

Abstract
The importance of mast cells in the development of the allergen-induced airway hyperreactivity and inflammation associated with asthma remains controversial. We found that genetically mast cell–deficient WBB6F1-W/Wv mice that were sensitized to ovalbumin (OVA) without adjuvant, then challenged repetitively with antigen intranasally, exhibited much weaker responses in terms of bronchial hyperreactivity to aerosolized methacholine, lung tissue eosinophil infiltration, and numbers of proliferating cells within the airway epithelium than did identically treated WBB6F1-+/+ normal mice. However, W/Wv mice that had undergone selective reconstitution of tissue mast cells with in vitro–derived mast cells of congenic +/+ mouse origin exhibited airway responses that were very similar to those of the +/+ mice. By contrast, W/Wv mice that were sensitized with OVA emulsified in alum and challenged with aerosolized OVA exhibited levels of airway hyperreactivity and lung tissue eosinophil infiltration that were similar to those of the corresponding +/+ mice. Nevertheless, these W/Wv mice exhibited significantly fewer proliferating cells within the airway epithelium than did identically treated +/+ mice. These results show that, depending on the “asthma model” investigated, mast cells can either have a critical role in, or not be essential for, multiple features of allergic airway responses in mice.
Keywords: eosinophil, epithelium, hyperresponsiveness, tissue remodeling, T lymphocyte
Introduction
Asthma is an increasingly prevalent and often severe disease characterized by chronic inflammation of the airways and heightened airway reactivity to stimuli of bronchoconstriction 1 2 3 4 5. Patients can exhibit the disorder when exposed to very small amounts of antigen, indicating that local amplification mechanisms can contribute significantly to inflammation in this setting 2 3 4 5 6 7. Tissue mast cells can be activated to release potent mediators of inflammation by antibody-dependent mechanisms, and can respond to very low doses of specific antigen 5 7. Although mast cells thus would appear to be well suited to amplify local inflammatory responses to antigen challenge, there is considerable debate about the importance of these cells in the expression of the features of chronic asthma 7 8 9 10 11 12 13 14 15 16 17 18.
For example, there is no question that features of chronic asthma, including airway hyperreactivity to methacholine and eosinophil infiltration (a characteristic of the chronic inflammation associated with asthma [2–7]), can be elicited in mouse models in the absence of IgE antibodies 8 12, all antibodies 9 10 11, or mast cells 14 15 16 17 18. However, it is also true that airway hyperreactivity can be induced in mice by IgE- 13 19 and mast cell–dependent mechanisms 19.
Can these apparently discordant findings be reconciled? We have hypothesized that mast cells may be critical in asthma because of their ability to amplify inflammation through the release of diverse proinflammatory mediators, and have proposed that this function may be especially important in settings that otherwise may favor the development or expression of relatively weak immunological responses 7. It is well known that many patients with asthma can express dramatic pathophysiological responses to challenge with low doses of specific antigen 20 21. However, many protocols used to induce “asthma models” in mice use sensitization with antigen mixed with an artificial adjuvant such as alum; this can promote a strong “nonspecific” antibody response, or have other effects that may influence features of the asthma model. Therefore, we used genetically mast cell–deficient and congenic normal mice to investigate the role of mast cells in two models of asthma: a conventional model using sensitization with alum, and a model that omitted this adjuvant.
Materials and Methods
Mice.
Genetically mast cell–deficient WBB6F1-KitW/KitW-v (W/Wv) mice and the congenic normal WBB6F1-+/+ (+/+) mice were purchased from The Jackson Laboratory. Adult W/Wv mice ordinarily contain <1.0% of the number of dermal mast cells present in the skin of the congenic normal (+/+) mice, and have no detectable mature mast cells in the respiratory system or other anatomical sites 22 23 24. Mice were kept in community cages at light periods of 12 h, and were fed water and mouse chow ad libitum. Age-matched animals were used in all individual experiments, including those comparing groups of W/Wv mice, congenic +/+ mice, and mast cell–reconstituted W/Wv mice (see below). All animal care and experimentation was conducted in accord with current National Institutes of Health and Stanford University Institutional Animal Care and Use Committee guidelines.
Mast Cell Reconstitution.
