

Abstract
The variable (V), (diversity [D]), and joining (J) region recombinases (recombination activating genes [RAGs]) can perform like transposases and are thought to have initiated development of the adaptive immune system in early vertebrates by splitting archaic V genes with transposable elements. In cartilaginous fishes, the immunoglobulin (Ig) light chain genes are organized as multiple VJ-constant (C) clusters; some loci are capable of rearrangement while others contain fused VJ. The latter may be key to understanding the evolutionary role of RAG. Are they relics of the archaic genes, or are they results of rearrangement in germ cells? Our data suggest that some fused VJ genes are not only recently rearranged, but also resulted from RAG-like activity involving hairpin intermediates. Expression studies show that these, like some other germline-joined Ig sequences, are expressed at significant levels only early in ontogeny. We suggest that a rejoined Ig gene may not merely be a sequence restricting antibody diversity, but is potentially a novel receptor no longer tied to somatic RAG expression and rearrangement. From the combined data, we arrived at the unexpected conclusion that, in some vertebrates, RAG is still an active force in changing the genome.
Keywords: V(D)J recombination, rearrangement, immunoglobulin genes, light chain, germline joining
Introduction
The lymphocyte antigen receptors encoded by Ig and TCR genes bind foreign ligands after clonal selection from a vast repertoire of combining sites. The diversity of the combining sites is generated in part by a mechanism that joins different gene segments in a combinatorial fashion, and in the process somatically deletes and sometimes inverts DNA in the lymphocyte. This mechanism and the antigen receptors have so far been found only in jawed vertebrates, e.g., cartilaginous fishes such as sharks and skates being representatives of the oldest of them 1. The unique rearrangement mechanism stirs interest in the origin and evolution of the Ig and TCR gene systems, which have not been identified in hagfish or lamprey, the living representatives of the oldest vertebrates. The apparent absence, too, of the recombination activating genes (RAG)1-1 and RAG-2, which encode the enzymes mediating gene rearrangement, suggests that these, and the genes for the antigen receptors, coevolved in an ancestral jawed vertebrate. Recently, it has been found that the RAG enzymes not only cause DNA excision, but can also mediate reintegration of the excised DNA 2 3, both processes through a biochemical mechanism akin to integrases and transposases. These findings support a hypothesis that the Ig and TCR gene segments, which are all separated by interspersed noncoding DNA with specific recombination signal sequences (RSS) adjacent to the exon joining ends, were generated after the RAG-mediated insertion of a mobile sequence into a prototype Ig gene, rending it into two gene segments, V and J (see Fig. 1, and references 2, 4, 5).
Figure 1.

Hypothetical scheme for evolution of Ig L chain genes. The precursor Ig gene underwent gene duplication, generating the V and C exons. Sometime in or before the early Devonian, >400 Myr BP, a mobile DNA sequence was inserted in the V exon, splitting it into V and J gene segments. Dark arrows on the DNA insert indicate the RSS. Gene duplication of the individual genes in tandem generated the overall L chain Ig gene arrangement in tetrapods, whereas duplication of V, J, and C genes produced the multiple cluster organization common in cartilaginous fishes. Based on diagram from reference 4.
In the course of evolution, the ancestral RAG transposase acquired specialized function in vertebrates, excising mobile sequences in developing lymphocytes. The gene segments thus targeted underwent extensive duplication and reorganization, generating the TCR chains and the H and L chains of antibodies. By and large, in tetrapods Ig and TCR genes are organized as tandem arrays of V, followed by (D and) J gene segments, and by a few C region genes (Fig. 1). In bony fishes, the Ig H chain genes are similarly organized 6 7. However, the Ig L chain genes in bony fishes, as well as the Ig H and L chain genes in cartilaginous fishes, are organized as multiple clusters each containing VH, D, JH, CH, and VL, JL, CL genes, respectively 8 9 10 11. This divergence of gene arrangement among vertebrates suggests that sharks and skates carry one of the organizational alternatives—perhaps the most ancient one—developed early in the evolution of the Ig genes.
In sharks and skates, many clusters contain Ig genes that are rearranged in the germline, sometimes as they can be in lymphocytes 10 12 13. L chain genes of any one type of the three so far reported have been found either all in split germline configuration or all as joined VJ (see Fig. 2, bottom). In contrast, the H chain gene clusters contain rearranged, partially rearranged, or nonrearranged V, D, and J genes 12 13 14. In comparing the H chain joined and split sequences, it was difficult to dissect the nature of the joining events because of the highly varied sequences at the junctions. The process by which these joined genes were created, whether RAG, terminal deoxynucleotidyl transferase (TdT), or the RSS were actually involved, is unclear.
Figure 2.
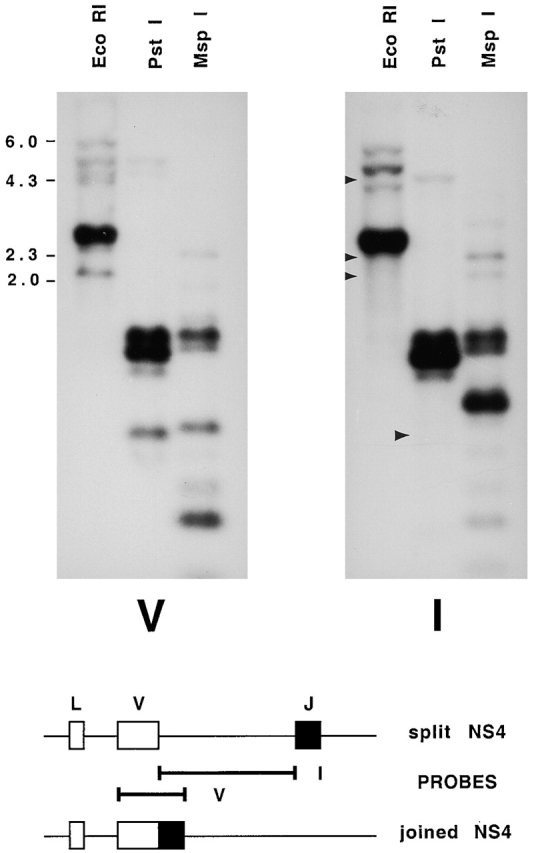
Genomic Southern blot hybridized with probes to the coding and intervening regions of NS4. Top, erythrocyte DNA from shark Y was digested with EcoRI, electrophoresed on a 1.2% agarose gel, and transferred onto a nylon filter. The filters were hybridized with radiolabeled DNA sequences from the V region of NS4 (V) and the intervening sequence between V and J (I). Arrows in the EcoRI lane point to the location of V-hybridizing bands at 2.1, 2.4, and 4.6 kb that were not detected by the I probe. These were cloned and contained germline-rearranged NS4 genes. Arrow in the PstI lane points to a fragment of 727–744 bp containing joined genes R4, R7, R18, R6, and R19, predicted from DNA sequence in Fig. 3 (dots over positions 79 and ∼816); accordingly, this fragment does not hybridize to I probe. The slower migrating bands in the same lanes at ∼1,150, 1,250, and 1,400 bp correspond to sizes expected with the presence of an intervening sequence of ∼500 bp; these bands accordingly hybridized with both V and I probes. Molecular size markers from λ phage DNA digested with HindIII are shown. The PstI fragment sizes were calculated from another gel; not shown. Bottom, location of V and I probes from cloned split (S5) and joined (R4) genes.
