

Abstract
Multiple sclerosis is a demyelinating disorder of the central nervous system (CNS), in which an immune attack directed against myelin constituents causes myelin destruction and death of oligodendrocytes, the myelin-producing cells. Here, the efficacy of nerve growth factor (NGF), a growth factor for neurons and oligodendrocytes, in promoting myelin repair was evaluated using the demyelinating model of experimental allergic encephalomyelitis (EAE) in the common marmoset. Surprisingly, we found that NGF delayed the onset of clinical EAE and, pathologically, prevented the full development of EAE lesions. We demonstrate by immunocytochemistry that NGF exerts its antiinflammatory effect by downregulating the production of interferon γ by T cells infiltrating the CNS, and upregulating the production of interleukin 10 by glial cells in both inflammatory lesions of EAE and normal-appearing CNS white matter. Thus, NGF, currently under investigation in human clinical trials as a neuronal trophic factor, may be an attractive candidate for therapy of autoimmune demyelinating disorders.
Keywords: multiple sclerosis, nonhuman primate, interferon γ, interleukin 10, growth factors
Introduction
Multiple sclerosis (MS) is a chronic, relapsing-remitting disease of the central nervous system (CNS) white matter characterized pathologically by perivenular infiltration of mononuclear cells and macrophages, demyelination with astrocyte proliferation and gliosis, and death of the oligodendrocytes 1. Based in part on analogy with the disease model experimental allergic encephalomyelitis (EAE), MS is thought to be an autoimmune disorder mediated by autoaggressive T cells that recognize myelin antigens in the context of class II HLA molecules 2. However, because neither the putative antigens that trigger these autoimmune responses nor the precise mechanisms responsible for demyelination in human MS have been unequivocally identified, the design of therapies that aim to prevent myelin destruction remains a challenge 2.
Nerve growth factor (NGF), a pleiotropic cytokine of the neurotrophin family, promotes the biosynthesis of myelin by oligodendrocytes in the CNS 3 4 and by Schwann cells in the peripheral nervous system 5. These myelin-promoting effects of NGF appear to be mediated by the high affinity NGF receptor TrkA rather than by the low affinity NGF receptor p75NGFR 6 7. In addition to its role in myelination, neuronal growth, and CNS differentiation 8, NGF is known to have pleiotropic effects on various cells of the immune system 9. The immunomodulatory properties of NGF, and its trophic effects on myelin-producing cells, provide a framework for the use of NGF in the treatment of immune-mediated demyelinating disorders. Here, we report that therapy with recombinant human (rh)NGF protects against CNS inflammation and demyelination in a nonhuman primate model of EAE, the common marmoset Callithrix jacchus. This antiinflammatory effect of NGF has not been previously reported, and appears to be mediated by an alteration of the balance between Th1 and Th2 cytokines within the CNS.
Materials and Methods
Animals and Treatment Groups.
C. jacchus marmosets were maintained in a primate colony at the University of California, San Francisco, and were cared for in accordance with all institutional guidelines. Phlebotomy and cerebrospinal fluid (CSF) collection were performed under anesthesia in accordance with standard approved protocols. For intracerebroventricular administration of treatments, a sterile 36-gauge polycarbonate cannula secured inside a stainless steel brain cannulation device (Alzet) was stereotaxically implanted into the right lateral cerebral ventricle under anesthesia with isoflurane (coordinates: AP 6 mm, L 1.25 mm, V 7 mm, relative to bregma 10). The cannula was secured to the skull with miniature bone screws and dental acrylic cement, and connected by a 28-gauge polyvinyl tubing tunneled under the skin to an osmotic minipump (model 2004; Alzet) delivering 0.9% NaCl and implanted subcutaneously on the right flank. 7–14 d after surgery, on the day when EAE was induced, subcutaneous treatment minipumps containing either rhNGF or cytochrome c (CYT) as a placebo (Boehringer) at a concentration of 1 mg/ml were substituted. The minipumps delivered 6 μl/d, and due to the dead volume of the tubing connecting the minipump and the brain cannula, drugs were delivered beginning 7 d after immunizations and until completion of the experiments.
Induction and Assessment of EAE.
EAE was induced by active immunization with 100 μg of a recombinant protein corresponding to the extracellular domain of rat MOG (rMOG) in combination with CFA and Bordetella pertussis as described previously 11. Clinical signs of EAE were recorded daily by an observer blinded to the treatment groups using a grading scale specifically developed for marmosets in our laboratory (Table ). 28 d after immunization, animals were killed under deep phenobarbital anesthesia by intracardiac perfusion with PBS followed by either 4% buffered paraformaldehyde or 3% paraformaldehyde/0.2% glutaraldehyde. Brain hemispheres, optic nerves, and spinal cord were removed and dissected in 1-mm-thick consecutive blocks, which were paraffin-embedded for routine histology and/or cryopreserved and stored at −80°C for immunocytochemistry. Pathological scores were assigned to paired sections throughout the entire neuraxis, using a grading scale to quantitate inflammation and demyelination: 0, normal; +, rare (1–3/section) perivascular cuffs with minimal demyelination; ++, 3–10 perivascular cuffs/section accompanied by moderate demyelination; +++, widespread perivascular cuffing, extensive demyelination with large confluent lesions.
