

Abstract
The mechanisms that regulate B cell memory and the rapid recall response to antigen remain poorly defined. This study focuses on the rapid expression of B cell memory upon antigen recall in vivo, and the replenishment of quiescent B cell memory that follows. Based on expression of CD138 and B220, we reveal a unique and major subtype of antigen-specific memory B cells (B220−CD138−) that are distinct from antibody-secreting B cells (B220+/−CD138+) and B220+CD138− memory B cells. These nonsecreting somatically mutated B220− memory responders rapidly dominate the splenic response and comprise >95% of antigen-specific memory B cells that migrate to the bone marrow. By day 42 after recall, the predominant quiescent memory B cell population in the spleen (75–85%) and the bone marrow (>95%) expresses the B220− phenotype. Upon adoptive transfer, B220− memory B cells proliferate to a lesser degree but produce greater amounts of antibody than their B220+ counterparts. The pattern of cellular differentiation after transfer indicates that B220− memory B cells act as stable self-replenishing intermediates that arise from B220+ memory B cells and produce antibody-secreting cells on rechallenge with antigen. Cell surface phenotype and Ig isotype expression divide the B220− compartment into two main subsets with distinct patterns of integrin and coreceptor expression. Thus, we identify new cellular components of B cell memory and propose a model for long-term protective immunity that is regulated by a complex balance of committed memory B cells with subspecialized immune function.
Keywords: antigen-specific immunity, immunological memory, B lymphocyte memory, B lymphocyte subset, mice
Introduction
The memory B cell response is typically characterized by the accelerated appearance of Ag-specific Ab in response to specific rechallenge with Ag 1 2 3. It is useful to consider Ag-specific memory B cell development in four broad phases 4: (a) induction of B cell memory occurs within the germinal center (GC) reaction; (b) maintenance of B cell memory begins with the exit of long-lived memory B cells from the GC reaction and maintains the memory B cell compartment in readiness for Ag rechallenge; (c) expression of B cell memory occurs upon Ag rechallenge and is an emergent cellular and humoral activity focused on Ag clearance; and (d) replenishment of the memory B cell compartment after Ag rechallenge is critical for long-term protective immunity. Although the systemic phenomenon of B cell memory is readily demonstrated in vivo, the nature and extent of cellular heterogeneity in the Ag-specific memory B cell compartment remains poorly resolved.
B cell memory is induced upon primary Ag challenge and is thought to be a product of the GC reaction 5 6 7 8. The specialized and dynamic microenvironment of the GC reaction coordinates affinity maturation of Ag-expanded B cells through reiterative cycles of somatic hypermutation and Ag-driven selection 9 10 11. In this manner, GCs regulate Ag-specific clonal maturation and subsequent entry into the long-lived memory B cell compartment 12 13. The initiation and maintenance of the GC reaction depends on a multitude of factors that have received a great deal of recent attention 4. The regional organization in the spleen before Ag stimulation 14 15 16 and chemokine-driven lymphoid circuitry 17 18 is vital to effective formation of GCs. T cell help 19 and CD40–CD40 ligand interactions greatly influence the initiation 20 21 22 and function 23 24 of the GC reaction. Although less is known of the mechanisms that drive positive selection in the GC reaction, native Ag clearly plays a role 25 26 and is most likely trapped within GC as immune complexes 27 28 29. Nevertheless, positively selected Ag-specific B cells that successfully exit the GC reaction constitute the beginnings of the long-lived memory B cell compartment.
Affinity-matured memory B cells are maintained in vivo in at least two different forms: memory response precursors and long-lived Ab-secreting B cells 4. Memory response precursors are typically defined as IgM−IgD−B220+ Ag-binding B cells that do not secrete Ab until rechallenge with Ag 13 30 31. These B220+ memory B cells can express mutated Ag receptors and are found in late primary and secondary responses 9 10 11 32 33 34. Long-lived Ab-secreting B cells 35 36 37 can also be considered part of the memory B cell compartment 38 39 40. These long-lived B cells are characterized by their ability to spontaneously secrete Ab directly ex vivo. They typically secrete high-affinity Abs with evidence of somatic hypermutation and Ag-driven selection in their expressed Ig 41 42. Thus, memory B cells are most reliably characterized as persistent Ag-binding B cells that display evidence of somatic hypermutation in their expressed Ig genes.
The murine model offers the greatest opportunity to quantitatively analyze de novo memory B cell development. An ideal response for direct ex vivo analysis of Ag-specific B cells is the immune response in C57BL/6 mice to the hapten (4-hydroxy-3-nitrophenyl)acetyl (NP [43, 44]). NP-specific B cells can be visualized directly in situ 9 45 46 or through NP binding by flow cytometry 11 32 33 34 41 47 48. The vast majority of NP-specific B cell clones from a primary response express B cell receptors (BCRs) with the VH186.2 H chain coupled to Vλ1 L chain 49 50. Somatic mutation proceeds in the GC microenvironment 9 10 and is seen in NP-specific B cells as early as day 6 after initial priming 51. Most NP-binding cells express mutated H and L chain genes by the end of the second wk after priming and can be visualized upon Ag rechallenge in vivo 9 11 32 33. Direct access to NP-specific B cells through flow cytometry provides a reliable means to analyze NP-specific B cell memory directly ex vivo.
This study focuses on the rapid expression of B cell memory upon Ag recall, and replenishment of the quiescent memory B cell compartment. We identify three distinct subtypes of NP+IgD− memory responders that emerge rapidly upon Ag rechallenge and persist at different levels into the quiescent memory compartment. All three subtypes express somatically mutated λ1 L chain genes, with Ab-secreting activity found only in the B220+/− CD138+ compartment. Previously described B220+ memory B cells appear as a B220+CD138− memory B cell subset with the greatest proliferative capacity on adoptive transfer with Ag. Upon Ag recall, the B220+ memory B cells rapidly produce a third and major B220−CD138− memory B cell subset. This new B220−CD138− memory subset also expands rapidly on recall, dominates the NP-specific memory response in both the spleen and bone marrow, and persists long term to comprise the majority of quiescent memory B cells. The B220− memory compartments are stable self-replenishing B cells with a high propensity to form Ab-secreting cells. The pattern of BCR coreceptor expression suggests that these memory cells have unique mechanisms for modulating Ag responses in vivo. Two further subtypes of B220− memory B cells can be defined based on CD11b and surface Ig (sIg) isotype expression (CD11b++IgG+ and CD11b+IgE+). These studies provide the first quantitative assessment of the extensive cellular and functional heterogeneity within the Ag-specific memory B cell compartment.
Materials and Methods
Isolation of NP-specific Memory B Cells.
6–10-wk old, specific pathogen-free, female C57BL/6 mice (The Jackson Laboratory) were immunized with 400 μg NP-KLH intraperitoneally in Ribi adjuvant (Ribi ImmunoChem Research). At least 8 wk after the initial injection, the animals were rechallenged with an identical immunization. The Duke University Institutional Animal Care and Use Committee approved this study. Spleen and bone marrow were harvested at various time points, and single cell suspensions were prepared as described previously 52 53.
Cell suspensions were labeled for flow cytometry at 2.0 × 108 cells/ml on ice for 45 min with predetermined optimal concentrations of fluorophore-labeled mAbs (purchased from PharMingen unless otherwise noted) or Ag and in combinations outlined in each figure legend. The following Abs were used for staining cells: Cy5PE-H129.19 (anti-CD4), Cy5PE–53-6.7 (anti-CD8), Cy5PE-F4/80 (antimacrophage; Caltag Laboratories), Texas red 11.26 (anti-IgD), allophycocyanin (APC)-NP (4-hydroxy-5-iodo-3-nitrophenyl), PE-281.2 (anti-CD138/syndecan), PE-6B2 (anti-CD45R/B220), PE-23G3 (anti-IgE; Southern Biotechnology Associates), FITC-6B2 (anti-CD45R/B220), FITC–30-F11 (anti-CD45/leukocyte common Ag), FITC-1D3 (anti-CD19), FITC-M1/70 (anti-CD11b/Mac-1), FITC–goat anti–mouse IgG (Southern Biotechnology Associates), FITC-HM79b (anti-CD79/Igβ), and FITC-JC5.1 (anti-λ1 L chain). After staining, cells were washed twice in PBS with 5% FCS. Cells were then resuspended in 2 μg/ml propidium iodide (PI) for dead cell exclusion in PBS, with 5% FCS for analysis.
Samples were analyzed using a modified dual laser FACStarPLUS™ (Becton Dickinson) capable of simultaneous seven parameter acquisition and fluorescence overlap compensation across lasers. Files were acquired using CELLQuest™ software (Becton Dickinson) and analyzed using FlowJo software (Tree Star). Profiles are presented as 5% probability contours with outliers.
Assay for Specific Ab.
Enzyme-linked immunospot (ELISPOT) for detection of NP-specific IgG was performed as described previously 11 34. 96-well multiscreen membrane filtration plates (Millipore) were coated for 4 h with 50 μl/well of NP-BSA (25 μg/ml) in PBS, washed, and incubated for 2 h with 100 μl of RPMI 1640 supplemented with 5% FCS. Cells were sorted directly into the wells and incubated for 18 h at 37°C in 5% CO2. The wells were washed before the addition of 50 μl/well of horseradish peroxidase goat anti–mouse IgG (Southern Biotechnology Associates) for 4 h. ELISPOTs were developed using 100 μl/well of filtered 3-1mino-9-ethyl carbazole substrate (Sigma Chemical Co.) in 0.1 M sodium acetate buffer, pH 5.0, 0.03% H2O2. The plates were washed with water, and positives were scored manually using a dissection microscope. Each assay was done in triplicate from at least three separate animals.
ELISA for detection of NP-specific IgG was performed as described previously 11 33. Flexible U-bottomed 96-well plates (Costar) were coated for 4 h with 50 μl/well of NP-BSA conjugate (25 μg/ml) and washed. Samples were added in PBS with 0.1% Tween 20, 0.3% (w/v) skim milk powder, and 1% FCS, held at room temperature for 4 h, washed before the addition of 50 μl/well of horseradish peroxidase conjugates of goat anti–mouse IgG (Southern Biotechnology Associates), and held for 4 h before washing and the addition of 100 μl/well of enzyme substrate 2,2′-azinobis(3-ethylbenzthiazoline) (Sigma Chemical) at 55 mg/ml in 0.1 M sodium citrate, pH 4.5, and 0.1% H2O2. Absorbance read at 405 nm.
Single Cell Repertoire Analysis.
cDNA synthesis was performed as follows: single B cells with the appropriate surface phenotype were sorted for repertoire analysis using the automatic cell-dispensing unit attached to the FACStarPLUS™ (Becton Dickinson) and Clone-Cyt™ software (Becton Dickinson). Each cell was sorted into an oligo d(T)-primed 5 μl cDNA reaction mixture as described previously (reference 53; 4 U/ml MLV reverse transcriptase [RT; GIBCO BRL] with recommended 1× RT buffer, 0.5 nM spermidine [Sigma Chemical Co.], 100 μg/ml BSA [Boehringer Mannheim], 10 ng/ml oligo d(T) [Becton Dickinson], 200 μM each dNTP [Boehringer Mannheim], 1 mM dithiothreitol [Promega], 220 U/ml RNAsin [Promega], 100 μg/ml Escherichia coli tRNA [Boehringer Mannheim], and 1% Triton X-100 set up in low-profile 72-well microtiter trays [Robbins Scientific]), held immediately at 37°C for 90 min, and then stored at −80°C until further use.
Nested PCR.