In this study, genes of one L chain type, NS4, have been isolated from the nurse shark Ginglymostoma cirratum. They are homologues of the type III L chains in the horned shark 15 16. Whereas the type III genes are reported to be all split 13, we have identified clones of NS4 V and J genes existing as both germline-joined and nonjoined (see Fig. 2). This mixed combination in one L chain type is a novel finding; furthermore, the NS4 genes are so similar that it is possible to explore not only the evolution among the various joined genes, but also their relationship to the split genes. The probable steps of the germline joining events were reconstructed, and these were compared with the joints in cDNA rearrangements obtained from lymphocytes of the same individual. Our data strongly suggest that the joined genes are products of a germline V(D)J RAG-mediated recombination event; this genomic evidence supports RAG-like activity occurring in germ cells.
Materials and Methods
Genomic Libraries.
Whole blood was obtained from nurse shark Y, and the cells were centrifuged through a Ficoll gradient to separate erythrocytes from PBLs. Genomic DNA was isolated from both populations of cells, and RNA was extracted from the PBLs. DNA from the RBCs was digested with EcoRI and fractionated on a 1% low melt agarose gel. DNA fragments corresponding to 6–4- and 3–2-kb ranges were isolated and separately ligated into λ ZapII phage arms (Stratagene). 2–2.5 × 105 bacteriophages from each library were screened with probes from the V region of NS4 and NS3. 90 phages from the 3–2-kb library hybridized with NS4 probe, and among them R4, R6, R18, RE18, R19, and all the “S” clones were identified. R7 was isolated from shark Y PBL genomic DNA library; we believe that R7 is a germline-joined gene because three independent clones were isolated, and two more sequences identical to R7 were isolated from RBC DNA from an unrelated animal, shark A. Each phage isolate was analyzed by PCR using primers to the leader (NSL) and J segment (JL5) of NS4 to determine whether the cloned gene was germline-rearranged as VJ. DNA was extracted from phage lysates and digested with EcoRI to determine the insert size. The selected phages were purified, and the plasmid component of λ ZapII was excised and subjected to the dideoxy sequencing technique 17.
PCR.
Oligonucleotide primers to the NS4 leader sequence (NSL) and the JL5 were synthesized by GIBCO BRL (NSL, 5′-GATTTCICAYATICARCT-3′; JL5, 5′-CTTGGTTCCTTTACCGAA-3′, where I is inosine, Y is T/C, and R is G/A), and used in PCR under the following cycling conditions: 96°C for 1 min, 46°C for 2 min, 72°C for 2 min for 39 cycles, and a final cycle with the 72°C step extended to 15 min. DNA templates consisted of 100–500 ng genomic DNA per 0.25-ml reaction, or of denatured phage lysate not exceeding 17% of the final volume.
Reverse transcription (RT)-PCR assays were performed as described previously 18. Oligo dT was used for priming first strand synthesis in total RNA from PBLs of shark Y. RT-PCR products were cloned into pGEM-T-Easy (Promega).
Probes.
A joined VJ NS4 sequence of 314 bp was initially obtained from shark Y genomic DNA by amplification with primers NS4A (5′-ATCACIATGACICARTC-3′) in framework region (FR)1 and JL5 in the J segment 16; the sequence was later identified as part of R4 in the genomic library. The germline-joined NS3 sequence is published 16 and was also amplified using primers in FR1 19 and J. The I probe containing intervening sequence between V and J was obtained from split NS4 gene S5 (similar in structure to S1 or S10), which contained a HincII site in the CDR3 in the 3′ region of the V and a SacI site in the RSS of the J gene end. S5 was subjected to restriction endonuclease digestion (New England Biolabs), and the ∼500-bp DNA fragment was isolated from a low melt gel, using Geneclean II (Bio101), and radiolabeled (Prime-It II; Stratagene). Hybridization and blotting procedures have been described elsewhere 20.
Animals.
Nurse sharks were captured off the coast of the Florida Keys, and blood was obtained from the caudal sinus into heparinized syringes. Blood cells from one individual, shark Y, were separated into erythrocyte and PBL fractions, which were used in the cloning and expression studies described under Genomic Libraries and PCR above. DNA extracted from either erythrocytes or nonficolled blood cells was obtained from other individuals.
Phylogenetic Analyses.
Nucleotide alignments were constructed manually. In rare instances of difficulty in alignment, homology assignments were informed by comparing the derived amino acid sequences. PAUP*4.0beta4 (Phylogenetic Analysis Using Parsimony [*and Other Methods] v4.0b2a; D.L. Swofford, Sinauer Associates) was used with five character and taxon partitions (data sets) of nucleotide or amino acid characters and taxa: (A) the coding regions only minus the first three codons not present in h6 (all taxa); (B) the coding regions and intron (NS4 family genes only); (C) the entire nucleotide alignment (NS4 genes, except S31 and S32); (D) only the first and second positions of the coding regions (all taxa); and (E) the derived amino acid alignment, except the first three residues (all taxa). Methods used to evaluate trees in searches were maximum parsimony (MP), minimum evolution (ME), and minimum likelihood (ML) as implemented in PAUP*. The neighbor-joining algorithm (NJ) was also used to estimate phylogenies. In MP analyses, characters were weighted equally; transformations were either weighted equally or transversions were weighted twice that of transitions. Gaps in the alignment were either ignored (coded as missing) or each gap was counted as a single insertion/deletion (indel) event, regardless of the number of sites covered by the gap. Before ME, NJ, and ML analyses, Modeltest 3.0 21 was used with PAUP* to test a variety of models in a hierarchical fashion to find the simplest model that fit the data not significantly worse than more complex (generalized) models. The model identified and used for tree searches by the distance and likelihood methods was K80+Γ (the two-parameter model of Kimura 22 with an assumed transition/transversion rate ratio of 1.2134, corrected for rate variation across sites with a gamma distribution shape parameter of 0.3301, estimated as four discrete rate categories). In analyses with ≤11 taxa, exhaustive searches were made, except when the ML method was used. In all other analyses, heuristic branch-swapping (tree-bisection-reconnection, TBR) searches were performed, with starting trees obtained in 1,000 replicates of random sequence additions. Additionally, in parsimony searches, the 3,000 best trees were kept and used for additional branch-swapping searches. For evolutionary reconstructions based on the inferred phylogenetic networks, rooting of the nurse shark NS4 phylogeny employed the outgroup method with all sequenced genes of the homologous type III family of horned shark and representatives from the more divergent NS3/type II and NS5/type I families. This rooting could only be used for data sets A, D, and E. However, analyses with these data sets revealed that SE7 is the most basally diverged member of the NS4 family; thus, SE7 was used for rooting with data sets B and C. For a final estimation of branch lengths, the ML method was used with the HKY85 model 23 with base frequencies and transition/transversion ratio estimated from the data, corrected for invariance and rate variation across sites by a gamma distribution approximated as four discrete rate categories (with parameters estimated from the data by PAUP*).
Statistical Analyses.
Support for different branches in the estimated trees was evaluated by bootstrapping (with replacement), jackknifing (50% deletion), decay index (“Bremer support index”; 24). Except with ML (where 100 replications were used), 2,000 bootstrap and jackknife replications were used for each analysis. Sets of ∼2,900–3,200 trees collected during tree searches were compared statistically with the KH 25 26 test, as implemented in PAUP*. For each such analysis, a strict consensus of unrejected trees was calculated to identify relationships that remained fully resolved.
Dating.
