

Abstract
Chemokines provide signals for activation and recruitment of effector cells into sites of inflammation, acting via specific G protein–coupled receptors. However, in vitro data demonstrating the presence of multiple ligands for a given chemokine receptor, and often multiple receptors for a given chemokine, have led to concerns of biologic redundancy. Here we show that acute cardiac allograft rejection is accompanied by progressive intragraft production of the chemokines interferon (IFN)-γ–inducible protein of 10 kD (IP-10), monokine induced by IFN-γ (Mig), and IFN-inducible T cell α chemoattractant (I-TAC), and by infiltration of activated T cells bearing the corresponding chemokine receptor, CXCR3. We used three in vivo models to demonstrate a role for CXCR3 in the development of transplant rejection. First, CXCR3-deficient (CXCR3−/−) mice showed profound resistance to development of acute allograft rejection. Second, CXCR3−/− allograft recipients treated with a brief, subtherapeutic course of cyclosporin A maintained their allografts permanently and without evidence of chronic rejection. Third, CXCR+/+ mice treated with an anti-CXCR3 monoclonal antibody showed prolongation of allograft survival, even if begun after the onset of rejection. Taken in conjunction with our findings of CXCR3 expression in rejecting human cardiac allografts, we conclude that CXCR3 plays a key role in T cell activation, recruitment, and allograft destruction.
Keywords: receptors, chemokine, transplantation, rejection, CXC chemokine receptor 3
Introduction
Mononuclear cell recruitment, one of the hallmarks of allograft rejection 1 2, is thought, conceptually, to follow the now classic sequence of leukocyte rolling along vascular endothelium, followed by stimulus-dependent attachment, triggering and transmigration, and directed migration along a chemotactic gradient 3. However, the chemokine-dependent phase of leukocyte recruitment, especially the mechanisms leading to graft infiltration by host T cells, which are key to allograft rejection, is still the least understood, with little in vivo data available 4. Indeed, in contrast to most other areas of medicine, knowledge of the involvement of chemokine pathways is somewhat better established in humans than in experimental animals due to a paucity of appropriate tools for in vivo studies, leading to insufficient mechanistic and interventional studies 5 6.
Our preliminary data 7 from the blinded molecular and immunohistologic evaluation of endomyocardial biopsies from patients with cardiac allografts showed that acute rejection was associated with intragraft expression of CXC chemokine receptor 3 (CXCR3 [8, 9]) and its ligands, which include IFN-γ–inducible protein of 10 kD (IP-10; reference 10), monokine induced by IFN-γ (Mig [11, 12]), and IFN-inducible T cell α chemoattractant (I-TAC [9, 13]). These clinical findings have led to our serial analysis of intragraft chemokine and chemokine receptor expression using MHC fully mismatched mouse cardiac allografts, including expression of CXCR3 and its ligands during cardiac rejection. Our studies demonstrate that compared with control CXCR3+/+ mice, CXCR3−/− mice show significantly delayed, or in some cases an absence of, acute or chronic rejection, and that anti-CXCR3 mAb can reverse the course of developing rejection such that targeting of CXCR3 may prove to be of clinical therapeutic significance.
Materials and Methods
Generation of CXCR3−/− Mice.
A mouse 129 genomic library (Genome Systems) was screened with mouse CXCR3 cDNA 14. A 13-kb genomic fragment containing exon 2 of the gene was used to construct the targeting vector; a 2.5-kb HINDIII and XbalI fragment was deleted and replaced by PGK-neo. This mutant fragment was subcloned into pPNT for double selection with G418 and gancyclovir. The targeted vector was linearized and electroporated into ESI-1 embryonic stem cells (Genome Systems). The correctly targeted event was screened by Southern blotting. Two targeted cell lines were injected into blastocysts derived from C57BL/6 mice. Chimeric males were bred to BALB/c females to yield germline transmission of the targeted allele. Mice used in this study were backcrossed at least six generations onto the C57BL/6 strain.
Transplantation.