Table 1.
Expanded Disability Status Scale for Marmoset EAE
| Function | Disability score | Maximal score | |
|---|---|---|---|
| 1 | Alertness | 0: normal; 1: reduced; 2: lethargic | 2 |
| 2 | Spontaneous mobility | 0: normal; 1: mild slowing; 2: marked slowing; 3: absent | 3 |
| 3 | Tremor | 0: none; 1: moderate; 2: severe | 2 |
| 4 | Tone | 0: normal; 1: mildly reduced; 2: markedly reduced; 3: absent | 12 |
| 5 | Motor (grip) | 0: normal; 1: mildly reduced; 2: markedly reduced; 3: absent | 12 |
| 6 | Sensory | ||
| Light touch | 0: normal; 1: reduced; 2: absent | 8 | |
| Pain | 0: normal; 1: reduced; 2: absent | 8 | |
| 7 | Eye movements | 0: normal; 1: abnormal | 1 |
| 8 | Vision (including pupillary reflex) | 0: normal; 1: abnormal; 2: absent | 2 |
| 9 | Vocalization | 0: normal; 1: changed | 1 |
| 10 | Bladder function | 0: normal; 1: abnormal | 1 |
| 11 | Other signs | 0: normal; 1: abnormal | 1 |
The total score is derived by adding the scores for each system. The maximal score for the scale is 45.
Immunocytochemistry.
Sections of brain and spinal cord (5–30 μm) were pretreated with 1% H2O2 in Tris-Cl buffer, and blocked with 5% normal goat serum (Vector Laboratories) and 0.3% Triton X-100 for 60 min. Sections were incubated in sequence with the primary antibodies overnight at 4°C, an appropriate biotinylated secondary antibody, and avidin–horseradish peroxidase complex (Vectastain ABC; Vector Laboratories), then developed with 3, 3o-diamino-benzidine (Sigma-Aldrich) and counterstained with hematoxylin (Research Genetics). The primary antibodies and their working dilutions were as follows: for NGF receptors, rabbit anti–rat TrkA (RTA), 1:1,000; rabbit anti–mouse p75NGFR (Rex), 1:1,000 (both provided by Dr. Louis Reichardt, University of California, San Francisco); rabbit anti–human TrkA (Sc 118; Santa Cruz Biotechnology, Inc.), 1:1,000; mouse anti–human p75NGFR (Boehringer), 1:1,000; for cytokines, mouse anti–monkey IFN-γ (MD1; U-Cytech), 1:50; and mouse anti–human IL-10 (MCA B-S10; Instruchemie), 1:100. Slides stained for IFN-γ and IL-10 were pretreated by overheating in a microwave for retrieval of antigens. For each cytokine, four consecutive slides on two sections from each animal were stained, which included an average of 12 ± 10 (mean ± SD) inflammatory infiltrates per slide; quantitation of infiltrates and positively stained cells was done using a grid of 250 × 250 μm.
Bioassay for rhNGF.
CSF levels of rhNGF were determined using a specific bioassay of cell survival in primary cultures of neural crest–derived chicken embryonic sensory neurons 12. In brief, dorsal root ganglia neurons were extracted from embryos, cultured in F14 medium with 10% horse serum and antibiotics, and then seeded onto poly-ornithine/laminin–coated wells in microtiter plates in the presence or absence of known concentrations of rhNGF, or aliquots of CSF (10–50 μl). Neuronal survival was assessed after 48 h by counting the number of surviving neurons with neurite elongation under a phase–contrast microscope. Neurotrophic activity was expressed as one half of the maximal stimulation index (EC50), e.g., 100 pg/ml for rhNGF.
Assessment of Peripheral Immune Reactivity.
T cell proliferative responses were performed using a standard 3H[thymidine] incorporation assay with 2 × 105 freshly isolated marmoset or human PBMCs 11 incubated with the following: no antigen; rMOG, 10 μg/ml; PHA, 2 μg/ml; killed Staphylococcus aureus Cowan strain (SAC), 1:10,000; LPS, 20 ng/ml; or rhNGF, 100 ng/ml. Stimulation indices were calculated as the ratio of 3H[thymidine] incorporation in stimulated to unstimulated (medium alone) wells. Serum antibody responses were tested by ELISA in 96-well plates coated with 1 μg rMOG/well using 100 μl of serum dilutions of C. jacchus sera and 100 μl of peroxidase-conjugated anti–monkey IgG (1:4,000; Sigma-Aldrich) 11. Plates were developed with o-phenylenediamine-peroxidase substrate and read at 490 nm in a Vmax ELISA reader (Molecular Devices).
Flow Cytometry.
Staining for flow cytometry studies was performed after blockade of Fc receptors with PBS plus 1% BSA and 10% goat serum (Vector Laboratories). Cells were stained with the following antibodies and the appropriate isotype controls: 20 μl unconjugated goat anti–human TrkA or mouse anti–human p75NGFR antibody followed by FITC-conjugated anti–goat or anti–mouse IgG (Jackson ImmunoResearch Laboratories); double staining was performed with PE-conjugated anti-CD4 (Beckman-Coulter), CD20 (Immunotech), or CD14 (Beckman-Coulter). Samples were analyzed in a Becton Dickinson FACScan™ apparatus, and data analysis was done by subtracting isotype control percentage from experimental percentage.