We assumed that the point of divergence among the NS4 and type III genes corresponded to the point of divergence between the two shark species. The earliest point at which rearrangements were likely to have occurred was then calculated as the fraction of the total length from the species divergence to the most closely related functional gene where each rearranged gene diverged. Only the nonrearranged, functional genes S31 and S10 were used so as to eliminate artefactual inflation of rate estimates due to increased rates in pseudogenes. Using only the most closely related functional genes for calibrating the molecular clock also reduces the error due to rate differences among genes. This proportion was then applied to the species divergence date between nurse shark and horned shark, assumed to be 180 million years before present (Myr BP), as estimated from fossil data 27, to derive the earliest date at which germline rearrangements could have occurred.
Results
Isolation of NS4 L Chain Genes.
A probe to the NS4 V region revealed a series of hybridizing EcoRI-digested DNA fragments by genomic Southern blotting: ∼6.0, 5.1, 4.6, and 4.3 kb, many bands at 2.8–2.65, 2.4, and 2.1 kb (Fig. 2, panel V). To isolate these sequences, two genomic libraries were constructed from size-selected erythrocyte DNA fragments of 6–4 and 3–2 kb. In the latter library, 90 phages were selected and PCR analysis was performed with primers to the leader and J gene segment. Nine phages gave NS4-hybridizing PCR fragments of ∼550 bp, four of ∼1,000 bp, and the rest ∼1,078 bp. This last, largest group consisted of nonrearranged (“split”) NS4 genes with a leader intron of 186 bp and an intervening sequence of ∼500 bp (502–524 bp) separating the V and J gene segments. All EcoRI inserts were in the 2.6–2.8-kb range. The second group of four phages also consisted of split genes, with a shorter V-J intervening sequence, and the insert size was 2.5 kb; the only one analyzed was S32.
The nine phages that produced PCR fragments of 550 bp all contained germline-rearranged NS4 genes. The EcoRI insert sizes varied, six being ∼2.1 kb, two 2.4 kb, and one 4.6 kb. A probe (I) consisting of the intervening sequence between V and J hybridized to all but these EcoRI fragments (Fig. 2, panel I; arrows in lane 1). A side-by-side comparison of the cloned joined gene of 4.6 kb (RE18) showed that it comigrated with the same band on a genomic Southern blot (not shown); both hybridized with V and not I probe.
Hybridization of these filters with the probe of another L chain type, NS3, identified three hybridizing phages that all contained DNA with similar restriction enzyme sites. In previous genomic Southern analyses, there appeared to be at least two copies of NS3 in the 3–2-kb region. Based on these results, and on comparisons of the NS4 signal from dilutions of total EcoRI-digested DNA with that of the selected 3–2-kb fraction, we estimate that 200,000 phages with 3–2-kb inserts represented ∼2 genomes of nurse shark DNA, assuming a genome size of 6–8 × 109 bp. Thus, in this region there are at least 45 NS4 genes, 3–5 of which are germline rearranged. In the 6–4-kb range there are five to six NS4 genes, at least one of which is germline-joined (Fig. 2); the latter (RE18) was cloned in the 3–2-kb library. Thus, our estimates suggest a total of 50 NS4 L chain genes in shark Y, 10% of which is not in the split germline configuration.
Six unique germline-joined NS4 genes were identified; some may be alleles, two (R6, R19) are pseudogenes. Except for RE18, all were obtained in duplicate from the screening of three independent genomic libraries prepared from shark Y, and thus assuredly originate from germline sequence. No split gene was identified in any cloned EcoRI fragment of 2.1 kb, consistent with the absence of I probe hybridization to the genomic DNA (Fig. 2).
The nucleotide sequences are shown in Fig. 3 and compared with nonjoined genes S1, S10, S31, and S32. The J gene segments of the latter sequences are also shown separated by slashes that indicate exclusion of the RSS and the intervening sequence. The predicted amino acid sequences are shown in Fig. 4.
Figure 3.



Nucleotide sequences of NS4 genes. The sequences of cloned NS4 genes containing the leader, V, and J exons are compared with the first sequence, R4. Dots indicate identity, dashes indicate gaps. R4, R7, R18, RE18, R6, and R19 are germline rearranged as VJ. S1, S10, S31, and S32 are not joined, and their V gene segments are shown up to but not including the RSS. Slashes indicate that the intervening sequence and the RSS on either side are left out. These sequences recommence with the coding region of the J gene segments and the 3′ noncoding sequence. Regulatory signals as the octamer and splice sites are boldfaced, and their locations are based on homology with other vertebrate L chain genes. The location of the 3′ splice signal of J is derived from NS4 full-length cDNA sequence, where the amino acid preceding the first residue of the constant region is arginine (reference 15), so that the last nucleotide in J is adenyl. The coding region is underlined; the leader sequence is split by an intron. The beginning of the V region is indicated by an asterisk over the first base. The division of the V region into FR and CDR is indicated (reference 19). The two PstI sites (CTGCAG) referred to in the Fig. 2 legend and text are indicated by bold dots.
Figure 4.

Derived amino acid sequences of NS4 genes. The amino acid sequences of most of the cloned NS4 genes shown in Fig. 2 are compared with R4. Dots indicate identity, dashes indicate gaps. R19 and S32 contain stop codons (asterisks), and one nucleotide was left out of CDR3 in R19 in order to obtain a reading frame that is comparable to the other sequences (indicated by a hyphen). R6 carried too many disruptions and is not shown.
Germline Origin of Joined NS4 Genes.
There are two PstI sites in both R and S sequences, at position 79 between octamer and leader, and at positions 806–833 after J gene segment (Fig. 3, bold dots). Thus, the sequences R4, R7, R18, R6, and R19, but not RE18, are expected to produce fragments of 727–754 bp when digested with PstI. In genomic Southern blots, such a band was observed with V probe and not with I probe (Fig. 2, panel I; arrow in second lane). Split sequences such as S1 and S10 would contain in addition an intervening sequence of ∼500 bp, and the predicted PstI fragments would be 1,200 bp. Such corresponding genomic DNA PstI fragments hybridized with both V and I probes (Fig. 2, both panels, second lanes; see legend).
Additional experiments demonstrating the germline origin of the R genes, in particular R4/R7, are shown in Fig. 5. DNA samples from seven wild nurse sharks were analyzed by genomic Southern blotting. The signal from the EcoRI 2.1-kb fragment, from which only joined genes have been identified, was present in all individuals and identical in intensity in the erythrocyte and PBL lanes of shark Y (Fig. 5 A, V, lanes 6 and 7), showing that contribution of somatically rearranged NS4 sequence, even in the PBL DNA, was not visible compared with the germline-rearranged genes.
Figure 5.


