CIS was degraded in 16 h after withdrawal of anti-CD3. (d) RT-PCR analysis of mRNA from CD4 T cells activated with anti-CD3 for different periods of time showing the kinetics of CIS, SOCS2, and IL-2 induction. Primers specific for G3PDH confirmed that equal amounts of RNA were present in each sample.
Since CIS can be induced by various cytokines, such as IL-2, IL-3, and erythropoietin (EPO; 5, 9), the expression of CIS in TCR-activated T cells could result from endogenously produced IL-2. To address this possibility, RNA extracted from CD4 T cells stimulated by anti-CD3 for different periods of time was analyzed for IL-2 expression by reverse transcription (RT)-PCR. In contrast to the early induction of CIS expression, IL-2 was detected only after anti-CD3 stimulation for >1 h (Fig. 1 d), which is well correlated with the results from mice with a fluorescent marker for IL-2 gene activation 16. In addition, CIS expression at a late time point of anti-CD3 stimulation in the presence of cycloheximide or cyclosporin A was examined by Northern blotting. The amount of induced CIS mRNA was not significantly altered by the protein synthesis inhibitor, but was reduced by the treatment of cyclosporin A (Fig. 1 b, lanes 6–8). This may indicate that in addition to STAT5, a calcium-dependent pathway is also involved in the regulation of CIS expression. Taken together, these results suggest that the immediate induction of CIS expression after TCR stimulation is transcriptionally regulated by TCR signaling, but not by autocrine IL-2.
Selective Expression of SOCS Family Members in TCR-activated T Cells.
The SOCS protein family consists of at least eight members (CIS, SOCS1–7; references 5 6 7 8, 17). To determine whether other members of the SOCS protein family apart from CIS are also induced in TCR-activated T cells, resting CD4 T cells were stimulated with anti-CD3 for different periods of time. After TCR stimulation, RNA was extracted and analyzed by RT-PCR for the expression of the four close homologues of CIS, SOCS1, SOCS2, SOCS3, and SOCS5 16. Selective expression of certain SOCS family members was revealed. Similar to CIS, SOCS2 was a TCR-induced immediate early gene indicative of a potential overlapping function with CIS in TCR signaling (Fig. 1 d). SOCS1, SOCS3, and SOCS5 were not induced by TCR stimulation (data not shown), suggestive of distinct functions of SOCS proteins in activated T cells.
Expression of CIS in CD4 T Cells of CD4-CIS Transgenic Mice Promotes TCR-mediated T Cell Activation.
To assess the physiological role of CIS protein in T cell function, we generated CIS transgenic mice in which CIS expression was driven by a CD4 promoter 13. After constructing the transgenic construct, we sequenced the CD4 promoter as well as CIS cDNA. The sequence was identical to the published sequence 5. The expression of the CIS transgene in CD4 T cells was verified by RT-PCR with one primer from the construct-derived CD4 sequence and the other primer derived from the CIS cDNA (Fig. 2 a), and by Western blotting with anti-CIS antiserum (Fig. 2 b). The molecular mass of transgenic CIS corresponded well to previously published results 5. Of the three transgenic lines used in this study, the level of transgene expression was highest in line 3, intermediate in line 8, and low in line 12. Expression of the CIS transgene in CD4 T cells was also directly detected by immunohistochemistry with anti-CIS antiserum (Fig. 2 c). Expression of the CIS transgene modestly reduced the number of thymocytes, but did not alter the normal profiles of thymocyte subpopulations (data not shown). The size and subset proportions of the peripheral T cell pool were unaffected. LNs were normal in size, the number of splenic T cells was not significantly increased, and the ratio of CD4 to CD8 cells was normal (Fig. 2 d).
Figure 2.

Expression of CIS transgene in CD4 T cells in CIS trangenic mice. (a) RT-PCR and (b) Western blot analysis of the CIS transgene expression in CD4 T cells showing expression of CIS transgene in three different transgenic lines. (c) Immunostaining of CD4 T cells with anti-CIS antibody showing expression of CIS transgene. (d) FACS® analysis of spleen cells showing normal profiles of T cell subpopulations.