Statistical Analysis.
Clinical and histological scores in the treatment groups were compared with the Mann-Whitney sum rank test, using StatView II software (Abacus). Quantitative immunohistochemistry for IFN-γ and IL-10 was analyzed by the Student's t test. A P value ≤ 0.05 was considered significant. Results are expressed as mean ± SD.
Results and Discussion
NGF Inhibits the Development of EAE in C. jacchus Marmosets.
Beginning 7 d after induction of EAE, C. jacchus marmosets were randomly assigned to treatment with either rhNGF or CYT as placebo administered via the intracerebroventricular cannula. In this experimental design, we chose to use an intracranial route to ensure accurate delivery of the drug into the CNS. This resulted in sustained elevated concentrations of rhNGF in the CSF of all rhNGF-treated animals (2–300 ng/ml), as measured by a specific bioassay.
Consistent with previous experience in MOG-sensitized marmosets 11, six placebo (CYT)-treated animals developed moderate to severe clinical signs of EAE beginning 10–16 d after immunization. Clinical signs in most of these animals progressed by the end of the study (day 28 after immunization). By contrast, a total of six rhNGF-treated marmosets showed either no clinical signs or a markedly attenuated disease course (Table ). In those rhNGF-treated animals that developed clinical signs, the onset of EAE was delayed compared with controls (day 21 ± 0.9 vs. day 11 ± 0.4 after immunization, P = 0.02). Maximal clinical scores observed averaged 12.5 ± 2.3 in controls vs. 2.6 ± 0.9 in rhNGF-treated animals (P = 0.01).
Table 2.
Clinical Findings in rhNGF- and Placebo-treated Animals
| Onset of EAE | ||||
|---|---|---|---|---|
| Animal | Day pi | Score | Maximal clinical score | (Day pi) |
| rhNGF group | ||||
| 26.30.92 | 28 | 2 | 2 | (28) |
| 26.29.94 | 14 | 3 | 6.5 | (28) |
| UC5.95 | 20 | 3.5 | 4 | (27) |
| 101 | 19 | 2 | 2.5 | (18) |
| 231.90 | – | 0 | 0 | (–) |
| FL 5.96 | 21 | 1 | 1 | (21) |
| Placebo group | ||||
| 26.17.94 | 17 | 11 | 14 | (17) |
| 26.33.93 | 10 | 2 | 21 | (28) |
| 157.94 | 7 | 2 | 5 | (19) |
| 198.94 | 7 | 2 | 14.5 | (28) |
| FL 3.95 | 15 | 1 | 7.5 | (28) |
| FL 6.96 | 14 | 1 | 13 | (19) |
pi, post immunization.
Neuropathological evaluation confirmed the observed clinical protection. Placebo-treated animals displayed typical large perivascular infiltrates comprised of mononuclear cells and macrophages accompanied by extensive concentric demyelination with gliosis widely distributed throughout the CNS. In rhNGF-treated marmosets, the number and size of the inflammatory infiltrates were reduced and demyelination was minimal (Fig. 1, and Table ). Although differences in severity and in the affected CNS regions existed between individual marmosets, the disease burden was greatly reduced in animals treated with rhNGF compared with controls. In both treatment groups, the amounts of demyelination and inflammation were concordant. Thus, treatment with rhNGF inhibited the development of inflammatory demyelination in marmosets.
Figure 1.

Representative neuropathological findings in placebo- (A) and rhNGF-treated (B) animals at the completion of the study (day 28 after immunization). Gross anatomical views of coronal sections through the temporal lobe illustrate multiple demyelinating plaques disseminated throughout the subcortical white matter in the control. In the rhNGF-treated animal, the density of plaques is markedly reduced. The paraffin-embedded sections were stained with Luxol Fast blue/periodic acid-Schiff.
Table 3.
Neuropathological Findings
| CNS region | |||||||
|---|---|---|---|---|---|---|---|
| Animal | Subcortical white matter | Corpus callosum | Optic chiasm, nerves, and tracts | Brain stem, cerebellum | Cervical spinal cord | Thoracic spinal cord | Lumbar spinal cord |
| rhNGF group | |||||||
| 26.30.92 | ++ | + | ++ | 0–+ | + | + | 0 |
| 26.29.94 | + | 0–+ | 0–+ | 0 | 0 | 0–+ | 0–+ |
| UC5.95 | 0 | + | 0 | 0 | + | 0 | 0 |
| 101 | 0 | 0 | 0 | 0 | + | 0 | 0 |
| 231.90 | + | 0 | + | + | + | + | + |
| FL 5.96 | + | ++ | + | 0 | 0 | 0 | 0 |
| Placebo group | |||||||
| 26.17.94 | +++ | ++ | +++ | ++ | +++ | +++ | ++ |
| 26.33.93 | +–++ | +–++ | +++ | +–++ | +++ | +++ | ++–+++ |
| 157.94 | 0 | 0 | +++ | 0 | ++ | 0 | 0 |
| 198.94 | + | 0 | + | 0 | +++ | ++–+++ | ++ |
| FL 3.95 | 0 | + | ++ | + | ++ | 0 | 0 |
| FL 6.96 | 0 | + | 0 | 0 | ++ | 0 | 0 |
Paired CNS sections were scored using a combined scale for inflammation and demyelination (0 to +++; see Materials and Methods).