Germline origin of rearranged NS4 genes. (A) EcoRI-digested DNA from seven wild nurse sharks hybridized with V and I probes. Lanes 1–3, DNA from RBC fraction of ficolled blood of three individuals; lanes 4 and 5, DNA from whole blood (WB) of two sharks; lanes 6 and 7, DNA from erythrocyte (RBCs) fraction (lane 6) and PBL fraction (lane 7) of ficolled whole blood from shark Y. The genomic DNA libraries were generated from these samples (see Materials and Methods). Lane 8, DNA from erythrocytes from shark B. The DNA samples were digested with EcoRI, electrophoresed together on one 1% agarose gel, and transferred to a nylon filter; the filter was cut in two and one half was hybridized to probe V, the other to probe I (see legend to Fig. 2). Arrows indicate bands at 4.6, 2.4, and 2.1 kb that hybridize to V but not to I. The band at 5.4 kb is variable in hybridization to I probe among individuals. When the V and I films were superimposed, the bands at 2.4 kb detected by V probe are distinct from the faint bands at ∼2.5 kb detected by I probe. (B) Identification of R4/R7 sequences in genomic DNA. The DNA from shark Y obtained from erythrocytes (lanes 1 and 2) and from PBLs (lanes 3 and 4) was digested with EcoRI and ClaI (lanes 2 and 4) or EcoRI alone (lanes 1 and 3). Hybridization with V probe showed that some of the EcoRI fragments containing NS4 V genes carried ClaI sites as well. Arrows point to ClaI and EcoRI-digested fragments that hybridize with V probe at 1,550, 1,450, 1,150, and 980 bp. The markers used are λ HindIII and φX174 HaeIII, at 4.36, 2.3, 2.0, 1.35, 1.08, and 0.872 kb. The interpretation of the digestion patterns is shown in the diagram. Top, split NS4 genes at 2.6 and 2.7 kb such as S1 and S31 with ClaI site in CDR2. Bottom, joined NS4 genes at 2.1 kb such as R4 and R7 with ClaI site in CDR2. Joined NS4 genes such R6 and R19 at 2.1 kb are not expected to be affected by ClaI, so the 2.1-kb signal in lanes 2 and 4 is reduced but not abolished.
There is a ClaI site in CDR2 of the R4 and R7 sequences (Fig. 3); restriction enzyme analyses confirmed that digestion with ClaI yielded fragments of ∼1,150 and 980 bp. Bands of this size hybridizing with V probe were observed in genomic DNA digested with EcoRI and ClaI (Fig. 5 B, lanes 2 and 4, arrows). R6 and R19 are on 2.1-kb EcoRI fragments but do not contain ClaI sites (Fig. 5 B, diagram on right). Accordingly, the signal from 2.1-kb fragments is reduced but not abolished in the EcoRI-ClaI–digested DNA.
Phylogenetic Analyses.
The NS4 coding sequences, together with the type III L chain homologues in horned shark and representatives from other L chain types, were subjected to several different methods of phylogenetic estimation. Only one tree appeared as a best tree in all of the analyses (Fig. 6). The various L chain types grouped together as NS4/type III, NS3/type II, and NS5/type I, suggesting that these are the respective orthologs in the two species. The nurse shark NS4 gene family is significantly supported as a monophyletic clade separate from the homologous type III gene family of the horned shark, indicating that gene expansion and diversification occurred independently in the two species lineages.
Figure 6.
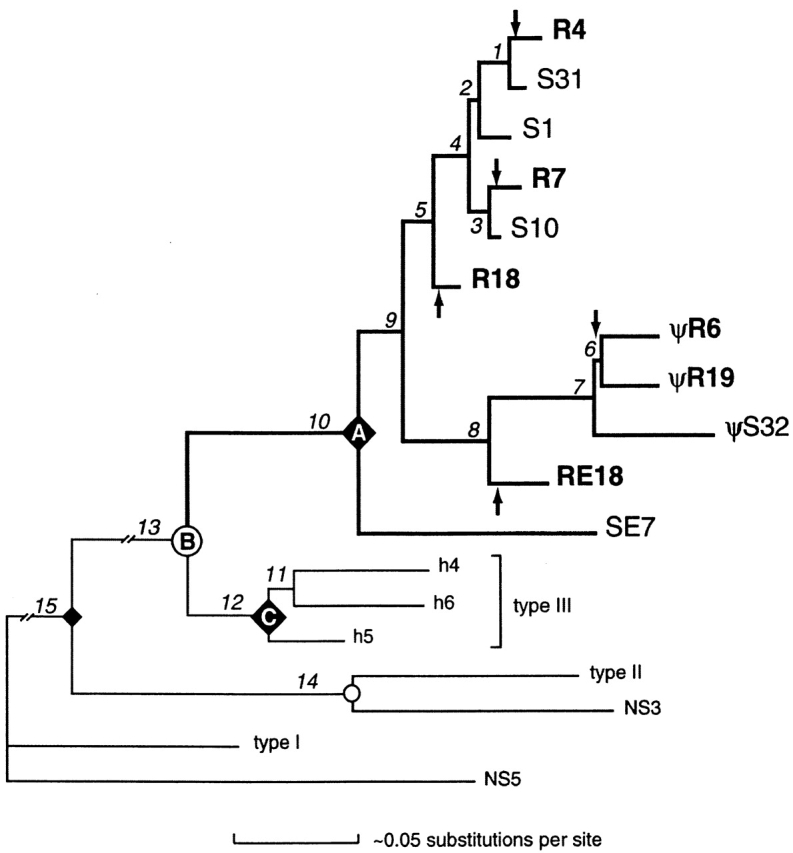
Phylogenetic tree of shark L chain V sequences. The relationships among the nurse shark NS4 family of genes are emphasized by bold branches. Other L chain types from nurse shark (NS3 and NS5) and horned shark (types I–III) were included in the analysis and used to root the tree. Branch lengths were estimated by likelihood using the HKY85+I+Γ model (reference 23), correcting for invariance and rate variation across sites as detailed in Materials and Methods. Nodes representing presumed speciation events are indicated as circles; e.g., node B corresponds to the divergence between all nurse shark NS4 genes and all horned shark type III genes. Nodes representing the most recent common ancestor of all members within a particular gene family are designated by ♦; e.g., all the NS4 genes derived from a common ancestral gene at node A and all the type III genes derived from a common ancestral gene at node C. Arrows designate branches in which germline rearrangements in NS4 genes most likely occurred. Lengths of internal branches (numbered segments) are approximately proportional to the estimated accumulation of nucleotide substitutions per site (scale at bottom), except for branches 13 and 15, which are ∼10 times as long and are truncated in the figure. The most strongly supported branches are 1, 4, 7, 8, 10, 12, 13, 14, and 15. For example, in one of many parsimony jackknife analyses of only the coding regions (assuming transversions were weighted twice that of transitions, ignoring alignment gaps, with 2,000 replications of a random selection of 50% of the nucleotide characters), each branch appeared in the following percentages of replicates (heuristic searches with 1,000 random taxon additions each): 1, 88%; 2, 61%; 3, 71%; 4, 82%; 5, 66%; 6, 51%; 7, 97%; 8, 86%; 9, 61%; 10, 95%; 11, 31%; 12, 98%; 13, 100%; 14, 100%; and 15, 100%. In a similar analysis that included noncoding regions (and excluded taxa for which these regions were not sequenced), jackknife values were (2,000 replications of exhaustive searches): 1+2, 37%; 3, 47%; 4, 75%; 5, 50%; 6, 98%; and 7+8, 94%. Horned shark cDNAs h4, h5, and h6 (available from EMBL/GenBank/DDBJ under accession nos. L25561, L25562, and L25563, respectively) were isolated by cross-hybridization with NS4 sequence. Based on separate phylogenetic analyses of V and C regions, type III was identified as a homologue of NS4 (reference 13); this conclusion is also supported by the current phylogram. All available type III sequences grouped strongly together, as did all the representative NS4 sequences (germline-rearranged R7, R4, R18, R6, R19, and RE18, and split S1, S10, S31, S32, and SE7), suggesting gene diversification after the species diverged. Pseudogenes are indicated. Other sequences are from different L chain types in horned shark (type I, X15316; type II, L25560) and nurse shark (NS3 [reference 16], NS5 [reference 39]). SE7 is the most basally divergent member of the NS4 gene family identified to date, and its sequence is not shown in Fig. 3.