First round of PCR: 2 μl of cDNA from each single cell cDNA reaction was used in a 25-μl reaction to amplify the λ L chain. The first-round PCR reaction mix consisted of 2 U/ml Taq polymerase with the recommended 1× reaction buffer (Promega), 0.1 mM of each dNTP (Boehringer Mannheim), 1 mM MgCl2, 0.8 μM primer LAMext3(sense) (5′-TACTCTCTCTCCTGGCTCTCAGCTC-3′), and 0.8 μM primer LAMext3(anti) (5′-GTTGTTGCTCTGTTTGGAAGGCTGG-3′). Each reaction set begins with 95°C for 5 min, then 40 cycles of 95°C for 15 s, 50°C for 45 s, and 72°C for 90 s, and ending with 72°C for 5 min. Second round of PCR: 1 μl of the first-round PCR product was used for an additional 25-μl amplification reaction using primers nested medially to the primers used in the first round of PCR (2 U/ml Taq polymerase with the recommended 1× reaction buffer [Promega], 0.1 mM of each dNTP [Boehringer Mannheim], 1 mM MgCl2, 0.8 μM primer LAM.sens[int] [5′-CCATTTCCCAGGCTGTTGTG-3′), and 0.8 μM primer LAM.anti(int) (5′-CTCCATACCCTGAGTGACAG-3′]). Each reaction set begins with 95°C for 5 min, then 40 cycles of 95°C for 15 s, 50°C for 45 s, and 72°C for 90 s, and ending with 72°C for 5 min. At least 2 negative cDNA samples were processed per 10 single cell samples. The negatives were interspersed with positives to control for contamination during sample preparation.
DNA Sequencing.
5 μl of second-round PCR product was run on a 1.5% agarose gel to screen for positives (single bands of the right size). PCR product was then separated from primers using a CL-6B Sepharose (Amersham Pharmacia Biotech) column. The PCR product was then directly sequenced (4 μl of PCR product, 4 μl Dye Terminator Ready Reaction Mix [Perkin Elmer Corp.], 1.6 pmol primer [LAM.seq 5′-GGCTGTTGTGACTCAGGAAT-3′]) using a linear amplification protocol for 25 cycles of 96°C for 10 s, 50°C for 5 s, and 60°C for 4 min on a 9600 GeneAmp PCR system (Perkin Elmer Corp.). Samples were separated on a 6.5% acrylamide gel after ethanol precipitation of sequencing reaction products, run on an ABI 373 sequencing system, and processed using ABI Prism sequence 2.1.2 for collection and analysis (Perkin Elmer Corp.).
Adoptive Transfer.
Unfractionated spleen cells (4 × 107) from C57BL/6 mice 4 or 42 d after secondary immunization with 400 μg NP-KLH in Ribi adjuvant were injected in 100 μL PBS into the tail vein of 6–10-wk-old C57BL/6 recombination activating gene 1 (Rag-1)–deficient mice (The Jackson Laboratory), with or without intraperitoneal injection of 50 μg NP-KLH (no adjuvant). For “exclusion transfer,” 5–8 × 106 spleen cells were sorted by flow cytometry to exclude any NP+IgD− cells and all CD138+ cells and then injected as a transfer of “background” cells (NP− spleen). 2–6 × 104 purified PI−CD4−CD8−F4/80−NP+IgD−CD138− B cells that were either B220+ or B220− were added to 5–8 × 106 NP− spleen cells. Purity of sorted populations was then reanalyzed before and after mixing with the NP− spleen cells immediately before transfer with purities of >95–98% for the subsets transferred. Spleens were harvested from the recipient mice 5 d after transfer and processed as described above.
Results
NP-specific Memory B Cells in the Spleen.
Using flow cytometry, we quantify directly the Ag-binding B cells from C57BL/6 mice immunized with the hapten-protein conjugate NP-KLH in the Ribi adjuvant system. The change to Ribi adjuvant (nonpolarizing for T cell help) for both priming and recall induces NP-specific Ab of all IgG isotypes and IgE to high levels in the serum of immunized mice (data not shown). We first focus on the emergent splenic memory response during the first 2 wk after intraperitoneal rechallenge with Ag. Ag-binding cells were clearly visualized in the spleen and bone marrow as PI− CD4−, CD8−, and F4/80− cells that have lost sIgD and bind NP-labeled APC (NP-APC; Fig. 1A iii). Ag-specific expansion of NP+IgD− cells was significant by day 3 in the spleen, and rapidly reaches peak levels by day 4 (Fig. 1A iv, top). This rate of NP-specific cellular expansion is 3–4 d faster than seen after initial priming 8 wk earlier (11; our unpublished results). Adjuvant-only controls indicate that NP-binding cells only emerge in response to the presence of NP-KLH (Fig. 1 A). Similar results were obtained using carrier alone and an irrelevant protein (data not shown; 11). Decline in the number of NP+IgD− cells in the spleen is already evident by the end of the second wk, but is clearly diminished by day 42 after recall (Fig. 1 A). The day 42 time point is representative of the quiescent memory compartment when the active GC reaction and the exaggerated humoral recall response have both resolved. The replenished memory compartment at this time point contains at least 100-fold more NP-specific cells than were detectable before rechallenge. Therefore, accelerated Ag-specific cellular expansion and the development of a larger quiescent memory compartment are two major cellular consequences of Ag rechallenge.
Figure 1.
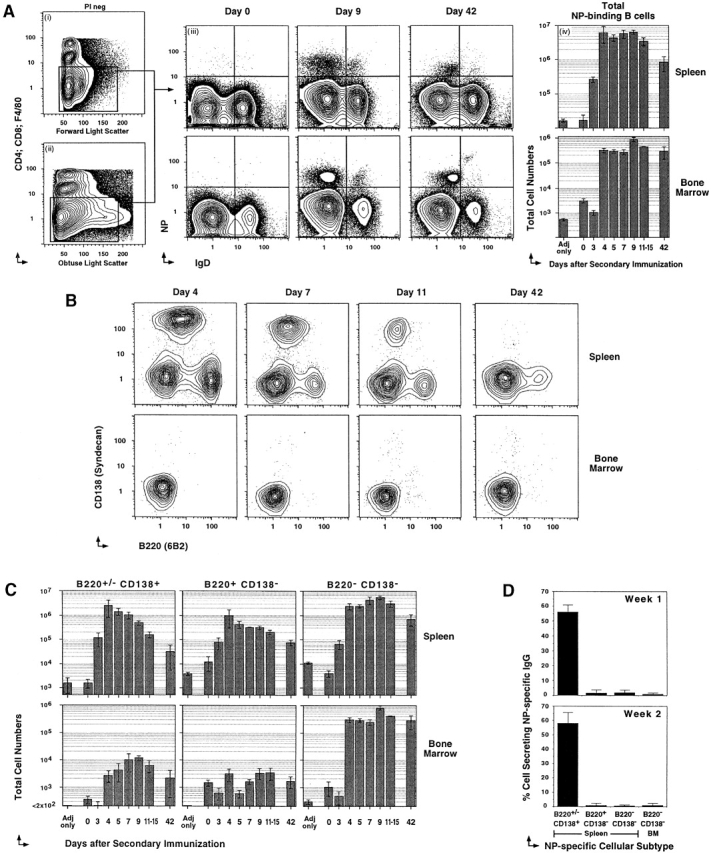
Three major subsets of NP-specific memory B cells. Previously primed C57BL/6 mice, rechallenged with 400 μg NP-KLH in Ribi adjuvant, were killed at various times after recall to label spleen and bone marrow cells for five-color flow cytometry. (A, i and ii) PI labeling is excluded at time of acquisition. One example of light scatter parameters and Cy5PE labeling (anti-CD4, anti-CD8, and anti-F4/80) used to exclude T cells, macrophage, and cells that nonspecifically label with Abs. Light scatter gates are broad to include B cell blasts. (A iii) Examples of IgD (Texas red 11.26) and NP (NP-APC) labeling on the Cy5PE− cells. Days after recall are displayed above each pair of panels. The top panels are spleen, and the lower are bone marrow. Each upper left quadrant delineates fluorescence intensities of cells regarded as NP+IgD−. (A iv) Cellular dynamics of the total NP-binding B cell compartment. Frequencies of NP-binding IgD− B cells obtained by flow cytometry, and total cell numbers from the spleen (top) and bone marrow (bottom) of each animal at the time of killing were used to estimate the change in total NP-binding cells during the course of the recall response. Varying numbers of single animals were used for each time point (n = 3–7), with mean ± SEM of total cells displayed. (B) Examples of CD138 (PE-281.2) and B220 (FITC–RA3-6B2) levels on NP-specific B cells (CD4−CD8−F4/80−IgD−NP+) of the spleen (top) and bone marrow (bottom). The day after Ag administration is displayed above each panel. (C) Cellular dynamics of the three NP-binding B cell subsets, with the phenotype displayed above each panel for the spleen (top) and bone marrow (bottom). Mean ± SEM of total cells is displayed across time after recall (n = 3–7 for each time point). (D) CD4,− CD8,− and F4/80− NP+IgD− B cells with the phenotype B220+/−CD138+, B220+CD138−, and B220−CD138− B cells from the spleen and bone marrow (BM) of the secondary response were sorted at 50 cells per well into NP-specific IgG-revealing ELISPOT assays. Positives were scored manually under a dissection microscope. Each assay was done in triplicate from at least three separate animals.
Three Phenotypically Distinct Memory B Cell Subsets in the Spleen.
Using five-color flow cytometry, we can directly phenotype the NP+IgD− compartment. Because cell surface phenotype can provide clues to function, we initially subtype the NP-specific cells using Abs to B220 (6B2, anti–B cell isoform of CD45R) and CD138 (281.2, anti-syndecan). A major subset of B220+/−CD138+ NP-specific memory responders clearly dominates the early phase of the splenic memory response, then declines rapidly during the first 2 wk (Fig. 1B and Fig. C, top panels). The B220+/− CD138+ subset is consistently represented at very low levels into the quiescent phase of the splenic response (day 42). A second subset of NP-specific memory responders expressed high levels of B220 and no CD138 (Fig. 1 B, top). These memory responders also reached peak levels early (day 4), but gradually declined during the first 2 wk after rechallenge (Fig. 1 C, top). A small compartment of B220+CD138− memory B cells also persisted into the quiescent phase. The third major subset of NP+IgD− memory responders expressed negligible levels of both B220 and CD138. These Ag-binding memory responders emerged in the spleen as rapidly as the other two subsets, but clearly dominated the Ag-specific compartment by the end of the first wk after rechallenge (Fig. 1B and Fig. C, top panels). The quiescent memory compartment of the spleen was also dominated by far greater numbers of the B220−CD138− memory cells (75–85% of NP+IgD− cells at day 42). Thus, the NP-specific memory responders emerge as three phenotypically distinct NP+IgD− subsets that all persist into the quiescent phase with the B220−CD138− subset clearly dominating by the end of the first wk after recall.
B220−CD138− NP-specific Memory Cells Dominate the Bone Marrow Compartment.
The long-lived Ab-secreting memory B cell compartment resides mainly in the bone marrow 38 39 40 41 42. These previous studies have focused on the bone marrow compartment after initial priming. To characterize the memory response, we next quantified the extent and kinetics of Ag-binding cells that emerged in the bone marrow after recall (Fig. 1 A, bottom panels). There was evidence of a decline in the basal levels of NP-binding cells at day 3, followed by a rapid rise by day 4 that reached maximal levels by day 9 (Fig. 1A iv, bottom). The numbers of NP+IgD− cells remained remarkably stable between the end of the second wk after rechallenge and into the quiescent memory compartment (day 42). Strikingly, the B220−CD138− subset comprised of the vast majority of NP+IgD− cells in the bone marrow throughout all stages of the emergent response and into the quiescent phase (>95–98% across all time points; Fig. 1 B, bottom). The B220+/−CD138+ memory responder subset is consistently present but poorly represented in the bone marrow, whereas the B220+CD138− subset is equally low but more sporadic than its CD138+ counterpart (Fig. 1B and Fig. C, bottom panels). Therefore, the NP-specific memory response in the bone marrow is dominated by the B220−CD138− subset that emerges and persists long term as a significant reservoir of quiescent memory B cells.
Ab-secreting Activity Assorts into the B220+/−CD138+ NP-binding Compartment.
CD138 reliably labels Ab-secreting B cells in the primary NP-specific response 11 34. To further evaluate the function of the three different NP-binding subsets, we sorted equal numbers of purified NP-binding subsets into ELISPOT assays for production of NP-specific IgG. In the absence of any stimulus in vitro, the ELISPOT estimates the frequency of cells secreting Ab in vivo. All NP-specific IgG Ab-secreting activity assorted into the splenic B220+/−CD138+ memory B cell compartment during the first 2 wk after rechallenge (Fig. 1 D). Although the B220+ and B220− NP-binding compartment expanded significantly on rechallenge, cells with this phenotype did not produce Ab during the first 2 wk after recall. In the bone marrow, the dominant NP-binding compartment (B220−) did not produce Ab during this same period (Fig. 1 D). Thus, NP-binding cells that differentiate into Ab-secreting cells during the memory response in vivo express the B220+/−CD138+ phenotype.