Cardiac allografting (15; n = 10/group) was performed using BALB/c (H-2d) donors and fully MHC-mismatched C57BL/6 (H-2b) recipients; B6 mice were CXCR3−/− (F6) versus littermate or commercial CXCR3+/+ mice (The Jackson Laboratory). Cyclosporine A (CsA; Sigma-Aldrich) was dissolved in olive oil and administered daily (10 mg/kg intraperitoneally [15]), and mAb therapy, using reagents described below, involved intraperitoneal injection of 500 μg every second day; in each case, therapy was begun at transplantation and stopped at 14 d after transplant. In additional experiments, the effects of mAb administration beginning at day 4 after transplant were also tested.
Immunopathology.
Histologic evaluation was undertaken using hematoxylin and eosin–stained paraffin sections. Infiltrating cells were detected by immunoperoxidase staining of cryostat sections with rat anti–mouse mAbs (BD PharMingen [16, 17]), plus quantitative morphometry 15. Preparation and use of the CXCR3 probe for in situ hybridization was performed as described 15.
Cellular and Molecular Studies.
Spleen cells were stimulated with Con A and cultured in IL-2 for 8–12 d to generate CXCR3+ T cell blasts 8 for use in chemotaxis and flow cytometry studies 18. MLR assays were performed as described 15. Northern blot analyses of T cells and T cell clones used CD3 mAb–activated spleen cells supplemented with IL-12 plus anti–IL-4 mAb for Th1 differentiation, or IL-4 plus anti–IL-12 and anti–IFN-γ mAbs for Th2 differentiation 15. Northern blots and RNase protection assays for cytokines, chemokines, and their receptors (BD PharMingen) were undertaken, with normalization to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or L32 genes 15 18. Rat anti–mouse CXCR3 mAb (4C4, IgM) was generated by immunization with CXCR3+ L1.2 cell transfectants, detection of binding to transfectants by flow cytometry, and screening for inhibition of chemotaxis of transfectants and T cell blasts to mouse recombinant CXCR3 ligands, as described 18.
Results and Discussion
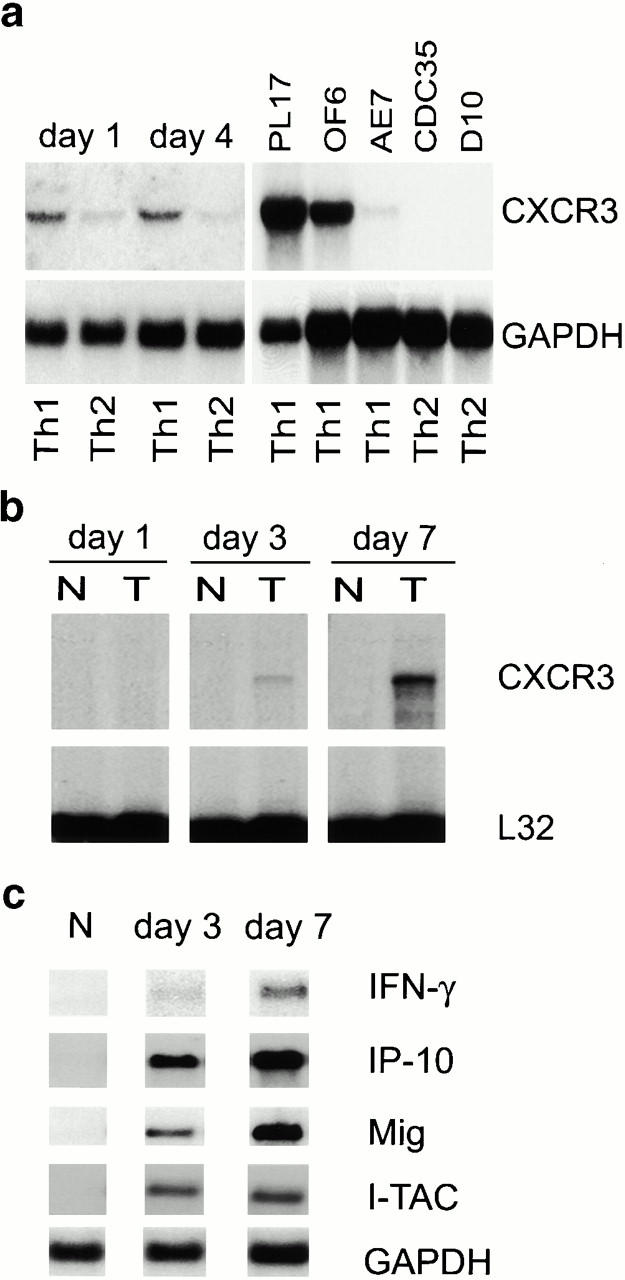
As allograft rejection is a T lymphocyte–dependent event 2, we undertook Northern blot analysis of CXCR3 expression by primary T cells and T cell clones in culture, and assessed CXCR3 involvement in allograft rejection in vivo. Freshly isolated mouse splenocytes lacked CXCR3 expression, but upon activation and culture in conditions that promote Th cell differentiation, Th1 cells expressed CXCR3 mRNA (Fig. 1 a). Established Th1 clones (PL17 and OF6), but