Three groups of NS4 genes emerged, clade I (S1, S10, S31, R4, R7, and R18), clade II (S32, R6, R19, RE18), and clade III (SE7), and the joined genes grouped with split genes. Assuming that once L chain genes were joined in the germline they cannot be reverted to split genes, it is estimated that there were at least five independent rearrangement events (Fig. 6, arrows) in the evolutionary lineage giving rise to the extant nurse shark genes. Accordingly, because S1, S31, SE7, and S10 are not joined, it is most parsimonious to infer that the ancestor at node A and indeed at all internal nodes of the tree, with the possible exception of the ancestor of R6 and R19, must have been nonrearranged. For instance, if R7 and R4 were closest relatives, we would not have been able to exclude the possibility that they shared a rearranged ancestral gene, but as it is clear that S31, a split gene, is the closest relative of R4, R7 and R4 arose from independent joining events.
When Did the Germline Recombination Occur?
The most closely related pairs of joined and split genes are R7/S10 and R4/S31 (Fig. 3). The similarity of germline-rearranged genes to the split ones suggests that R4, for instance, may have arisen from a gene cluster similar to S31. Dating the divergence of R4 from S31 would give the earliest time at which the R4 germline rearrangement could have occurred. The rooting of the NS4 phylogeny is described in Materials and Methods, and the R4/S31 divergence point was calculated as a proportion of total branch length (0.129/0.134) to S31 from the species divergence point (node B) at 180 Myr BP 27; that is, 180 Myr − [180 Myr (0.129/0.134)], 7 Myr BP. Thus, R4 is estimated to have diverged from S31 at least 7 Myr BP, and the recombination event creating the R4 joined gene occurred some time between 0 and 7 Myr BP. The estimated dates of the other rearranged genes are 11 Myr BP for R7/S10 and 49 Myr BP for R18/S10. The earliest date of origin of RE18 was measured as a fraction of the length between RE18 terminus and node B, which was found to be 38 Myr BP.
Molecular Events in Germline Joining.
Fig. 7 shows two possibilities (a and b) for the recombination events generating the R7 joint. In both cases, the 3′ flank of the R7 gene segment coding region is assumed to have been the same length as in S10, and the 5′ flank of the R7 J gene segment is assumed to have been the same length as other clade I NS4 J gene segments so far analyzed (Fig. 7, bottom).
Figure 7.

Derivation of the joints in NS4 germline-rearranged genes. The V and J flanks of split genes are shown without RSS. The possible processing events producing the joints of germline-joined R7, R4, RE18, R6, R19, and R18 are shown aligned with the most similar nonjoined NS4 coding ends isolated in these studies. Assuming that the split ancestor of R7 resembled S10, the R7 joint could have been produced by (a) cleavage at the RSS and relegation of blunt ends, or (b) cleavage at the RSS, formation of hairpin at V and J ends, single-stranded nick occurring two or more nucleotides from end, processing resulting on P region (underlined), and deletion of two bases from the J flank. Pathway R7 (a) does not preclude hairpin formation, but there is no processing evidence for it. Under cDNA, germline split S10 is compared with cDNA sequences obtained by RT-PCR from shark Y that share similar CDR3. The analysis of the cDNA joints suggest P region in both sequences. The sequences in the cDNA joints that do not appear to belong to either V or J may be N region. R4 is similar to R7. RE18 joint could have resulted from deletion in the V coding end (a), or J coding end (b), or both (c). The flanks of nonjoined ancestor of R6, R19, and R18 could have resembled S32, in which case deletion only from J end occurred during recombination (a). In all other possibilities, nucleotides were removed from either end. Examples of coding flanks from NS4 split genes are included for comparison; the RSS resemble those from other vertebrates. Clade I refers to the cluster of genes including R4, R7, and R18, clade II to that including R6, R19, and RE18 (see Fig. 6); clade III genes are characterized by divergent FR3 and CDR3 of 24 bp; complete sequences are not shown. Asterisk indicates split gene is pseudogene. S32 is shown in Fig. 3 and Fig. 4; S15 has TGA in FR2; S5 sequence is not shown but carries TGA in FR3, a GT–AT splice signal combination in leader intron, and noncanonical RSS heptamer sequences.
One possibility (a) is that the R7 J 5′ flank was different from the S10 J flank in the first two bases. The recombination proceeded by cleavage of DNA adjacent to the RSS, and joining of the blunt coding ends was without any nucleotide addition or loss. The second possibility (b) is that after removal of the intervening sequence, hairpin coding ends were formed. Single-strand nicking on both the flanks occurred at least two nucleotides from the ends, and subsequent processing resulted in a two-base P region at the V end and a two-base deletion of the J gene end.
The second possibility strongly suggests that the germline recombination occurred by V(D)J rearrangement mechanisms described in lymphocytes, as the coding ends would have undergone an intermediate hairpin stage to achieve the R7 joint. By comparison, P region is also found occasionally in NS4 cDNA sequences cloned from shark Y. In the examples shown, cDNA 6 and cDNA 23, which are similar in CDR3 to S10, both contain P and probably N regions. Even if the cDNAs were rearrangements to a J segment different from the one found closest to S10, some of the nucleotides in CDR3 still appear to be nontemplated.
Analysis of the joints of R4, R18, RE18, R6, and R19 also suggest that the recombination events may have entailed modification or removal of nucleotides from either flank similar to R7 (Fig. 7). The most obvious difference from cDNA is that in somatically rearranged sequences, there are stretches of several bases that may be N region. Therefore, germline rearrangement of the NS4 L chain gene segments appears to occur much as it does in lymphocytes, and in the R sequences there is little evidence of TdT activity.
Although our phylogenetic analyses pointed to the NS4 joined genes being products of ancestral split genes rather than the other way around, additional supporting evidence comes from examination of the coding sequence on either side of the RSS in the split genes, showing no indication of the characteristic integration site duplication of 3–5 bp that occurs in V(D)J-mediated RSS insertions 2 3 9.
Germline-rearranged NS4 in Shark Population.
The germline-rearranged NS4 genes R4 and R7 carry a ClaI site in CDR2, and in the CDR3 of R4 there is also a BsgI site. Erythrocyte DNA from seven outbred nurse sharks was subjected to PCR using the NSL and JL5 primers. As the 550-bp fragments amplify far more efficiently than the 1-kb nonrearranged genes 16, after 40 cycles almost the only PCR product strongly hybridizing to V probe was that from the rearranged genes and this was found in all 7 samples (not shown). When digested with ClaI (402 and 156 bp), or ClaI and BsgI (402, 131, and 25 bp), all samples contained a subpopulation of DNA with ClaI sites, and at least four of the samples contained PCR fragments with restriction endonuclease sites similar to R4. R18, another joined gene defined by a unique EcoRV site in FR3 in combination with the absence of a universal AclI site in FR3, was also detected in all shark samples. Moreover, PCR-generated sequences identical to genes R4 and R7 were cloned from shark A and genes R4 and R18 cloned from shark B.
All the animals in Fig. 5 have also been analyzed for polymorphism at the MHC class I genes, and no two samples show identical RFLP (data not shown). The levels of MHC class I and class II polymorphism in this population of sharks are as high as those found in mouse and human populations (28; and Flajnik, M.F., unpublished data). Thus, the presence of germline-joined NS4 genes in each animal tested is not the result of a recent bottleneck. In combination with results shown in Fig. 5 A, we conclude that most of the germline-rearranged genes are widespread in the nurse shark population in the Florida Keys, and that the recombination events that generated them do not occur frequently in the germ cells.