Selective reconstitution of mast cells in mast cell–deficient WBB6F1-W/Wvmice (The Jackson Laboratory) was carried out by the method described by Martin et al. 19, with slight modification. Suspended bone marrow cells from WBB6F1-+/+ normal mice were cultured in WEHI-3–conditioned medium (containing IL-3) for 4–5 wk, at which time the cell populations were composed of >95% immature mast cells, as assessed by staining with toluidine blue. 5 × 106 bone marrow–derived cultured mast cells (BMCMCs) were infused via the tail vein into each W/Wv mouse, and the recipients (BMCMC→W/Wv mice) were studied at least 28 wk later. Reconstitution of W/Wv mice with BMCMCs did not repair their anemia (as determined by hematocrit performed shortly before the date of study), confirming the selectivity of the mast cell reconstitution 19 23 24. OVA-sensitized, OVA-challenged BMCMC→W/Wv mice had 4.2 ± 2.9 mast cells/mm2 of peritracheal tissue (versus none or 19.2 ± 3.0 for W/Wv or +/+ mice, respectively) and 9.0 ± 2.1 mast cells/cm2 of lung tissue (versus none or 0.4 ± 0.1 for W/Wv or +/+ mice, respectively).
Immunization and Airway Challenge with Antigen.
Some mice were immunized by intraperitoneal injection of 10 μg OVA (Sigma-Aldrich) in 0.1 ml saline on each of seven alternate days. 40 d after the beginning of the sensitization period, groups of mice (8–10 mice per group per experiment) received the first of three intranasal challenges, each 3 d apart, of either 20 μl of saline (as a control) or 20 μl of saline containing 2, 20, or 200 μg of OVA. Mice were assessed for airway hyperresponsiveness (AHR) and airway inflammation 24 h after the last intranasal challenge. To induce an apparently mast cell–independent model of asthma, other groups of mice were immunized by intraperitoneal injection of 20 μg of OVA emulsified in 2.25 mg alum (Imject®Alum; Pierce Chemical Co.) in a total volume of 200 μl on days 1 and 14 15. These mice were challenged by the airways with OVA (1% in saline) for 20 min on days 28, 29, and 30 by ultrasonic nebulization 15, and were assessed 24 h after the last OVA exposure for AHR and airway inflammation.
Measurement of Airway Reactivity.
24 h after the final OVA challenge, bronchial reactivity to aerosolized methacholine was measured using the Buxco whole body plethysmograph (Buxco Electronics) as described by Hamelmann et al. 25. In brief, unrestrained conscious mice were placed in whole body plethysmographic chambers and, after 5 min of stabilization, dose response curves to aerosolized methacholine were generated. Increasing concentrations of methacholine were aerosolized for 3 min each, and mean airway bronchoconstriction readings, as assessed by Enhanced Respiratory Pause (PenH), were obtained over 10-min periods. PenH can be conceptualized as the phase shift of the thoracic flow and the nasal flow curves; increased phase shift correlates with increased respiratory system resistance. PenH is calculated by the formula PenH = (Te/RT −1) × PEF/PIF, where Te is expiratory time, RT is relaxation time, PEF is peak expiratory flow, and PIF is peak inspiratory flow.
Histology.
Mice were killed by CO2 inhalation, then lungs were inflated with 1 ml of air and removed. The right lung of each mouse was fixed in 10% neutral buffered formalin, whereas the left lung and trachea were fixed in Carnoy's fixative. For analysis of lung eosinophils, formalin-fixed tissues were embedded in paraffin, and 5-μm sections were stained with Congo red 26. Total numbers of eosinophils per lung tissue section and perivascular eosinophils were counted at ×400 magnification, and numbers were expressed per mm2. For analysis of lung mast cells, 5-μm sections from Carnoy's or formalin-fixed tissues were stained with Alcian blue/safranin.
Detection of 5-Bromo-2′-Deoxyuridine+ Cells.
In some experiments, mice received (+)-5-bromo-2′-deoxyuridine (BrdU; 100 mg/kg) 1 h before sacrifice 27. BrdU+ cells were detected by standard immunohistochemical procedures using an anti-BrdU alkaline phosphatase F(ab′)2 fragment (diluted to 0.3 U/ml in blocking buffer for 1 h; Boehringer); tissues were developed with Fast Red for 15 min, then counterstained with Mayer's hematoxylin for 20 min. BrdU+ cells within the tracheal or bronchiolar epithelium were counted, and numbers were expressed per mm of epithelium.