not Th2 clones (CDC35 and D10), also expressed CXCR3 mRNA (Fig. 1 a). Heterotopic cardiac allografts in the fully MHC-mismatched BALB/C to C57BL/6 combination survive 7–8 d 17. RNase protection assays showed that normal hearts and isografts lacked CXCR3 mRNA, whereas allografts showed progressive CXCR3 mRNA expression, peaking at day 6 after transplant, just before end-stage rejection (Fig. 1 b). CXCR3 mRNA expression closely followed intragraft mRNA levels of the three known CXCR3 ligands IP-10, Mig, and I-TAC, plus IFN-γ (Fig. 1 c). Expression of CXCR3 mRNA was localized to infiltrating leukocytes by in situ hybridization (Fig. 1 d), and CXCR3+ mononuclear cell infiltration was confirmed by immunohistology (Fig. 1 d). These results show that activated T cells, especially Th1 cells, express CXCR3 and infiltrate cardiac allografts in conjunction with graft expression of the ligands IP-10, Mig, and I-TAC 19.
Figure 1.
Analysis of CXCR3 expression by activated T cells in vitro and during graft rejection. (a) Northern blot analysis of CXCR3 expression by primary T cells activated under polarizing conditions and by Th1 cell clones. (b) RNase protection assays of CXCR3 expression in serial cardiac transplants (T) showing rejection by day 7 but not corresponding native hearts (N). (c) Northern blots showing serial allograft but not native (N) heart expression of IFN-γ–induced CXCR3 ligands IP-10, Mig, and I-TAC. (d) Graft CXCR3 mRNA (blue, left) expression is restricted to focal round cells (sense control, bottom left); immunoperoxidase at high power (brown, right) shows CXCR3 protein expression by infiltrating mononuclear leukocytes (IgM control, bottom right). Bars, 100 and 10 μm, respectively.

To establish the role of CXCR3 expression in allograft rejection, we used homologous recombination to disrupt exon 2 of the X chromosome–linked 20 CXCR3 gene (Fig. 2 a). Mice heterologous and homozygous for the CXCR3 mutation were normal in appearance, growth, and fertility. CXCR3 deficiency was transmitted in a Mendelian fashion, as identified by Southern blot analysis of tail DNA (Fig. 2 b). T cell blasts from homozygous CXCR3-deficient (CXCR3−/−) mice lacked CXCR3 protein expression (Fig. 2 c), and failed to respond in chemotaxis assays to IP-10 (Fig. 2 d) or Mig, indicating that CXCR3 is the sole functional receptor for these chemokines. With relevance to subsequent in vivo studies, T cell blasts from CXCR3+/+ but not CXCR3−/− mice were also labeled using the CXCR3-specific mAb (Fig. 2 e).
Figure 2.