Expression of Germline-joined Genes.
R4 and R18 are distinguishable from other NS4 sequences by the restriction enzyme analyses described above. RT-PCR was performed on PBL RNA from shark Y, using primers NSL and JL5, and no significant amounts of either R4 or R18 sequence could be detected in PCR products of the expected cDNA size of ∼350 bp. 40 cDNA joints were examined, and none matched those of the R genes. However, analyses of spleen RNA from neonatal shark pups showed the presence of both; this was confirmed by isolation of these sequences from pup spleen and pup epigonal cDNA libraries (data not shown).
Discussion
The Origin of Germline-joined Ig Genes.
When germline-rearranged Ig genes were discovered in the horned shark 12, it was initially not clear whether these sequences were the products of recombination in germ cells, or an archaic form of Ig genes from whence was derived gene segments capable of recombination. Litman and coworkers subsequently demonstrated that while the horned shark L chain type I genes were in germline-split configuration, the homologue in skates was entirely germline joined 13 29. It appeared probable, but not conclusive, that joining or splitting event(s) occurred after divergence of Batoidea, at least 200 Myr BP 27. If the joined genes originated from split ones, certain questions arise. Is the recombination mechanism in germ cells similar to that described in lymphocytes? Does Ig gene recombination occur frequently, and do these joined genes have a selected function? If the process is really mediated by RAG enzymes present in germ cells, is the excised DNA reinserted, allowing the chance of creating another split gene?
While the mixture of rearranged and nonrearranged genes has been reported for horned shark H chain genes 12, such a combination in one L chain type is the first thus described. Unlike horned shark H chain genes, the joined and split NS4 V genes are very closely related in sequence (up to 99% identity), and phylogenetic analysis was possible. An ancient gene duplication giving rise to three NS4 clades occurred within the nurse shark lineage soon after species divergence. Joined genes appeared in two clades, and except for R6/R19, a closer relationship in each case to split genes suggests five independent recombination events.
In this study, the six nurse shark joined sequences identified are found to be the results of rearrangement in the germline occurring at several different points in time. The most closely related joined and split genes, S31 and R4, diverged 7 Myr BP and the recombination events creating the joined R4 gene therefore occurred between 0 and 7 Myr BP. The RAG transposon is thought to have entered the genome of jawed vertebrates >400 Myr BP 2 5; our calculations suggest that RAG-like activity, as evidenced by recent germline-joining, may be ongoing.
Rearrangement in Germ Cells by RAG-like Enzymes.
The close identity of S10 and R7, especially at CDR3, also permitted an analysis of the R7 joint, under the assumption that it arose from a gene similar to S10. Our analyses of the R7 joint and others suggest that the joining process may have involved the RAG recombinase enzymes. RAG studies performed in the mammalian systems have shown that RAG-1 and RAG-2 recognize the RSS and mediated the cleavage of DNA adjacent to the RSS 30 31 32; the resulting double-stranded breaks on the coding sides of the RSS formed hairpin structures 33. RAG proteins participated in nicking the hairpins 34, and the opened coding ends lost or acquired additional sequence before joining, generating N and P regions 35 36. Whereas N region is generated by TdT, P region is acquired as a result of these RAG activities. The possibility that some nucleotides in the R4 and R7 joints are P region suggests a hairpin intermediate like that found in the RAG-mediated pathway. The deletion of nucleotides, which occurred in the shark 5′ flank of J segment, is also a characteristic of V(D)J rearrangement. In the joint of each joined gene, no part of the sequence is attributable to the shark RSS (classical heptamers and nonamers); they have been completely excised as if recognized by the recombinase. These observations, and similar processing of the coding ends in the shark cDNA, support the idea that the genes were joined and the intervening DNA excised by recombinase enzymes, as in lymphocytes 37.
Genomic Southern analyses of a second shark B (Fig. 5 A, lane 8) suggest that it too carried NS4 joined genes on EcoRI-digested fragments of 4.6 kb (RE18), 2.4 kb (R6/R19), and 2.1 kb (R4/R7/R18). This observation and the cloning of identical R4, R7, and R18 sequences from other sharks suggest that Ig germline gene rearrangement is not a frequent event, but that once established in the population, the presence of the joined gene is stable.
Antigen Receptors without Rearrangement.
In cases where all the identified genes of an isotype are joined, as are L chain genes in sandbar shark 38, in skate 13 29, and in nurse shark NS3 16 39, cDNA sequences have been isolated. However, in the case of mixed split and joined genes of one isotype, as in H chain genes in the horned shark, expression of the germline-joined H chain genes was not detected 40, and this cast doubt on the biological significance of the joined and partially joined H chain genes.
In the NS4 L chain type, four of the six joined genes are rearranged in frame and are potentially functional; all of the joined genes have the same octamer motif and other regulatory sequences as the split genes. Like Litman and coworkers, we were not able to detect significant amounts of germline-joined Ig message in adults, although the major L chain mRNA in nurse shark is the NS4 type 39. However, we also tested nurse shark pups and have detected expression of the two germline-joined NS4 genes that we were screening for, R4 and R18.
Do the joined genes have a special function? The pups not only express germline-joined NS4 genes but also a germline-joined H chain that constitutes more than half of the H chains in serum Ig. Adult nurse sharks, in contrast, express little such message (Flajnik, M.F., unpublished data). The coupling of the germline-joined NS4 L chain and the novel H chain isotype in shark pup lymphocytes presents the possibility that some B cells could differentiate and express antigen receptors independently of RAG activity.
The germline joining of particular Ig gene segments may have occurred fortuitously, but once established in the population, such genes may have evolved specialized function, as suggested by the similarly restricted expression of the newly discovered joined H chain. Seen from this viewpoint, a rejoined Ig gene is not merely a sequence restricting antibody diversity, but can evolve potentially as a novel receptor no longer tied to the rearrangement process.
Our hypothesis does not yet have a direct example, but the case of L chain surrogate V-pre-B is suggestive. From sequence homology and its ability to combine with H chain, V-pre-B is a homologue of L chains 41. It could have been an ancient rejoined gene, although there is little sequence homology with J (or RSS) in its extended 3′ end, but it is clearly a V region sequence that no longer has the function in common with other antibody V regions, and because rearrangement is not needed for its role, it is independent of RAG. In other words, V-pre-B is an Ig sequence that no longer requires rearrangement, and its function has likewise evolved away conventional L chain.
A different example of specialized function may be the role evolved by the H chain pseudogenes in the chicken; these are germline-fused, VD sequences that serve as templates for H chain gene conversion. This is discussed in the following section.
Role of RAG in Evolution of Antigen Receptors.
We propose that germ cell recombination of Ig genes are infrequent events, but that they may still occur, altering the germline content in nurse sharks. We suggest that the RAG proteins are responsible for generating germline-joined Ig genes. Are there RAG enzymes in the oocyte or sperm? RAG-1 was detected by RT-PCR in zebrafish ovary 42. RAG-2 was found in Xenopus oocytes by Northern blotting 43. However, the problem in the fish studies is that the origin of the RAG signal cannot be conclusively identified as being not from blood-borne cells; only in situ hybridization will establish this reliably.