Measurement of OVA-specific and Total IgE and IgG1 Antibodies.
Sera were obtained 1 d before antigen challenge in mice that had been sensitized with OVA alone or with OVA emulsified in alum (on days 39 or 27 after sensitization, respectively). OVA-specific IgE and IgG1 levels were measured by ELISA according to the method described by Spergel et al. 28. Total serum IgE and IgG1 levels were measured by ELISA (BD PharMingen).
Statistical Analyses.
Differences in responses in various groups of mice were tested for significance by analysis of variance (ANOVA) for AHR, or by the unpaired Student's t test (two tailed) for all other features of the models. After determining that responses of individual groups of mice of specific genotype and sensitization and challenge protocol did not differ significantly in replicate experiments, the results were pooled for statistical analyses and for presentation in Fig. 1, Fig. 2, and Fig. 4. Note that, in mice of a given W/Wv or +/+ genotype, there were no significant age-related differences in any tested features of the response (AHR, eosinophil or BrdU+ cell numbers, or IgE or IgG1 responses) in the older mice that were used for experiments that included a BMCMC→W/Wv group, compared with the younger mice that were used in the other experiments.
Figure 1.

Airway responses to aerosolized methacholine. (A) OVA-sensitized W/Wv mice (open symbols) that were challenged three times intranasally with either saline (⋄) or with 2 μg (□), 20 μg (○), or 200 μg (▵) of OVA were significantly less responsive to aerosolized methacholine than were identically treated WBB6F1-+/+ control mice (filled symbols; *P < 0.05). Significant differences from mice of the same genotype that were challenged with saline (⋄, W/Wv; ♦, +/+) are indicated by crosses (P < 0.05). (B) Naive W/Wv mice, +/+ mice, or W/Wvmice that were reconstituted with BMCMCs (BMCMC→W/Wv mice) were tested with aerosolized methacholine before (○, W/Wv; •, +/+; ▪, BMCMC→W/Wv) or after (▵, W/Wv; ▴, +/+; ★, BMCMC→W/Wv) three intranasal challenges with saline. Note the different PenH scale in B, compared with those in A, C, and D. (C) Airway responses to aerosolized methacholine in OVA-challenged W/Wv mice that were sensitized with OVA emulsified in alum (○) were not significantly different from those of identically treated WBB6F1-+/+ mice (•). Significant differences from mice of the same genotype challenged with saline (⋄, W/Wv; ♦, +/+) are indicated by crosses (P < 0.05). (D) W/Wv mice that were reconstituted systemically with BMCMCs (BMCMC→W/Wv) 6 mo before OVA sensitization and challenge (▾) exhibited PenH responses to aerosolized methacholine that were not significantly different from those of identically treated WBB6F1-+/+ mice (•), whereas identically treated mast cell–deficient W/Wv mice (○) had significantly lower PenH responses (*P < 0.05 versus +/+ or BMCMC→W/Wv groups). Significant differences from mice of the same type challenged with saline (⋄, W/Wv; ♦, +/+; ★, BMCMC→W/Wv are indicated by crosses (P < 0.05) ||: P < 0.05 versus values for saline-challenged W/Wv or +/+ mice. Standard errors in A–D are not shown, but were ≤12.6, 11.9, 11.7, or 18% of the mean values in A (n = 22 per group), B (n = 15 per group), C (n = 10 per group), and D (n = 10 per group), respectively. QED, every other day.
Figure 2.