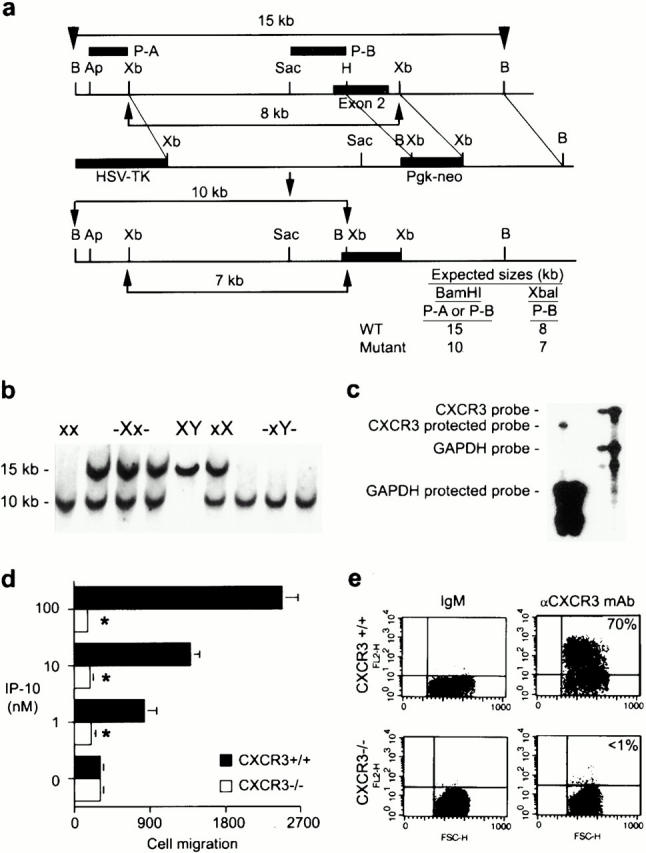
Targeted disruption of the murine CXCR3 gene. (a) Wild-type (WT) allele, targeted vector, and mutated allele of mouse CXCR3 gene. The wild-type gene contains exon 2 of the receptor (black rectangle) whereas in the mutant, most of exon 2 (800 bp of coding region) was deleted and replaced by neomycin resistance gene driven by the phosphoglycerate kinase (Pgk) promoter. (b) Southern blot analysis of tail DNA from littermates. As a BamHI site was introduced with the neo gene, a 10-kb mutant band (x) instead of a 15-kb wild-type band (X) was observed when DNA was digested by BamHI and hybridized with probe A or probe B. (c) CXCR3 expression was assessed by RNase protection assay in wild-type and homozygote littermate animals. (d) The lack of chemotaxis in vitro to IP-10 by T cell blasts from CXCR3−/− versus CXCR3+/+ mice. Asterisks indicate significantly reduced chemotaxis compared with wild-type response (P < 0.001, Mann-Whitney U test). (e) The lack of CXCR3 membrane expression by T cell blasts from CXCR3−/− versus CXCR3+/+ mice (flow cytometry).
CXCR3−/− mice had normal T cell–proliferative responses upon mitogen stimulation (Fig. 3 a), but decreased MLR responses (Fig. 3 b). The addition of anti-CXCR3 mAb reduced the MLR of wild-type mice (Fig. 3 c), but had no effect on mitogen responses (data not shown). In vivo, CXCR3−/− mice showed profoundly decreased alloresponses (Fig. 3 d). Whereas CXCR3+/+ mice rejected BALB/c cardiac allografts within 1 wk, CXCR3−/− mice accepted corresponding allografts for a mean of 58 ± 3 d (P < 0.001); no benefit was seen when hearts from CXCR3−/− mice were used as donor organs in BALB/c mice. Targeting of CXCR3 resulted in strong synergy with the immunosuppressive agent, CsA, consistent with data that CsA does not affect CXCR3 ligand induction 15 21. 2 wk of therapy with CsA (10 mg/kg) prolonged allograft survival by only 3 d in wild-type recipients, but induced permanent engraftment (>100 d, P < 0.001) in CXCR3−/− mice (Fig. 3 d); evaluation of the latter allografts harvested at day 100 after transplant showed normal morphology, with a lack of graft leukocyte infiltration, myocardial injury, or evidence of transplant arteriosclerosis (data not shown). Use of the CXCR3 mAb in CXCR3+/+ mice significantly prolonged allograft survival when used from the time of transplantation, and was also effective when commenced once rejection had begun, i.e., at day 4 after transplant (Fig. 3 e). These findings demonstrate a profound effect of targeting CXCR3 on the development of cardiac allograft rejection.