Several considerations dictated the choice of the C. jacchus model of EAE for this study. First, this nonhuman primate species is best suited to test the efficacy of the human NGF protein, due to potential species-related differences in the expression of the NGF receptors TrkA and p75NGFR 13. Second, pathological features of rMOG-induced EAE in C. jacchus include moderate CNS inflammation, an early demyelinating component, and significant remyelination, all characteristics found in human MS. Third, the mechanisms of demyelination in C. jacchus EAE and MS are similar and involve vesicular disruption of myelin mediated by autoantibodies that recognize MOG 14. Treatment with rhNGF was begun 7 d after immunization, presumably at a stage of EAE where immune responses against the immunizing antigen had already developed. We had hypothesized that in this experimental design the effect of treatment, if any, would be to promote clinical recovery and remyelination in the rhNGF-treated animals. Surprisingly, rhNGF prevented the initial development and delayed the onset of clinical signs of EAE, and suppressed both the inflammatory and demyelinating components of CNS pathology. Because most of the rhNGF-treated animals displayed only minimal demyelination, it was not possible to assess differences in remyelination.
Based on in vitro observations in rodents, several growth factors with trophic and survival effects on oligodendrocytes have been considered for therapy in demyelinating disorders, including insulin-like growth factor (IGF)-1, glial growth factor (GGF)-2, platelet-derived growth factor, ciliary neurotrophic factor, and fibroblast growth factor 15. In vivo, both IGF-1 and GGF-2 15 16 have been shown to be capable of reducing disease severity in murine EAE; however, exacerbation of disease has also been described with IGF-1 17. It is important to recognize that, due to the interspecies differences in the biological effects of growth factors and the differential expression of their specific receptors, information derived from rodent studies may not be applicable to humans. Our study clearly demonstrates that NGF protects against CNS inflammatory and demyelinating disease in a nonhuman primate species that is known to develop EAE lesions that are indistinguishable from acute lesions of MS 14. Although NGF has been shown to ameliorate experimental allergic neuritis 18, to our knowledge this growth factor has not been tested in rodent EAE and it is possible that its protective effect is restricted to primate species.
Mechanisms Responsible for the Protective Effects of NGF.
Expression of NGF receptors in the CNS of marmosets was studied by immunocytochemistry, using antibodies against human or rat surface antigens. Both the high affinity NGF receptor (TrkA) and the low affinity NGF receptor (p75NGFR) were detected in marmoset basal forebrain neurons, indicating that these antibodies fully cross-reacted with C. jacchus NGF receptors (Fig. 2 A). In CNS white matter, widespread expression of TrkA and p75NGFR was detected in cells that had the morphologic appearance of astrocytes and/or microglial cells (Fig. 2 B) and in oligodendrocytes (not shown). In addition, a proportion of the mononuclear cells and macrophages comprising the inflammatory infiltrates of EAE lesions were positive for TrkA and p75NGFR (Fig. 2 C). Expression of NGF receptors on marmoset immune cell subsets was studied by FACS® analysis, using double staining of PBMCs. Both the TrkA receptor and the p75NGFR were widely expressed in CD4+ T cells (52 and 67%, respectively), monocytes (57 and 56%), and in a small subpopulation of B cells (6 and 9%) (data not shown). Thus, in this nonhuman primate system, glial and inflammatory cells within the CNS and a large proportion of immune cells in the periphery expressed NGF receptors, raising the possibility that several regulatory pathways may have participated in the protective effect of NGF on EAE.
Figure 2.
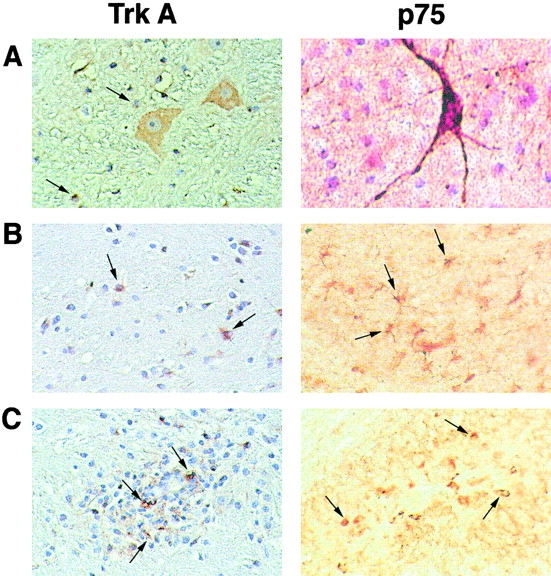
Characterization of NGF receptors in marmoset brain by immunohistochemistry. Staining for TrkA is shown at the left (5-μm paraffin-embedded section), and for p75NGFR at the right (30-μm-thick section stained in flotation). (A) Both NGF receptors are present in cholinergic neurons of the basal forebrain. Arrows show satellite oligodendrocytes stained for TrkA (left). (B) Arrows indicate some of the TrkA- and p75NGFR-positive glial cells (astrocytes or microglia) in normal white matter. (C) In CNS inflammatory infiltrates, strong staining for both NGF receptors is also apparent on many mononuclear cells and macrophages (arrows). Original magnifications: ×400.