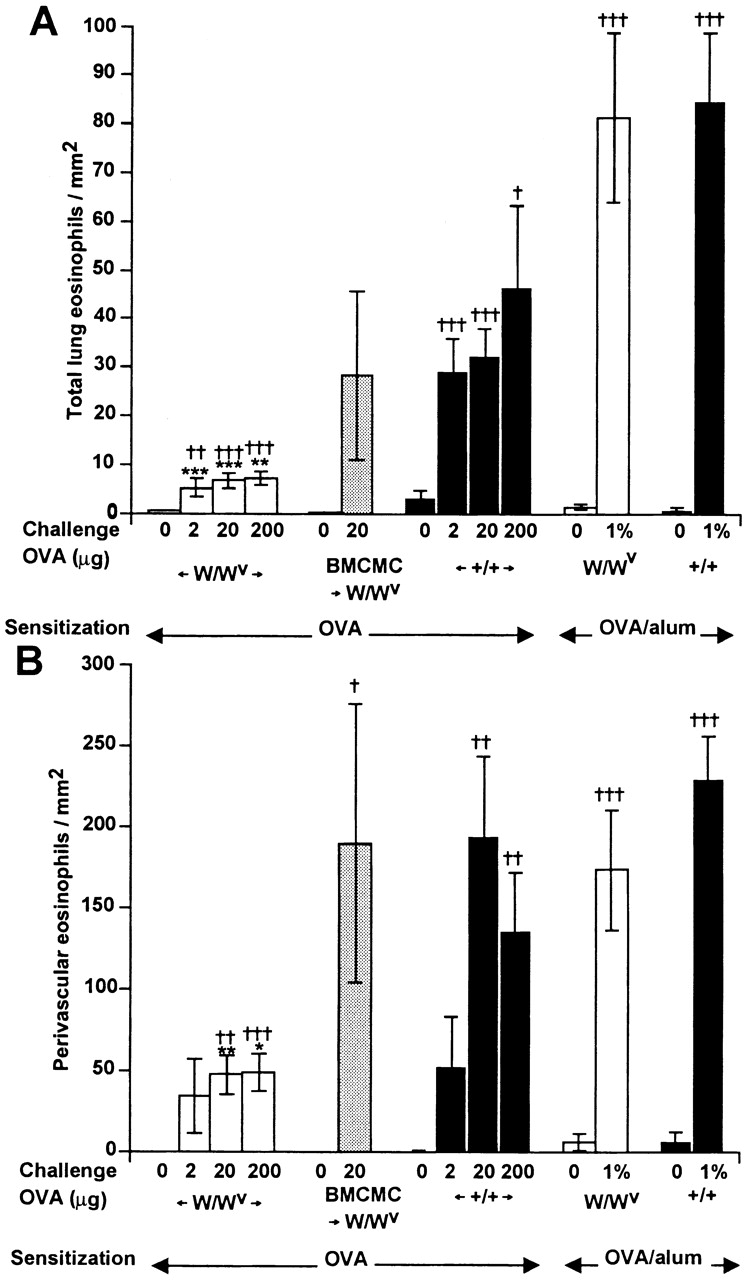
Lung tissue eosinophil infiltration. (A) Total numbers of eosinophils/mm2 of lung tissue. (B) Eosinophils/mm2 of perivascular tissue. Statistical significance was determined using the unpaired Student's t test (two tailed). *P < 0.05, ** P < 0.01, and *** P < 0.001 versus values for identically treated WBB6F1-+/+ mice; † P < 0.05, †† P < 0.01, and ††† P < 0.001 versus values for identically sensitized but saline-challenged mice (shown as mice challenged with “0” OVA) of the same genotype. Numbers of mice per group: n = 10 for BMCMC →W/Wv and n = 10 for W/Wvor +/+ in the OVA/alum groups, and n = 24–32 for W/Wv or +/+ in the OVA groups.
Figure 4.
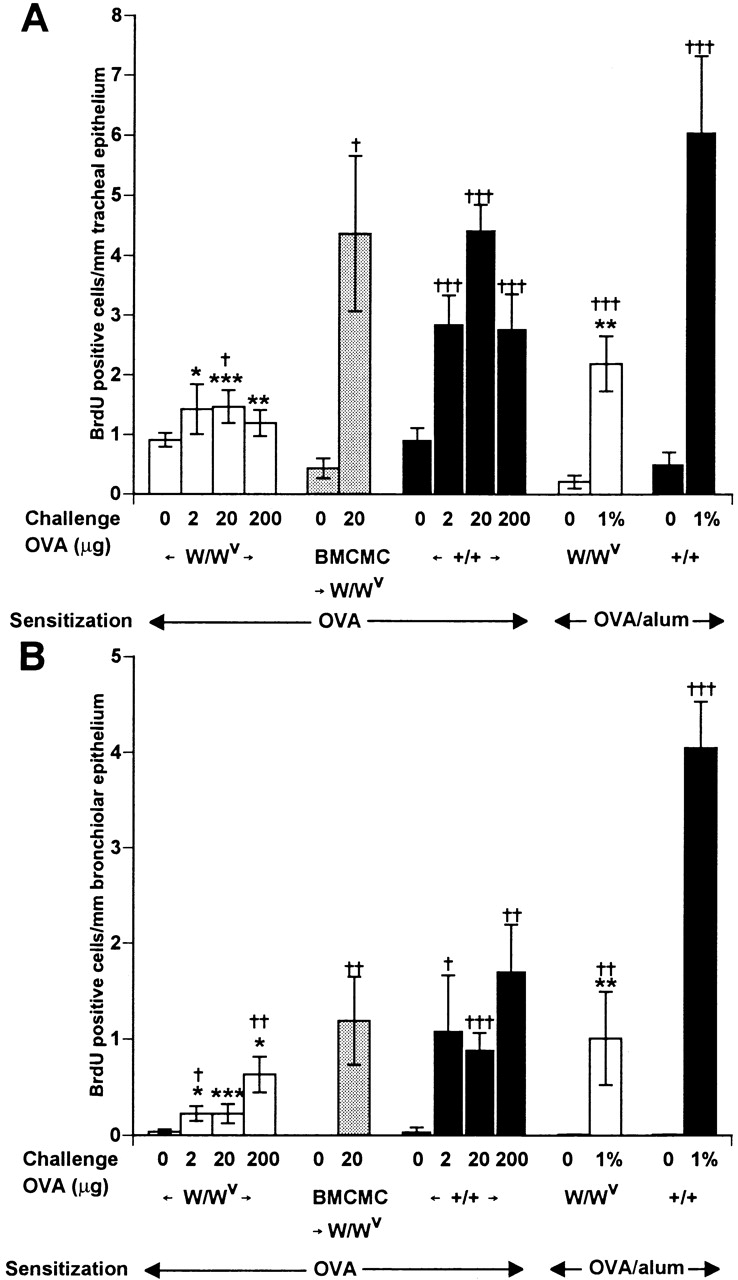
BrdU+ cells within airway epithelium. (A) BrdU+ cells/mm of tracheal epithelium. (B) BrdU+ cells/mm of bronchiolar epithelium. 10 bronchioles per section were examined and the average number of BrdU+ cells within the epithelial layer was calculated. *P < 0.05, **P < 0.01, and ***P < 0.001 versus values for identically treated WBB6F1-+/+ mice; † P < 0.05, †† P < 0.01, and ††† P < 0.001 versus values for identically sensitized but saline-challenged mice (shown as mice challenged with “0” OVA) of the same genotype. Numbers of mice per group are as in Fig. 2.
Results and Discussion
Mast Cells Can Enhance Allergen-induced Airway Reactivity to Methacholine.