Figure 3.
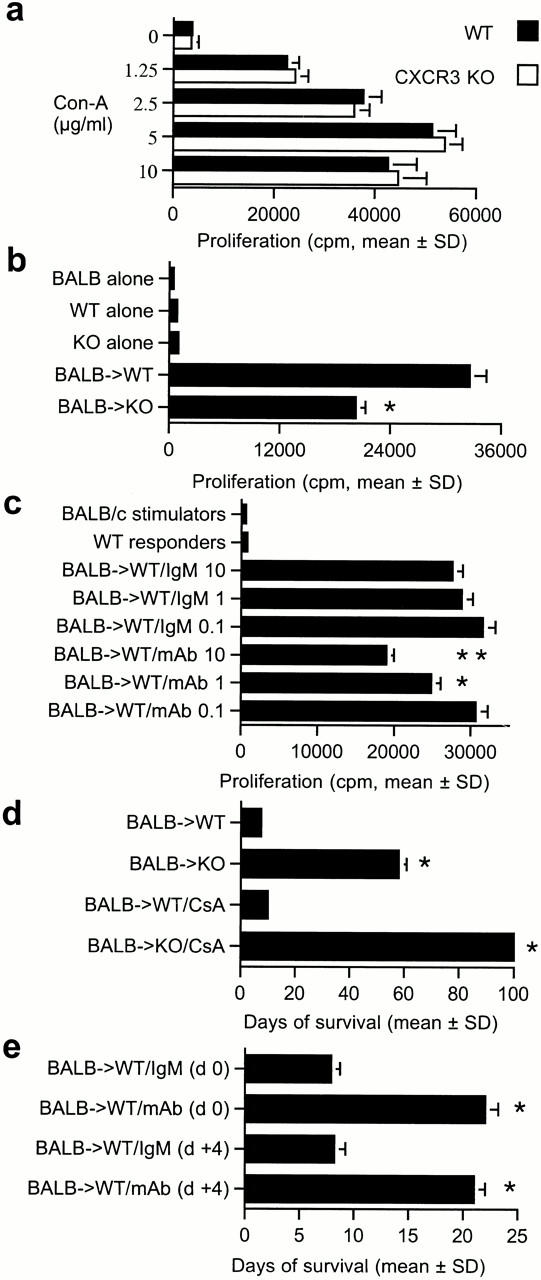
In vitro and in vivo effects of CXCR3 targeting. (a) CXCR3−/− knockout (KO) mice have (a) normal mitogen responses but (b) diminished alloreactivity (MLR); bars show the mean ± SD for 12 wells. Asterisks indicate significantly decreased proliferation compared with wild-type (WT) responses (P < 0.001, Mann-Whitney U test). (c) Dose-dependent inhibition of MLR by anti-CXCR3 mAb; bars indicate the mean ± SD for 12 wells, mAb final concentrations expressed in μg/ml. Asterisks indicate significantly decreased proliferation using CXCR3 mAb compared with cells treated with IgM (*P < 0.005, **P < 0.001, Mann-Whitney U test). (d) Prolongation of cardiac allograft survival in knockout versus wild-type recipients, and permanent engraftment when knockout recipients received 14 d of CsA. Bars indicate the mean ± SD for six mice. Asterisks indicate significantly increased prolongation of allograft survival compared with the respective control group (P < 0.001, Mann-Whitney U test). (e) Anti-CXCR3 mAb prolongs cardiac graft survival whether begun pretransplant (day 0) or once rejection has begun (day 4); bars indicate the mean ± SD for six mice. Asterisks indicate significantly increased prolongation of allograft survival compared with the respective control group (P < 0.001, Mann-Whitney U test).