Does germline rearrangement occur whenever the Ig genes are organized in clusters? In bony fishes, the H chain gene organization is like mammals but the L chain genes are in clusters. In catfish, where there are an estimated 60 L chain clusters, no rearranged genes have been amplified by PCR (Hsu, E., unpublished results), but they may just have escaped detection. It is also possible that the inverted orientation of the catfish L chain V genes 44 45 hinders the success of the rare joining event, as such a rearrangement would require reintegration of the excised DNA by ligation at two sites. However, a second duplicated H chain locus with nonfunctional C region exons was found in catfish, and the H chain gene segments, which are in one transcriptional orientation, are rearranged in the germline as VDJ 46.
In the chicken, antibody diversification is largely created by rearrangement of a single functional V gene at the H or L chain locus, followed by gene conversion modifications, using sequence from multiple upstream pseudogenes as templates 47 48 49 50. The H chain pseudogenes are discernibly germline-joined VD sequences. Our interpretation would be that an original VD sequence could have been generated by RAG-mediated recombination in the germ cells; the fused VD underwent gene duplication, coevolving with the dominance of the gene conversion mechanism. A supporting observation is that while rearrangements of VD alone are not normally found in lymphocytes, they are present as such in the germline in shark 12. The existence of VD and VDD joined shark genes suggest that recombination of Ig gene segments in germ cells are distinguishable from the differentiation signals of lymphocytes.
The current notion is that the RAG proteins can perform like transposases and may have initiated the development of the adaptive immune system in early vertebrates by splitting V genes with transposable elements 2 5. We suggest that with the discovery of recently rejoined Ig L chain genes and the restricted expression of these and a germline-joined H chain gene, RAG in some vertebrates could be a force in evolution still.
Acknowledgments
In memory of Charles M. Steinberg. We wish to thank Drs. Chris Roman and Louis Du Pasquier for their comments and criticism, and Dr. John Maisey of the American Museum of Natural History for clarifying elasmobranch phylogeny. We also thank Dr. Churchill McKinney for her help with blood sampling.
This work was supported in part by National Science Foundation grant MCB 9723203 and National Institutes of Health grant RR06603.
Footnotes
Abbreviations used in this paper: FR, framework region; Myr BP, million years before present; RAG, recombination activating gene; RSS, recombination signal sequence(s); RT, reverse transcription; TdT, terminal deoxynucleotidyl transferase.
References
- Du Pasquier L., Flajnik M.F. Origin and evolution of the vertebrate immune system. In: Paul W.E., editor. Fundamental Immunology. 4th ed. Lippincott-Raven; Philadelphia: 1999. pp. 605–650. [Google Scholar]
- Agrawal A., Eastman Q.M., Schatz D.G. Transposition mediated by RAG1 and RAG2 and its implications for the evolution of the immune system. Nature. 1998;394:744–751. doi: 10.1038/29457. [DOI] [PubMed] [Google Scholar]
- Hiom K., Melek M., Gellert M. DNA transposition by the RAG1 and RAG2 proteinsa possible source of oncogenic translocations. Cell. 1998;94:463–470. doi: 10.1016/s0092-8674(00)81587-1. [DOI] [PubMed] [Google Scholar]
- Sakano H., Hüppi K., Heinrich G., Tonegawa S. Sequences at the somatic recombination sites of immunoglobulin light-chain genes. Nature. 1979;280:288–294. doi: 10.1038/280288a0. [DOI] [PubMed] [Google Scholar]
- Thompson C.B. New insights into V(D)J recombination and its role in the evolution of the immune system. Immunity. 1995;3:531–539. doi: 10.1016/1074-7613(95)90124-8. [DOI] [PubMed] [Google Scholar]
- Amemyia C.T., Litman G.W. Complete nucleotide sequence of an immunoglobulin heavy chain gene and analysis of immunoglobulin gene organization in a primitive teleost species. Proc. Natl. Acad. Sci. USA. 1990;87:811–815. doi: 10.1073/pnas.87.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaffari S.H., Lobb C.J. Organization of immunoglobulin heavy chain constant and joining region genes in the channel catfish. Mol. Immunol. 1992;29:151–159. doi: 10.1016/0161-5890(92)90096-g. [DOI] [PubMed] [Google Scholar]
- Daggfeldt A., Bengtén E., Pilström L. A cluster type organization of the loci of the immunoglobulin light chain in Atlantic cod (Gadus morhua L.) and rainbow trout (Oncorhynchus mykiss Walbaum) indicated by nucleotide sequences of cDNAs and hybridization analysis. Immunogenetics. 1993;38:199–209. doi: 10.1007/BF00211520. [DOI] [PubMed] [Google Scholar]
- Hinds K.R., Litman G.W. Major reorganization of immunoglobulin VH segmental elements during vertebrate evolution. Nature. 1986;320:546–549. doi: 10.1038/320546a0. [DOI] [PubMed] [Google Scholar]
- Hohman V.S., Schuchman D.B., Schluter S.F., Marchalonis J.J. Genomic clone for the sandbar shark l light chaingeneration of diversity in the absence of gene rearrangement. Proc. Natl. Acad. Sci. USA. 1989;90:9882–9886. doi: 10.1073/pnas.90.21.9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamblott M.J., Litman G.W. Genomic organization and sequences of immunoglobulin light chains in a primitive vertebrate suggest coevolution of immunoglobulin gene organization. EMBO (Eur. Mol. Biol. Organ.) J. 1989;8:3733–3739. doi: 10.1002/j.1460-2075.1989.tb08549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokubu F., Litman R., Shamblott M.J., Hinds K., Litman G.W. Diverse organization of immunoglobulin VH gene loci in a primitive vertebrate. EMBO (Eur. Mol. Biol. Organ.) J. 1988;7:3413–3422. doi: 10.1002/j.1460-2075.1988.tb03215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rast J.P., Anderson M., Ota T., Litman R.T., Margittai M., Shamblott M.J., Litman G.W. Immunoglobulin light chain class multiplicity and alternative organizational forms in early vertebrate phylogeny. Immunogenetics. 1994;40:83–99. doi: 10.1007/BF00188170. [DOI] [PubMed] [Google Scholar]
- Yoder J.A., Litman G.W. Immune-type diversity in the absence of somatic rearrangement. Curr. Top. Microbiol. Immunol. 2000;248:271–282. doi: 10.1007/978-3-642-59674-2_12. [DOI] [PubMed] [Google Scholar]
- Greenberg A.S., Steiner L., Kasahara M., Flajnik M.F. Isolation of a shark immunoglobulin light chain cDNA clone encoding a protein resembling mammalian κ light chainsimplications for the evolution of light chains. Proc. Natl. Acad. Sci. USA. 1993;90:10603–10607. doi: 10.1073/pnas.90.22.10603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.S., Greenberg A., Hsu E. Evolution and somatic diversification of immunoglobulin light chains. Curr. Top. Microbiol. Immunol. 2000;248:285–300. doi: 10.1007/978-3-642-59674-2_13. [DOI] [PubMed] [Google Scholar]
- Sanger F., Nicklen S., Coulson A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M., Hsu E., Marcuz A., Courtet M., Du Pasquier L., Steinberg C. What limits affinity maturation of antibodies in Xenopus—the rate of somatic mutation or the ability to select mutants? EMBO (Eur. Mol. Biol. Organ.) J. 1992;11:4337–4347. doi: 10.1002/j.1460-2075.1992.tb05533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabat, E.A., T.T. Wu, H.M. Perry, K.S. Gottesman, and C. Foeller. 1991. Sequences of Proteins of Immunological Interest. 5th ed. US Department of Health and Human Services, Washington, D.C. xiii–xvii.