As it has been found that CIS negatively regulates cytokine receptor signaling, we anticipated that CIS expression in T cells would inhibit T cell activation mediated by TCR. To examine the functionality of CD4 T cells from CIS transgenic mice, responses to TCR stimulation were analyzed by incubation of purified CD4 T cells with anti-CD3 antibody. In contrast to the inhibitory effect of CIS in cytokine-induced activation, CD4 T cells from spleens of CIS transgenic mice expressed remarkably enhanced proliferative responses to anti-CD3 stimulation (Fig. 3 a). CD4 T cells from spleens of normal mice exhibited only minor proliferative responses towards the anti-CD3 stimulation (Fig. 3 a). In agreement with previous reports 18, costimulation of normal CD4 T cells with anti-CD28 in addition to anti-CD3 induced pronounced proliferation (Fig. 3 a). Moreover, this type of costimulated response was significantly stronger in CD4 T cells expressing the CIS transgene than in CD4 T cells from normal mice (Fig. 3 a). A strongly enhanced proliferative response to PMA plus ionomycin stimulation was also detected in CD4 T cells from CIS transgenic mice compared with CD4 T cells from normal mice (Fig. 3 a). To exclude the effect of IL-2 produced by activated T cells, the proliferative response to anti-CD3 stimulation was also measured in the presence of anti–IL-2 antibodies. The antibodies did not affect the increased proliferative response of CIS-expressing CD4 T cells to TCR stimulation (Fig. 3 a). Previously, it was reported that the IL-2–stimulated proliferative response was reduced slightly in T cells expressing CIS 10. In this study, a very small decrease of the proliferative response to 100 U/ml IL-2 was observed, which may be due to a partial inhibition of STAT5 activated described below. These results suggested that CIS might be directly involved in TCR signaling. Moreover, when stimulated with anti-CD3, CD4 T cells expressing the CIS transgene secreted twice the amount of IL-2 and IFN-γ compared with CD4 T cells of normal mice (Fig. 3 b), further strengthening the evidence of increased activation of CD4 T cells from CIS transgenic mice.
Figure 3.


Elevated CIS expression promoted T cell proliferation and cytokine production. (a) Proliferation assay of CD4 T cells was performed after 3-d stimulation with indicated stimulators showing increased responses of CD4 T cells from CIS transgenic mice. (b) ELISA of IL-2 and IFN-γ (assay kit; PharMingen), showing increased expression of IL-2 and IFN-γ in the CD4 T cells from CIS transgenic mice after anti-CD3 stimulation for 3 d compared with normal mice.
CIS Prolongs Survival of CD4 T Cells After TCR Activation.
In addition to an enhanced proliferative T cell response to TCR stimulation in vitro, anti-CD3–activated CD4 T cells from CIS transgenic mice also survived longer after stimulation than did CD4 T cells from normal mice (Fig. 4 a). To further confirm the viability of activated CD4 cells, CD4 cells from spleens of CIS transgenic and normal mice were first stimulated with anti-CD3 for 3 d, then the anti-CD3 was removed on day 4. After removal of anti-CD3, cells were maintained in normal medium for 7 d. On day 10, cells were restimulated with anti-CD3. The proliferation of CD4 cells was measured at different time points after stimulation (Fig. 4 b). Significantly, after removal of the anti-CD3 stimulus, the CIS-expressing CD4 T cells, but not CD4 T cells from normal mice, could be readily reactivated by TCR stimulation after 7 d (Fig. 4 b). These results suggest that CIS homeostatically regulates the activated T cells by decreasing TCR-driven apoptosis.
Figure 4.


Prolonged survival of TCR-activated T cells from CIS transgenic mice. (a) TCR-mediated cell death. Concentrations of viable cells were determined in a hemocytometer, and viability was judged by trypan blue exclusion. (b) Reactivation of surviving CD4 T cells after withdrawal of the stimulator, anti-CD3, showing that CIS expression could increase activation and prolong survival of T cell in the absence of stimulator. Anti-CD3 stimulation is indicated by black bars.
CIS Expression Enhances T Cell Responses to Antigen In Vivo.
To test whether the augmented response to TCR activation in vitro translated into an enhanced T cell response to antigen in vivo, we stimulated CIS transgenic mice with the superantigen SEA. A superantigen–MHC complex is recognized by particular TCR Vβ elements regardless of their TCR-α partner 14; therefore, they can be used to measure a specific T cell proliferative response to antigen. SEA activates T cells expressing TCR Vβ3 14. Splenic CD4 T cells bearing Vβ3 were quantified over a period of 2 wk. In both normal and CIS transgenic mice, the response of CD4 TCR Vβ3 cells peaked on days 2–3. However, the expansion of Vβ3 cells was about twofold higher in the CIS transgenic mice on day 3 (Fig. 5). The peak response in normal mice dropped rapidly and was back to basal level by day 7 (Fig. 5), due to apoptotic death of activated T cells 14. The CD4 TCR Vβ3 cells in the transgenic mice declined slowly, remaining in excess for 2 wk after immunization (Fig. 5). These results indicate a functional role of CIS in the regulation and maintenance of the immune response to antigen in vivo.
Figure 5.
