

Abstract
The Epstein-Barr virus (EBV)-encoded nuclear antigen EBNA1 is critical for the persistence of the viral episome in replicating EBV-transformed human B cells. Therefore, all EBV-induced tumors express this foreign antigen. However, EBNA1 is invisible to CD8+ cytotoxic T lymphocytes because its Gly/Ala repeat domain prevents proteasome-dependent processing for presentation on major histocompatibility complex (MHC) class I. We now describe that CD4+ T cells from healthy adults are primed to EBNA1. In fact, among latent EBV antigens that stimulate CD4+ T cells, EBNA1 is preferentially recognized. We present evidence that the CD4+ response may provide a protective role, including interferon γ secretion and direct cytolysis after encounter of transformed B lymphocyte cell lines (B-LCLs). Dendritic cells (DCs) process EBNA1 from purified protein and from MHC class II–mismatched, EBNA1-expressing cells including B-LCLs. In contrast, B-LCLs and Burkitt's lymphoma lines likely present EBNA1 after endogenous processing, as their capacity to cross-present from exogenous sources is weak or undetectable. By limiting dilution, there is a tight correlation between the capacity of CD4+ T cell lines to recognize autologous B-LCL–expressing EBNA1 and DCs that have captured EBNA1. Therefore, CD4+ T cells can respond to the EBNA1 protein that is crucial for EBV persistence. We suggest that this immune response is initiated in vivo by DCs that present EBV-infected B cells, and that EBNA1-specific CD4+ T cell immunity be enhanced to prevent and treat EBV-associated malignancies.
Keywords: Epstein-Barr virus, Epstein-Barr virus nuclear antigen 1, CD4+ T cell, cross-presentation, dendritic cells
Introduction
EBV is a human gamma herpesvirus with a tropism for B lymphocytes 1. More than 95% of the adult population carries EBV as a lifelong asymptomatic infection. Nevertheless, EBV has strong growth-transforming capacities 2. Each of its three latency programs gives rise to specific tumors originating from B cells or other cell types. As exemplified by EBV-transformed B cells (B lymphocyte cell lines [B-LCLs]) or lymphoproliferative disease, the latency III phenotype is characterized by the expression of nine gene products: six EBV nuclear antigens (EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNALP) and three latent membrane proteins (LMP1, LMP2A, and LMP2B) 3. In latency II, EBV-associated malignancies like Hodgkin's lymphoma, nasopharyngeal carcinoma, T cell lymphoma, gastric carcinoma, and uterine leiomyosarcoma, three specific EBV genes, EBNA1, LMP1, and LMP2, are maintained 3. Burkitt's lymphoma exemplifies the EBV latency I phenotype. Only the EBNA1 protein is expressed in these transformed B cells.
EBNA1, LMP1, and LMP2 are probably critical for tumorigenesis, inducing cell proliferation as well as resistance to apoptosis 4. EBNA1 binds as a dimer to the viral origin of replication and ensures episomal replication during B cell growth 5 6 7 8. The COOH-terminal part of LMP1 can act as a direct oncogene 9 10 11 by mimicking CD40-mediated B cell activation 12 13 14 15. Thus, LMP1 engages signaling proteins for the TNF receptor family 16 17 and Janus kinase 3 18, ultimately leading to nuclear factor κB 17 and AP-1 (Jun/Jun or Jun/Fos) 19 induction. In addition, LMP1 expression protects against apoptosis by induction of bcl-2 20 21. Instead, LMP2 mimics B cell receptor signaling, constitutively engaging syk and lyn, protein tyrosine kinases 22. In Burkitt's lymphoma, only EBNA1 seems to be required, as transformation is achieved by additional mechanisms probably involving c-myc uncoupling through chromosomal translocation 2. However, the increased incidence of B cell neoplasia in EBNA1 transgenic mice argues for an involvement of this EBV product in transformation even in latency I malignancies 23.
How then is transformation to latency I and II malignancies avoided in most carriers of EBV? Immunity to EBNA1 a priori could provide resistance to transformed cells, but it has proven difficult to detect specific T cell responses to this essential protein for EBV persistence. In fact, EBNA1 blocks its own processing for MHC class I presentation 24. This has been attributed to a deficit in proteasomal processing, caused by the NH2-terminal Gly/Ala (GA) repeat domain 25 26. A similar GA stretch prevents IκBα degradation by the proteasome 27. Other EBV latency gene products are the focus of a strong MHC class I–restricted CTL response, especially EBNA3A, EBNA3B, and EBNA3C 28. However, EBNA3 proteins are not expressed in most of the EBV-associated tumors mentioned above, and instead are expressed in transformed cultured lines (B-LCLs) and lymphoproliferative syndromes in immunosuppressed patients. CD8+ CTL responses to tumor-associated LMP1 29 and LMP2 30 proteins have only occasionally been detected.
It has been repeatedly apparent that the development 31 32 33 and maintenance 34 35 36 37 of effective CD8+ CTLs are dependent on CD4+ T cell help. Recognition of EBV products by CD4+ T cells has not been investigated in the same detail as the CD8+ T cell response 38. Only two EBV-specific CD4+ T cell clones have been described before 39 54. While the EBNA2-specific CD4+ T cell clone recognized HLA-DQ–matched B-LCLs, the EBNA1-specific CD4+ T cell clone only killed targets after exogenous loading with recombinant EBNA1 protein. Therefore, it was suggested that EBNA1 prevents its own endogenous presentation onto MHC class II, and that CD4+ T cell recognition of EBNA1 does not mediate protective immunity against EBV-associated malignancies.
Dendritic cells (DCs) are potent APCs for CD4+ and CD8+ T cell immunity 40 41. Therefore, we used DCs to search for CD4+ T cell responses to individual latent EBV products. For EBNA1, we delivered the antigen as recombinant protein 42 43 44 45, in recombinant vaccinia virus (vv) constructs, and by coculturing with B-LCLs. We have uncovered a strong CD4+ T cell response to EBNA1, as monitored by T cell activation and proliferation, IFN-γ secretion, and CTL activity. Paralleling the above results with one EBNA2-specific CD4+ T cell clone, we demonstrate that the EBNA1-specific CD4 responses—generated routinely from adult blood samples—recognize HLA-DR–matched B-LCLs and therefore could provide resistance to EBV infection and EBV-associated malignancy.
Materials and Methods
Cell Lines.
The EBV-transformed B cell lines LRM (HLA-A2, -B44, -DRB1*0401, -DQA1*03, -DQB1*0301, and -DP4) 46, LG2 (HLA-DRB1*0101, -DQA*0101, -DQB1*0501, -DPA1*0101, and -DPB1*0201) 47, and newly generated B-LCL and the Burkitt's lymphoma lines Ramos, EBV−, and Daudi, reverted to latency III (obtained from American Type Culture Collection), were cultured in RPMI 1640/10% FCS/glutamine/gentamicin. LCL-JT (HLA-DRB1*0301 and -DRB1* 1301), LCL-BM (HLA-A1, -A3, -B7, -B8, -Cw6, -Cw7, -DR4, -DRw14, -DRw52, -DRw53, and -DQw3), LCL-DC (HLA-A2, -A24, -B38, -B46, -Cw1, -Cw7, -DRB1*1502, -DRB1*0901, -DRB4*01, -DRB5*0101, -DQB1*0502, and -DQB1*0303), LCL-BC (HLA-DRB1*0401, -DRB1*0701, DRB4*01, -DQB1* 0302, and -DQB1*0201), and buffy coat–derived B-LCLs were generated by culturing PBMCs of healthy donors with supernatant of the marmoset cell line B95.8 in RPMI 1640/20% FCS/glutamine/gentamicin/1 μg/ml cyclosporin A. The rabbit RK13 and monkey BSC40 kidney cell line was grown in DMEM/15% FCS/glutamine/gentamicin.
DC and PBMC Preparations.
Leukocyte concentrates (buffy coats) from the New York Blood Center, as well as whole blood from lab donors, served as sources of PBMCs isolated by density gradient centrifugation on Ficoll-Paque (Amersham Pharmacia Biotech). CD2+ PBMCs were separated by rosetting with neuraminidase-treated (Calbiochem) sheep RBCs (Colorado Serum Company) followed by red cell lysis with 1.66% ammonium chloride. Where indicated, CD8+ or CD4+ T cells were depleted with Leu2a or OKT8 (for CD8) or HP2/6 (for CD4) antibodies, followed by incubation with sheep anti–mouse IgG Dynabeads and a magnetic particle concentrator, MCP-1 (Dynal). Positive selection for CD4+ PBMCs was performed using anti-CD4 MicroBeads, MS+/RS+ columns, and MiniMACS separator (Miltenyi Biotec). DCs were generated from CD2− PBMCs as described 48. 106 CD2− PBMCs/ml were plated in 6-well plates with RPMI 1640/1% single donor plasma/glutamine/gentamicin. 100 μl medium was replaced at days 2, 4, and 6. Recombinant human (rh)IL-4 and rhGM-CSF were added to a final concentration of 1,000 U/ml at days 0, 2, 4, and 6. On day 7, the floating immature DCs were transferred to new plates at 3 × 105 cells/ml and half of the medium was replaced by monocyte-conditioned medium to mature the DCs for 2 d. DCs and T cells were used fresh or after cryopreservation.