To assess whether treatment with rhNGF modified the response of the peripheral immune system to the immunizing antigen, MOG-induced proliferative responses in PBMCs and serum anti-MOG antibody titers were monitored at days 0, 14, and 28 of the study. MOG-specific proliferative responses and anti-MOG antibody titers were similar in rhNGF- and placebo-treated animals (Fig. 3), indicating that peripheral T cell and B cell responses were not affected by intracerebral administration of NGF. Additional experiments using PBMCs and macrophages isolated from normal subjects and stimulated with PHA, SAC, or LPS in the presence or absence of exogenous rhNGF (1–1,000 ng/ml) confirmed that rhNGF did not interfere with either proliferative responses or macrophage reactivity. Finally, the expression of NGF receptors on subsets of PBMCs was not modified by preincubation with exogenous rhNGF at concentrations ranging from 1 to 1,000 ng/ml (data not shown). Taken together, these data suggest that treatment with NGF inhibited the development of EAE through interference with effector mechanisms of EAE pathology rather than through modulation of the induction phase of EAE.
Figure 3.
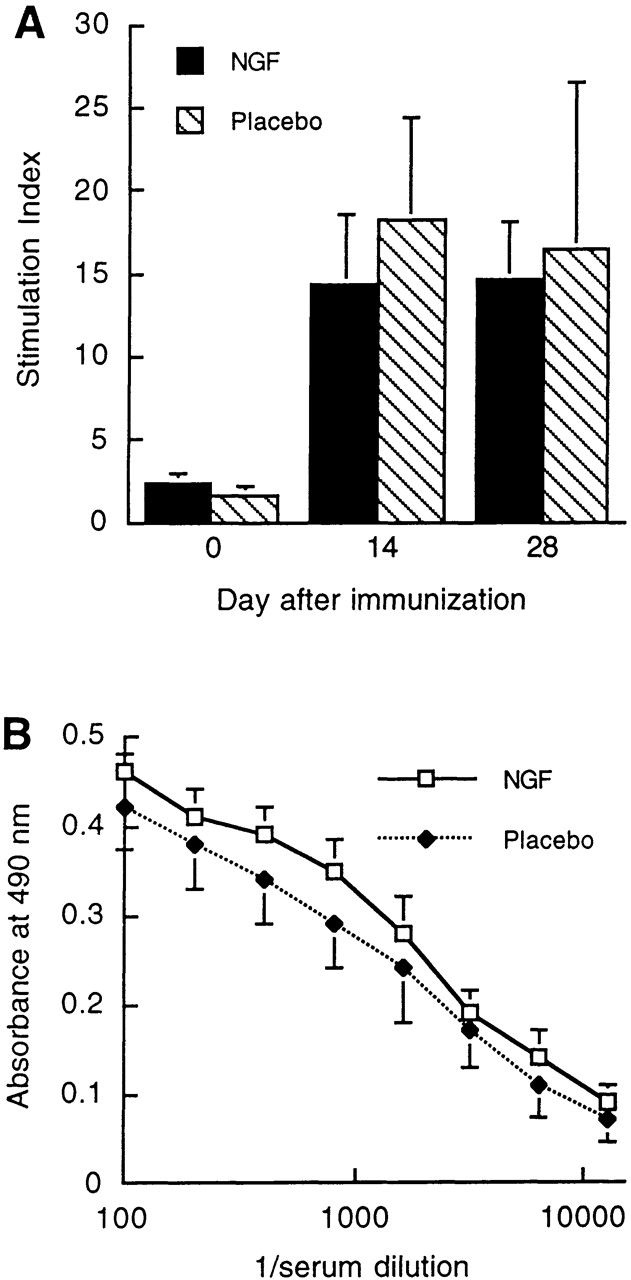
Serial measurements of T cell and antibody reactivity against MOG during the course of EAE in placebo- and rhNGF-treated marmosets. (A) MOG-specific T cell proliferative responses in marmoset PBMCs at days 0, 14, and 28 after immunization. (B) Serum anti-MOG antibody titers measured by ELISA at day 28 after immunization. The differences between placebo- and rhNGF-treated animals were not statistically significant.