Histologic analysis of allografts harvested at day 5 after transplant showed that in contrast to control grafts, grafts in CXCR3−/− or CXCR3 mAb–treated CXCR3+/+ mice were well preserved (Fig. 4, a–d). Compared with control grafts, allografts in CXCR3−/− recipients had significantly reduced numbers of CD4+ and CD8+ T cells and macrophages, and no IL-2R+ (CD25+) cells indicative of immune activation (Fig. 4 e). Allografts in CXCR3−/− recipients contained decreased IFN-γ mRNA (Fig. 4 f), and decreased expression of the chemokines macrophage inflammatory protein (MIP)-1β and regulated upon activation, normal T cell expressed and secreted (RANTES; Fig. 4 g). Consistent with the latter findings, allografts in CXCR3−/− mice showed decreased expression of the corresponding chemokine receptors CCR1, CCR2, and CCR5, produced by T cells and macrophages (Fig. 4 h).
Figure 4.
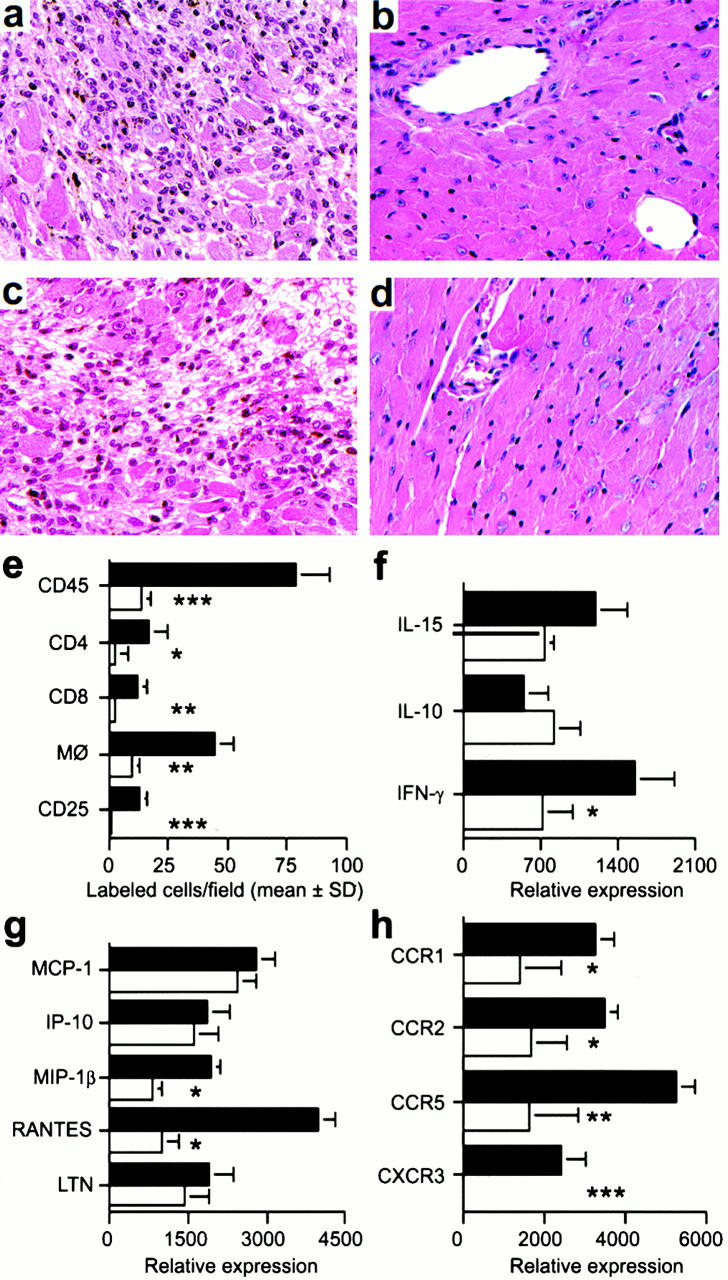
Mechanisms underlying beneficial effects of targeting CXCR3 in allograft recipients. (a) Histology showing acute rejection with extensive mononuclear cell infiltration and myocyte necrosis in CXCR3+/+ recipients (day 7 after transplant). (b) The lack of histologic evidence of rejection in CXCR3−/− recipients (day 7). (c) CXCR3+/+ allograft recipients treated with IgM underwent acute rejection by day 7. (d) Normal appearance of allografts in CXCR3+/+ allograft recipients treated with anti-CXCR3 mAb (day 7). (a–d) Original magnification: ×125. (e) Immunohistology showed significant reduction in recruitment of intragraft CD45+ cells (all leukocytes), CD4 and CD8 T cell subsets, macrophages, and IL-2R+ (CD25+) cells in CXCR3−/− (white bars) versus CXCR3+/+ recipients (black bars) at day 7 after transplant. Data (mean ± SD) are from counting 20 consecutive fields/graft and 3 grafts/group; asterisks indicate significantly reduced cell numbers versus controls (*P < 0.05, **P < 0.01, ***P < 0.005). (f, g, and h) The results of RNase protection assays of the same set of grafts (mean ± SD, four to six grafts/bar), with statistical analysis by the Mann-Whitney U test. CXCR3−/− recipients show decreased mRNA levels of intragraft: (f) IFN-γ (*P < 0.05); (g) MIP-1β and RANTES (both *P < 0.005); and (h) the chemokine receptors CCR1 (*P < 0.05), CCR2 (*P < 0.05), CCR5 (**P < 0.01), and CXCR3 (***P < 0.005). MCP-1, monocyte chemoattractant protein 1; LTN, lymphotactin.
In conclusion, targeting of CXCR3 markedly decreases the tempo and severity of allograft rejection in vivo. This effect is associated with significantly decreased intragraft accumulation of activated T cells producing IFN-γ and consequent impairment of chemokine-dependent recruitment of multiple effector cell types, suggesting a rationale for targeting of CXCR3, in synergy with conventional immunosuppression, in clinical transplantation, and potentially in the management of acute allograft rejection.