Fig. 1 A shows airway responses to aerosolized methacholine in genetically mast cell–deficient WBB6F1-W/Wv mice and congenic normal (+/+) mice that were sensitized with OVA without adjuvant and then challenged three times intranasally with either saline or 2, 20, or 200 μg of OVA, each 3 d apart. In each of four separate experiments, W/Wv mice were significantly less responsive to aerosolized methacholine, as assessed by PenH readings, than were the identically treated congenic WBB6F1-+/+ mice.
Note that the lower sensitivity to methacholine of W/Wv mice in comparison to the congenic normal mice was detectable even in OVA-sensitized mice that were challenged intranasally with saline rather than antigen (Fig. 1 A). We reported previously that the “baseline” airway responses to intravenous methacholine in anesthetized, tracheotomized naive W/Wv mice were very similar to those in the congenic +/+ mice 19. In this study, assessment of baseline PenH responses to aerosolized methacholine in nonanesthetized mice indicated that naive W/Wv and congenic +/+ mice had virtually identical airway responsiveness to methacholine (Fig. 1 B). Moreover, these responses were altered minimally, if at all, in naive mice that had been challenged intranasally with saline three times before methacholine challenge (Fig. 1 B).
Taken together, the results shown in Fig. 1A and Fig. B indicate that in +/+ but not in W/Wv mice, sensitization with OVA resulted in an enhancement of AHR to methacholine that was detectable ∼1 mo after the end of the period of antigen sensitization. Although the explanation for this interesting finding is not yet clear, the underlying mechanisms appear to be at least in part mast cell dependent (see below).