- Lee A., Desravines S., Hsu E. IgH diversity in an individual with only one million B lymphocytes. Dev. Immunol. 1993;3:211–222. doi: 10.1155/1993/17249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D., Crandall K.A. MODELTESTtesting the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- Kimura M. A simple method for estimating evolutionary rate of base substitution through comparative studies of nucleotide sequences. J. Mol. Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Hasegawa M., Kishino H., Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985;21:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Bremer K. Branch support and tree stability. Cladistics. 1994;10:295–304. [Google Scholar]
- Kishino H., Hasegawa M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in Hominoidea . J. Mol. Evol. 1989;29:170–179. doi: 10.1007/BF02100115. [DOI] [PubMed] [Google Scholar]
- Templeton A.R. Phylogenetic inference from restriction endonuclease cleavage site maps with particular reference to the humans and apes. Evolution. 1983;37:221–244. doi: 10.1111/j.1558-5646.1983.tb05533.x. [DOI] [PubMed] [Google Scholar]
- Cappetta H., Duffin C., Zidek J. Chondrichthyes. In: Benton M.J., editor. The Fossil Record. 2nd ed. Chapman and Hall; London: 1993. pp. 593–609. [Google Scholar]
- Kasahara M., McKinney E.C., Flajnik M.F., Ishibashi T. The evolutionary origin of the MHCpolymorphism of class IIA genes in the cartilaginous fish. Eur. J. Immunol. 1993;23:2160–2165. doi: 10.1002/eji.1830230917. [DOI] [PubMed] [Google Scholar]
- Anderson M.K., Shamblott M.J., Litman R.T., Litman G.W. Generation of immunoglobulin light chain gene diversity in Raja erinacea is not associated with somatic rearrangement, an exception to a central paradigm of B cell immunity. J. Exp. Med. 1995;182:109–119. doi: 10.1084/jem.182.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz D.G., Oettinger M.A., Baltimore D. The V(D)J recombination activating gene, RAG-1. Cell. 1989;59:1035–1048. doi: 10.1016/0092-8674(89)90760-5. [DOI] [PubMed] [Google Scholar]
- Oettinger M.A., Schatz D.G., Gorka C., Baltimore D. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science. 1990;248:1517–1523. doi: 10.1126/science.2360047. [DOI] [PubMed] [Google Scholar]
- McBlane J.F., van Gent D.C., Ramsden D.A., Romeo C., Cuomo C.A., Gellert M., Oettinger M.A. Cleavage at a V(D)J recombination signal requires only RAG1 and RAG2 proteins and occurs in two steps. Cell. 1995;83:387–395. doi: 10.1016/0092-8674(95)90116-7. [DOI] [PubMed] [Google Scholar]
- Roth D.B., Menetski J.P., Nakajima P.B., Bosma M.J., Gellert M. V(D)J recombinationbroken DNA molecules with covalently sealed (hairpin) coding ends in scid mouse thymocytes. Cell. 1992;70:1–9. doi: 10.1016/0092-8674(92)90248-b. [DOI] [PubMed] [Google Scholar]
- Shockett P.E., Schatz D.G. DNA hairpin opening mediated by the RAG1 and RAG2 proteins. Mol. Cell. Biol. 1999;9:4159–4166. doi: 10.1128/mcb.19.6.4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alt F.W., Baltimore D. Joining of immunoglobulin heavy chain gene segmentsimplications from a chromosome with evidence of three D-JH fusions. Proc. Natl. Acad. Sci. USA. 1982;79:4118–4122. doi: 10.1073/pnas.79.13.4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafaille J.J., DeCloux A., Bonneville M., Takagaki Y., Tonegawa S. Junctional sequences of T cell receptor genesimplications for αβ T cell lineages and for a novel intermediate of V-(D)-J joining. Cell. 1989;59:859–870. doi: 10.1016/0092-8674(89)90609-0. [DOI] [PubMed] [Google Scholar]
- Lewis S.M. The mechanism of V(D)J joininglessons from molecular, immunological, and comparative studies. Adv. Immunol. 1994;56:27–150. doi: 10.1016/s0065-2776(08)60450-2. [DOI] [PubMed] [Google Scholar]
- Schluter S.F., Hohman V.S., Edmundson A.B., Marchalonis J.J. Evolution of immunoglobulin light chainscDNA clones specifying sandbar shark constant regions. Proc. Natl. Acad. Sci. USA. 1989;86:9961–9965. doi: 10.1073/pnas.86.24.9961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg A.S. Evolution of the antigen receptor family. Ph.D. thesis 1994. The University of Miami; Miami: pp. 313 pp [Google Scholar]
- Litman G.W., Rast J.P., Shamblott M.J., Haire R.N., Hulst M., Roess W., Litman R.T. Phylogenetic diversification of immunoglobulin genes and the antibody repertoire. Mol. Biol. Evol. 1993;10:60–72. doi: 10.1093/oxfordjournals.molbev.a040000. [DOI] [PubMed] [Google Scholar]
- Kudo A., Melchers F. A second gene, Vpre-B, in the lambda 5 locus of the mouse, which appears to be selectively expressed in pre-B lymphocytes. EMBO (Eur. Mol. Biol. Organ.) J. 1987;6:2267–2272. doi: 10.1002/j.1460-2075.1987.tb02500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett C.E., Cherry J.J., Steiner L.A. Characterization and expression of the recombination activating genes (rag1 and rag2) of zebrafish. Immunogenetics. 1997;45:394–404. doi: 10.1007/s002510050221. [DOI] [PubMed] [Google Scholar]
- Greenhalgh P., Oleson C.E., Steiner L.A. Characterization and expression of recombination activating genes (RAG-1 and RAG-2) in Xenopus laevis . J. Immunol. 1993;151:3100–3110. [PubMed] [Google Scholar]
- Ghaffari S.H, Lobb C.J. Structure and genomic organization of immunoglobulin light chain in the channel catfish. J. Immunol. 1993;151:6900–6912. [PubMed] [Google Scholar]
- Ghaffari S.H., Lobb C.J. Structure and genomic organization of a second class of immunoglobulin light chain genes in the channel catfish. J. Immunol. 1997;159:250–258. [PubMed] [Google Scholar]
- Ghaffari S.H., Lobb C.J. Structure and genomic organization of a second cluster of immunoglobulin heavy chain gene segments in the channel catfish. J. Immunol. 1999;162:1519–1529. [PubMed] [Google Scholar]
- Reynaud C.A., Anquez V., Dahan A., Weill J.C. A single rearrangement event generates most of the chicken immunoglobulin light chain diversity. Cell. 1985;40:283–291. doi: 10.1016/0092-8674(85)90142-4. [DOI] [PubMed] [Google Scholar]
- Reynaud C.A., Anquez V., Grimal H., Weill J.C. A hyperconversion mechanism generates the chicken light chain preimmune repertoire. Cell. 1987;48:379–388. doi: 10.1016/0092-8674(87)90189-9. [DOI] [PubMed] [Google Scholar]
- Reynaud C.A., Dahan A., Anquez V., Weill J.C. Somatic hyperconversion diversifies the single VH gene of the chicken with a high incidence on the D region. Cell. 1989;40:283–291. doi: 10.1016/0092-8674(89)90879-9. [DOI] [PubMed] [Google Scholar]
- Thompson C.B., Neiman P.E. Somatic diversification of the chicken immunoglobulin light chain gene is limited to the rearranged variable gene segment. Cell. 1987;48:369–378. doi: 10.1016/0092-8674(87)90188-7. [DOI] [PubMed] [Google Scholar]