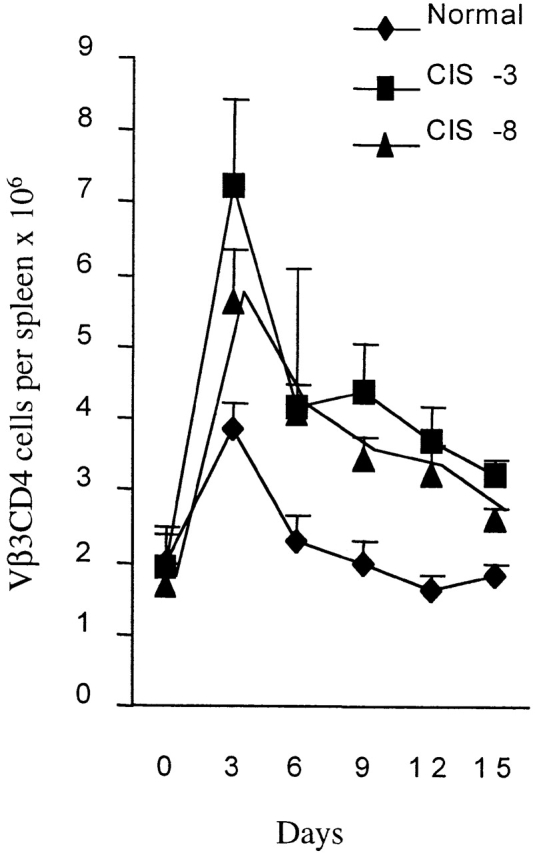
CIS-expressing Vβ3 CD4 T cells exhibited increased responsiveness to antigen in vivo. Vβ3-CD4 T cell responses to SEA were analyzed by FACS® after stimulation of mice with SEA (10 μg/ml intraperitoneally), showing increased and prolonged proliferative T cell response in CIS transgenic mice.
CIS Regulates MAP Kinase Activation in T Cells after TCR Stimulation.
Seeking the molecular mechanisms of CIS function in TCR signaling, both proximal and later events downstream of TCR signaling were analyzed in CIS transgenic CD4 T cells. The most proximal consequences of TCR activation, as indicated by phosphorylation of ZAP-70, were similar in normal and CIS transgenic CD4 T cells (Fig. 6 a). In contrast, more downstream events such as the activation of MAP kinases ERK and JNK were significantly increased upon anti-CD3 stimulation in CIS transgenic T cells (Fig. 6 b). The peak of JNK and ERK activation was at 10 min in both wild-type and CIS transgenic T cells. However, according to mean PhosphorImage® values of phosphorylated c-Jun or MBP, the level of JNK activity was increased more than sevenfold in CIS transgenic T cells after 10 min stimulation (Fig. 6 c). Similarly to JNK, ERK activity was increased in CIS transgenic T cells, but only about threefold (Fig. 6 c). These results indicate that induction of MAP kinase activation is the mechanism by which CIS regulates TCR activation.
Figure 6.

CIS induced strong MAP kinase activity. (a) Tyrosine phophorylation assay of ZAP-70 in response to TCR stimulation showed similar activation of ZAP-70 in CD4 T cells from normal and CIS transgenic mice. (b) Kinase assays of JNK and ERK from anti-CD3–activated CD4 T cells showing increased JNK and ERK activation in CIS transgenic CD4 T cells. The results were representative of three experiments with similar results. (c) Augmentation of the activity was calculated as the ratio between the mean PhosphorImage® values of stimulated samples and the mean values from unstimulated samples.
CIS Interacts with PKCθ in the Activated T Cells from Normal Mice.
To further understand the mechanisms of CIS function in T cell activation, coprecipitation experiments using anti-CIS antiserum were performed with normal CD4 T cells stimulated with anti-CD3 for 16 h. In activated CD4 T cells, a coprecipitated protein with a molecular mass of 80 kD was obtained (Fig. 7 a). The coprecipitated species was PKCθ confirmed by blotting of coprecipitates with anti-PKCθ antibody (Fig. 7 a). Since PKCθ is predominantly expressed in T cells and has the ability to stimulate AP-1 transcriptional activity by regulating JNK 19, the association of CIS with PKCθ may be important for regulation of the PKCθ activation of T cells. CIS was mostly studied in its role as a negative regulator of cytokine signaling, especially via the STAT5 pathway 10 20. Therefore, we analyzed STAT5 activation in normal and CIS-expressing CD4 T cells after anti-CD3 or IL-2 activation. STAT5 phosphorylation was not detected in anti-CD3–stimulated T cells either normal or CIS-expressing (Fig. 7 b). The IL-2–induced STAT5 activation was analyzed by stimulation of anti-CD3 preactivated T cells with 100 U/ml IL-2. Although the phosphorylation of STAT5 was detected in both normal and CIS-expressing T cells, the level of STAT5 phosphorylation in CIS-expressing T cells was slightly decreased (Fig. 7 b). This partial inhibition of STAT5 phosphorylation in CIS-expressing T cells is consistent with a previous report 20. Thus, the function of CIS association with PKCθ may be distinct from the inhibitory effect of CIS on STAT5 activation, and may be important in regulation of TCR signaling.
Figure 7.