In contrast to the results obtained in mice that were sensitized to OVA without alum and then challenged intranasally, W/Wv mice that were sensitized with 20 μg of OVA emulsified in alum and then challenged with a 1% OVA solution by aerosol for 20 min on each of three consecutive days were not significantly less responsive to aerosolized methacholine than were the identically treated WBB6F1-+/+ wild-type mice (Fig. 1 C). These results are thus in accord with those reported by Takeda et al.(15), who previously investigated this model of sensitization and challenge in W/Wv and +/+ mice. Moreover, in contrast to the results we obtained in the other model of sensitization and challenge, the responses to methacholine in W/Wv or +/+ mice that had been sensitized with OVA in alum and then challenged by aerosol with saline were not significantly different (Fig. 1 C). Presumably, the lack of significant AHR to methacholine in the saline-challenged, OVA-sensitized +/+ mice shown in Fig. 1 C (in contrast to the saline-challenged OVA-sensitized +/+ mice shown in Fig. 1 A) reflects the differences in the details of sensitization and/or the different routes of saline challenge (intranasal versus aerosol) in the two models of OVA sensitization and challenge.
To assess the extent to which the differences between the responses of W/Wv and +/+ mice shown in Fig. 1 A represented consequences of the mast cell deficiency of the W/Wvmice, as opposed to other effects of their mutations at c-kit, we repeated the experiments in three groups of age-matched mice: mast cell–deficient W/Wv mice, congenic normal (+/+) mice, and W/Wv mice that had undergone selective repair of their mast cell deficiency (BMCMC→W/Wv mice). We found that mast cell–deficient W/Wv mice that were selectively reconstituted with BMCMCs ∼6 mo before OVA sensitization and challenge were significantly more responsive to aerosolized methacholine than were the identically treated W/Wv mice; indeed, these mast cell–reconstituted W/Wv mice gave responses to OVA that were statistically indistinguishable from those of the identically treated WBB6F1-+/+ mice (Fig. 1 D).
Mast Cells Contribute to Persistent AHR to Methacholine in OVA-sensitized Mice Challenged with Saline Instead of Allergen.
In confirmation of the results shown in Fig. 1 A, we found that OVA-sensitized W/Wv mice that were challenged with saline rather than antigen were substantially less responsive to methacholine than were the identically treated +/+ mice (Fig. 1 D). However, OVA-sensitized, saline-challenged BMCMC→W/Wv mice exhibited responses to methacholine that were intermediate between, and significantly different from those of either the mast cell–deficient W/Wv mice or the congenic normal +/+ mice.
It is of interest that the AHR to methacholine observed in OVA-sensitized, OVA-challenged +/+ mice or BMCMC→W/Wv mice was statistically indistinguishable, whereas the AHR to methacholine detected in OVA-sensitized, saline-challenged BMCMC→W/Wv mice was significantly less than that observed in the corresponding +/+ mice. There are at least two possible explanations for the latter result: (a) the AHR to methacholine observed in OVA-sensitized, saline-challenged +/+ mice is only partially dependent on mast cells, with the remaining component(s) reflecting mast cell–independent consequences of the c-kit mutations in W/Wv mice; and (b) the phenomenon is entirely mast cell dependent, but the weaker expression of the response in the BMCMC→W/Wv mice reflects differences between these mice and the +/+ mice in the numbers, anatomical distribution, phenotype, or function of the adoptively transferred mast cells versus the native mast cell populations of +/+ mice. Given the practical constraints of this model system, distinguishing between these two alternatives may not be easy.
It will be important to elucidate the mechanisms that contribute to persistent AHR after allergen sensitization. As described below (see Fig. 2), few or no eosinophils were detectable in the airways of OVA-sensitized +/+ mice that had been challenged with saline rather than OVA. However, it is possible that the 2-wk period of sensitization by intraperitoneal injection of OVA every other day induced airway inflammation that had resolved (at least as could be assessed by standard histological examination) by the time of saline challenge ∼1 mo later, but that this transient inflammation had long-lasting effects on AHR. We have already reported that systemic IgE-dependent activation of mast cells by a single intravenous administration of anti-IgE to naive mice can induce AHR to intravenous challenge with methacholine 19. Perhaps (a) OVA-specific IgE and/or IgG1 antibodies, present during the latter part of the 2-wk period of OVA sensitization, provided sufficient priming of airway mast cells to permit these cells to release mediators in response to the final injection(s) of OVA at the end of the series of seven injections of antigen, and (b) such repeated, low-level release of mast cell mediators can result in a persistent state of AHR to methacholine. However, it is not immediately apparent what specific mast cell–dependent mechanisms might account for the persistence of the AHR for ∼1 mo after the last administration of antigen.