Cytokines produced by T cells may profoundly influence the course of autoimmune disorders 19. In rodents and in marmosets, CNS inflammation results from the priming of Th1 cells that secrete proinflammatory cytokines such as TNF-α and IFN-γ. Conversely, EAE can be inhibited by immunosuppressive cytokines (IL-4, IL-5, IL-10, and TGF-β) produced by Th2 cells (for a review, see reference 2). In the current study, we examined the expression of IFN-γ and IL-10 within the CNS of placebo- and NGF-treated marmosets by immunocytochemistry. Results are presented in Fig. 4 and Table . In agreement with the scores of standard histological analysis, placebo-treated controls had significantly more infiltrates than rhNGF-treated animals (20.2 ± 9.0 vs. 7.3 ± 8.0, respectively). Consistent with a Th1 response, IFN-γ was expressed at high levels by infiltrating inflammatory cells in these controls. In marked contrast, IFN-γ was mostly absent from perivascular inflammatory cuffs in rhNGF-treated marmosets (Fig. 4A and Fig. B). An opposite pattern was seen for the expression of IL-10, which was markedly upregulated in inflammatory infiltrates of rhNGF-treated animals compared with controls. Quantitative analysis confirmed that these differences between the two treatment groups were significant, with the exception of the absolute number of IL-10–positive inflammatory cells per infiltrate (Table ). This was because IL-10 was expressed in lesions from controls, which included more inflammatory cells than infiltrates from rhNGF-treated animals. However, the percentage of IL-10–positive cells per infiltrate was increased in rhNGF-treated marmosets (Table ). In addition to inflammatory lesions, IL-10 was distinctly expressed at high levels in glial cells of normal-appearing white matter in NGF-protected marmosets. These cells had the morphology of astrocytes, although activated microglia cannot be formally excluded, and did not express IFN-γ (Fig. 4C and Fig. D; Table ). Taken together, these results indicate that the protective effect of NGF was mediated through local modulation of the intracerebral network of cytokines produced by inflammatory cells of the perivascular infiltrates, and by glial cells in the unaffected CNS white matter.
Figure 4.
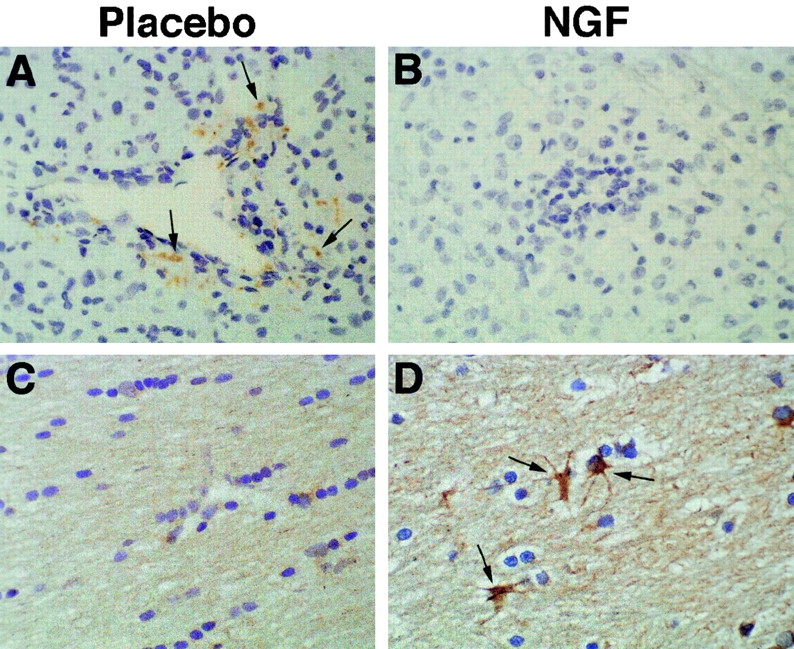
Effects of rhNGF on cytokine production within the CNS of nonhuman primates. Representative sections from placebo-treated (left) and rhNGF-treated (right) marmosets. (A and B) Staining for IFN-γ demonstrates the presence of this cytokine in inflammatory mononuclear cells in the center of a perivascular infiltrate from a placebo-treated animal (some designated by arrows); complete suppression is evident in an infiltrate from an rhNGF-treated animal. (C and D) Staining for IL-10, demonstrating strong upregulation of this cytokine in astrocytes of the corpus callosum (normal white matter) in an rhNGF-treated animal. Original magnifications: ×400.
Table 4.
Quantitative Immunohistochemistry of Cytokines in the CNS of rhNGF- and Placebo-treated Animals
| Perivascular infiltrates | Normal white matter | ||||
|---|---|---|---|---|---|
| Cytokine examined | No. of infiltrates analyzed/cm2 | No. of stained mononuclear cells/grid | (Percentage of total) | No. of stained glial cells/grid | No. of stained glial cells/grid |
| IFN-γ | |||||
| Placebo | 5.1 ± 2.4 | 14.0 ± 13.0 | (14.6 ± 14.0) | 0 | 0 |
| rhNGF | 1.3 ± 1.5 | 1.8 ± 2.0 | (0.3 ± 0.7) | 0 | 0 |
| P value | 0.004 | 0.020 | (0.020) | – | – |
| IL-10 | |||||
| Placebo | 5.3 ± 2.6 | 7.3 ± 0.1 | (4.8 ± 4.0) | 1.5 ± 1.3 | 1.1 ± 1.6 |
| rhNGF | 3.1 ± 3.8 | 10.0 ± 1.74 | (23.3 ± 16.0) | 24.1 ± 12.0 | 10.1 ± 2.1 |
| P value | 0.050 | 0.400 | (0.036) | 0.007 | <0.0005 |
The production of NGF is increased in a variety of CNS disorders, including brain injury 20, acute rodent EAE 21, and during the relapsing phase of MS 22. These conditions are associated with the presence of cytokines that stimulate the production of NGF by astrocytes, such as IL-1, TNF-α 23, IL-4, and IL-5 24, which perhaps represents a physiological response to tissue injury 25. Our observation that NGF in turn can modulate cytokine expression by inflammatory and glial cells in EAE underlines the importance of this molecule as a potent autocrine, antiinflammatory cytokine produced within the blood–brain barrier. The effect of NGF on IL-10 expression appears similar to that reported for the neuroregulin GGF-2 16, and may be a common immunomodulatory pathway for several CNS growth factors. In diseases like EAE and MS, presentation of myelin antigens to T cells is dependent on the expression of class II HLA molecules on astrocytes and macrophages/microglia, which is upregulated by IFN-γ 26 and downregulated by IL-10 27. Thus, a possible effector mechanism for the protective action of NGF could be decreased HLA class II expression within the CNS, either indirectly through its effects on IFN-γ and IL-10 production or by a direct effect on astrocytes 28. Indeed, further investigations are needed in this primate system with close phylogenetic similarity to humans, to determine whether the effects of NGF on CNS cytokine production provide long-term benefit in a chronic experimental design. In light of recent evidence suggesting a role for neuronal damage as a mechanism participating in the pathogenesis of MS lesions 29 30, NGF or agonist compounds could represent a novel therapeutic alternative for demyelinating disorders that combines the benefits of neuroprotection and modulation of CNS autoimmunity.