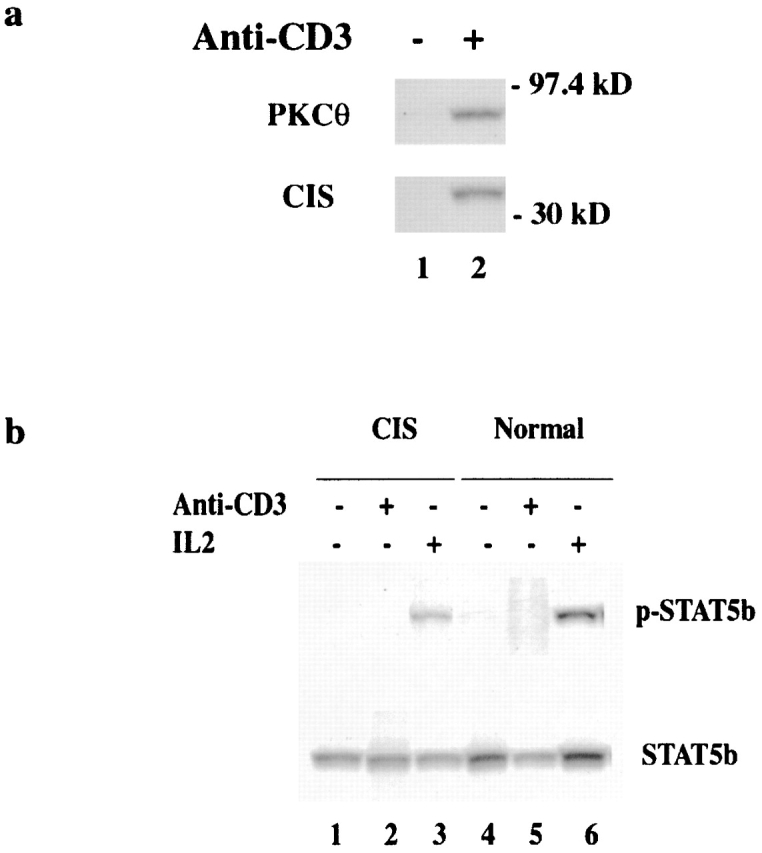
CIS interacts with activated PKCθ in activated normal T cells, and expression of CIS slightly modulates IL-2–induced STAT5 activation in activated T cells. (a) CD4 T cells from spleen of normal mice were incubated without (lane 1) or with anti-CD3 (lane 2) for 16 h. The activated CD4 T cells were lysed, and cleared lysates were precipitated with anti-CIS antiserum. The precipitates were separated on 10% SDS-PAGE and sequentially blotted by anti-PKCθ and anti-CIS antibodies, respectively. (b) The activation of STAT5 was directly analyzed in CD4 T cells after 15 min incubation with 5 mg/ml anti-CD3 and in CD4 T cells that were prestimulated for 48 h with 5 μg/ml anti-CD3 and then incubated for 15 min with 200 U/ml IL-2. The STAT5 was precipitated with anti-STAT5 antiserum and blotted by an antibody specific for phosphorylated STAT5. Western blot with anti-STAT5 antiserum showed similar amounts of STAT5 in all samples tested.
Discussion
Members of the SOCS protein family were discovered as negative regulators of cytokine signaling by inhibition of the Jak-STAT pathway 5 6 7 8. CIS is the first discovered SOCS protein and an immediate early gene induced by IL-2, IL-3, and EPO 5 9. CIS was demonstrated to act as a negative feedback loop for inhibition of IL-3– and EPO-activated Jak-STAT5 signaling by blocking the interaction of STAT5 and its docking sites on the cytoplasmic domain of the receptor chain 5. Forced expression of CIS inhibited IL-3–induced proliferation of M1 cells and inhibited EPO-dependent tyrosine phophorylation of STAT5 9. The function of CIS in other signaling pathways is unknown.
By differential display analysis of TCR-induced transcripts in T cells, we found that CIS is a TCR-induced immediate early gene. The expression of CIS in activated T cells was dependent on the presence of TCR stimulation, suggestive of an important role for CIS in the TCR signaling. This was demonstrated by a functional study of CIS-expressing T cells from CIS transgenic mice. In contrast to the inhibitory effect in IL-3 and EPO signaling, expression of CIS strongly promoted T cell proliferation, cytokine production, and prolonged survival of activated T cells. The regulation of TCR signaling correlates with the CIS-mediated activation of MAP kinases, indicated by the interaction of CIS with PKCθ in TCR-stimulated normal CD4 T cells as well as the significantly increased JNK and ERK activities in the CIS-expressing T cells after TCR stimulation.
Analysis of SOCS protein expression in TCR-stimulated T cells revealed selective induction of CIS and SOCS2 proteins, indicative of specificity of CIS in regulation of TCR signaling and the potential overlapping of function between CIS and SOCS2. The selective induction of SOCS family proteins by cytokines has also been observed. Thus, CIS and SOCS1, but not SOCS2 or SOCS3, were induced by IL-6 in the M1 cell line 6. These results together with ours suggest that despite the structural similarity among SOCS proteins, they selectively participate in different signaling pathways.