Vaccinia Virus Stock Generation and Infection of DCs.
Recombinant vv were expanded in rabbit RK13 and titrated on monkey BSC40 kidney cells. Mature DCs were infected at a multiplicity of infection (MOI) of 2 for 1 h at 37°C and washed three times. The efficiency of infection was checked after 6–12 h by FACS® as described using intracellular staining of a vaccinia early protein of 29 kD with the VV1-6B6 antibody 49.
Generation of CD4+ T Cell Lines and Clones.
CD8−CD2+ or CD4+ PBMCs were stimulated with mature DCs at a ratio of 30:1 (T/DC). For the CM171198 cell line, T cells of the healthy donor CM (HLA-A*0201, -A*6801, -B*4402, -B*0702, -C*0501, -C*0702, -DRB1*1501, -DRB1*0401, -DRB5*01, -DRB4*01, -DQB1*0602, and -DQB1*0301) were stimulated for 4 wk with vvEBNA1ΔGA-infected autologous mature DCs with weekly restimulations including autologous CD2+ PBMCs as feeders, and then alternating with the EBV-transformed HLA-DR–matched cell line LRM or vvEBNA1ΔGA-infected DCs. The CM110199 cell line was generated from T cells of the donor CM by using DCs that had been incubated with 10 μM of recombinant EBNA1 protein, added at day 7 together with the maturation stimulus. Purified rEBNA1 from Escherichia coli and baculovirus/insect cell expression systems were alternatively used 42 45. Where indicated, E. coli–derived DNA-C or PCNA were used as control proteins (eControl). After 7–14 d of stimulation in DMEM plus 5% human serum (HS), 50 U/ml rIL-2 was supplemented. The EBNA1-specific T cell line 090199.6 from leucocyte concentrates was established similarly from CD4+ PBMCs. For stimulations with the autologous B-LCLs, CD2+ PBMCs of the healthy donor JT were stimulated for 14 d with the irradiated autologous LCL-JT at a B cell to T cell ratio of 1:10 in DMEM plus 5% HS plus 10 U/ml IL-2 (Lymphocult). Where indicated, CD4+ or CD8+ T cells were depleted. EBNA1-specific CD4+ CTLs were cloned under limiting dilution conditions by stimulating 105, 3 × 104 and 104 CD8−CD2+ PBMCs from leukocyte concentrates or 30, 10, and 3 CM110199 cells with 104, autologous or HLA-DR–matched B-LCLs in 96-well plates for 14 d with one restimulation at day 7. IL-2 was added to the cultures during the restimulation to a final concentration of 10 U/ml (Lymphocult). Microcultures were tested in split well 51Cr-release assays against autologous DCs infected with vvEBNA1ΔGA or vvTK−, autologous B-LCLs, or LCL721.221. As <30% of the wells developed CTLs or IFN-γ–secreting cells, it is >90% probable that the responding wells represent clones 50. The BC cell line was generated separating EBNA1-specific CD4+ T cells after stimulation with vvEBNA1ΔGA-infected DCs using the IFN-γ secretion assay according to the manufacturer's instructions (Miltenyi Biotec).
FACS® Analysis of Stimulated CD4+ T Cell Populations and PBMCs.
Mature DCs were infected with recombinant vv at an MOI of 2, or with influenza virus (PR8, Puerto Rico/8/34; Spafas, Inc.) at an MOI of 0.5 for 1 h at 37°C in RPMI 1640 HS. DCs were washed twice, and 3 × 103 were added to 105 CD8− CD2+ PBMCs in 96-well plates for 7 d. The cultures were restimulated with irradiated (3,000 rads) 105 PBMCs and 3 × 103 DCs per well and incubated for an additional 7 d. At day 14, cultures were stained for 30 min on ice with 1 μl Simultest CD4-FITC/CD8-PE (Becton Dickinson) and analyzed on a FACScan™ (Becton Dickinson). CD56 antibody staining (BD PharMingen) used PE–goat anti–mouse IgG antibody (Biosource International) as secondary. PBMCs were typed for HLA-DR4 using HLA-DR4 antibody (Accurate) as primary and FITC–goat anti–mouse IgG antibody (Biosource International) as secondary.
Enzyme-linked Immunospot Assay for IFN-γ–secreting Cells.
Enzyme-linked immunospot (ELISPOT) assays were performed as described previously 51. MAHA S45 plates (Millipore) were coated with anti–IFN-γ antibody 1-D1K (Mabtech). Plates were blocked with DMEM plus 5% HS. Afterwards, 105 responder T cells and 3 × 103 stimulator DCs or 104 B-LCLs were added per well and incubated for 1–2 d. Where indicated, DCs, Ramos, or LCL-BC cells were cocultured with twofold excess of allogeneic vv expressing EBNA1 (vvEBNA1ΔGA), nonstructural protein 1 (vvNS1), or influenza matrix protein (vvMP)-infected DCs or allogeneic B-LCLs before cocultivation with T cells in the ELISPOT plates. For DCs, this cocultivation was performed during maturation. Then the plates were incubated with biotinylated anti-IFN-γ antibody 7-B6-1 (Mabtech). Afterwards, preassembled avidin-peroxidase complex Vectastain ABC kit (Vector Laboratories) was added. Spots were developed by addition of stable DAB (Research Genetics). Plates were washed three times with water and air dried. Spot-forming cells (SFCs)/105 cells were counted using a stereomicroscope (mean counts of triplicates). Where indicated, anti–HLA-DR antibody L243 52 or anti–HLA-A, -B, -C antibody B-H9 (Biosource International) was added to 5 μg/ml.
Proliferation Assays.
105 responder T cells were incubated with 3 × 103 to 104 stimulator DCs for 5 d in DMEM plus 5% HS. 1 μCi [3H]thymidine was added/well overnight, harvested by an automatic device (Skatron), and counted in a Betaplate 1205 (LKB Wallac). Counts represent mean values of triplicates.
51Cr-Release Assay.
Targets were labeled with 50 μCi Na2 51CrO4 for 45 min at 37°C. Labeled targets were incubated for 4 h with CTLs in RPMI/10% FCS/2 mM glutamine. The supernatant was harvested using a Skatron harvesting system, and radioactivity was measured in a γ counter (1470 Wizard; LKB Wallac). Percent specific lysis was calculated as ([cpm experimental well − cpm spontaneous release]/[cpm maximum release − cpm spontaneous release]) ×100%. Spontaneous release was determined by incubating the labeled targets with medium, and maximum release was determined by incubating targets in 1% Triton X-100 solution.
Results
CD4+ T Cells Consistently Recognize EBNA1.
To evaluate adult CD4+ T cell responsiveness to EBV latency gene products, CD8−CD2+ PBMC were stimulated for 2 wk with autologous DCs, separately infected with recombinant vv constructs expressing the EBV latent antigens EBNA1, EBNA3A, EBNA3B, EBNA3C, LMP1, and LMP2. For EBNA1, we also delivered the antigen as recombinant protein (rEBNA1) 42 45. Responses were assessed by the presence of enlarged CD4+ T cells (“blasts”) after 2-wk-long stimulations with DCs. In the first week, one of a panel of vvEBV recombinants was used to stimulate the CD4+ T cells. Then the cultures were divided in two and restimulated for a second week with the original recombinant vv or with vvTK− as control. We looked for blastogenesis specific to the EBV recombinant that stimulated the CD4+ cultures in the first week.
All 10 donors showed strong responses to vvEBNA1 (Table and Table , and Fig. 1A and Fig. B). The response to the negative control (vvTK−) were weak (Fig. 1 E) in all but one donor, excluded from the Tables. All donors responded to influenza-infected DCs as a positive control (Fig. 1 F). CD4 T cell responses by the 10 donors to the other vvEBV constructs were detected less consistently: EBNA3B (5/10), EBNA3A (1/10), EBNA3C (1/10), and LMP1 (6/10) (Table ). To ensure that all the recombinant vv infected a comparable proportion of the mature DCs, the intracellular expression of the 29-kD vaccinia early protein was measured by FACS®. Reproducibly, 40–60% of DCs were infected with the different vv (data not shown). The reliability of the CD4+ recognition of EBNA1 was also evident in an ELISPOT assay for IFN-γ secretion, where EBNA1 was the EBV latency gene most frequently recognized (Table ).
Table 1.