The Importance of Mast Cells as Amplifiers of Allergen-induced AHR Can Vary Markedly Depending on the “Asthma Model” Studied.
Taken together, our data indicate that in an asthma model that omits alum as an adjuvant and employs intranasal challenge with antigen, mast cells can significantly enhance AHR to methacholine (Fig. 1A and Fig. D). Moreover, this mast cell–dependent effect on the AHR to methacholine can be detected even in OVA-sensitized mice that have been challenged with saline rather than antigen (Fig. 1A and Fig. D). By contrast, in an asthma model that employs alum as an adjuvant during sensitization and uses aerosol challenge with antigen, the slight difference in AHR to methacholine in antigen-challenged W/Wv and +/+ mice was not statistically significant (Fig. 1 C), nor did such OVA/alum-sensitized mice exhibit significant differences in their responses to methacholine after saline challenge (Fig. 1 C).
In a report that appeared while this study was in review, Kobayashi et al. 29 found that a mast cell–dependent component of AHR to intravenous acetylcholine chloride could be detected 24 h after the final antigen challenge in mice that had been sensitized with OVA/alum (10 μg OVA absorbed to 1 mg of alum intraperitoneally on days 0 and 5) and challenged 12 and 16 d later with aerosolized OVA (5 mg/ml for 10 min). The differences between the results with OVA/alum-immunized mice in the study by Kobayashi et al. 29 and in our experiments may reflect the differences in the details of OVA sensitization and challenge—we used higher doses of OVA for sensitization (20 μg with 2.25 mg alum intraperitoneally on days 1 and 14), as well as a different protocol for antigen challenge (1% OVA by aerosol for 20 min on days 28, 29, and 30)—and/or other differences in the two studies. For example, Kobayashi et al. 29 measured airway responses to intravenous challenge in anesthetized, artificially ventilated mice, whereas we measured airway responses to aerosol challenge in nonanesthetized, spontaneously breathing mice. Indeed, when Kobayashi et al. 29 tested a stronger protocol of OVA challenge (10 mg/ml for 30 min three times a day on days 12, 14, 16, and 20), they detected no significant differences in the AHR of +/+ and W/Wv mice. Nevertheless, the work of Kobayashi et al. 29 and our study are in accord in showing that relatively “weak” protocols of antigen sensitization and challenge can reveal an important contribution of mast cells to antigen-induced AHR to cholinergic stimulation that may be masked under other circumstances.
Mast Cells Can Enhance Allergen-induced Increases in Numbers of Airway Eosinophils and BrdU+ Cells.
To investigate whether mast cells also contributed to chronic inflammation of the airways, we examined two features of the response: (a) numbers of recruited eosinophils, and (b) numbers of cells in the affected airways that had incorporated the thymidine analogue BrdU, which is indicative of cell proliferation 26. In accord with the results of Takeda et al. 15, we found that OVA challenge induced the infiltration of roughly equivalent numbers of eosinophils, whether quantified in the entire lung tissue (Fig. 2 A) or in the perivascular regions (Fig. 2 B), in W/Wv or +/+ mice that had been sensitized with OVA and alum. Notably, Kobayashi et al. 29 also detected no significant difference in the extent of eosinophil recruitment (as assessed by quantification of eosinophils in bronchoalveolar lavage fluid) in W/Wv or +/+ mice that were subjected to protocols of sensitization with OVA/alum and antigen challenge that differed from those used in our study.
By contrast, we found that OVA challenge induced minimal eosinophil infiltration in W/Wvmice that had been sensitized without alum, but the responses in the identically treated +/+ mice, especially in the perivascular regions, approached those in the OVA/alum-sensitized animals (Fig. 2A and Fig. B). In mice sensitized to OVA without adjuvant, responses to challenge with 20 μg of OVA were essentially identical in mast cell–reconstituted W/Wv mice and in +/+ controls (Fig. 2A and Fig. B; Fig. 3 A).
Figure 3.