Molecular Characterization of Memory B Cells.
The λ L chain is dominantly expressed in primary and memory responses to NP-KLH 11 33 45. We next screened all three NP-binding subtypes for expression of this L chain. Only ∼5% of all naive B cells in C57BL/6 mice express the λ L chain (Fig. 2 A, top), whereas all three NP-binding B cell subsets in the spleen contained 90–95% λ+ cells (Fig. 2 A, bottom three histograms). Levels of λ expression were similar on all three NP+IgD− subsets, and were consistent with the loss of sIgD at the time of original Ag experience. The frequency of λ+ cells and the level of λ expression on the NP+IgD− cells in the bone marrow was similar to that seen in the spleen (data not shown). The λ expression offers further evidence that all three subpopulations are NP-specific B cells. The absence of κ L chain expression on these λ+ NP-binding B cells also argues against an FcR-mediated binding of Ab in vivo that leads to artificial labeling with NP-APC for flow cytometry (naive NP− B cells, κ mean fluorescence index [MFI] = 57; λ+ cells, κ MFI = 10; NP+ cells in spleen, κ MFI = 12; NP+ cells in bone marrow, κ MFI = 13).
Figure 2.
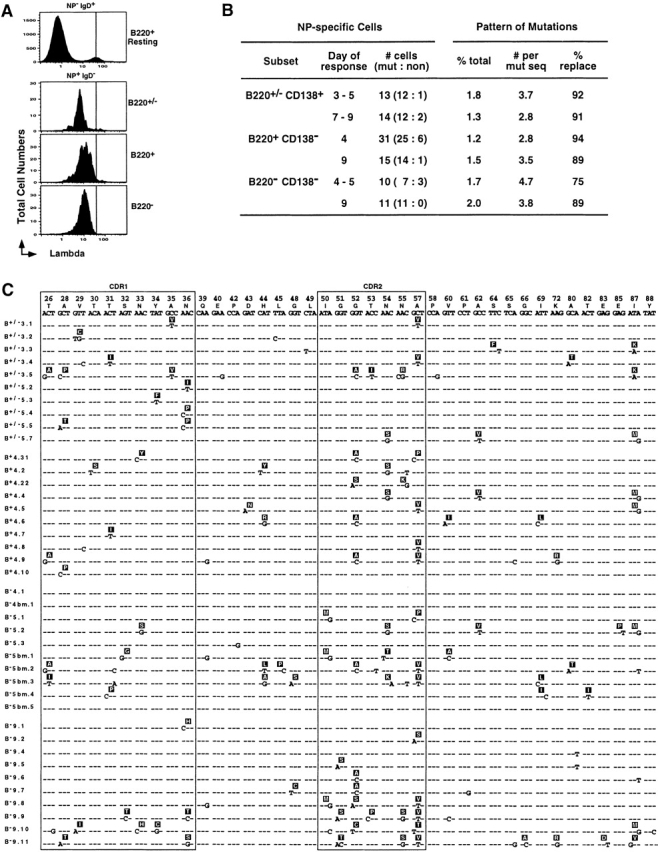
All three NP-specific subsets express somatically mutated Vλ1 L chains. (A) Representative histograms of λ1 L chain expression (FITC-JC5) on CD4−, CD8−, and F4/80− cells that were also NP−IgD+B220+ (first panel), NP+IgD−B220+/− (second panel), NP+IgD−B220+ (third panel), and NP+ IgD−B220− (fourth panel). (B) A summary of the extent and pattern of somatic hypermutation in the λ1 L chain expressed by the three major subsets of NP-specific B cells (PI−CD4− CD8−F4/80−NP+IgD−). Single NP-specific B cells from each of the three subpopulations of cells (as defined in the legend to Fig. 1) at different time points of the recall response were sorted into cDNA reaction mix, subjected to two separate rounds of PCR for λ L chain and PCR products, sequenced as described in Materials and Methods. Day of response refers to time after rechallenge; # cells refers to the number of λ L chains sequenced from individual cells and the proportion that were mutated versus nonmutated; % total represents the percentage of the total base pairs sequenced that were mutated (reliable sequence from position 26–88 for all samples), with the average number of mutations per mutated sequence summarized in the next column; and the % replacement mutations in the CDR1 and 2 are presented in the final column. (C) A representative set of Vλ gene nucleotide and predicted amino acid sequence obtained from individual NP-specific B cells. Only the nucleotides differing from the germline V-J sequence are presented: dashes represent identity; CDR 1 and 2 are boxed. The cells are grouped into four categories, with each representing sequence from individual cells with the phenotype of the following: the upper panel is NP+IgD− B220+/−CD138+ B cells (labeled B+/−) from days 3 and 5; the second panel is NP+IgD−B220+CD138− B cells (B+) from day 4; the third panel is NP+IgD−B220−CD138− B cells (B−) from the spleen or bone marrow (bm) from day 4 and 5; and the bottom panel is NP+IgD−B220−CD138− B cells (B−) from day 9 spleen.
Although all three subsets were NP-specific B cells, it was not clear that they were all memory B cells. One cardinal hallmark for B cell memory is the expression of somatically-mutated Ig with evidence for Ag-driven selection. We next sorted single cells from each of the three major subsets for RT-PCR and DNA sequence analysis directed at the λ L chain. PCR products from individual reactions were then sequenced to assay for somatic mutation. All three subsets of NP-specific memory responders emerged similarly mutated by days 3 or 4, and did not significantly alter in extent of mutation by day 9 after rechallenge (Fig. 2B and Fig. C). A high frequency of replacement mutations in hypervariable regions 1 and 2 was also consistent with Ag-driven selection. These changes most likely occurred during the induction phase of memory after initial Ag priming for all three subsets. These data argue against the adsorption of NP-binding Ab on the cells labeled as NP-specific by flow cytometry. NP-specific cells in the bone marrow had similar levels of λ L chain mutations (Fig. 2 C). The efficiency of obtaining a PCR product across the three subsets was 60% for CD138+ cells, 30% for B220+ B cells, and 15% for B220− B cells (range from 5–50% for the latter). We attributed these differences to varied mRNA levels, and they are similar to differences observed between TCR-α and TCR-β chains from single Ag-specific T cells 52 53. Therefore, all three subtypes of NP-specific memory responders not only express the mRNA for the appropriate L chain, but also express it in a somatically mutated form with evidence for Ag-driven selection.
Proliferative Capacity and Ab-secreting Potential of Quiescent Memory B Cells.
Although the B220+/−CD138+ memory subset was mainly Ab-secreting cells (Fig. 1 D), the function of the NP-specific B220+ and B220− memory responders and their relationship to each other was still not clear. The only definitive way to address function was through adoptive transfer and challenge with Ag. We first focused on day 42 after recall in the quiescent memory phase and adoptively transferred unfractionated spleen cells (4.0 × 107 cells) with soluble Ag into Rag-1–deficient mice. 5 d later, all three NP-specific subpopulations of memory responders were recovered from the recipient spleens (Fig. 3 A). The majority of NP-specific cells recovered were the B220−CD138− memory subset (48%), with fewer B220+/−CD138+ (30%) and B220+CD138− (22%) cells present (Fig. 3 D). The recovery of NP-specific B cells was 250-fold greater, and the production of NP-specific IgG 300-fold higher in the presence of Ag than without (Fig. 3 E). Interestingly, the B220+CD138− subset was the only fraction to survive in the absence of Ag, although cell numbers were very low (Fig. 3 A). These data indicate that cell expansion after transfer was not due to the absence of lymphocytes in the Rag-1–deficient recipients but was driven by Ag. Hence, Rag-1–deficient mice can support the survival and expansion of all three subtypes of NP-specific quiescent memory B cells and promote the production of NP-specific IgG.
Figure 3.
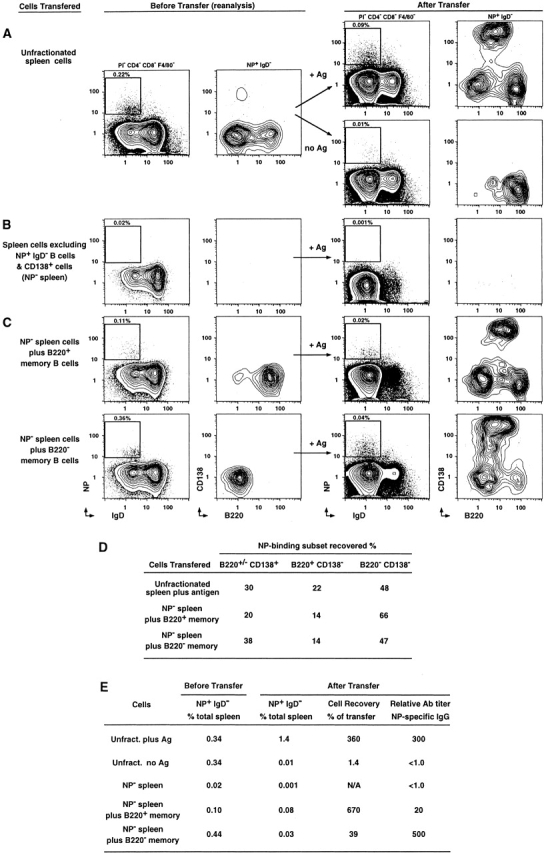
Adoptive transfer of purified quiescent B220+ and B220− memory B cells. (A) 4 × 107 unfractionated spleen cells from animals 42 d after rechallenge were injected into the tail vein of Rag-1–deficient animals, with or without intraperitoneal injection of 50 μg NP-KLH (no adjuvant). The first panel represents the NP and IgD profiles of PI−, CD4−, CD8−, and F4/80− spleen cells before transfer (percentage of NP-specific cells in total spleen cells is indicated above the insert). The second panel represents the CD138 and B220 profiles of the NP-specific cells in these animals. The third panel represents the NP and IgD profiles of the PI−, CD4−, CD8−, and F4/80− spleen cells from Rag-1–deficient mice 5 d after transfer. The fourth panel represents the CD138 and B220 profiles of these NP-specific cells (+ or − Ag as labeled). (B) The first two panels present reanalysis of spleen cells from day 42 memory mice, excluding NP+IgD− and all CD138+ cells at the time of sorting. 5–8 × 106 spleen cells sorted this way (NP− spleen) were injected into the tail vein (with Ag intraperitoneally as above), and spleen cells from Rag-1–deficient recipients analyzed 5 d after transfer (third and fourth panel). (C) 2–6 × 104 purified NP+IgD−CD138− that were either B220+ (>95% purified; top) or B220− (>99% purified; bottom) were added to 5–8 × 106 NP− spleen cells before adoptive transfer. The first two panels in each set display reanalysis of cell populations to be transferred after addition of the purified memory subset. The third panel displays (PI−, CD4−, CD8−, and F4/80− NP and IgD cells in Rag-1–deficient recipients 5 d after transfer, with the frequency of NP+IgD− cells in the total spleen indicated above inset. The last panel displays CD138 and B220 profiles for NP+IgD− spleen cells in the Rag-1–deficient recipients 5 d after transfer. (D) Mean of two adoptive transfer experiments displaying the proportion of the three subsets recovered after transfer. The level of NP+ events after transfer of the NP− spleen cells alone was negligible, so they are not included in this table. (E) Summary of cell recovery and Ab production during time of transfer (n = 2 or 3 animals). Cell recovery was calculated as the percent of NP-specific cells recovered over the number transferred. N/A indicates not applicable, as the number of NP-specific cells transferred and recovered was negligible. The relative titer of NP-specific Ab is displayed as the titer of serum that produced an OD of 1.4 OD units (maximum of 4.0 OD units).