Percentages of Blasting CD4+ T Lymphocytes upon Stimulation with DCs Infected with Recombinant Vaccinia-EBV Viruses
| Donor no. | vvTK− | vvEBNA1 | vvEBNA3A | vvEBNA3B | vvEBNA3C | vvLMP1 | vvLMP2A | vvBMLF1 | Influenza |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4 | 17 (2) | 22 (8) | 11 (3) | ND | 23 (3) | 8 (4) | 3 (3) | 17 (6) |
| 2 | 2 | 15 (3) | 6 (2) | 17 (8) | 8 (3) | 8 (2) | 3 (2) | 0 (0) | 12 (3) |
| 3 | 0 | 11 (6) | 0 (0) | 19 (8) | 0 (0) | 30 (18) | 0 (0) | 0 (0) | 28 (10) |
| 4 | 1 | 11 (6) | 1 (1) | 12 (7) | 2 (1) | 14 (10) | 1 (0) | 1 (0) | 35 (6) |
| 5 | 1 | 15 (4) | 1 (1) | 4 (2) | 3 (1) | 6 (1) | 1 (1) | 0 (1) | 13 (10) |
| 6 | 2 | 8 (3) | ND | 2 (2) | 1 (2) | 5 (1) | 3 (3) | ND | 15 (7) |
| 7 | 3 | 11 (6) | 2 (2) | 2 (2) | 1 (1) | 3 (2) | 3 (1) | 3 (3) | 34 (11) |
| 8 | 1 | 12 (4) | 0 (0) | 10 (2) | 2 (1) | 8 (9) | 2 (2) | 9 (8) | 7 (3) |
| 9 | 2 | 8 (0) | 1 (0) | 3 (0) | 4 (2) | 18 (5) | 5 (0) | 4 (4) | 2 (1) |
| 10 | 5 | 18 (3) | 5 (3) | 3 (1) | 17 (3) | 8 (2) | 4 (1) | 5 (2) | 25 (3) |
Table 2.
Number of IFN-γ–producing CD4+ T Lymphocytes upon Stimulation with DCs Infected with Recombinant Vaccinia-EBV Viruses
| Donor no. | vvTK− | vvEBNA1 | vvEBNA3A | vvEBNA3B | vvEBNA3C | vvLMP1 | vvLMP2A | vvBMLF1 | Influenza |
|---|---|---|---|---|---|---|---|---|---|
| 5 | 7 ± 1 | 23 ± 1(7 ± 4) | 5 ± 4(7 ± 4) | 2 ± 1(5 ± 1) | 6 ± 2(5 ± 1) | 4 ± 1(2 ± 1) | 10 ± 4(6 ± 1) | 1 ± 1(2 ± 1) | 79 ± 3(9 ± 1) |
| 6 | 3 ± 1 | 88 ± 8 (37 ± 1) | ND | 5 ± 1(4 ± 2) | 3 ± 1(2 ± 2) | 4 ± 2(3 ± 1) | 6 ± 3(2 ± 2) | ND | 83 ± 3(27 ± 6) |
| 7 | 1 ± 1 | 16 ± 1(4 ± 1) | 4 ± 2(2 ± 2) | 2 ± 1(3 ± 0) | 2 ± 0(3 ± 0) | 2 ± 2(2 ± 1) | 6 ± 4(1 ± 0) | 4 ± 2(5 ± 1) | 77 ± 2(5 ± 1) |
| 8 | 3 ± 1 | 36 ± 1(6 ± 2) | 2 ± 1(1 ± 0) | 25 ± 4(1 ± 1) | 1 ± 1(2 ± 2) | 64 ± 2(5 ± 2) | 6 ± 2(2 ± 1) | 18 ± 1(2 ± 1) | 83 ± 1(3 ± 1) |
| 9 | 2 ± 1 | 17 ± 1(1 ± 1) | 2 ± 1(2 ± 2) | 5 ± 2(2 ± 2) | 8 ± 4(2 ± 1) | 25 ± 5(1 ± 1) | 10 ± 3(3 ± 1) | 6 ± 2(2 ± 1) | 15 ± 4(4 ± 1) |
| 10 | 5 ± 1 | 48 ± 3 (15 ± 3) | 15 ± 2(6 ± 4) | 13 ± 3(4 ± 3) | 108 ± 3(9 ± 5) | 38 ± 1 (10 ± 3) | 14 ± 1(7 ± 2) | 5 ± 2(3 ± 1) | 225 ± 5(8 ± 4) |
Figure 1.
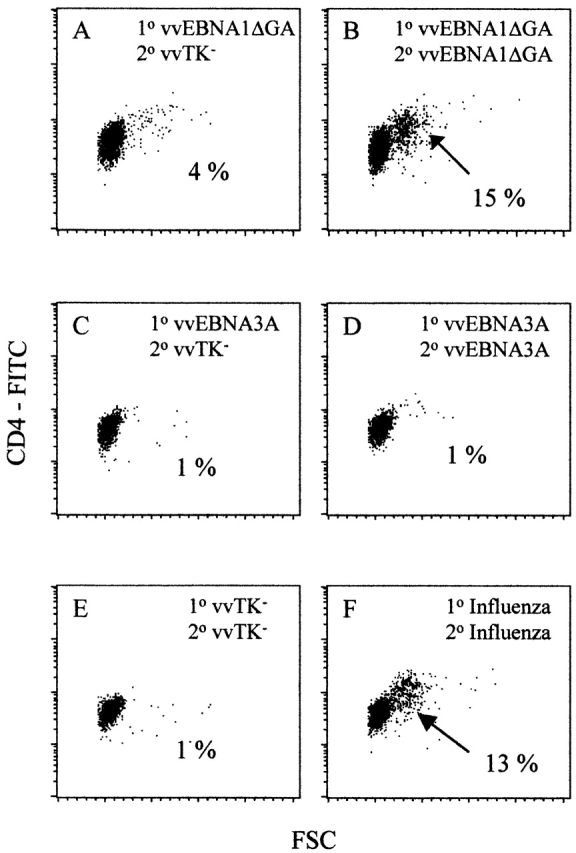
EBNA1 is recognized by CD4+ T cells from healthy EBV carriers. Blast formation (large forward scatter [FSC], x-axis) by CD4+ T cells (CD4-FITC, y-axis) was monitored by flow cytometry. Cultures of CD2+CD8− T cells were stimulated with autologous DCs infected with vv constructs. (A) Culture stimulated with vvEBNA1ΔGA-infected DCs and restimulated with vvTK−-infected DCs. (B) Blasting of a culture stimulated with vvEBNA1ΔGA-infected DCs and restimulated with vvEBNA1ΔGA-infected DCs. (C) Culture stimulated with vvEBNA3A-infected DCs and restimulated with vvTK−-infected DCs. (D) T cells were stimulated and restimulated with vvEBNA3A-infected DCs. (E) Culture stimulated and restimulated with vvTK−-infected DCs to evaluate the background of vaccinia stimulation. (F) CD2+CD8− T cells responding to influenza virus–infected DCs as positive control. All cultures were prepared from the same donor (no. 5 in Table and Table ). Percentage of blasted subpopulations (arrows) are indicated. 1°, first stimulation; 2°, restimulation.
We regard these CD4+ T cell responses to EBNA1 to reflect priming by EBV infection of the blood donors in vivo, as we did not see blastogenesis in 2 wk if we stimulated neonatal T cells from cord blood specimens with EBNA1 (Fig. 2). The fetal CD4+ T cells performed similarly to adult CD4+ T cells in MLR proliferation assays, and the fetal DCs were likewise capable of eliciting strong MLR proliferation of adult CD4+ T cells (Fig. 2 A). However, none of the fetal samples were able to recognize EBV or influenza products in IFN-γ ELISPOT assays (Fig. 2). CD4+ T cells from adult controls recognized vvEBNA1ΔGA, vvEBNA3B, vvLMP1, and influenza-infected autologous DCs in the same assay (Fig. 2).
Figure 2.

Adult EBV-specific CD4+ T cells are primed in vivo. CD4+ T cells from cord blood (C) and adult leukocyte concentrates (B) were stimulated with autologous DCs infected with various recombinant vv or influenza virus as a control for 14 d. The reactivities of the cultures were tested in IFN-γ ELISPOT assays using autologous DCs infected with the same recombinant vv as targets (TK−, vvTK−; E1, vvEBNA1ΔGA; E2, vvEBNA2; E3B, vvEBNA3B; E3C, vvEBNA3C; L1, vvLMP1; L2, vvLMP2A; and B1, vvBMLF1). In a primary immune reaction, exemplified by alloreactivity (shown for cord blood sample, #2 in A), adult and fetal DCs and T cells functioned comparably.
CD4+ T Cells Recognize EBNA1 in an MHC Class II–restricted Fashion.
We verified that our donors showed HLA class II diversity, as only two expressed HLA-DR4 (data not shown). To establish that MHC class II products were presenting EBNA1, we generated T cell lines, initially, from an HLA-DR4+ donor. We then assessed reactivity of the lines with DCs infected with recombinant vv expressing EBNA1 or pulsed with soluble EBNA1 protein. One line, CM171198, was derived from CD8−CD2+ PBMCs stimulated alternatively with autologous DCs infected with vvEBNA1ΔGA or the DR4-matched B-LCL LRM. The vvEBNA1 construct was deleted of the GA repeat that blocks MHC class I presentation and also reduces expression of EBNA1. Another line, CM110199, was stimulated with DCs charged during their final maturation with recombinant EBNA1 protein expressed either in E. coli (eEBNA1) or in a baculovirus/insect cell system (bEBNA1). After 1 mo of culture, both lines were predominantly CD4+ T cells, 90% in CM171198 and 76% in CM110199, with CD56+ NK cells being the main contaminant (not shown).