Acknowledgments
We thank Drs. Barbara Barres and Scott Zamvit for helpful suggestions and discussions; Drs. Ken Kunsuke, Shuzo Sato, and Gregory Timmel for help developing stereotaxic procedure in marmosets; William Hyun for advice in flow cytometry; and Peter Bacchetti for advice in statistics.
This work was supported in part by Boehringer-Mannheim GmbH, Penzberg, Germany, which also provided rhNGF; the National Institutes of Health (grant AI43073-06); and the Nancy Davis Center Without Walls. P. Villoslada was a postdoctoral fellow of the Spanish Ministry of Health (FIS 96/5122 and 97/5459). C.P. Genain is a Harry Weaver Neuroscience Scholar from the National Multiple Sclerosis Society.
References
- Raine C. Demyelinating diseases. In: Davis R., Robertson D., editors. Textbook of Neuropathology. 3rd ed. Williams & Wilkins; Baltimore: 1997. pp. 627–714. [Google Scholar]
- Hohlfeld R. Biotechnological agents for the immunotherapy of multiple sclerosis. Principles, problems and perspectives. Brain. 1997;120:865–916. doi: 10.1093/brain/120.5.865. [DOI] [PubMed] [Google Scholar]
- Althaus H., Kloppner S., Schmidt-Schultz T., Schwartz P. Nerve growth factor induces proliferation and enhances fiber regeneration in oligodendrocytes isolated from adult pig brain. Neurosci. Lett. 1992;135:219–223. doi: 10.1016/0304-3940(92)90440-i. [DOI] [PubMed] [Google Scholar]
- Cohen R., Marmur R., Norton W., Mehler M., Kessler J. Nerve growth factor and neurotrophin-3 differentially regulate the proliferation and survival of developing rat brain oligodendrocytes. J. Neurosci. 1996;16:6433–6442. doi: 10.1523/JNEUROSCI.16-20-06433.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urschel B., Hulsebosch C. Schwann cell-neuronal interactions in the rat involve nerve growth factor. J. Comp. Neurol. 1990;296:114–122. doi: 10.1002/cne.902960107. [DOI] [PubMed] [Google Scholar]
- Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. Annu. Rev. Neurosci. 1995;18:223–253. doi: 10.1146/annurev.ne.18.030195.001255. [DOI] [PubMed] [Google Scholar]
- Carter B., Lewin G. Neurotrophins live or let diedoes p75NTR decide? Neuron. 1997;18:187–190. doi: 10.1016/s0896-6273(00)80259-7. [DOI] [PubMed] [Google Scholar]
- Lewin G., Barde Y. Physiology of the neurotrophins. Annu. Rev. Neurosci. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R., Skaper S., Dal Toso R., Petrelli L., Leon A. Nerve growth factorfrom neurotropin to neurokine. Trends Neurosci. 1996;19:514–520. doi: 10.1016/S0166-2236(96)10058-8. [DOI] [PubMed] [Google Scholar]
- Stephan H., Baron G., Schwerdtfeger W. The brain of the common marmoset (Callithrix jacchus). A stereotaxic atlas 1980. Springer-Verlag; Berlin/Heidelberg/New York: pp. 93 pp [Google Scholar]
- Genain C.P., Nguyen M.H., Letvin N.L., Pearl R., Davis R.L., Adelman M., Lees M.B., Linington C., Hauser S.L. Antibody facilitation of multiple sclerosis–like lesions in a non human primate. J. Clin. Invest. 1995;96:2966–2974. doi: 10.1172/JCI118368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barde Y., Edgar D., Thoenen H. Sensory neurons in culturechanging requirements for survival factors during embryonic development. Proc. Natl. Acad. Sci. USA. 1980;77:1199–1203. doi: 10.1073/pnas.77.2.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo F., Holtzman D., Grimes M., Mobley W. Nerve growth factoractions in the peripheral and central nervous systems. In: Fallon J., Loughlin S., editors. Neurotrophic Factors. Academic Press; New York: 1992. pp. 209–256. [Google Scholar]
- Genain C., Cannella B., Hauser S., Raine C. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat. Med. 1999;5:170–175. doi: 10.1038/5532. [DOI] [PubMed] [Google Scholar]
- Webster H.D. Growth factors and myelin regeneration in multiple sclerosis. Mult. Scler. 1997;3:113–120. doi: 10.1177/135245859700300210. [DOI] [PubMed] [Google Scholar]