Acknowledgments
We thank N. Shemmeri for immunopathology and D. Soler and D. Picarella for production of CXCR3 mAb.
This work was supported by grants from the National Institutes of Health to W.W Hancock and C.G. Gerard.
References
- Hancock W.W. Analysis of intragraft effector mechanisms associated with human renal allograft rejectionimmunohistological studies using monoclonal antibodies. Immunol. Rev. 1984;77:61–84. doi: 10.1111/j.1600-065x.1984.tb00718.x. [DOI] [PubMed] [Google Scholar]
- Hall B.M., Dorsch S.E. Cells mediating allograft rejection. Immunol. Rev. 1984;77:31–59. doi: 10.1111/j.1600-065x.1984.tb00717.x. [DOI] [PubMed] [Google Scholar]
- Springer T.A. Traffic signals for lymphocyte recirculation and leukocyte emigrationthe multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- Hancock W.W., Gao W., Faia K.L., Csizmadia V. Chemokines and their receptors in allograft rejection. Curr. Opin. Immunol. 2000;12:511–516. doi: 10.1016/s0952-7915(00)00130-8. [DOI] [PubMed] [Google Scholar]
- Murphy P.M., Baggiolini M., Charo I.F., Hebert C.A., Horuk R., Matsushima K., Miller L.H., Oppenheim J.J., Power C.A. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol. Rev. 2000;52:145–176. [PubMed] [Google Scholar]
- Zlotnik A., Yoshie O. Chemokinesa new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- Exeni A., Melter M., Fang J., Hancock W.W., Ganz P., Briscoe D. Expression of chemokine receptors in human cardiac allografts Transplantation. 69 2000. 453a(Abstr [Google Scholar]
- Loetscher M., Gerber B., Loetscher P., Jones S.A., Piali L., Clark-Lewis I., Baggiolini M., Moser B. Chemokine receptor specific for IP-10 and Migstructure, function, and expression in activated T-lymphocytes. J. Exp. Med. 1996;184:963–969. doi: 10.1084/jem.184.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole K.E., Strick C.A., Paradis T.J., Ogborne K.T., Loetscher M., Gladue R.P., Lin W., Boyd J.G., Moser B., Wood D.E. Interferon-inducible T cell alpha chemoattractant (I-TAC)a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J. Exp. Med. 1998;187:2009–2021. doi: 10.1084/jem.187.12.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luster A.D., Ravetch J.V. Biochemical characterization of a gamma interferon–inducible cytokine (IP-10) J. Exp. Med. 1987;166:1084–1097. doi: 10.1084/jem.166.4.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber J.M. HuMiga new human member of the chemokine family of cytokines. Biochem. Biophys. Res. Commun. 1993;192:223–230. doi: 10.1006/bbrc.1993.1403. [DOI] [PubMed] [Google Scholar]