The CD4+ T cell lines recognized DCs that were infected with vvEBNA1ΔGA or exposed to recombinant EBNA1 (Fig. 3A and Fig. B). Reactivity could be measured as IFN-γ secretion (ELISPOT assays, Fig. 3 A) or by proliferation (Fig. 3 B). The T cell responses were blocked by an anti–HLA-DR antibody, L243, but not anti–HLA class I antibody, B-H9 (Fig. 3 A). In addition to DCs charged with EBNA1, the CM171198 cell line recognized EBV-transformed B-LCLs without further addition of antigen (Fig. 3A and Fig. B, bottom). In B-LCLs, only full-length EBNA1 is expressed at detectable levels 24. This implies that full-length EBNA1, as expressed endogenously by B-LCLs, can be processed on MHC class II molecules for CD4+ T cell recognition. The B-LCLs had to be matched at the DR4 allele to trigger T cell function. Thus, DR4+ B-LCLs (LRM and LCL-BM) induced proliferation, but DR4− cells (LG2 and LCL-DC) did not (Fig. 3A and Fig. B, bottom).
Figure 3.

Recognition of EBNA1 by a T cell line from an HLA-DR4+ donor stimulated with DCs loaded with soluble EBNA1 protein or a vaccinia-EBNA1 construct. (A) Top, IFN-γ SFCs/105 cells upon stimulation with DCs loaded with recombinant bEBNA1 protein (DC + bEBNA1) or without loading (DC). Bottom, Spot formation upon incubation with vvTK−-infected DCs (DC + vvTK−) or vvEBNA1ΔGA-infected DCs (DC + vvEBNA1ΔGA). The MHC restriction is analyzed using the antibodies L243, anti–HLA-DR (+L243), and B-H9, anti–HLA class I (+B-H9), for blocking. In addition, spot formation upon stimulation with the HLA-DR4+ B-LCL LRM (LRM) versus the HLA-DR4− B-LCL LG2 (LG2) is shown. (B) Proliferative responses of the CM171198 cell line to various EBNA1 expressing targets. Top, APCs were DCs loaded with rEBNA1 expressed in E. coli (eEBNA1), or baculovirus/insect cells (bEBNA1), or DCs with a vv construct expressing EBNA1 (vvEBNA1ΔGA) or vector alone (vvTK−). Bottom, the APCs were B-LCLs sharing the HLA-DR4 allele (LRM and LCL-BM) with the CM171198 cell line, or mismatched for HLA-DR4 (LG2 and LCL-DC).
EBNA1-specific CD4+ T Cells Kill B-LCLs.
To determine if EBNA1 was an antigen for CD4+ CTLs, we stimulated CD8−CD2+ PBMCs from an HLA-DR4− donor, JT, with irradiated autologous B-LCLs (expressing all known latent EBV antigens [3]) for 2 wk. In parallel, the B-LCLs were used to stimulate bulk CD2+ and CD4− CD2+ T cells. The content of the stimulated T cell populations was determined by FACS®. CD8-depleted responders were enriched for CD4+ cells, CD4-depleted responders were enriched for CD8+ cells, and the bulk T cells had a CD4+/CD8+ ratio of 1:2 (see Fig. 5, top). All contained ∼25% CD56+ NK cells (not shown).
Figure 5.
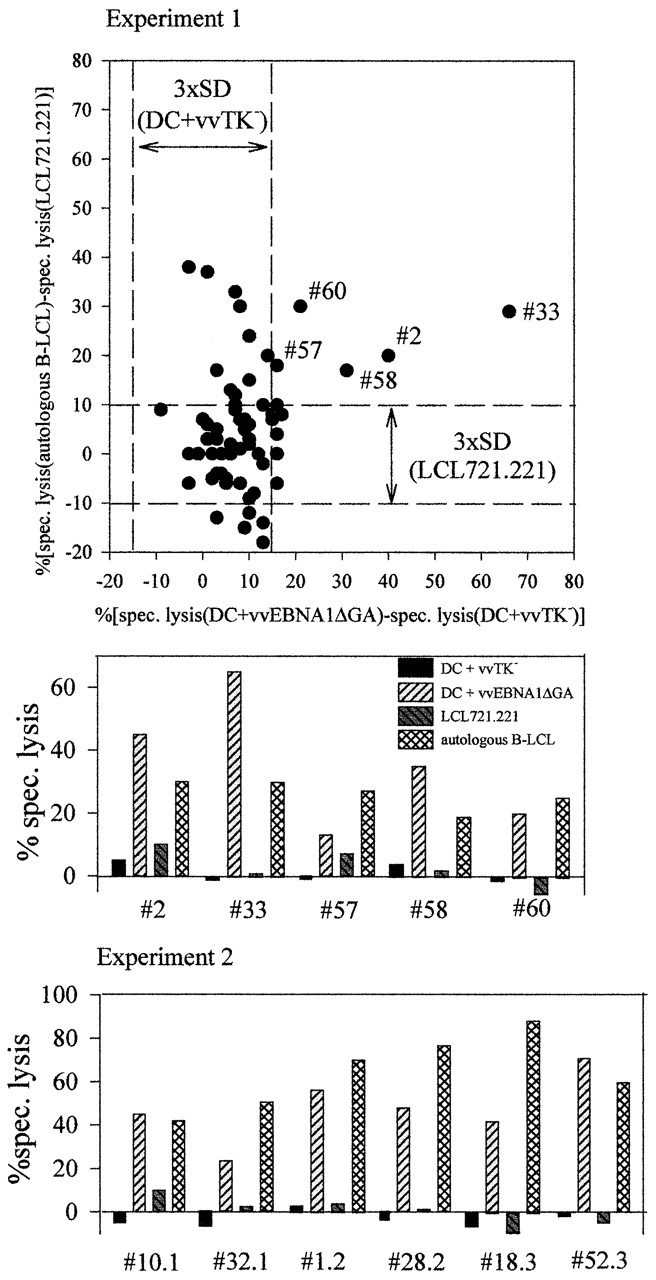
EBNA1-specific CD4+ CTL sublines kill B-LCLs. B-LCLs were generated from two leukocyte concentrates. Cryopreserved CD8−CD2+ PBMCs were then stimulated for 2 wk with the autologous B-LCLs under limiting dilution. Then the wells were split and tested in 51Cr-release assays against vvEBNA1ΔGA- or vvTK−-infected DCs as well as autologous B-LCLs and LCL721.221, an HLA class I− NK target. The dot plot shows for the first experiment, as an example, the CTL specificities that could be detected above three times the SD (3×SD) of the unspecific targets. Only CTLs exclusively recognizing autologous B-LCLs or recognizing autologous B-LCLs and vvEBNA1ΔGA-infected autologous DCs could be detected. All 11 sublines that recognized EBNA1-expressing DCs also demonstrated specific killing of autologous EBV-transformed B cells.
51Cr-release assays showed that the three populations of stimulated T cells killed autologous B-LCLs, with less recognition of the T2 cell line (Fig. 4, bottom left). As expected, the killing of autologous B-LCLs was completely blocked by L243 anti–HLA-DR antibody when CD4+-enriched populations, but not CD8+-enriched cultures, were tested (Fig. 4, bottom left). However, the CD8-enriched cultures repeatedly developed stronger cytolytic activity than CD4-enriched cultures. Killing by the bulk T cells was partially inhibited by the L243 antibody, but the block was ≤50% in our three experiments.
Figure 4.

HLA-DR restriction and EBNA1 recognition by T cell subsets. Different populations of PBMCs (CD2+, CD8−CD2+, or CD4−CD2+) were stimulated for 2 wk with autologous B-LCL. Top, the effectors were analyzed by FACS® for CD4 and CD8 expression. Bottom left, cytolysis of autologous B-LCL (LCL-JT) in the presence or absence of 5 μg/ml L243, anti–HLA-DR antibody (LCL-JT + L243). T2 cells are targets for NK cells. Bottom right, lytic activity against autologous B-LCL (LCL-JT) compared with autologous DCs pulsed with E. coli–derived control protein (eControl), E. coli–derived EBNA1 (eEBNA1), or baculovirus/insect cell–derived EBNA1 (bEBNA1). % spec. lysis, percent specific lysis.
EBNA1-specific CTL function was also assessed using DC targets that had been infected with vvEBNA1ΔGA or recombinant eEBNA1 and bEBNA1 proteins (Fig. 4). CD4+-enriched T cells lysed EBNA1-pulsed DCs. In contrast, CD8+-enriched cultures and bulk T cells were able to kill LCLs, but not the EBNA1-pulsed DCs (Fig. 4, bottom right).