To transfer purified subsets of nonsecreting NP-specific memory B cells, we needed to provide a source of Ag-specific T cell help. We next sorted spleen cells from day 42 after recall excluding all CD4−, CD8−, and F4/80− NP+IgD− B cells before adoptive transfer into Rag-1–deficient mice (Fig. 3 B). We also excluded all CD138+ cells to minimize the transfer of sIg− plasma cells. This “background” NP− spleen transfer aims at providing the requisite T cell help without contributing to the cellular or humoral NP response in recipients. 5 d after transfer with Ag, the recipient mice showed a significant reconstitution of IgD+B220+ B cells (Fig. 3 B) and CD4 and CD8 T cells in the spleen (data not shown). Importantly, very few NP-specific cells were transferred in the NP− spleen cells, and negligible numbers of NP-specific B cells were recovered after transfer in recipient (Fig. 3 E). In addition, negligible NP-specific IgG was detected in the recipient serum (Fig. 3 E). We next sorted 4–6 × 104 of either memory B cell subset (B220+ or B220−), mixed each purified memory subset with the background populations of NP− spleen cells (5–8 × 106 cells total), and reanalyzed the samples immediately before transfer (Fig. 3 C; first two panels in each row). 5 d after transfer, recipient spleens were analyzed for the frequency of NP-specific B cells, the balance of the three memory B cell subsets, and the production of NP-specific IgG in vivo (Fig. 3C–E).
5 d after transfer, B220+ NP-specific memory cells produced mostly B220−CD138− NP-specific cells (66%), with some B220+/−CD138+ Ab-secreting B cells (20%) and a few B220+CD138− cells (14%; Fig. 3C [top] and D). B220− NP-specific memory cells produced mostly B220−CD138− cells (47%) and B220+/−CD138+ Ab-secreting cells (38%) and few B220+CD138− cells (14%; Fig. 3C [bottom] and D). Thus, the pattern of cellular differentiation on transfer was consistent with progressive linear differentiation between the B220+, B220−, and CD138+ memory B cells. This progression of differentiation appears dependent on Ag in this short-term adoptive transfer assay. Interestingly, cell recovery after transfer of the B220+ cells was 15-fold higher than after transfer of the B220− cells (670% vs. 39%; Fig. 3 E), In striking contrast, the amount of Ab in the recipient serum after 5 d was 25-fold greater after transfer of the B220− cells than the B220+ cells (500 vs. 20 relative titer; Fig. 3 E). These data would suggest that the B220+ memory B cells retain greater proliferative capacity than their downstream B220− memory B cell counterparts. The ability of the B220− memory B cells to produce greater amounts of Ab during the same span of time indicates a greater degree of commitment towards plasma cell differentiation than the B220+ cells. These functional data are consistent with the linear differentiation of B220+ memory B cells into B220− memory B cells that in turn terminally differentiate into Ab-secreting cells.
Cellular Differentiation during the Emerging Memory Response.
To further test this model of linear differentiation, we turned to the emerging phase of the memory response. Using an identical experimental design to the day 42 transfers, we transferred unfractionated spleen cells from animals 4 d after recall into Rag-1–deficient mice (Fig. 4 A). 5 d after transfer, all three subsets of memory responders were present in the recipient spleens; however, exogeneously added Ag was not required for subset reconstitution (Fig. 4A, Fig. D, and Fig. E). In the absence of added Ag, only 1% cell recovery was achieved, compared with 60% with Ag (Fig. 4 E). The presence of added Ag appeared more necessary for cell recovery than for production of Ab (Fig. 4 E). In the unfractionated spleen cell transfer at day 4, a high number of terminally differentiated Ab-secreting cells are present in the transfer, and some level of Ag is most likely also transferred at this acute stage of the memory response. Nevertheless, as with day 42, all NP activity was excluded with the exclusion sorting procedure described previously and resulted in no detectable NP-specific cell expansion or serum IgG production 5 d after transfer (Fig. 4B and Fig. E). We also transferred 50 μl of serum from mice 9 d after recall into Rag-1–deficient recipients, then assayed for appearance of NP-binding cells 24 h after transfer to see if there were any cells in the recipients that could trap the NP-specific Ab and then score as NP-binding B cells after transfer. An average of 1:108,000 events (± 20,000; n = 3, ± SEM) in the spleen and 1:500,000 events in the bone marrow (all three animals were identical) argues against nonspecific FcR-mediated binding.
Figure 4.
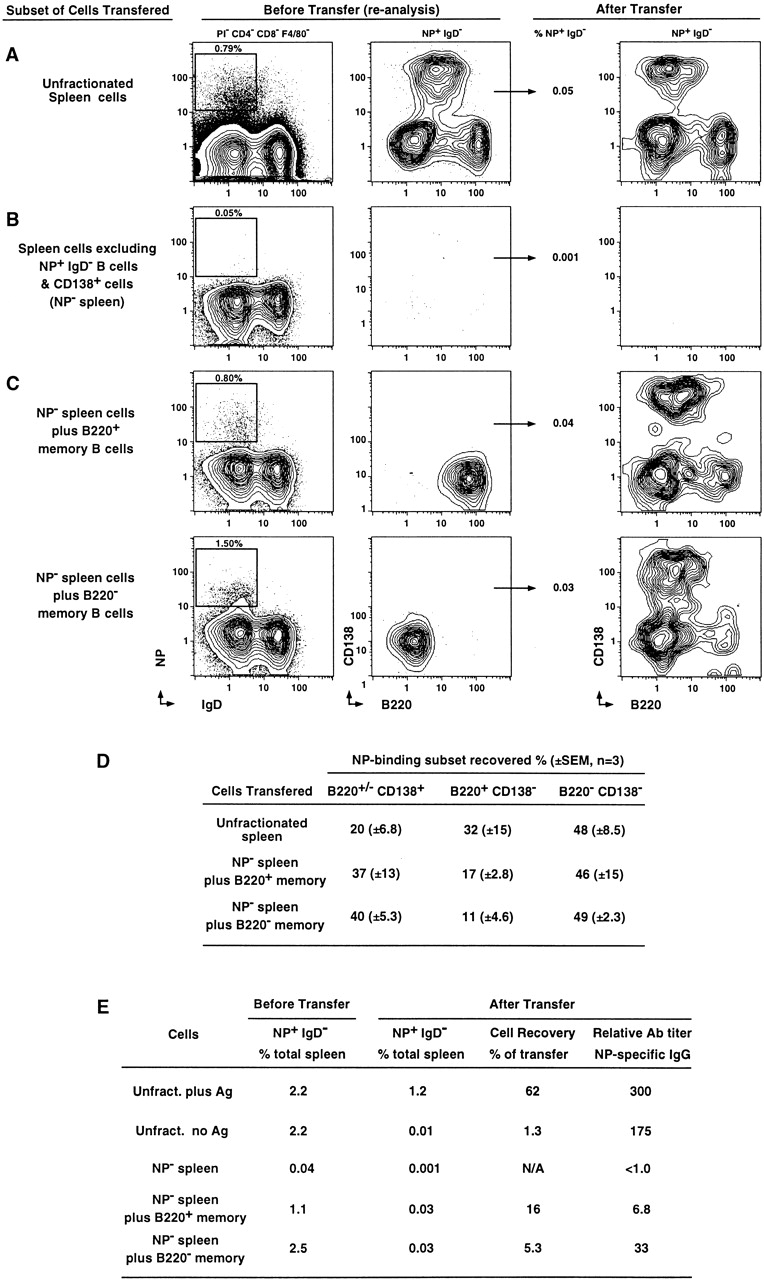
Adoptive transfer of purified emergent B220+ and B220− memory B cells. (A) 4 × 107 unfractionated spleen cells from animals 4 d after rechallenge were injected into the tail vein of C57BL/6 Rag-1–deficient animals by intraperitoneal injection with 50 μg NP-KLH (no adjuvant). The first panel represents the NP and IgD profiles of the spleen cells that were transferred. The second panel represents the CD138 and B220 profiles of the NP+IgD− cells in these animals (percentage of NP-specific cells in total spleen cells before transfer is indicated above inset). The percentage of NP+IgD− cells after transfer (of total spleen cells in recipient) is displayed in the next column, and the third panel represents the CD138 and B220 profiles of the NP+IgD− cells in the spleens of the Rag-1–deficient mice 5 d after transfer. (B) The first two panels present reanalysis of spleen cells from day 4 memory mice, excluding NP+IgD− and all CD138+ cells at the time of sorting. 5–8 × 106 spleen cells sorted this way (NP− spleen) were injected into the tail vein (with Ag intraperitoneally as above), and spleen cells from Rag-1–deficient donors analyzed 5 d after transfer (third panel). (C) 2–6 × 104 purified NP+IgD−CD138− that were B220+ (top) or B220− bottom) were added to 5–8 × 106 NP− spleen cells before adoptive transfer. The first two panels in each set are the reanalysis of the cell mixture immediately before transfer, and in the last panel are the CD138 and B220 profiles of the NP+IgD−spleen cells in the Rag-1–deficient recipients 5 d after transfer. (D) Mean ± SEM from three adoptive transfer experiments displaying the proportion of the three subsets recovered after transfer. The level of NP+ events after transfer of the NP− spleen cells alone was negligible, so they are not included in this table. (E) Summary of cell recovery and Ab production during time of transfer (n = 2 or 3 animals). Cell recovery was calculated as the percent of NP-specific cells recovered divided by the number transferred. N/A indicates not applicable, as the number of NP-specific cells transferred and recovered was negligible. The relative titer of NP-specific Ab is displayed as the titer of serum that produced an OD of 1.4 OD units (maximum of 4.0 OD units).
Compared with the day 42 transfer, the same general pattern of cellular differentiation was observed after transfer of purified memory B cell subsets 4 d after rechallenge. When purified B220+ memory responders were transferred, the major cellular products were NP-specific B220− (46%) and CD138+ cells (37%), with few remaining B220+ cells (17%; Fig. 4C and Fig. D). When B220− memory responders were transferred, mostly B220− cells were produced (49%), with similar numbers of CD138+ cells (40%) and a minority of B220+ cells (11%; Fig. 4C and Fig. D). However, total cell recovery and Ab production was lower after transfer of day 4 memory responders than after transfer of the quiescent memory B cells (Fig. 4 E). These differences may reflect different growth requirements for NP-specific memory cells in the emergent compared with quiescent phase in vivo. Nevertheless, the B220+ memory responders demonstrated slightly higher proliferative capacity than their B220− counterparts, and the B220− cells produced slightly higher NP-specific IgG upon transfer (Fig. 4 E). Thus, it appears that the cellular differentiation pattern displayed after transfer of the day 42 quiescent memory cells may already be established by day 4 of the memory response.
B220−CD138− Memory B Cells Express a Unique Cell Surface Phenotype.
Consistent with a stable change in differentiation between the B220+ and B220− memory B cells, a change in cell surface phenotype may indicate different means for responding to Ag and differential activation requirements. CD45 modulates BCR signal transduction and appears downregulated on both B220− memory B cells and CD138+ Ab-secreting B cells. The mAb RA3-6B2 (used in the current studies) recognizes the B cell isoform of CD45 (B220) through its expression of exon A and specific carbohydrate residues 54. In contrast, the mAb 30-F11 recognizes CD45 regardless of isoform. Using 30-F11 expression, all NP-specific memory B cell subsets appear to express CD45 to similar levels (Fig. 5, left). Thus, the B220− (RA3-6B2−) and CD138+ memory B cells have an isoform or glycosylation variant of CD45, suggesting alternate ligands or functions for this molecule across different memory B cell subsets.
Figure 5.