- Liao F., Rabin R.L., Yannelli J.R., Koniaris L.G., Vanguri P., Farber J.M. Human Mig chemokinebiochemical and functional characterization. J. Exp. Med. 1995;182:1301–1314. doi: 10.1084/jem.182.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rani M.R.S., Foster G.R., Leung S., Leaman D., Stark G.R., Ransohoff R.M. Characterization of beta-R1, a gene that is selectively induced by interferon beta (IFN-beta) compared with IFN-alpha. J. Biol. Chem. 1996;271:22878–22884. doi: 10.1074/jbc.271.37.22878. [DOI] [PubMed] [Google Scholar]
- Lu B., Humbles A., Bota D., Gerard C., Moser B., Soler D., Luster A.D., Gerard N.P. Structure and function of the murine chemokine receptor CXCR3. Eur. J. Immunol. 1999;29:3804–3812. doi: 10.1002/(SICI)1521-4141(199911)29:11<3804::AID-IMMU3804>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Gao W., Topham P.S., King J.A., Smiley S.T., Csizmadia V., Lu B., Gerard C.J., Hancock W.W. Targeting of the chemokine receptor CCR1 suppresses development of acute and chronic cardiac allograft rejection. J. Clin. Invest. 2000;105:35–44. doi: 10.1172/JCI8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock W.W., Sayegh M.H., Zheng X.G., Peach R., Linsley P.S., Turka L.A. Costimulatory function and expression of CD40 ligand, CD80, and CD86 in vascularized murine cardiac allograft rejection. Proc. Natl. Acad. Sci. USA. 1996;93:13967–13972. doi: 10.1073/pnas.93.24.13967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock W.W., Buelow R., Sayegh M.H., Turka L.A. Antibody-induced transplant arteriosclerosis is prevented by graft expression of anti-oxidant and anti-apoptotic genes. Nat. Med. 1998;4:1392–1396. doi: 10.1038/3982. [DOI] [PubMed] [Google Scholar]
- Topham P.S., Csizmadia V., Soler D., Hines D., Gerard C.J., Salant D.J., Hancock W.W. Lack of chemokine receptor CCR1 enhances Th1 responses and glomerular injury during nephrotoxic nephritis. J. Clin. Invest. 1999;104:1549–1557. doi: 10.1172/JCI7707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A., Morita K., Engeman T.M., Koga S., Vapnek E.M., Hobart M.G., Fairchild R.L. Early expression of interferon-gamma inducible protein 10 and monokine induced by interferon-gamma in cardiac allografts is mediated by CD8+ T cells. Transplantation. 2000;69:1147–1155. doi: 10.1097/00007890-200003270-00020. [DOI] [PubMed] [Google Scholar]
- Soto H., Wang W., Strieter R.M., Copeland N.G., Gilbert D.J., Jenkins N.A., Hedrick J., Zlotnik A. The CC chemokine 6Ckine binds the CXC chemokine receptor CXCR3. Proc. Natl. Acad. Sci. USA. 1998;95:8205–8210. doi: 10.1073/pnas.95.14.8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazanet M.M., Neote K., Hughes C.C.W. Expression of IFN-inducible T cell alpha chemoattractant by human endothelial cells is cyclosporin A-resistant and promotes T cell adhesionimplications for cyclosporin A-resistant immune inflammation. J. Immunol. 2000;164:5383–5388. doi: 10.4049/jimmunol.164.10.5383. [DOI] [PubMed] [Google Scholar]