To begin to determine if individual CD4+ T cells could lyse DCs pulsed with EBNA1, as well as B-LCLs expressing EBNA1 endogenously, we used autologous B-LCLs to isolate CD4+ CTLs by limiting dilution from cryopreserved T cells. The DCs, T cells, and autologous B-LCLs were derived from leukocyte concentrates. All 11 limiting dilution sublines that killed DCs in an EBNA1-dependent fashion also killed autologous B-LCLs, and we did not find any clone that killed the DCs and not the B-LCLs (Fig. 5). This indicates a tight correlation between recognition of epitopes expressed by B-LCLs and recognition of DCs that have been pulsed with EBNA1. As we were studying cells obtained under limiting dilution conditions, with <30% recognizing B-LCLs, it is >90% likely that individual clones were responsible for killing DCs and B-LCLs, although formal cloning experiments will be required 50. Recognition of DCs infected with the vvTK− control vector or LCL721.221, an HLA class I–negative NK target, was poor in all 11 sublines (Fig. 5). Therefore, CD4+ T cells can lyse autologous B-LCLs, and one target very likely is EBNA1.
EBNA1 Is Recognized on B-LCLs after Endogenous Processing.
As we were observing EBNA1-specific CD4+ cells that make Th1 cytokines and exert cytolytic activity upon encountering transformed B-LCLs, it was important to establish that this new and potentially protective mechanism would operate on EBNA1 that was processed endogenously by transformed B cells. The alternative would be that in our cultures some cells were dying and were being reprocessed via an exogenous or endocytic pathway in a fraction of LCLs. We are not aware of inhibitors that efficiently and selectively block processing of EBNA1 from an endogenous or exogenous route.
Therefore, we first compared the capacity of DCs (as a positive control) and the EBV− Burkitt's lymphoma cell line, Ramos, to present EBNA1 through an exogenous pathway, either rEBNA1 protein or EBNA1 expressed by allogeneic B-LCLs (Fig. 6 A). Because of MHC class II mismatching, the allogeneic B-LCLs could not directly present EBNA1 to T cell lines that had been selected for IFN-γ secretion upon stimulation with vvEBNA1ΔGA-infected autologous DCs. The Ramos cell line as well as the autologous DCs could present vvEBNA1ΔGA to an EBNA1-specific CD4+ T cell line from a donor matched at HLA-DR7 to Ramos (Fig. 6 A). In contrast, only the DCs, and not the Ramos Burkitt's lymphoma cells, presented exogenous rEBNA1 and EBNA1 from allogeneic LCLs (having ∼20% trypan blue–positive or dead cells; Fig. 6 A).
Figure 6.

DCs are efficient, and B-LCLs inefficient, in presenting EBNA1 by an exogenous pathway. (A) The EBNA1-specific HLA-DRB*0701+CD4+ T cell line BC, selected for IFN-γ secretion upon stimulation with vvEBNA1ΔGA-infected autologous DCs, responds comparably to vvEBNA1ΔGA-infected autologous DCs and vvEBNA1ΔGA-infected HLA-DRB*0701+ Ramos cells. DCs, but not Ramos cells, present recombinant EBNA1 proteins and allogenic LCL-JT. (B) The EBNA1-specific CD4+ T cell line 090199.6 recognized autologous DCs either infected with vvEBNA1DGA, or cocultured with infected allogeneic (allo) DCs. Semiallogeneic (semiallo) DCs were recognized irrespective of coculturing with autologous DCs.
DCs also presented EBNA1 from additional exogenous sources. In Fig. 6 B, DCs presented EBNA1 from four different allogeneic DC preparations infected with vvEBNA1ΔGA, presumably because infection with vv is cytotoxic for some of the infected DCs 49. As vv-infected DCs cannot produce virus particles 49 53, coinfection could not be responsible for the observed EBNA1 transfer from one DC to another. A fifth semiallogeneic DC preparation presented vvEBNA1ΔGA directly to the T cell line (Fig. 6 B).
Although there is already evidence in the literature that Burkitt's lymphoma cells are comparably efficient to B-LCLs in presenting soluble antigen on MHC class II 54, we wanted to show that both types of lymphoma also share the inability to cross-present antigen from cocultured cells. Therefore, we infected mismatched DCs and EBV latency III type Daudi cells with recombinant vv expressing influenza NS1 or MP, and then looked for cross-presentation of these influenza products by autologous B-LCLs or DCs, to CD4+ T cells of an influenza-reactive donor. Again, the DCs could present antigen from the allogeneic vvNS1- or vvMP-infected cells, but the B-LCLs could not (Fig. 7). As a positive control, we showed that autologous B-LCLs could present NS1 (Fig. 7 A) and MP (Fig. 7 B) to CD4+ T cells when directly infected with vvNS1 or vvMP, respectively. Coculturing B-LCLs with allogeneic infected B-LCLs for 2 d apparently did not transfer sufficient antigen amounts to the matched B-LCLs to trigger IFN-γ secretion by CD4+ T cells. In contrast NS1- or MP-expressing allogeneic B-LCLs could be efficiently processed by DCs and presented to CD4+ T cells.
Figure 7.
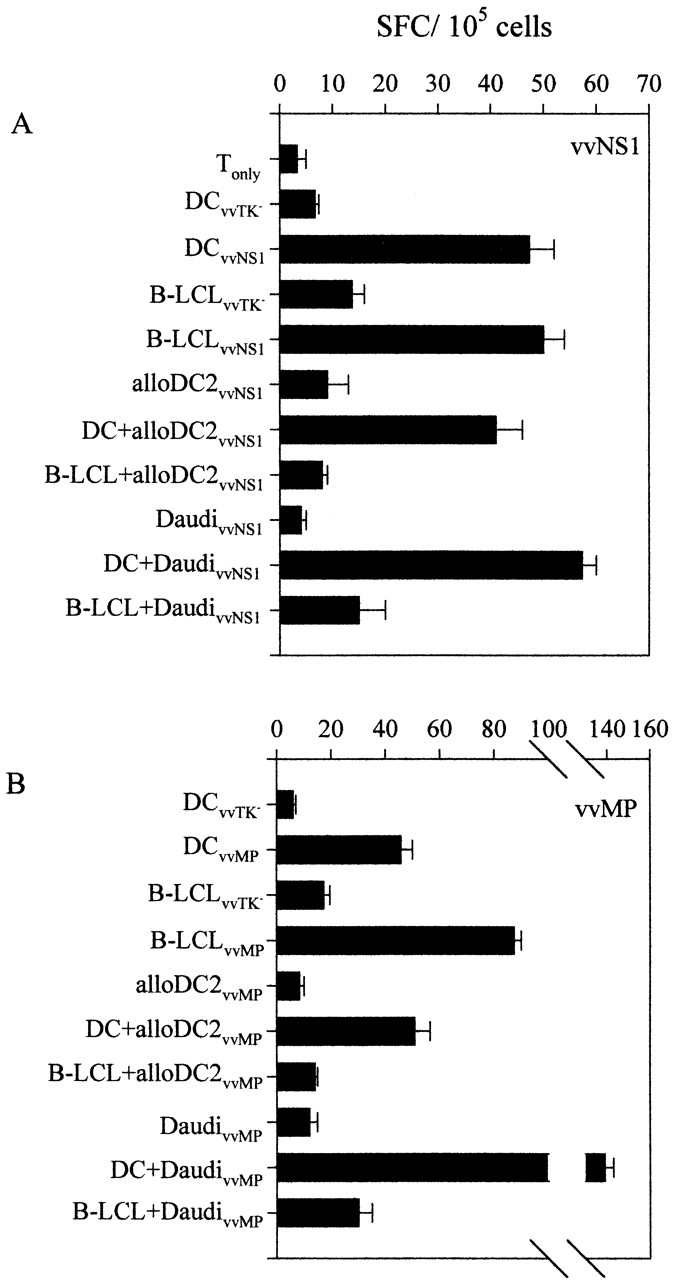
Influenza antigens are efficiently cross-presented by DCs, but not B-LCLs. Allogeneic DCs (alloDC2) and the latency III type Burkitt's lymphoma cell line Daudi were infected with recombinant vv expressing either (A) NS1 (vvNS1), or (B) influenza MP (vvMP). Then DCs and B-LCLs were cultured with maturing DCs or B-LCLs derived from another healthy adult donor for 2 d at a ratio of vv-infected cells to presenting cells of 2:1 (DC+/B-LCL+). CD2+CD8− PBMCs autologous to the presenting cells were tested in IFN-γ ELISPOT assays. As controls, the presenting DCs or B-LCLs were also directly infected with vvNS1 and vvMP or vvTK− as control (DCvvTK−/DCvvNS1/DCvvMP or B-LCLvvTK−/B-LCLvvMP/B-LCLvvNS1).
This series of experiments indicates that DCs efficiently cross-present on MHC class II antigens (EBNA1, NS1, and MP) from allogeneic DCs or B-LCLs, but that transformed B cells (B-LCLs or Burkitt's lymphoma cells) are weak or inactive in this respect. The findings are consistent with prior publications showing that B-LCLs can present soluble proteins that bind to their Ig receptor, but not by a nonspecific exogenous route 55. Therefore, CD4 T cell recognition of EBNA1 in B-LCLs very likely represents recognition of the endogenously processed protein that is critical for EBV-induced transformation.