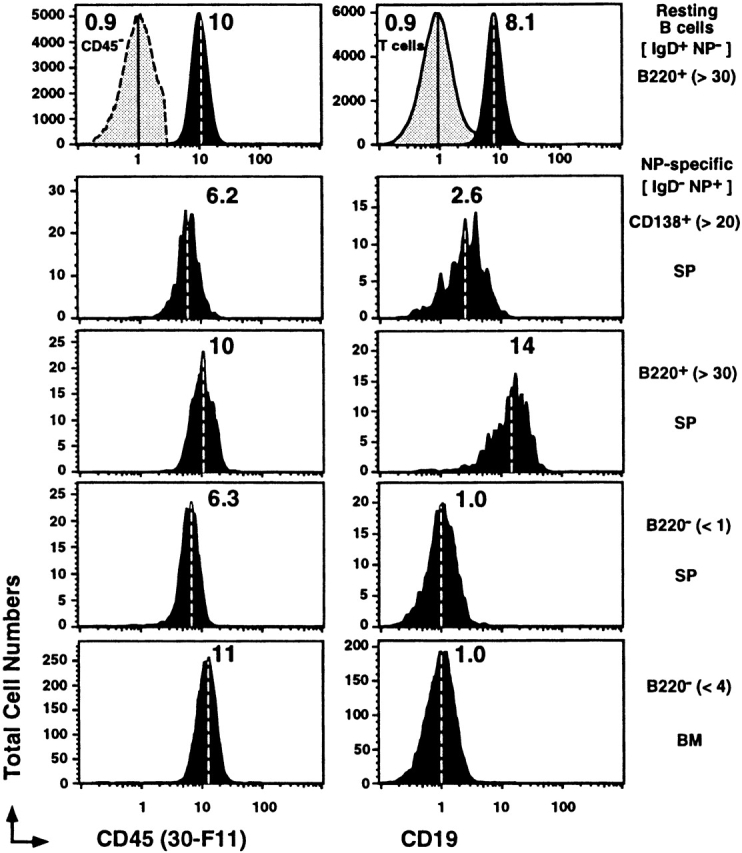
B220−CD138− memory B cells express an alternate form of CD45 and no CD19. Representative histograms for CD45 (FITC–30-F11) and CD19 (FITC-1D3) on CD4−, CD8−, and F4/80− cells that were also IgD+NP−B220+ B cells (first row), IgD−NP+CD138+ (second row), IgD−NP+B220+ (third row), and IgD−NP+B220− cells from the spleen (fourth row) and bone marrow (fifth row). Where pertinent, levels of labeling for “T cells” (the majority of Cy5-PE+ are CD4 and CD8 T cells), CD45−IgD−NP−B220− cells are also displayed in the first row. All displays are from the same animal at day 9 of the recall response and are representative of three to eight animals. The dashed lines within each histogram and the number associated depict the geometric mean fluorescence for the reagent presented.
CD19 is another typical B cell lineage marker known to positively regulate BCR signal transduction and decrease on differentiation to Ab-secreting cells 55. Resting B cells and NP-specific B220+ memory cells express CD19 to similar levels (Fig. 5, right). However, CD19 is downregulated on the NP-specific B220− memory B cell compartment. Expression of CD19 is two- to threefold lower than that expressed on the B220+/−CD138+ Ab-secreting memory compartment, and is similar to background levels on T cells (Fig. 5, right). The absence of CD19 may indicate hyperresponsiveness to BCR signal transduction. Consistent with different Ag-receptor signaling potentials, CD22, CD23 (the low affinity FcεR), CD24, and CD38 were also absent on the B220− memory B cells, whereas CD43 was markedly upregulated (data not shown). The absence of CD5 (data not shown) indicated that the NP-specific B220− subset did not belong to the B1a B cell subtype 56. Overall, the cell surface phenotype for the B220− memory B cells is unique and predicts a very different mechanism of Ag receptor–mediated signal transduction.
Two Major Subsets of B220− Memory B Cells.
Although coreceptor expression offers clues to Ag responsiveness, integrin expression highlights differences in the interaction with the microenvironment, and perhaps unique migration patterns. CD11b expression not only distinguishes the B220− memory B cells from the CD138+ and B220+ memory B cells, but also divides the B220− memory B cell compartment into two major subsets (Fig. 6 A). CD11b expression on the NP-specific B220− memory compartment is best viewed in the bone marrow (where the B220− B cells dominate) against sIgE expression (Fig. 6 A; bottom right). The NP-specific B220− B cells clearly divide into two major subsets, CD11b++IgE− and CD11b+IgE+. These two subsets are also present within the NP-specific cells of the spleen (Fig. 6 A; middle right panel). When CD11b and IgE labeling is paired separately with B220 and CD138 in splenic samples, the CD11b++ and IgE+ cells appear clearly contained within the NP-specific B220− memory compartment (data not shown). Both the CD11b++ and IgE+ B220− subsets are isotype switched and negative for sIgM (data not shown). Although there is a high level of background labeling with anti-IgG on naive B cells and the IgE+ subset (Fig. 6 B), the CD11b++IgE− subset express sIgG at 20-fold higher levels, comparable to levels on B220+ and CD138+ NP-specific cells (Fig. 6 B). The sIgE+ cells are not secreting NP-specific IgE as assessed by an ELISPOT assay (data not shown). Both NP-specific B220− subsets are present throughout the memory response and into the quiescent memory phase; however, the balance of cells within each subset is variable (data not shown). Nevertheless, sIgE expression is only found in the B220− compartment and provides a clear example of commitment to subspecialized function among the long-lived B220− memory B cells.
Figure 6.
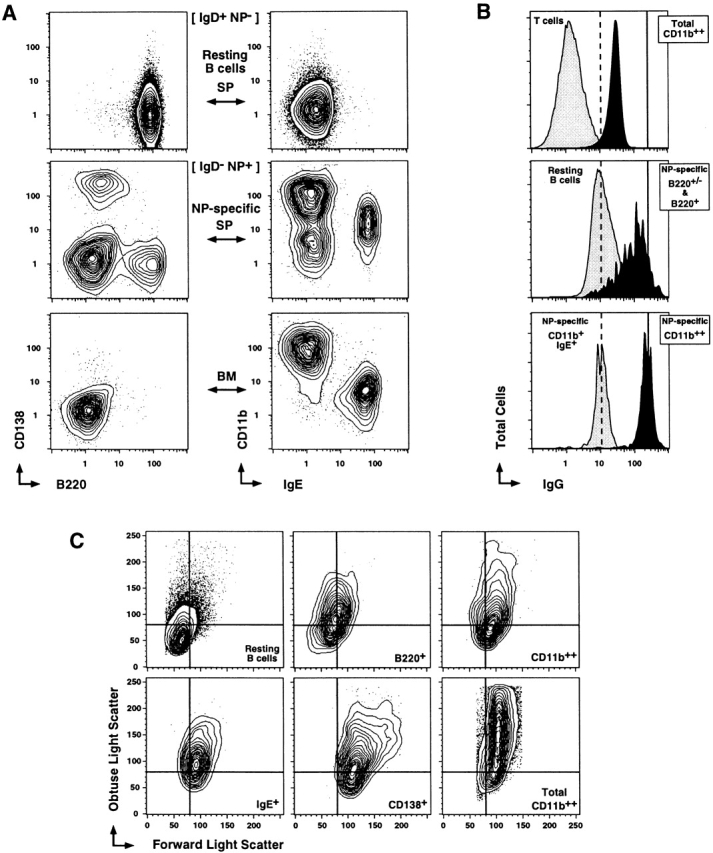

Two major subsets of NP-specific B220− memory B cells. (A) Representative profiles of CD138 and B220 (first column) and CD11b and IgE (second column) on CD4−, CD8−, and F4/80− cells that were also NP−IgD+ (first row), NP+IgD− spleen (second row), and NP+IgD− bone marrow (third row). (B) Histograms for anti-IgG labeling on total CD11b++NP−IgD− (top, black), small forward/side scatter Cy5PE+ cells (mostly T cells; top, gray), NP−IgD+B220+ (resting B cells; middle, gray), NP+IgD−CD11b− (NP-specific B220+/− and B220+; middle, black), NP+IgD−CD11b++ (bottom, black), and NP+IgD−CD11b+IgE+ (bottom, gray). Cells were labeled with PE–anti-IgG, blocked with normal mouse serum, and then labeled with mAbs as described. (C) Representative profiles for forward and obtuse light scatter on CD4−, CD8−, and F4/80− cells that were also NP−IgD+ B220+ (resting B cells; top left), NP+ IgD−CD138−B220+ (B220+; top middle), NP+IgD−CD11b++IgE− (CD11b++; top right), NP+IgD−CD11b+IgE+ (IgE+; bottom left), NP+IgD−CD138+B220+/− (CD138+; bottom middle), and CD11b++ NP−IgD− (total CD11b++; bottom right). (A–C) Profiles are from day 9 of the recall response and are representative of three to nine animals. (D) TEM of CD4−, CD8−, and F4/80− NP+IgD−CD11b++IgE− spleen cells from day 9 recall response. 1.2 × 105 cells were sorted into 2% glutaraldehyde and processed for TEM by Duke Comprehensive Cancer Facility. Images are representative of at least 100 individual CD11b++ NP-specific cells scanned.
The light scatter properties of the four different memory responders at day 9 after recall are displayed in Fig. 6 C. All NP-specific memory B cell subsets have higher obtuse light scatter and are larger by forward light scatter than resting B cells, but have lower obtuse light scatter than total CD11b++ cells in the spleen. Thus, the CD11b++ B cells are not representative of all CD11b++ cells in the spleen (which are mostly macrophage). To further investigate the morphology of this unique memory B cell compartment, we selected CD11b++ memory cells 9 d after recall for transmission electron micrographs (TEMs; Fig. 6 D). Many of the CD11b++ cells scanned had typical resting lymphoid ultrastructure (∼20%; n = 100), with rounded nucleus and scant cytoplasm (Fig. 6 D, i). Another fraction appeared lymphoblastoid (∼15%), with a more ovoid nucleus, loose chromatin, and more abundant cytoplasm (Fig. 6 D, ii). The majority of NP-specific CD11b++ cells (∼65%) contained the more rounded nucleus typical of a lymphocyte, but with a notch (Fig. 6 D, iii). Thus, the B220− memory B cells share many typical physical characteristics of lymphocyte, with some unique ultrastructural features in the CD11b+ subtype.
CD79b Expression Divides B220− Memory B Cell Compartment.
Finally, we address the expression of the BCR signaling complex subunit CD79b (Igβ) across all NP-binding B cells (Fig. 7). High levels of CD79b are expressed on resting B cells, the B220+ memory B cells, and the CD11b++IgE− memory B cell subset. The level of expression appears progressively lower between the resting state B220+ and B220−CD11b++ cells, but to levels 10-fold above background. Nevertheless, the CD79b expression represents the first typical and characteristic B cell Ag expressed on the CD11b++IgE− subset. In contrast, the NP-specific CD138+ and IgE+ subsets express CD79b levels only marginally above background (Fig. 7; T cells). The low levels of CD79b on Ab-secreting cells may be consistent with terminal differentiation and the reduced need to respond to Ag. The loss of CD79b on the nonsecreting IgE+ cells is not understood, but indicates very different Ag responsiveness across different Ig isotypes in the B220− memory B cell compartment. Thus, cellular potentials on transfer, levels of coreceptor and integrin expression, pattern of Ig isotype, and signaling subunits assort NP-specific memory B cells into four separate cellular compartments, each with unique roles in the maintenance of long-term protective immunity.
Figure 7.
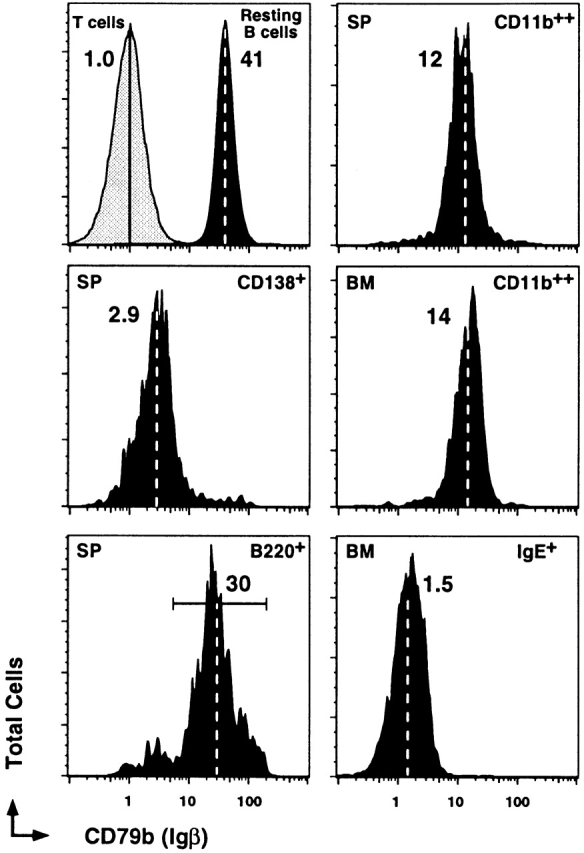
CD79b expression on NP-specific memory B cells. Representative histograms for CD79b on spleen CD4−, CD8−, and F4/80− cells that were also NP−IgD+B220+ B cells (first column, top), IgD−NP+CD138+ (first column, middle), IgD−NP+B220+ (first column, bottom), IgD−NP+CD11b++ (second column, top), bone marrow IgD−NP+CD11b++ (second column, middle), and IgD−NP+IgE− (second column, bottom). Levels of labeling for T cells (small forward/side scatter, Cy5-PE+ cells) are also displayed in the first column, top. All displays are from day 9 recall response and are representative of eight animals. The dashed lines within each histogram and the number associated depict the geometric mean fluorescence intensity for CD79b.