CIS is one of the STAT5-regulated genes. Results from STAT5a/b mutant mice showed that the expression of CIS in the thymus was dependent on the STAT5 proteins 21. Furthermore, two STAT5 binding sites were discovered within the CIS gene promoter 22. Therefore, CIS has been proposed to be a feedback modulator of STAT5. Its expression is induced by STAT5 and it negatively modulates STAT5 activation. Interestingly, it was found that activated STAT5 interacts with the TCR complex and stimulates T cell proliferation 23. However, this was the reverse of the recent findings, in which the activation of STAT5 specifically does not occur through the TCR signaling 24. We could not detect the STAT5 activation after TCR stimulation in T cells isolated from spleen of normal or CIS transgenic mice. This may be due to the difference of primary cells used in this study and cell lines used in the previous report 23. Even if STAT5 activation is induced after TCR stimulation, the strong enhancement of the proliferative response to TCR stimulation in CIS-expressing T cells suggested that the function of CIS in T cell activation may not associate with STAT5. Suppression of STAT5 functions by CIS was also shown by the study of the CIS transgenic mice under the control of a β-actin promoter 10. From that study, in addition to the inhibition of STAT5 activation in liver and mammary glands by the expression of CIS transgene, it inhibited IL-2–induced STAT5 activation in T cells 10. Moreover, consistent with our finding, anti-CD3 plus anti-CD28 was shown to induce CIS and TCR stimulation of CIS transgenic T cells, resulting in an augmented, rather than decreased, IL-2Rα expression 10. Given these results, the authors suggested that CIS might be involved in TCR signaling in a STAT5-independent manner 10. Interestingly, in β-actin promoter–driven CIS transgenic mice, the inhibition of STAT5 activation by the CIS transgene did not significantly affect the proliferation of T cells towards the IL-2 stimulation 10. This does not correlate with the findings from STAT5 knockout mice, in which the cell cycle progression of T cells induced by both anti-CD3 and IL-2 was completely abolished 21. Conflicting results with regard to inhibition of STAT5 activation by CIS were reported with a CIS-transfected T cell line 25. It was found that IL-2–induced tyrosine phosphorylation of STAT5b was markedly reduced in a T cell line expressing SOCS3 but not CIS 25. In our CIS trasgenic mice, the STAT5 activation after IL-2 stimulation was only partially inhibited by the expression of CIS transgene. We also found that the IL-2–induced proliferation of CIS-expressing T cells was not significantly altered. The systemic expression of CIS from β-actin promoter–driven transgenics may result in the developmental deficiency of certain Jak kinase function that may be required for STAT5 activation. This may not be the case in lineage-specific transgenics. Nevertheless, the positive regulation of TCR signaling and negative regulation of IL-2–induced STAT5 activation indicate a reciprocal function of CIS dependent on distinct stimuli.
In search for the mechanisms of CIS function in TCR signaling, we have examined proximal as well as downstream events of TCR signaling in CIS-expressing T cells after TCR stimulation. We could not detect the interaction of CIS with the TCR complex, ZAP-70, and Lck (data not shown). The activation of ZAP-70 after TCR stimulation was similar in normal and CIS-expressing T cells, indicating that CIS regulates downstream events of TCR signaling. Indeed, after TCR stimulation, a binding of CIS to PKCθ in normal T cells and a strong increase of MAP kinase activity in CIS-expressing T cells were demonstrated. Furthermore, the JNK activity was remarkably induced in CIS-expressing T cells and barely detected in normal T cells after TCR stimulation. ERK activity was also increased more than threefold in CIS-expressing T cells. On the basis of these results, we suggest that CIS is one of intermediate regulators that transmit a signal from TCR to MAP kinase pathways by regulation of the PKCθ.
PKCθ is preferentially expressed in T cells and selectively modulated during T cell activation 19 26 27. It stimulates the transcription factor AP-1 in T cells by activation of MAP kinase 19 26. The MAP kinases are essential for TCR-mediated activation by regulation of the AP-1 transcription factor composed of Jun, Fos, and activating transcription factor (ATF) subunits 28. Activated AP-1 regulates different target genes and thus executes distinct functions of activated T cells 24. Both ERK and JNK activation are required for T cell activation 28 29. However, while ligation of the TCR–CD3 complex activates ERK 30, the activation of JNK requires the occupation of both the TCR and CD28 31, resulting in optimal activation of T cells. The greatly enhanced JNK activation in CIS-expressing T cells may indicate an important role of CIS in the integration of TCR and CD28 signals. Interestingly, induction of functional inactivation or anergy in T cells correlates with the lack of ERK and JNK activation 32. After TCR activation, kinase cascades, regulated by multiple factors, activate the MAP kinases 28. Although several of these regulators have been identified, including the guanine nucleotide binding proteins p21ras, PKC 29, and also the Rho family GTPases, which activate the JNK 33, it is still largely unknown how the initial events after TCR activation regulate the activation of MAP kinase pathways. Several adapter proteins like Shc, Grb2, and SLP-76 have been shown to be involved in the regulation of MAP kinase activity 4. Grb2 was found to associate with LAT after phosphorylation of LAT by ZAP-70. Recruitment of Grb2 to LAT localizes Sos protein to Ras, resulting in Ras activation and subsequently activation of MAP kinase pathways 4. In parallel with the Grb protein family, the functions of the SOCS family proteins are also mediated by interaction of the SH2 domain of SOCS proteins to target molecules 3.