Discussion
EBNA1-specific CD4+ T Cell Immunity Exists in All Healthy Adults.
In an effort to define the CD4+ T cell repertoire for EBV latency gene products, we have uncovered a consistent CD4+ T cell response to EBNA1 presented by DCs and by autologous B-LCLs. The EBNA1-specific CD4+ T cells proliferate, secrete IFN-γ, kill targets, and can be readily propagated as EBNA1-specific, MHC class II-restricted lines. Other latency antigens (LMP1, LMP2, EBNA3A, EBNA3B, and EBNA3C; Table and Table ) can be recognized by CD4+ T cells, but less consistently. The CD4+ T cells described here are found in bulk cultures and kill B-LCLs without further addition of EBNA1. The results are in marked contrast to the prior literature that has described only a single EBNA1-specific CD4+ T cell clone, and it only killed B-LCL targets upon supplementation with exogenous EBNA1 39. The consistent detection of EBNA1 immunity in our studies could reflect the use of CD4+ T cells as responders, and DCs as APCs.
The consistent recognition of EBNA1 by CD4+ T cells is strikingly different from CD8+ T cells. CD8+ T cells, specific for EBNA1-derived MHC class I epitopes, can be found in blood, but they do not recognize autologous or HLA-matched B-LCLs 24. This indicates that the physiological levels of EBNA1 are not sufficiently processed onto MHC class I, nor do B-LCLs cross-present EBNA1 when B-LCLs die in culture. The block of endogenous processing of EBNA1 is well understood, i.e., the Gly/Ala repeat prevents efficient proteasomal degradation and therefore epitope generation for MHC class I presentation 25 26. In light of the efficient cross-presentation of antigens on MHC class I by DCs 56, it is likely that the observed EBNA1-specific CD8+ T cells are primed by DCs that process B-LCLs onto MHC class I. However, because of the Gly/Ala repeat, such CD8+ T cells would not see EBNA1 expressed by EBV-infected cells.
Evidence That Endogenous Processing and Presentation of Physiological Epitope Levels on B-LCLs Are Sufficient to Elicit EBNA1-specific CD4+ T Cell Immunity.
The EBNA1-specific CD4+ T cells described in this study recognize autologous B-LCLs by proliferation, IFN-γ secretion, and cytolysis. As these responses are MHC class II restricted, and because EBNA1 is expressed endogenously in B-LCLs, two routes of processing can be envisioned. Either EBNA1 is processed directly by the EBV-infected cell itself (endogenous pathway), or dying EBNA1-expressing B-LCLs are endocytosed and presented (indirect or exogenous or cross-presentation pathway). This does not necessarily mean that the processing machinery differs, but that in the first endogenous case the antigen is processed in the cell that synthesizes EBNA1, making it a more reliable target for protective immunity in humans. From our experiments, exogenous processing is an unlikely explanation for presentation of EBNA1 on B-LCLs, as antigen transfer from HLA-mismatched cells was not observed (Fig. 6). B-LCLs were previously shown to be poor APCs for exogenous proteins 55, 100–300 times less efficient than DCs. Similarly we found that the Ramos Burkitt's lymphoma line inefficiently processed rEBNA1 and cross-presented EBNA1 from cocultured B-LCLs to CD4+ effector cells (Fig. 6 A). Cross-presentation of influenza NS1 and MP by B-LCLs from vvNS1- or vvMP-infected DCs or EBV latency III type Daudi cells was also inefficient (Fig. 7). In contrast, DCs cross-presented antigen from B-LCLs or vvEBNA1ΔGA-infected DCs, and processed rEBNA1 (Fig. 6A and Fig. B). This was not restricted to EBNA1, as DCs also presented NS1 and MP from cocultured vvNS1- and vvMP-infected DCs and Daudi cells (Fig. 7). As a positive control for the competence of the lymphoma cells as APCs, we showed that direct infection of Ramos or B-LCL cells with recombinant EBNA1 or influenza vv led to CD4+ T cell responses. This implies that in healthy carriers, the DCs that are known initiators of immune responses 41 cross-present B-LCL–derived EBNA1 for priming of specific CD4+ T cells. Once activated, these CD4+ T cells may attack B cells that process EBNA1 endogenously and contribute to EBV-specific immunity.
The Gly/Ala Repeat Domain Does Not Influence MHC Class II Processing of EBNA1.
Our experiments confirm that EBNA1 can be processed onto MHC class II irrespective of the presence or absence of its Gly/Ala repeat. This domain inhibits proteasome-dependent processing for MHC class I 25 26. B-LCLs, the only reliable source of full-length EBNA1 24, readily present EBNA1 to specific CD4+ T cells (Fig. 2 Fig. 3 Fig. 4). Moreover, B-LCLs with full-length EBNA1 can be cross-presented by DCs (Fig. 5 B). Therefore, the proposal that EBNA1 prevents its endogeous MHC class II presentation 39 cannot be supported by our findings.
Evidence That CD4+ T Cell Immunity Could Contribute to the Control of EBV.
EBNA1-specific CD4+ T cells could provide direct resistance to EBV-transformed cells, through their cytokines and lytic function or by sustaining the CD8+ CTL response to other lymphoma-related EBV products such as LMP1 and LMP2. A good deal of circumstantial evidence for CD4+ T cell protection against gamma herpesviruses in vivo exists. (a) CTLs to EBV in the cottontop tamarin Sanguinis oedipus 57 are to a large extent MHC class II restricted 58. MHC class I–restricted, EBV-specific CTLs have yet to be found in this New World monkey, and this species lacks classical MHC class I (although it does express homologues of nonclassical class I genes like HLA-G and HLA-F; 59). (b) Gamma herpesvirus infection in mice by MHV-68 can be controlled by IFN-γ–secreting CD4+ T cells 60. (c) Control of the growth of EBV-transformed B cells by CD4+ T cells has been described in culture 61. (d) Early in HIV-1 infection, when CD4+ T cell counts are still high but CD4+ T cell function starts to be compromised, patients can develop Burkitt's lymphomas, expressing only the EBNA1 EBV latency gene product, rather than mononucleosis with all nine EBV latency gene products 62. (e) Impaired CD4+ responses are thought to be responsible for the EBV-induced infectious mononucleosis seen in X-linked lymphoproliferative disease patients who have a mutation or deletion in signalling lymphocyte activation molecule (SLAM)-associated protein (SAP), an inhibitor of the T cell costimulatory molecule SLAM or CDw150 63. This lack in SLAM function especially affects Th1 immunity, as SLAM engagement mediates increased IFN-γ secretion 64 65. Thus, EBNA1-specific Th1 cells are probably disabled in X-linked lymphoproliferative patients.
Our findings uncover an immune response that changes current thinking on immune surveillance against EBV. The data on CD4+ T cell responses to EBV latent antigens in bulk T cell preparations place EBNA1 at the top of the recognition hierarchy. Previously, the specificities of only two isolated CD4+ T cell clones have been described 39 54. Since only the EBNA2-specific CD4+ T cell clone recognized B-LCLs, the significance for CD4+ T cell recognition was questionable, particularly in the context of latency I and II programs of EBV transformation. However, the new data reveal a hierarchy of CD4+ T cell recognition that differs significantly from CD8+ T cell responses to EBV 28. EBNA3A, EBNA3B, and EBNA3C antigens dominate for CD8+ T cell recognition, but play a subdominant role for CD4+ T cells. In contrast, EBNA1, believed to be invisible to the immune system because of a block in its MHC class I presentation 24, is the main target of MHC class II–restricted CD4+ T cell responses. As we find these responses in all tested healthy adults, we suggest that EBNA1-specific CD4+ T cells provide resistance to the development of Burkitt's lymphomas, Hodgkin's, and other EBV-associated malignancies. Likewise, the new data suggest that EBNA1 be tested as an antigen to prevent and treat such malignancies.
Acknowledgments
We thank Dr. Björn Clausen for critically reading the manuscript, and Dr. B. Moss for the gift of recombinant vvNS1 and vvMP vaccinia viruses.
This work was supported by a grant from the Cancer Research Institute and by grant AZ40874 from the National Institute of Allergy and Infectious Diseases to R.M. Steinman. C. Münz and M. Subklewe were recipients of Deutsche Forschungsgemeinschaft postdoctoral fellowships (grants Mu1522/1-1 and Su197/1-1). K.L. Bickham is a National Institutes of Health Fellow of the Pediatric Scientist Development Program (grant K12-HD00850), and M. Subklewe was supported in addition by the Cure for Lymphoma Foundation.
Footnotes
M.G. Kurilla's present address is Dupont Pharmaceuticals Corporation, Experimental Station, Wilmington, DE 19880.