Discussion
In this study, we quantitatively analyze the emergence of Ag-specific memory B cell responders and the subsequent reestablishment of the quiescent memory B cell compartment. We reveal a novel B220− memory B cell compartment that constitutes a significant component of the memory response to Ag and forms the major cellular reservoir of B cell memory in both the spleen and the bone marrow. Upon Ag rechallenge, the B220− memory B cells rapidly self-replenish, but also differentiate into Ab-secreting cells with high efficiency. The B220+ memory B cells also function as memory response precursors, but mainly differentiate into B220− memory B cells upon Ag rechallenge. All B220− memory cells have isotype switched and divide broadly into two subsets, CD11b++IgG+ and CD11b+ IgE+. Both B220− subsets differ markedly in coreceptor expression from either naive B cells or B220+ memory B cells. This cell surface phenotype predicts differential activation requirements and provides unique means for regulating the response to Ag rechallenge in vivo. Taken together, these data highlight the complex organization of B cell memory into cellular compartments with distinct patterns of regulation and subspecialized immune function.
Expression of B Cell Memory.
We chose to analyze the emergent memory response to Ag as one way to quantify the activity of memory B cells directly ex vivo. Ideally, memory B cell expansion to Ag rechallenge would lead to the rapid formation of Ab-secreting cells and the replenishment (or preservation) of the memory B cell compartment. Ab-secreting cells emerge first in the T cell zones of the spleen during a primary response 5 57 and express high levels of CD138 11 34. In the memory response, NP+IgD−B220+/−CD138+ cells emerge rapidly to levels up to 10-fold greater than that seen after initial priming 11. B220+ memory B cells also appear in response to recall. Ag-binding isotype-switched B cells with this phenotype at late time points after initial priming can be regarded as quiescent memory B cells 13 30 31 58. After Ag recall in vivo, isotype-switched B cells with the B220+ phenotype cannot be distinguished from B cells that have initiated a secondary GC reaction 5 8 59 60. Nevertheless, accelerated expansion of both B220+/−CD138+ and B220+CD138− memory B cells is a major cellular consequence of Ag rechallenge.
The appearance of a major subset of B220−CD138− Ag-binding B cells was unexpected. These NP-binding B220− cells expanded rapidly and specifically in response to Ag rechallenge, expressed somatically-mutated BCRs, and were not secreting Ab in vivo. The adoptive transfer experiments 4 d after recall provide the clearest insights into the role of these B220− cells in the emerging memory response (Fig. 4). They arose from purified B220+CD138− precursors on transfer, but could also self-replenish with high efficiency when transferred on their own. Besides self-replenishment, they predominantly produced Ab-secreting cells but still retained some ability to produce B220+ memory cells on transfer.
Further insight into the distinct function of the B220− cells was offered through integrin and Ig isotype expression (Fig. 6). The B220− compartment divided into CD11b++ and CD11b+ subsets, both distinguishable from the other two memory subsets (CD11b−). There have been reports for expression of CD11b on both activated T cells 61 and subsets of splenic B cells 62. CD11b has also been described on the B-1 B cell lineage 56, at levels lower than on the sIgE− subset. CD11b/CD18 is also known as complement receptor type 3 and may provide an alternate coreceptor for the BCR 63 64. In our earlier studies, CD11b binding was used to exclude cells such as macrophages from the analyses of the NP-specific B cell response 11 33 34 and explains why we had not previously revealed the B220− memory subset. IgG expression was dominant in B220+, CD138+, and CD11b++ NP-specific subsets. In contrast, sIgE+ memory cells were only found in the B220−CD11b+ subset. Interestingly, IgE was not detected in the serum of recipient animals after adoptive transfer of day 4 or 42 memory B cells, suggesting that the secretion of this isotype is tightly regulated in vivo. Although the BCR signaling subunit was expressed at high levels in the CD11b++IgG+ compartment, it was absent in the IgE+ cells. Therefore, we describe two functionally distinct subtypes of the B220− memory B cells that emerge in response to Ag recall.
B Cell Memory in the Bone Marrow.
The predominance of the B220−CD138− memory B cells in the bone marrow was also unexpected. Much of the high affinity Ab that persists in the serum comes from long-lived Ab-secreting B cells that reside in the bone marrow 35 36 37 38 39 40 65. When quantified by flow cytometry or ELISPOT analysis, Ab-secreting B cells were present in the bone marrow at very low frequencies 41 42. These frequencies correspond well to the low frequencies of CD138+ B cells seen in the bone marrow in this analysis. Nevertheless, in the bone marrow, >95% of the Ag-specific compartment comprised of B220−CD138− nonsecreting B cells. These cells most likely migrate to the bone marrow from the splenic response to Ag and persist there at high levels to at least day 42 after recall. These cells may be the isotype-switched equivalent to the CD24low somatically-mutated IgM memory B cells that migrate to the bone marrow in humans 66. Upon adoptive transfer of NP-specific B cells from the spleen, B220−CD138− B cells do appear in the bone marrow (data not shown), arguing that migration can occur between these two compartments. The bone marrow and spleen may contain the appropriate microenvironment for the long-term survival of B220−CD138− memory B cell compartment. Thus, the bone marrow provides an alternative reservoir for B220− memory response precursors that may migrate into the spleen and expand upon Ag rechallenge.
Somatic Hypermutation in the Memory B Cell Compartment.
There was no obvious change in the pattern of somatic mutation in all three Ag-specific memory responders during the emergent memory response. Although lower than typically expected for the VH186.2 9 11 33, the level of mutation was significant and consistent in the Vλ1 L chain for all three memory populations, and was higher than reported previously for early in the primary response (day 12; reference 32). All three memory compartments emerged by days 3–4 in the spleen with already mutated L chain genes. However, the extent of mutation did not change by day 9. Although expected for the Ab-secreting compartment, this is surprising for the B220+CD138− memory response compartment that most likely constitutes a secondary GC reaction. It also suggests that the B220− memory compartment was expanded after recall with no further diversification and selection. This finding is consistent with the non-GC phenotype of the B220− compartment, and further implies that expansion of the novel memory compartment did not require a GC phase after recall. These data support the contention that memory B cells do not further diversify by somatic hypermutation upon Ag recall 67.
Replenishment of the Memory B Cell Reservoir.
The origin of memory B cells still remains controversial. Jacob and Kelsoe provide evidence for the branching of GC B cells and Ab-secreting B cells from common precursors and argue for a common clonal origin for Ag-specific effector cells and memory cells 46. In contrast, Klinman and colleagues present evidence for a preexisting CD24low lineage of B cells that act as precursors for the memory response, have a greater propensity to form GC and do not contribute substantially to the primary Ab response 68 69 70. As a further complication, entry into the memory compartment appears to be dictated by the affinity of the mutated BCR relative to its unmutated counterpart 71, and may be derived from only a small fraction of all GCs available 72. Thus, variability in origins and function of the memory B cells as well as heterogeneity in their localization and trafficking patterns may complicate the memory B cell compartment.
Our analysis of the quiescent memory phase (day 42 after recall) provides insight into the persistent memory B cell compartment. B220+ memory precursors do appear in our studies, and the activity of these cells on adoptive transfer helps to confirm earlier studies on B220+ memory B cells 13 30 31 58. However, the B220− memory B cell compartment has not been described previously. Some aspects of B220− memory B cell function are reminiscent of the CD24low memory lineage demonstrated by Klinman and colleagues 68 69 70. In the memory response, it is not possible to distinguish the functions of the CD24low memory B cells 70, as they appear to form GCs as well as Ab-secreting cells. Although Klinman and colleagues' more recent analysis of the memory response suggests multiple activities within the memory compartment, they associate all memory function with the CD24low phenotype 70. The B220− memory B cells appear to predominantly form Ab-secreting B cells, and may only form GCs in the absence of B220+ memory B cells. Further, the B220− NP-specific memory B cell subset lacks expression of CD24; however, the B220+ memory B cell compartment expresses higher levels of CD24 than found on naive B cells (data not shown), inconsistent with the notion that all memory is CD24low 70. The CD24low memory B cells also seem capable of accumulating somatic mutations during the recall response 70, an activity that is not apparent in the B220− memory compartment.
Atypical Coreceptor Expression on B220− Memory B Cells.
The atypical cell surface phenotype of the novel B220− memory B cell compartment predicts very different mechanisms for BCR-mediated signal transduction. Although it is clear that all three Ag-specific B cell populations express similar levels of CD45, they express different isoforms or glycosylation variants of the B220 molecule. CD45 positively regulates BCR signal transduction 73 74 75 and modulates positive and negative selection of self-reactive B cells 76. Although different isoforms of CD45 are thought to be expressed on memory T cell subsets, their functional consequence remains unclear. B220−CD19− B cells have been described by Wabl and colleagues 77 78 in the quasimonoclonal mouse model, although their function in this model remains unknown. In a more recent manuscript, these B220−CD19− B cells express mutated forms of the transgenic receptor, arguing for the memory status of these cells 79. Interestingly, the B220−CD19− B cells from these transgenic animals do not express CD11b, and hence may be distinct from the B220− memory B cells in this study. CD19 is another BCR response regulator, and is thought to positively impact on signal transduction 55. Although it is recognized that CD19 is downregulated on plasma cells, it was assumed that loss of CD19 is associated with the loss of sIg, and hence not required on this terminally differentiated cell type. The effect of CD19 downregulation on the B220− compartment remains unclear; however, it is clear that these atypical memory B cells can respond vigorously to Ag in the absence of CD19.
CD5 and CD22 are negative regulators of BCR signal transduction 80 81, and their absence may lead to hyperresponsiveness to Ag triggering in the B220− subsets. Similarly, CD43 ligation can induce cell cycle arrest and extend B cell survival in vitro 82 83. Thus, CD43 upregulation on the B220− memory compartment may play a role in the persistence of these cells in vivo. Expression of CD79b on the CD11b++ subset suggests that BCR signal transduction itself may be similar between the B220−CD11b++ and the B220+ memory compartments, but distinct in the B220−IgE+ compartment. CD21 expression also appears to assort between the B220−CD11b++ (intermediate CD21) and B220−IgE+ (CD21low; data not shown). Overall, the extensive changes in cell surface phenotype indicate a change in physiology required for the homeostatic maintenance of memory B cells, or it may indicate hypersensitivity to Ag recall and a heightened readiness to respond.
B Cell Memory as a Balance of Cellular Subsets.
Understanding and generating effective B cell memory underpins all efforts at vaccine design and has the potential to impact many aspects of fundamental and clinical immunology. Our studies demonstrate that B cell memory is organized into at least three cellular compartments. The three main memory B cell compartments appear to have decreasing potential for cellular differentiation. B220+ memory B cells have the highest potential for change, and rapidly produce B220− memory B cells with few Ab-secreting cells upon Ag rechallenge. B220− memory B cells self-replenish rapidly upon Ag rechallenge and produce Ab-secreting cells with high efficiency, but retain little capacity to produce B220+ memory B cells. This pattern of differentiation suggests that B220− memory B cells have an intermediate potential for change. In contrast, Ab-secreting cells are long lived, terminally-differentiated end stage cells with no potential for change. This schematic broadly indicates a model of linear differentiation, with B220+ memory B cells producing B220− memory B cells, which in turn produce CD138+ Ab-secreting cells. This model implies that B cell memory exists as a complex balance of stable precursors at differing stages of functional commitment. A parallel model exists for T cell differentiation in which Ag experience leads to functional precommitment within subsets of Ag-specific T cells 84 85 86 87. In the T cell compartment, precommitment allows for rapid expression of preferred effector functions such as cytokine production. Similar functional expediency may underpin subspecialization within the memory B cell compartment. Thus, promoting an effective balance of memory B cell subtypes after priming would be a critical means for regulating effective long-term protective immunity in vivo.
Acknowledgments
We thank Maria Karvelas, David Driver, and Garnett Kelsoe for constructive comments and discussions. We also thank Fred Finkelman for the kind gift of 11.26, and Lee Herzenberg for JC5.1. Thanks also to the Duke Comprehensive Cancer Center Flow Cytometry and Electron Microscopy Shared Resources.