The different functions of CIS in different signaling pathways may largely depend on the target proteins in CIS-expressing cells. The association of CIS with PKCu and increased MAP kinase activity in CIS transgenic T cells after TCR stimulation indicate that the function of CIS in regulation of the T cell activation may be distinct from its inhibitory effect for cytokine signaling.
Our data provide functional and biochemical evidence for CIS regulation of T cell activation. The SOCS protein family has been identified on the basis of its negative modulation of cytokine signaling. Germline mutation of the SOCS1 gene resulted in growth retardation and death within 3 wk after birth 34 35. Multiple organ apoptosis including in lymphocytes was observed in SOCS1-deficient mice 36, which was due to the inhibition of fatal neonatal actions of IFN-γ 36 37. More recently, the negative regulation of fetal liver hematopoiesis was discovered from the study of SOCS3-deficient mice 38 indicating that SOCS proteins have more diverse functions than as an inhibitor of cytokine signaling. Indeed, rapid induction of SOCS3 by growth hormone 39 and inhibition of Tec tyrosine kinase by SOCS1 have been demonstrated 40, suggesting that SOCS proteins exhibit broad functions across multiple signaling pathways. The presented results clearly show that the function of CIS is not limited to negative regulation. The bidirectional regulation of T cell activation and cytokine signaling by CIS may represent a novel mechanism for controlling both immune responses and homeostasis.
Acknowledgments
We would like to thank Dr. N. Killeen (University of California, San Francisco) for providing the CD4 promoter vector, p428.
This work was supported by the Swedish Medical Council (grant K99-06X-13033-01A), the Foundation for Strategic Research (SSF), and the Medical faculty, Lund University.
Footnotes
Abbreviations used in this paper: AP-1, activating protein 1; ATF, activating transcription factor; CIS, cytokine-induced SH2 protein(s); ERK, extracellular signal–regulated kinase; G3PDH, glyceraldehyde 3-phosphate dehydrogenase; GST, glutathione S-transferase; JNK, c-Jun NH2-terminal kinase; LAT, linker for activation of T cells; MAP, mitogen-activated protein; MBP, myelin basic protein; PKC, protein kinase C; SEA, staphylococcal enterotoxin A; SH2, Src homology 2; SLP-76, SH2 domain–containing leukocyte protein of 76 kD; SOCS, suppressor of cytokine signaling; STAT, signal transducer and activator of transcription.
References
- Weiss A., Littman D.R. Signal transduction by lymphocyte antigen receptors. Cell. 1994;73:209–212. doi: 10.1016/0092-8674(94)90334-4. [DOI] [PubMed] [Google Scholar]
- Wang R.L., Samelson L.E. Complex complexessignaling at the TCR. Immunity. 1996;5:197–205. doi: 10.1016/s1074-7613(00)80315-5. [DOI] [PubMed] [Google Scholar]
- Clements J.L., Boerth N.J., Lee J.R., Koretzky G.A. Intergration of T cell receptor-dependent signaling pathways by adapter proteins. Annu. Rev. Immunol. 1999;17:89–108. doi: 10.1146/annurev.immunol.17.1.89. [DOI] [PubMed] [Google Scholar]
- Rudd C.E. Adaptors and molecular scaffolds in immune cell signaling. Cell. 1999;96:5–8. doi: 10.1016/s0092-8674(00)80953-8. [DOI] [PubMed] [Google Scholar]
- Yoshimura A., Ohkubo T., Kiguchi T., Jenkins N.A., Gilbert D.J., Copeland N.G., Hara T., Miyajima A. A novel cytokine inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin-3 and erythropoietin receptors. EMBO (Euro. Mol. Biol. Organ.) J. 1995;14:2816–2826. doi: 10.1002/j.1460-2075.1995.tb07281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr R., Willson T.A., Viney E.M., Murray L.J., Rayner J.R., Jenkins B.J., Gonda T.J., Alexander W.S., Metcalf D., Nicola N.A. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- Endo T.A., Masuhara M., YoKouchi M., Suzuki R., Sakamoto H., Mitsui K., Matsumoto A., Tanimura S., Ohtsubo M., Missawa H. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387:921–924. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- Naka T., Narazaki M., Hirata M., Matsumoto T., Minamoto S., Aono A., Nishimoto N., Kajta T., Taga T., Yoshizaki K. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;387:924–929. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- Matsumoto A., Masuhara M., Mitsui K., Yokouchi M., Ohtsubo M., Misawa H., Miyajima A., Yoshimura A. CIS, a cytokine inducible SH2 protein, is a target of the JAK-STAT5 pathway and modulates STAT5 activation. Blood. 1997;89:3148–3154. [PubMed] [Google Scholar]