Abbreviations used in this paper: B-LCL, B lymphocyte cell line; DC, dendritic cell; EBNA, EBV nuclear antigen; ELISPOT, enzyme-linked immunospot; GA, Gly/Ala; HS, human serum; LMP, latent membrane protein; MOI, multiplicity of infection; MP, matrix protein; NS1, nonstructural protein 1; SFC, spot-forming cell; SLAM, signalling lymphocyte activation molecule; vv, vaccinia virus.
References
- Kieff E. Epstein-Barr virus and its replication. In: Fields B.N., Knipe D.M., Howley P.M., editors. Virology. Lippincott-Raven Publishers; Philadelphia: 1996. pp. 2343–2396. [Google Scholar]
- Klein G. Epstein-Barr virus strategy in normal and neoplastic B cells. Cell. 1994;77:791–793. doi: 10.1016/0092-8674(94)90125-2. [DOI] [PubMed] [Google Scholar]
- Rickinson A.B., Kieff E. Epstein-Barr virus. In: Fields B.N., Knipe D.M., Howley P.M., editors. Virology. Lippincott-Raven Publishers; Philadelphia: 1996. pp. 2397–2446. [Google Scholar]
- Gregory C.D., Dive C., Henderson S., Smith C.A., Williams G.T., Gordon J., Rickinson A.B. Activation of Epstein-Barr virus latent genes protects human B cells from death by apoptosis. Nature. 1991;349:612–614. doi: 10.1038/349612a0. [DOI] [PubMed] [Google Scholar]
- Bochkarev A., Barwell J.A., Pfuetzner R.A., Bochkareva E., Frappier L., Edwards A.M. Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein, EBNA1, bound to DNA. Cell. 1996;84:791–800. doi: 10.1016/s0092-8674(00)81056-9. [DOI] [PubMed] [Google Scholar]
- Shah W.A., Ambinder R.F., Hayward G.S., Hayward S.D. Binding of EBNA-1 to DNA creates a protease-resistant domain that encompasses the DNA recognition and dimerization functions. J. Virol. 1992;66:3355–3362. doi: 10.1128/jvi.66.6.3355-3362.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates J.L., Warren N., Sugden B. Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature. 1985;313:812–815. doi: 10.1038/313812a0. [DOI] [PubMed] [Google Scholar]
- Rowe M., Lear A.L., Croom-Carter D., Davies A.H., Rickinson A.B. Three pathways of Epstein-Barr virus gene activation from EBNA1-positive latency in B lymphocytes. J. Virol. 1992;66:122–131. doi: 10.1128/jvi.66.1.122-131.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye K.M., Izumi K.M., Mosialos G., Kieff E. The Epstein-Barr virus LMP1 cytoplasmic carboxy terminus is essential for B-lymphocyte transformation; fibroblast cocultivation complements a critical function within the terminal 155 residues. J. Virol. 1995;69:675–683. doi: 10.1128/jvi.69.2.675-683.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Liebowitz D., Wang F., Gregory C., Rickinson A., Larson R., Springer T., Kieff E. Epstein-Barr virus latent infection membrane protein alters the human B-lymphocyte phenotypedeletion of the amino terminus abolishes activity. J. Virol. 1988;62:4173–4184. doi: 10.1128/jvi.62.11.4173-4184.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Liebowitz D., Kieff E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell. 1985;43:831–840. doi: 10.1016/0092-8674(85)90256-9. [DOI] [PubMed] [Google Scholar]
- Busch L.K., Bishop G.A. The EBV transforming protein, latent membrane protein 1, mimics and cooperates with CD40 signaling in B lymphocytes. J. Immunol. 1999;162:2555–2561. [PubMed] [Google Scholar]
- Henriquez N.V., Floettmann E., Salmon M., Rowe M., Rickinson A.B. Differential responses to CD40 ligation among Burkitt lymphoma lines that are uniformly responsive to Epstein-Barr virus latent membrane protein 1. J. Immunol. 1999;162:3298–3307. [PubMed] [Google Scholar]
- Uchida J., Yasui T., Takaoka-Shichijo Y., Muraoka M., Kulwichit W., Raab-Traub N., Kikutani H. Mimicry of CD40 signals by Epstein-Barr virus LMP1 in B lymphocyte responses. Science. 1999;286:300–303. doi: 10.1126/science.286.5438.300. [DOI] [PubMed] [Google Scholar]
- Kilger E., Kieser A., Baumann M., Hammerschmidt W. Epstein-Barr virus-mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:1700–1709. doi: 10.1093/emboj/17.6.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosialos G., Birkenbach M., Yalamanchili R., VanArsdale T., Ware C., Kieff E. The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell. 1995;80:389–399. doi: 10.1016/0092-8674(95)90489-1. [DOI] [PubMed] [Google Scholar]
- Kieser A., Kaiser C., Hammerschmidt W. LMP1 signal transduction differs substantially from TNF receptor 1 signaling in the molecular functions of TRADD and TRAF2. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:2511–2521. doi: 10.1093/emboj/18.9.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gires O., Kohlhuber F., Kilger E., Baumann M., Kieser A., Kaiser C., Zeidler R., Scheffer B., Ueffing M., Hammerschmidt W. Latent membrane protein 1 of Epstein-Barr virus interacts with JAK3 and activates STAT proteins. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:3064–3073. doi: 10.1093/emboj/18.11.3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieser A., Kilger E., Gires O., Ueffing M., Kolch W., Hammerschmidt W. Epstein-Barr virus latent membrane protein-1 triggers AP-1 activity via the c-Jun N-terminal kinase cascade. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:6478–6485. doi: 10.1093/emboj/16.21.6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson S., Rowe M., Gregory C., Croom-Carter D., Wang F., Longnecker R., Kieff E., Rickinson A. Induction of bcl-2 expression by Epstein-Barr virus latent membrane protein 1 protects infected B cells from programmed cell death. Cell. 1991;65:1107–1115. doi: 10.1016/0092-8674(91)90007-l. [DOI] [PubMed] [Google Scholar]
- Rowe M., Peng-Pilon M., Huen D.S., Hardy R., Croom-Carter D., Lundgren E., Rickinson A.B. Upregulation of bcl-2 by the Epstein-Barr virus latent membrane protein LMP1a B-cell-specific response that is delayed relative to NF-kappa B activation and to induction of cell surface markers. J. Virol. 1994;68:5602–5612. doi: 10.1128/jvi.68.9.5602-5612.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell R.G., Wilson J.B., Anderson S.J., Longnecker R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity. 1998;9:405–411. doi: 10.1016/s1074-7613(00)80623-8. [DOI] [PubMed] [Google Scholar]
- Wilson J.B., Bell J.L., Levine A.J. Expression of Epstein-Barr virus nuclear antigen-1 induces B cell neoplasia in transgenic mice. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:3117–3126. [PMC free article] [PubMed] [Google Scholar]
- Blake N., Lee S., Redchenko I., Thomas W., Steven N., Leese A., Steigerwald-Mullen P., Kurilla M.G., Frappier L., Rickinson A. Human CD8+ T cell responses to EBV EBNA1HLA class I presentation of the (Gly-Ala)-containing protein requires exogenous processing. Immunity. 1997;7:791–802. doi: 10.1016/s1074-7613(00)80397-0. [DOI] [PubMed] [Google Scholar]
- Levitskaya J., Coram M., Levitsky V., Imreh S., Steigerwald-Mullen P.M., Klein G., Kurilla M.G., Masucci M.G. Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature. 1995;375:685–688. doi: 10.1038/375685a0. [DOI] [PubMed] [Google Scholar]
- Levitskaya J., Sharipo A., Leonchiks A., Ciechanover A., Masucci M.G. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. USA. 1997;94:12616–12621. doi: 10.1073/pnas.94.23.12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharipo A., Imreh M., Leonchiks A., Imreh S., Masucci M.G. A minimal glycine-alanine repeat prevents the interaction of ubiquitinated I kappaB alpha with the proteasomea new mechanism for selective inhibition of proteolysis. Nat. Med. 1998;4:939–944. doi: 10.1038/nm0898-939. [DOI] [PubMed] [Google Scholar]
- Steven N.M., Leese A.M., Annels N.E., Lee S.P., Rickinson A.B. Epitope focusing in the primary cytotoxic T cell response to Epstein-Barr virus and its relationship to T cell memory. J. Exp. Med. 1996;184:1801–1813. doi: 10.1084/jem.184.5.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna R., Burrows S.R., Nicholls J., Poulsen L.M. Identification of cytotoxic T cell epitopes within Epstein-Barr virus (EBV) oncogene latent membrane protein 1 (LMP1)evidence for HLA A2 supertype-restricted immune recognition of EBV-infected cells by LMP1-specific cytotoxic T lymphocytes. Eur. J. Immunol. 1998;28:451–458. doi: 10.1002/(SICI)1521-4141(199802)28:02<451::AID-IMMU451>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Lee S.P., Thomas W.A., Blake N.W., Rickinson A.B. Transporter (TAP)-independent processing of a multiple membrane-spanning protein, the Epstein-Barr virus latent membrane protein 2. Eur. J. Immunol. 1996;26:1875–1883. doi: 10.1002/eji.1830260831. [DOI] [PubMed] [Google Scholar]