This work was supported by a grant from the National Institutes of Health (AI40215) and by an Arthritis Foundation Biomedical Grant.
Footnotes
Abbreviations used in this paper: APC, allophycocyanin; BCR, B cell receptor; GC, germinal center; ELISPOT, enzyme-linked immunospot; MFI, mean fluorescence index; NP, (4-hydroxy-3-nitrophenyl)acetyl; NP-APC, NP-labeled APC; PI, propidium iodide; Rag-1, recombination activating gene 1; RT, reverse transcriptase; sIg, surface Ig; TEM, transmission electron micrograph.
References
- Eisen H.N., Siskind G.W. Variations in the affinity of antibodies during an immune response. Biochemistry. 1969;3:996–1008. doi: 10.1021/bi00895a027. [DOI] [PubMed] [Google Scholar]
- Siskind G.W., Benacerraf B. Cell selection by antigen in the immune response. Adv. Immunol. 1969;10:1–50. doi: 10.1016/s0065-2776(08)60414-9. [DOI] [PubMed] [Google Scholar]
- Coico R.F., Bhogal B.S., Thorbecke G.J. Relationship of germinal centers in lymphoid tissue to immunologic memory. VI. Transfer of B cell memory with lymph node cells fractionated according to their receptors for peanut agglutinin. J. Immunol. 1983;131:2254–2257. [PubMed] [Google Scholar]
- McHeyzer-Williams M.G., Ahmed R. B cell memory and the long-lived plasma cell. Curr. Opin. Immunol. 1999;11:172–179. doi: 10.1016/s0952-7915(99)80029-6. [DOI] [PubMed] [Google Scholar]
- MacLennan I.C.M., Gray D. Antigen-driven selection of virgin and memory B cells. Immunol. Rev. 1986;91:61–85. doi: 10.1111/j.1600-065x.1986.tb01484.x. [DOI] [PubMed] [Google Scholar]
- Ahmed R., Gray D. Immunological memory and protective immunityunderstanding their relation. Science. 1996;272:54–60. doi: 10.1126/science.272.5258.54. [DOI] [PubMed] [Google Scholar]
- Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381:751–758. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- McHeyzer-Williams M.G. Immune response decisions at the single cell level. Semin. Immunol. 1997;9:219–227. doi: 10.1006/smim.1997.0079. [DOI] [PubMed] [Google Scholar]
- Jacob J., Kelsoe G., Rajewsky K., Weiss U. Intraclonal generation of antibody mutants in germinal centres. Nature. 1991;354:389–392. doi: 10.1038/354389a0. [DOI] [PubMed] [Google Scholar]
- Berek C., Berger A., Apel M. Maturation of the immune response in germinal centers. Cell. 1991;67:1121–1129. doi: 10.1016/0092-8674(91)90289-b. [DOI] [PubMed] [Google Scholar]
- McHeyzer-Williams M.G., McLean M.J., Lalor P.A., Nossal G.J.V. Antigen-driven B cell differentiation in vivo. J. Exp. Med. 1993;178:295–307. doi: 10.1084/jem.178.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray D., Skarvall H. B-cell memory is short-lived in the absence of antigen. Nature. 1988;336:70–73. doi: 10.1038/336070a0. [DOI] [PubMed] [Google Scholar]
- Schittek B., Rajewsky K. Maintenance of B-cell memory by long-lived cells generated from proliferating precursors. Nature. 1990;346:749–751. doi: 10.1038/346749a0. [DOI] [PubMed] [Google Scholar]
- Matsumoto M., Mariathasan S., Nahm M.H., Baranyay F., Peschon J.J., Chaplin D.D. Role of lymphotoxin and the type I TNF receptor in the formation of germinal centers. Science. 1996;271:1289–1291. doi: 10.1126/science.271.5253.1289. [DOI] [PubMed] [Google Scholar]
- Fu Y.X., Huang G., Matsumoto M., Molina H., Chaplin D.D. Independent signals regulate development of primary and secondary follicle structure in spleen and mesenteric lymph node. Proc. Natl. Acad. Sci. USA. 1997;94:5739–5743. doi: 10.1073/pnas.94.11.5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koni P.A., Sacca R., Lawton P., Browning J.L., Ruddle N.H., Flavell R.A. Distinct roles in lymphoid organogenesis for lymphotoxins alpha and beta revealed in lymphotoxin beta-deficient mice. Immunity. 1997;6:491–500. doi: 10.1016/s1074-7613(00)80292-7. [DOI] [PubMed] [Google Scholar]
- Forster R., Mattis A.E., Kremmer E., Wolf E., Brem G., Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87:1037–1047. doi: 10.1016/s0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- Gunn M.D., Ngo V.N., Ansel K.M., Ekland E.H., Cyster J.G., Williams L.T. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt's lymphoma receptor-1. Nature. 1998;391:799–803. doi: 10.1038/35876. [DOI] [PubMed] [Google Scholar]
- Kallberg E., Jainandunsing S., Gray D., Leanderson T. Somatic mutation of immunoglobulin V genes in vitro. Science. 1996;271:1285–1289. doi: 10.1126/science.271.5253.1285. [DOI] [PubMed] [Google Scholar]
- Armitage R.J., Fanslow W.C., Strockbine L., Sato T.A., Clifford K.N., Macduff B.M., Anderson D.M., Gimpel S.D., Davis-Smith T., Maliszewski C.R. Molecular and biological characterization of a murine ligand for CD40. Nature. 1992;357:80–82. doi: 10.1038/357080a0. [DOI] [PubMed] [Google Scholar]
- Van den Eertwegh A.J., Noelle R.J., Roy M., Shepherd D.M., Aruffo A., Ledbetter J.A., Boersma W.J., Claassen E. In vivo CD40–gp39 interactions are essential for thymus-dependent humoral immunity. I. In vivo expression of CD40 ligand, cytokines, and antibody production delineates sites of cognate T–B cell interactions. J. Exp. Med. 1993;178:1555–1565. doi: 10.1084/jem.178.5.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Essen D., Kikutani H., Gray D. CD40 ligand-transduced co-stimulation of T cells in the development of helper function. Nature. 1995;378:620–623. doi: 10.1038/378620a0. [DOI] [PubMed] [Google Scholar]
- Gray D., Dullforce P., Jainandunsing S. Memory B cell development but not germinal center formation is impaired by in vivo blockade of CD40–CD40 ligand interaction. J. Exp. Med. 1994;180:141–155. doi: 10.1084/jem.180.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S., Hathcock K., Zheng B., Kepler T.B., Hodes R., Kelsoe G. Cellular interaction in germinal centers. Roles of CD40 ligand and B7-2 in established germinal centers. J. Immunol. 1995;155:556–567. [PubMed] [Google Scholar]
- Shokat K.M., Goodnow C.C. Antigen-induced B-cell death and elimination during germinal-centre immune responses. Nature. 1995;375:334–338. doi: 10.1038/375334a0. [DOI] [PubMed] [Google Scholar]
- Pulendran B., Kannourakis G., Nouri S., Smith K.G., Nossal G.J.V. Soluble antigen can cause enhanced apoptosis of germinal-centre B cells. Nature. 1995;375:331–334. doi: 10.1038/375331a0. [DOI] [PubMed] [Google Scholar]
- Tew J.G., Phipps R.P., Mandel T.E. The maintenance and regulation of the humoral immune responsepersisting antigen and the role of follicular antigen-binding dendritic cells as accessory cells. Immunol. Rev. 1980;53:175–201. doi: 10.1111/j.1600-065x.1980.tb01044.x. [DOI] [PubMed] [Google Scholar]
- Croix D.A., Ahearn J.M., Rosengard A.M., Han S., Kelsoe G., Ma M., Carroll M.C. Antibody response to a T-dependent antigen requires B cell expression of complement receptors. J. Exp. Med. 1996;183:1857–1864. doi: 10.1084/jem.183.4.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M.B., Goerg S., Shen L., Prodeus A.P., Goodnow C.C., Kelsoe G., Carroll M.C. Dependence of germinal center B cells on expression of CD21/CD35 for survival. Science. 1998;280:582–585. doi: 10.1126/science.280.5363.582. [DOI] [PubMed] [Google Scholar]
- Black S.J., van der Loo W., Loken M.R., Herzenberg L.A. Expression of IgD by murine lymphocytes. Loss of surface IgD indicates maturation of memory B cells. J. Exp. Med. 1978;147:984–996. doi: 10.1084/jem.147.4.984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K., Ishii R., Yamasaki K., Kishimoto T., Hardy R.R. Isolation of high-affinity memory B cellsphycoerythrin as a probe for antigen-binding cells. Proc. Natl. Acad. Sci. USA. 1987;84:1379–1383. doi: 10.1073/pnas.84.5.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford J.E., McHeyzer-Williams M.G., Lieber M.R. Analysis of individual immunoglobulin lambda light chain genes amplified from single cells is inconsistent with variable region gene conversion in germinal-center B cell somatic mutation. Eur. J. Immunol. 1994;24:1816–1822. doi: 10.1002/eji.1830240814. [DOI] [PubMed] [Google Scholar]
- McHeyzer-Williams M.G., Nossal G.J.V., Lalor P.A. Molecular characterization of single memory B cells. Nature. 1991;350:502–505. doi: 10.1038/350502a0. [DOI] [PubMed] [Google Scholar]
- Lalor P.A., Nossal G.J.V., Sanderson R.D., McHeyzer-Williams M.G. Functional and molecular characterization of single, (4-hydroxy-3- nitrophenyl)acetyl (NP)-specific, IgG1+ B cells from antibody-secreting and memory B cell pathways in the C57BL/6 immune response to NP. Eur. J. Immunol. 1992;22:3001–3011. doi: 10.1002/eji.1830221136. [DOI] [PubMed] [Google Scholar]
- Benner R., Hijmans W., Haaijman J.J. The bone marrowthe major source of serum immunoglobulins, but still a neglected site of antibody formation. Clin. Exp. Immunol. 1981;46:1–8. [PMC free article] [PubMed] [Google Scholar]
- Ho F., Lortan J.E., MacLennan I.C.M., Khan M. Distinct short-lived and long-lived antibody-producing cell populations. Eur. J. Immunol. 1986;16:1297–1301. doi: 10.1002/eji.1830161018. [DOI] [PubMed] [Google Scholar]
- Dilosa R.M., Maeda K., Masuda A., Szakal A.K., Tew J.G. Germinal center B cells and antibody production in the bone marrow. J. Immunol. 1991;146:4071–4077. [PubMed] [Google Scholar]
- Manz R.A., Thiel A., Radbruch A. Lifetime of plasma cells in the bone marrow. Nature. 1997;388:133–134. doi: 10.1038/40540. [DOI] [PubMed] [Google Scholar]
- Manz R.A., Lohning M., Cassese G., Thiel A., Radbruch A. Survival of long-lived plasma cells is independent of antigen. Int. Immunol. 1998;10:1703–1711. doi: 10.1093/intimm/10.11.1703. [DOI] [PubMed] [Google Scholar]
- Slifka M.K., Antia R., Whitmire J.K., Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8:363–372. doi: 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- Smith K.G., Light A., Nossal G.J.V., Tarlinton D.M. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:2996–3006. doi: 10.1093/emboj/16.11.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y., Dutta P.R., Cerasoli D.M., Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. V. Affinity maturation develops in two stages of clonal selection. J. Exp. Med. 1998;187:885–895. doi: 10.1084/jem.187.6.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makela O., Karjalainen K. Inherited immunoglobulin idiotypes of the mouse. Immunol. Rev. 1977;34:119–138. doi: 10.1111/j.1600-065x.1977.tb00370.x. [DOI] [PubMed] [Google Scholar]
- Reth M., Hammerling G.J., Rajewsky K. Analysis of the repertoire of anti-NP antibodies in C57BL/6 mice by cell fusion. I. Characterization of antibody families in the primary and hyperimmune response. Eur. J. Immunol. 1978;8:393–400. doi: 10.1002/eji.1830080605. [DOI] [PubMed] [Google Scholar]