- Cannella B., Hoban C., Gao Y., Garcia-Arenas R., Lawson D., Marchionni M., Gwynne D., Raine C. The neuregulin, glial growth factor 2, diminishes autoimmune demyelination and enhances remyelination in a chronic relapsing model for multiple sclerosis. Proc. Natl. Acad. Sci. USA. 1998;95:10100–10105. doi: 10.1073/pnas.95.17.10100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Racke A., Bittner P., Cross A., Carlino J., Racke M. Regulation of experimental allergic encephalomyelitis with insulin growth factor (IGF-1) and IGF-1/IGF-binding protein-3 complex (IGF/IGFBP3) J. Clin. Invest. 1998;101:1797–1804. doi: 10.1172/JCI1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer R., Zhang Y., Gehrmann J., Gold R., Thoenen H., Wekerle H. Gene transfer through the blood-nerve barrierNGF-engineered neuritogenic T lymphocytes attenuate experimental autoimmune neuritis. Nat. Med. 1995;1:1162–1166. doi: 10.1038/nm1195-1162. [DOI] [PubMed] [Google Scholar]
- Charlton B., Lafferty K. The Th1/Th2 balance in autoimmunity. Curr. Opin. Immunol. 1995;7:793–798. doi: 10.1016/0952-7915(95)80050-6. [DOI] [PubMed] [Google Scholar]
- Kossmann T., Hans V., Imhof H., Trentz O., Morgantikossmann M. Interleukin-6 released in human cerebrospinal fluid following traumatic brain injury may trigger nerve growth factor production in astrocytes. Brain Res. 1996;713:143–152. doi: 10.1016/0006-8993(95)01501-9. [DOI] [PubMed] [Google Scholar]
- De Simone R., Micera A., Tirassa P., Aloe L. mRNA for NGF and p75 in the central nervous system of rats affected by experimental allergic encephalomyelitis. Neuropathol. Appl. Neurobiol. 1996;22:54–59. doi: 10.1111/j.1365-2990.1996.tb00846.x. [DOI] [PubMed] [Google Scholar]
- Laudiero L.B., Aloe L., Levi-Montalcini R., Buttinelli C., Schilter D., Gillessen S., Otten U. Multiple sclerosis patients express increased levels of beta-nerve growth factor in cerebrospinal fluid. Neurosci. Lett. 1992;147:9–12. doi: 10.1016/0304-3940(92)90762-v. [DOI] [PubMed] [Google Scholar]
- Gadient R.A., Cron K.C., Otten U. Interleukin-1 beta and tumor necrosis factor-alpha synergistically stimulate nerve growth factor (NGF) release from cultured rat astrocytes. Neurosci. Lett. 1990;117:335–340. doi: 10.1016/0304-3940(90)90687-5. [DOI] [PubMed] [Google Scholar]
- Awatsuji H., Furukawa Y., Hirota M., Murakami Y., Nii S., Furukawa S., Hayashi K. Interleukin-4 and -5 as modulators of nerve growth factor synthesis/secretion in astrocytes. J. Neurosci. Res. 1993;34:539–545. doi: 10.1002/jnr.490340506. [DOI] [PubMed] [Google Scholar]
- Dugan L.L., Creedon D.J., Johnson E.M., Jr., Holtzman D.M. Rapid suppression of free radical formation by nerve growth factor involves the mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci. USA. 1997;94:4086–4091. doi: 10.1073/pnas.94.8.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierz W., Endler B., Reske K., Wekerle H., Fontana A. Astrocytes as antigen-presenting cells. I. Induction of Ia antigen expression on astrocytes by T cells via immune interferon and its effect on antigen presentation. J. Immunol. 1985;134:3785–3793. [PubMed] [Google Scholar]
- Williams K., Dooley N., Ulvestad E., Becher B., Antel J. IL-10 production by adult human derived microglial cells. Neurochem. Int. 1996;29:55–64. doi: 10.1016/0197-0186(95)00138-7. [DOI] [PubMed] [Google Scholar]
- Neumann H., Misgeld T., Matsumuro K., Wekerle H. Neurotrophins inhibit major histocompatibility class II inducibility of microgliainvolvement of the p75 neurotrophin receptor. Proc. Natl. Acad. Sci. USA. 1998;95:5779–5784. doi: 10.1073/pnas.95.10.5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp B., Peterson J., Ransohoff R., Rudick R., Mork S., Bo L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Steinman L. Multiple approaches to multiple sclerosis. Nat. Med. 2000;6:15–16. doi: 10.1038/71466. [DOI] [PubMed] [Google Scholar]