- Ridge J.P., Di Rosa F., Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- Bennett S.R., Carbone F.R., Karamalis F., Flavell R.A., Miller J.F., Heath W.R. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- Schoenberger S.P., Toes R.E., van der Voort E.I., Offringa R., Melief C.J. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- Cardin R.D., Brooks J.W., Sarawar S.R., Doherty P.C. Progressive loss of CD8+ T cell-mediated control of a gamma-herpesvirus in the absence of CD4+ T cells. J. Exp. Med. 1996;184:863–871. doi: 10.1084/jem.184.3.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalams S.A., Walker B.D. The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J. Exp. Med. 1998;188:2199–2204. doi: 10.1084/jem.188.12.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajac A.J., Blattman J.N., Murali-Krishna K., Sourdive D.J., Suresh M., Altman J.D., Ahmed R. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg E.S., Billingsley J.M., Caliendo A.M., Boswell S.L., Sax P.E., Kalams S.A., Walker B.D. Vigorous HIV-1-specific CD4+ T cell responses associated with control of viremia. Science. 1997;278:1447–1450. doi: 10.1126/science.278.5342.1447. [DOI] [PubMed] [Google Scholar]
- Rickinson A.B., Moss D.J. Human cytotoxic T lymphocyte responses to Epstein-Barr virus infection. Annu. Rev. Immunol. 1997;15:405–431. doi: 10.1146/annurev.immunol.15.1.405. [DOI] [PubMed] [Google Scholar]
- Khanna R., Burrows S.R., Steigerwald-Mullen P.M., Moss D.J., Kurilla M.G., Cooper L. Targeting Epstein-Barr virus nuclear antigen 1 (EBNA1) through the class II pathway restores immune recognition by EBNA1-specific cytotoxic T lymphocytesevidence for HLA-DM-independent processing. Int. Immunol. 1997;9:1537–1543. doi: 10.1093/intimm/9.10.1537. [DOI] [PubMed] [Google Scholar]
- Steinman R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- Banchereau J., Steinman R.M. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Zhang D., Frappier L., Gibbs E., Hurwitz J., O'Donnell M. Human RPA (hSSB) interacts with EBNA1, the latent origin binding protein of Epstein-Barr virus. Nucleic Acids Res. 1998;26:631–637. doi: 10.1093/nar/26.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean F.B., O'Donnell M. Two steps to binding replication origins? DNA-protein interactions. Curr. Biol. 1996;6:931–934. doi: 10.1016/s0960-9822(02)00629-2. [DOI] [PubMed] [Google Scholar]
- Frappier L., O'Donnell M. EBNA1 distorts oriP, the Epstein-Barr virus latent replication origin. J. Virol. 1992;66:1786–1790. doi: 10.1128/jvi.66.3.1786-1790.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frappier L., O'Donnell M. Overproduction, purification, and characterization of EBNA1, the origin binding protein of Epstein-Barr virus. J. Biol. Chem. 1991;266:7819–7826. [PubMed] [Google Scholar]
- Friede T., Gnau V., Jung G., Keilholz W., Stevanovic S., Rammensee H.G. Natural ligand motifs of closely related HLA-DR4 molecules predict features of rheumatoid arthritis associated peptides. Biochim. Biophys. Acta. 1996;1316:85–101. doi: 10.1016/0925-4439(96)00010-5. [DOI] [PubMed] [Google Scholar]
- Gorga J.C., Horejsi V., Johnson D.R., Raghupathy R., Strominger J.L. Purification and characterization of class II histocompatibility antigens from a homozygous human B cell line. J. Biol. Chem. 1987;262:16087–16094. [PubMed] [Google Scholar]
- Bender A., Sapp M., Schuler G., Steinman R.M., Bhardwaj N. Improved methods for the generation of dendritic cells from nonproliferating progenitors in human blood. J. Immunol. Methods. 1996;196:121–135. doi: 10.1016/0022-1759(96)00079-8. [DOI] [PubMed] [Google Scholar]
- Subklewe M., Chahroudi A., Schmaljohn A., Kurilla M.G., Bhardwaj N., Steinman R.M. Induction of Epstein-Barr virus-specific cytotoxic T-lymphocyte responses using dendritic cells pulsed with EBNA-3A peptides or UV-inactivated, recombinant EBNA-3A vaccinia virus. Blood. 1999;94:1372–1381. [PubMed] [Google Scholar]
- Taswell C., MacDonald H.R., Cerottini J.C. Clonal analysis of cytolytic T lymphocyte specificity. I. Phenotypically distinct sets of clones as the cellular basis of cross-reactivity to alloantigens. J. Exp. Med. 1980;151:1372–1385. doi: 10.1084/jem.151.6.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subklewe M., Chahroudi A., Bickham K., Kurilla M., Steinman R.M. Presentation of Ebstein-Barr virus latency antigens to CD8+, interferon-gamma-secreting, T lymphocytes. Eur. J. Immonol. 1999;29:3995–4001. doi: 10.1002/(SICI)1521-4141(199912)29:12<3995::AID-IMMU3995>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Lampson L.A., Levy R. Two populations of Ia-like molecules on a human B cell line. J. Immunol. 1980;125:293–299. [PubMed] [Google Scholar]
- Engelmayer J., Larsson M., Subklewe M., Chahroudi A., Cox W.I., Steinman R.M., Bhardwaj N. Vaccinia virus inhibits the maturation of human dendritic cellsa novel mechanism of immune evasion. J. Immunol. 1999;163:6762–6768. [PubMed] [Google Scholar]
- Khanna R., Burrows S.R., Thomson S.A., Moss D.J., Cresswell P., Poulsen L.M., Cooper L. Class I processing-defective Burkitt's lymphoma cells are recognized efficiently by CD4+ EBV-specific CTLs. J. Immunol. 1997;158:3619–3625. [PubMed] [Google Scholar]
- Sallusto F., Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 1994;179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert M.L., Pearce S.F., Francisco L.M., Sauter B., Roy P., Silverstein R.L., Bhardwaj N. Immature dendritic cells phagocytose apoptotic cells via alphavbeta5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J. Exp. Med. 1998;188:1359–1368. doi: 10.1084/jem.188.7.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary M.L., Epstein M.A., Finerty S., Dorfman R.F., Bornkamm G.W., Kirkwood J.K., Morgan A.J., Sklar J. Individual tumors of multifocal EB virus-induced malignant lymphomas in tamarins arise from different B-cell clones. Science. 1985;228:722–724. doi: 10.1126/science.2986287. [DOI] [PubMed] [Google Scholar]
- Wilson A.D., Shooshstari M., Finerty S., Watkins P., Morgan A.J. Virus-specific cytotoxic T cell responses are associated with immunity of the cottontop tamarin to Epstein-Barr virus (EBV) Clin. Exp. Immunol. 1996;103:199–205. doi: 10.1046/j.1365-2249.1996.d01-607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins D.I., Chen Z.W., Hughes A.L., Evans M.G., Tedder T.F., Letvin N.L. Evolution of the MHC class I genes of a New World primate from ancestral homologues of human non-classical genes. Nature. 1990;346:60–63. doi: 10.1038/346060a0. [DOI] [PubMed] [Google Scholar]
- Christensen J.P., Cardin R.D., Branum K.C., Doherty P.C. CD4+ T cell-mediated control of a gamma-herpesvirus in B cell-deficient mice is mediated by IFN-gamma. Proc. Natl. Acad. Sci. USA. 1999;96:5135–5140. doi: 10.1073/pnas.96.9.5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattner E.J., Elkon K.B., Yoo D.H., Tumang J., Krammer P.H., Crow M.K., Friedman S.M. CD40 ligation induces Apo-1/Fas expression on human B lymphocytes and facilitates apoptosis through the Apo-1/Fas pathway. J. Exp. Med. 1995;182:1557–1565. doi: 10.1084/jem.182.5.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine A.M. Acquired immunodeficiency syndrome-related lymphoma. Blood. 1992;80:8–20. [PubMed] [Google Scholar]
- Sayos J., Wu C., Morra M., Wang N., Zhang X., Allen D., van Schaik S., Notarangelo L., Geha R., Roncarolo M.G. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature. 1998;395:462–469. doi: 10.1038/26683. [DOI] [PubMed] [Google Scholar]
- Castro A.G., Hauser T.M., Cocks B.G., Abrams J., Zurawski S., Churakova T., Zonin F., Robinson D., Tangye S.G., Aversa G. Molecular and functional characterization of mouse signaling lymphocytic activation molecule (SLAM)differential expression and responsiveness in Th1 and Th2 cells. J. Immunol. 1999;163:5860–5870. [PubMed] [Google Scholar]
- Aversa G., Chang C.C., Carballido J.M., Cocks B.G., de Vries J.E. Engagement of the signaling lymphocytic activation molecule (SLAM) on activated T cells results in IL-2-independent, cyclosporin A-sensitive T cell proliferation and IFN-gamma production. J. Immunol. 1997;158:4036–4044. [PubMed] [Google Scholar]