- Jacob J., Kassir R., Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl) acetyl. I. The architecture and dynamics of responding cell populations. J. Exp. Med. 1991;173:1165–1175. doi: 10.1084/jem.173.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob J., Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3- nitrophenyl)acetyl. II. A common clonal origin for periarteriolar lymphoid sheath-associated foci and germinal centers. J. Exp. Med. 1992;176:679–687. doi: 10.1084/jem.176.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K.G., Weiss U., Rajewsky K., Nossal G.J.V., Tarlinton D.M. Bcl-2 increases memory B cell recruitment but does not perturb selection in germinal centers. Immunity. 1994;1:803–813. doi: 10.1016/s1074-7613(94)80022-7. [DOI] [PubMed] [Google Scholar]
- Smith K.G., Hewitson T.D., Nossal G.J.V., Tarlinton D.M. The phenotype and fate of the antibody-forming cells of the splenic foci. Eur. J. Immunol. 1996;26:444–448. doi: 10.1002/eji.1830260226. [DOI] [PubMed] [Google Scholar]
- Allen D., Cumano A., Dildrop R., Kocks C., Rajewsky K., Rajewsky N., Roes J., Sablitzky F., Siekevitz M. Timing, genetic requirements and functional consequences of somatic hypermutation during B-cell development. Immunol. Rev. 1987;96:5–22. doi: 10.1111/j.1600-065x.1987.tb00506.x. [DOI] [PubMed] [Google Scholar]
- Bothwell A.L., Paskind M., Reth M., Imanishi-Kari T., Rajewsky K., Baltimore D. Heavy chain variable region contribution to the NPb family of antibodiessomatic mutation evident in a gamma 2a variable region. Cell. 1981;24:625–637. doi: 10.1016/0092-8674(81)90089-1. [DOI] [PubMed] [Google Scholar]
- Weiss U., Zoebelein R., Rajewsky K. Accumulation of somatic mutants in the B cell compartment after primary immunization with a T cell-dependent antigen. Eur. J. Immunol. 1992;22:511–517. doi: 10.1002/eji.1830220233. [DOI] [PubMed] [Google Scholar]
- McHeyzer-Williams M.G., Davis M.M. Antigen-specific development of primary and memory T cells in vivo. Science. 1995;268:106–111. doi: 10.1126/science.7535476. [DOI] [PubMed] [Google Scholar]
- McHeyzer-Williams L.J., Panus J.F., Mikszta J.A., McHeyzer-Williams M.G. Evolution of antigen-specific T cell receptors in vivopreimmune and antigen-driven selection of preferred complementarity-determining region 3 (CDR3) motifs. J. Exp. Med. 1999;189:1823–1838. doi: 10.1084/jem.189.11.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson P., Maita A., Ng D.H.W. A family of leukocyte-specific cell surface glycoproteins Herzenberg L.A., Weir D.M., Herzenberg L.A., Blackwell C. Weir's Handbook of Experimental Immunology. Vol. II 1996. 62 Blackwell Science; Cambridge, MA: 1–62.16. [Google Scholar]
- Fujimoto M., Poe J.C., Inaoki M., Tedder T.F. CD19 regulates B lymphocyte responses to transmembrane signals. Semin. Immunol. 1998;10:267–277. doi: 10.1006/smim.1998.9999. [DOI] [PubMed] [Google Scholar]
- Kantor A.B., Herzenberg L.A. Origin of murine B cell lineages. Annu. Rev. Immunol. 1993;11:501–538. doi: 10.1146/annurev.iy.11.040193.002441. [DOI] [PubMed] [Google Scholar]
- Kelsoe G. Life and death in germinal centers (redux) Immunity. 1996;4:107–111. doi: 10.1016/s1074-7613(00)80675-5. [DOI] [PubMed] [Google Scholar]
- Herzenberg L.A., Black S.J., Tokuhisa T. Memory B cells at successive stages of differentiation. Affinity maturation and the role of IgD receptors. J. Exp. Med. 1980;151:1071–1087. doi: 10.1084/jem.151.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher E.C., Rouse R.V., Coffman R.L., Nottenburg C.N., Hardy R.R., Weissman I.L. Surface phenotype of Peyer's patch germinal center cellsimplications for the role of germinal centers in B cell differentiation. J. Immunol. 1982;129:2698–2707. [PubMed] [Google Scholar]
- Han S., Zheng B., Takahashi Y., Kelsoe G. Distinctive characteristics of germinal center B cells. Semin. Immunol. 1997;9:255–260. doi: 10.1006/smim.1997.0081. [DOI] [PubMed] [Google Scholar]
- McFarland H.I., Nahill S.R., Maciaszek J.W., Welsh R.M. CD11b (Mac-1)a marker for CD8+ cytotoxic T cell activation and memory in virus infection. J. Immunol. 1992;149:1326–1333. [PubMed] [Google Scholar]
- Borrello M.A., Phipps R.P. Fibroblast-secreted macrophage colony-stimulating factor is responsible for generation of biphenotypic B/macrophage cells from a subset of mouse B lymphocytes. J. Immunol. 1999;163:3605–3611. [PubMed] [Google Scholar]
- Zeger D.L., Osman N., Hennings M., McKenzie I.F., Sears D.W., Hogarth P.M. Mouse macrophage beta subunit (CD11b) cDNA for the CR3 complement receptor/Mac-1 antigen. Immunogenetics. 1990;31:191–197. doi: 10.1007/BF00211555. [DOI] [PubMed] [Google Scholar]
- Petty H.R., Todd R.F., III. Integrins as promiscuous signal transduction devices. Immunol. Today. 1996;17:209–212. doi: 10.1016/0167-5699(96)30013-3. [DOI] [PubMed] [Google Scholar]
- Slifka M.K., Matloubian M., Ahmed R. Bone marrow is a major site of long-term antibody production after acute viral infection. J. Virol. 1995;69:1895–1902. doi: 10.1128/jvi.69.3.1895-1902.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paramithiotis E., Cooper M.D. Memory B lymphocytes migrate to bone marrow in humans. Proc. Natl. Acad. Sci. USA. 1997;94:208–212. doi: 10.1073/pnas.94.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siekevitz M., Kocks C., Rajewsky K., Dildrop R. Analysis of somatic mutation and class switching in naive and memory B cells generating adoptive primary and secondary responses. Cell. 1987;48:757–770. doi: 10.1016/0092-8674(87)90073-0. [DOI] [PubMed] [Google Scholar]
- Linton P.L., Decker D.J., Klinman N.R. Primary antibody-forming cells and secondary B cells are generated from separate precursor cell subpopulations. Cell. 1989;59:1049–1059. doi: 10.1016/0092-8674(89)90761-7. [DOI] [PubMed] [Google Scholar]
- Linton P.J., Lo D., Lai L., Thorbecke G.J., Klinman N.R. Among naive precursor cell subpopulations only progenitors of memory B cells originate germinal centers. Eur. J. Immunol. 1992;22:1293–1297. doi: 10.1002/eji.1830220526. [DOI] [PubMed] [Google Scholar]
- Decker D.J., Linton P.J., Zaharevitz S., Biery M., Gingeras T.R., Klinman N.R. Defining subsets of naive and memory B cells based on the ability of their progeny to somatically mutate in vitro. Immunity. 1995;2:195–203. doi: 10.1016/s1074-7613(95)80092-1. [DOI] [PubMed] [Google Scholar]
- Vora K.A., Manser T. Altering the antibody repertoire via transgene homologous recombinationevidence for global and clone-autonomous regulation of antigen-driven B cell differentiation. J. Exp. Med. 1995;181:271–281. doi: 10.1084/jem.181.1.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A.H., Jena P.K., Wysocki L.J. Tracing the development of single memory-lineage B cells in a highly defined immune response. J. Exp. Med. 1996;183:2053–2063. doi: 10.1084/jem.183.5.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingel J.T., Thomas M.L. Evidence that the leukocyte-common antigen is required for antigen-induced T lymphocyte proliferation. Cell. 1989;58:1055–1065. doi: 10.1016/0092-8674(89)90504-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koretzky G.A., Picus J., Thomas M.L., Weiss A. Tyrosine phosphatase CD45 is essential for coupling T-cell antigen receptor to the phosphatidyl inositol pathway. Nature. 1990;346:66–68. doi: 10.1038/346066a0. [DOI] [PubMed] [Google Scholar]
- Kishihara K., Penninger J., Wallace V.A., Kundig T.M., Kawai K., Wakeham A., Timms E., Pfeffer K., Ohashi P.S., Thomas M.L. Normal B lymphocyte development but impaired T cell maturation in CD45-exon6 protein tyrosine phosphatase-deficient mice. Cell. 1993;74:143–156. doi: 10.1016/0092-8674(93)90302-7. [DOI] [PubMed] [Google Scholar]
- Cyster J.G., Healy J.I., Kishihara K., Mak T.W., Thomas M.L., Goodnow C.C. Regulation of B-lymphocyte negative and positive selection by tyrosine phosphatase CD45. Nature. 1996;381:325–328. doi: 10.1038/381325a0. [DOI] [PubMed] [Google Scholar]
- Cascalho M., Ma A., Lee S., Masat L., Wabl M. A quasi-monoclonal mouse. Science. 1996;272:1649–1652. doi: 10.1126/science.272.5268.1649. [DOI] [PubMed] [Google Scholar]
- Cascalho M., Wong J., Wabl M. VH gene replacement in hyperselected B cells of the quasimonoclonal mouse. J. Immunol. 1997;159:5795–5801. [PubMed] [Google Scholar]
- Cascalho M., Wong J., Brown J., Jack H., Steinberg C., Wabl M. A B220−, CD19− population of B cells in the peripheral blood of quasimonoclonal mice. Intl. Immunol. 2000;12:29–35. doi: 10.1093/intimm/12.1.29. [DOI] [PubMed] [Google Scholar]
- Bikah G., Carey J., Ciallella J.R., Tarakhovsky A., Bondada S. CD5-mediated negative regulation of antigen receptor-induced growth signals in B-1 B cells. Science. 1996;274:1906–1909. doi: 10.1126/science.274.5294.1906. [DOI] [PubMed] [Google Scholar]
- Tedder T.F., Tuscano J., Sato S., Kehrl J.H. CD22, a B lymphocyte-specific adhesion molecule that regulates antigen receptor signaling. Annu. Rev. Immunol. 1997;15:481–504. doi: 10.1146/annurev.immunol.15.1.481. [DOI] [PubMed] [Google Scholar]
- Dragone L.L., Barth R.K., Sitar K.L., Disbrow G.L., Frelinger J.G. Disregulation of leukosialin (CD43, Ly48, sialophorin) expression in the B-cell lineage of transgenic mice increases splenic B-cell number and survival. Proc. Natl. Acad. Sci. USA. 1995;92:626–630. doi: 10.1073/pnas.92.2.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misawa Y., Nagaoka H., Kimoto H., Ishii Y., Kitamura K., Tsunetsugu-Yokota Y., Shibuya M., Takemori T. CD43 expression in a B cell lymphoma, WEHI 231, reduces susceptibility to G1 arrest and extends survival in culture upon serum depletion. Eur. J. Immunol. 1996;26:2573–2581. doi: 10.1002/eji.1830261106. [DOI] [PubMed] [Google Scholar]
- Reiner S.L., Seder R.A. Dealing from the evolutionary pawnshophow lymphocytes make decisions. Immunity. 1999;11:1–10. doi: 10.1016/s1074-7613(00)80076-x. [DOI] [PubMed] [Google Scholar]
- Swain S.L., Hu H., Huston G. Class II-independent generation of CD4 memory T cells from effectors. Science. 1999;286:1381–1383. doi: 10.1126/science.286.5443.1381. [DOI] [PubMed] [Google Scholar]
- Bird J.J., Brown D.R., Mullen A.C., Moskowitz N.H., Mahowald M.A., Sider J.R., Gajewski T.F., Wang C.R., Reiner S.L. Helper T cell differentiation is controlled by the cell cycle. Immunity. 1998;9:229–237. doi: 10.1016/s1074-7613(00)80605-6. [DOI] [PubMed] [Google Scholar]
- Gett A.V., Hodgkin P.D. Cell division regulates the T cell cytokine repertoire, revealing a mechanism underlying immune class regulation. Proc. Natl. Acad. Sci. USA. 1998;95:9488–9493. doi: 10.1073/pnas.95.16.9488. [DOI] [PMC free article] [PubMed] [Google Scholar]