- Matsumoto A., Seki Y., Kubo M., Ohtsuka S., Suzuki A., Hayashi I., Tsuji K., Nakahata T., Okabe M., Yamada S. Suppression of STAT5 functions in liver, mammary glands, and T cells in cytokine-inducible SH2-containing protein 1 transgenic mice. Mol. Cell. Biol. 1999;19:6396–6407. doi: 10.1128/mcb.19.9.6396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang P., Pardee A. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science. 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- Li S.-L., Sjögren H.O., Hellman U., Pettersson R.F., Wang P. Cloning and functional characterization of a subunit of the transporter associated with antigen processing. Proc. Natl. Acad. Sci. USA. 1997;94:8708–8713. doi: 10.1073/pnas.94.16.8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada S., Scarborough J.D., Killeen N., Littman D. A lineage-specific transcriptional silencer regulates CD4 gene expression during T lymphocyte development. Cell. 1994;77:917–929. doi: 10.1016/0092-8674(94)90140-6. [DOI] [PubMed] [Google Scholar]
- Webb S.R., Gascoigne N.R.J. T-cell activation by superantigens. Curr. Opin. Immunol. 1994;6:467–475. doi: 10.1016/0952-7915(94)90129-5. [DOI] [PubMed] [Google Scholar]
- Hibi M., Lin A., Smeal T., Minden A., Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- Naramura M., Hu R.J., Gu H. Mice with a fluorescent marker for interleukin 2 gene activation. Immunity. 1998;9:209–216. doi: 10.1016/s1074-7613(00)80603-2. [DOI] [PubMed] [Google Scholar]
- Hilton D.J., Richardson R.T., Alexander W.S., Viney E.M., Willson T.A., Sprigg N.S., Starr R., Nicholson S.E., Metcalf D., Nicola N.A. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc. Natl. Acad. Sci. USA. 1997;95:114–119. doi: 10.1073/pnas.95.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahinian A., Pfeffer K., Lee K.P., Kundig T.M., Kishihara K., Wakeham A., Kawai K., Ohashi P.S., Thompson C.B., Mak T.W. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 1993;261:609–612. doi: 10.1126/science.7688139. [DOI] [PubMed] [Google Scholar]
- Werlen G., Jacinto E., Xia Y., Karin M. Calcineurin preferentially synergizes with PKC-theta to activate JNK and IL-2 promoter in T lymphocytes. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:3101–3111. doi: 10.1093/emboj/17.11.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aman M.J., Migone T.-S., Sasaki A., Ascherman D.P., Zhu M.-H., Soldaini E., Imada K., Miyajima A., Yoshimura A., Leonard W.J. CIS associates with the interleukin-2 receptor β chain and inhibits interleukin-2-dependent signaling. J. Biol. Chem. 1999;274:30266–30272. doi: 10.1074/jbc.274.42.30266. [DOI] [PubMed] [Google Scholar]
- Moriggl R., Topham D.J., Teglund S., Sexl V., Mckay C., Wang D., Hoffmeyer A., Deursen J.V., Sangster M.Y., Bunting K.D. Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity. 1999;10:249–259. doi: 10.1016/s1074-7613(00)80025-4. [DOI] [PubMed] [Google Scholar]
- Verdier F., Rabionet R., Gouilleux F., Beisenherz-Huss C., Varlet P., Muller O., Mayeux P., Lacombe C., Gisselbrecht S., Chretien S. A sequence of the CIS gene promoter interacts preferentially with two associated STAT5A dimersa distinct biochemical difference between STAT5A and STAT5B. Mol. Cell. Biol. 1998;18:5852–5860. doi: 10.1128/mcb.18.10.5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte T., Leitenberg D., Dittel B.N., al-Ramadi B.K., Xie B., Chin Y.E., Janeway C.A., Jr., Bothwell A.L.M., Bottomly K., Fu X.-Y. STAT5 interaction with the T cell receptor complex and stimulation of T cell proliferation. Science. 1999;283:222–225. doi: 10.1126/science.283.5399.222. [DOI] [PubMed] [Google Scholar]
- Moriggl R., Sexl V., Piekorz R., Topham D., Ihle J.N. Stat5 activation is uniquely associated with cytokine signaling in peripheral T cells. Immunity. 1999;11:225–230. doi: 10.1016/s1074-7613(00)80097-7. [DOI] [PubMed] [Google Scholar]
- Cohney S.J., Senden D., Cacalano N.A., Yoshimura A., Mui A., Migone T.S., Johnston J.A. SOCS-3 is tyrosine phophorylated in response to interleukin-2 and suppresses STAT5 phophorylation and lymphocyte proliferation. Mol. Cell. Biol. 1999;19:4980–4988. doi: 10.1128/mcb.19.7.4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baier-Bitterlich G., Uberall F., Bauer B., Fresser F., Wachter H., Grunicke H., Utermann G., Altman A., Barier G. Protein kinase C-theta isoenzyme selective stimulation of the transcription factor complex AP-1 in T lymphocytes. Mol. Cell. Biol. 1996;16:1842–1850. doi: 10.1128/mcb.16.4.1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monks C.R., Kupfer H., Tamir I., Barlow A., Kupfer A. Selective modulation of protein kinase C-theta during T-cell activation. Nature. 1997;385:83–86. doi: 10.1038/385083a0. [DOI] [PubMed] [Google Scholar]
- Su B., Karin M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr. Opin. Immunol. 1996;8:402–411. doi: 10.1016/s0952-7915(96)80131-2. [DOI] [PubMed] [Google Scholar]
- Cantrell D. T cell antigen receptor signal transduction pathways. Annu. Rev. Immunol. 1996;14:259–274. doi: 10.1146/annurev.immunol.14.1.259. [DOI] [PubMed] [Google Scholar]
- Schwartz R. H. Costimulation of T lymphocytesthe role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell. 1992;71:1065–1068. doi: 10.1016/s0092-8674(05)80055-8. [DOI] [PubMed] [Google Scholar]
- Su B., Jacinto E., Hibi M., Karin M., Ben-Neriah Y. JNK is involved in signal integration during costimulation of T lymphocytes. Cell. 1994;77:727–736. doi: 10.1016/0092-8674(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Li W., Whaley C.D., Mondino A., Mueller D.L. Blocked signal transduction to the ERK and JNK protein kinases in anergic CD4+ T cells. Science. 1996;271:1272–1276. doi: 10.1126/science.271.5253.1272. [DOI] [PubMed] [Google Scholar]
- Hill C.S., Wynne H.J., Treisman R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell. 1995;81:1159–1170. doi: 10.1016/s0092-8674(05)80020-0. [DOI] [PubMed] [Google Scholar]
- Naka T., Matsumoto T., Narazaki M., Fujimoto M., Morita Y., Ohsawa Y., Saito H., Nagasawa T., Uchiyama Y., Kishimoto T. Accelerated apoptosis of lymphocytes by augmented induction of Bax in SSI-1 (STAT-induced STAT inhibitor-1) deficient mice. Proc. Natl. Acad. Sci. USA. 1998;95:15577–15582. doi: 10.1073/pnas.95.26.15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr R., Metcalf D., Elefanty A.G., Brysha M., Willson T.A., Nicola N.A., Hilton D.J., Alexander W.S. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc. Natl. Acad. Sci. USA. 1998;95:14395–14399. doi: 10.1073/pnas.95.24.14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander A.S., Starr R., Fenner J.E., Scott C.L., Handman E., Sprigg N.S., Corbin J.E., Cornish A.L., Darwiche R., Owczarek C.M. SOCS1 is a critical inhibitor of interferon γ signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- Marine J.-C., Topham D.J., McKay C., Wang D., Parganas E., Stravopodis D., Yoshimura A., Ihle J.N. SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell. 1999;98:609–616. doi: 10.1016/s0092-8674(00)80048-3. [DOI] [PubMed] [Google Scholar]
- Marine J.-C., McKay C., Wang D., Topham D.J., Parganas E., Nakajima H., Pendeville H., Yasukawa H., Sasaki A., Yoshimura A. SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell. 1999;98:617–627. doi: 10.1016/s0092-8674(00)80049-5. [DOI] [PubMed] [Google Scholar]
- Adams T.E., Hansen J.A., Starr R., Nicola N.A., Hilton D.J., Billestrup N. Growth hormone preferentially induces the rapid, transient expression of SOCS3, a novel inhibitor of cytokine receptor signaling. J. Biol. Chem. 1997;273:1285–1287. doi: 10.1074/jbc.273.3.1285. [DOI] [PubMed] [Google Scholar]
- Ohya K.-i., Kajigaya S., Yamashita Y., Miyazato A., Hatake K., Miura Y., Ikeda U., Shimada K., Ozawa K., Mano H. SOCS-1/JAB/SSI-1 can bind to and suppress Tec protein-tyrosine kinase. J. Biol. Chem. 1997;272:27178–27182. doi: 10.1074/jbc.272.43.27178. [DOI] [PubMed] [Google Scholar]
- Li S.-L., Kaaya E., Ekman M., Ordonez C., Feichtinger H., Putkonen P., Böttiger D., Biberfeld G., Biberfeld P. Thymus histopathology in relation to SIVsm infection of cynomolgus monkeys. J. AIDS. 1995;9:1–10. [PubMed] [Google Scholar]