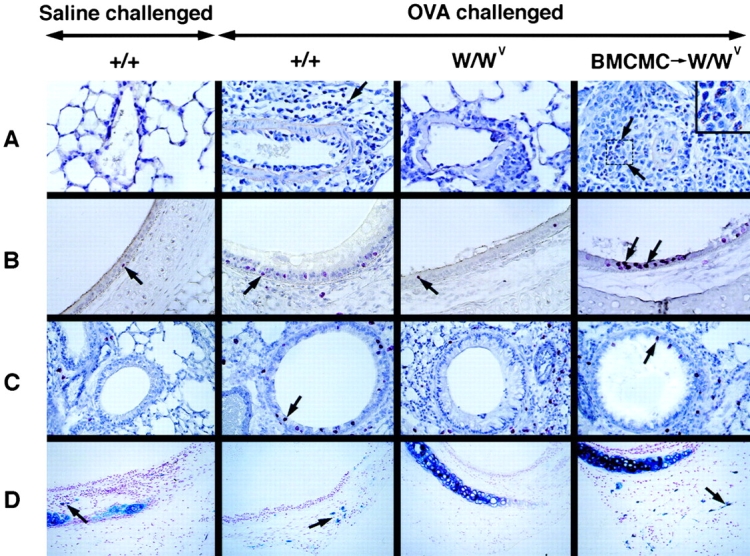
Histological analysis of lung inflammation. (A) Perivascular inflammation, with eosinophils (some eosinophils are indicated by arrows); Congo red stain. Original magnification: ×400. The inset in A (BMCMC→W/Wv) shows a higher-magnification view of the area indicated by the two arrows in order to illustrate eosinophils (granulocytes with red cytoplasm). Original magnification in inset: ×800. (B) BrdU+ cells (red, some indicated with arrows) within the tracheal epithelium. Immunohistochemistry was developed with Fast Red, with Mayer's hematoxylin counterstain. (C) BrdU+ cells (red, some indicated with arrows) within the bronchiolar epithelium. (D) Mast cells (Alcian blue–positive cells, some indicated with arrows) in the trachea. Original magnification in B–D: ×200.
Many studies have indicated that the airway epithelium in subjects with asthma exhibits injury and repair, and that this process is associated with proliferating epithelial cells 30 31 32. In our experiments, histological examination indicated that BrdU was incorporated into cells in the tracheal (Fig. 3 B) and bronchiolar (Fig. 3 C) epithelium, as well as into cells outside of the airways (Fig. 3A and Fig. B). By morphological criteria, the BrdU+ cells in the epithelium appeared to include both epithelial cells and some infiltrating mononuclear leukocytes. Although establishing the specific identity of the various types of BrdU+ cells in this model will require additional studies, it is clear that mast cells are important in regulating the total number of BrdU+ cells in the airway epithelium (Fig. 4). Notably, and in contrast to our findings with AHR to methacholine (Fig. 1) or eosinophil infiltration (Fig. 2), OVA-challenged W/Wv mice exhibited significantly reduced numbers of BrdU+ cells compared with identically-treated +/+ mice after OVA sensitization either with or without alum. As with the two other features of asthma that we assessed in this model, in mice sensitized to OVA without adjuvant, numbers of BrdU+ cells after challenge with 20 μg of OVA were statistically indistinguishable in mast cell-reconstituted W/Wv mice and congenic wild-type mice (Fig. 4A and Fig. B).
W/Wv and +/+ Mice Develop Similar IgE and IgG1 Responses to Antigen Sensitization.
W/Wv, +/+, and BMCMC→W/Wv mice developed remarkably similar IgE and IgG1 antibody responses to OVA sensitization (Table ). However, mice sensitized with alum developed slightly lower levels of OVA-specific IgG1, but much higher levels of total IgG1, than did mice sensitized without alum (Table ).
Table 1.
OVA-specific and Total IgE and IgG1 Levels in the Serum
| OVA-specific Ig | Total Ig levels | ||||
|---|---|---|---|---|---|
| Genotype | Sensitization | IgE | IgG1 | IgE | IgG1 |
| (ng/ml) | (μg/ml) | (μg/ml) | |||
| W/WV | Naive | 2.30 | 0.06 | 0.18 | 1.05 |
| +/+ | Naive | 3.18 | 0.05 | 0.15 | 1.07 |
| W/WV | OVA/alum | 64.8 | 0.65 | 0.43 | 35.6 |
| +/+ | OVA/alum | 59.4 | 0.65 | 0.56 | 64.2 |
| W/WV | OVA | 80.8 | 0.68 | 0.54 | 9.30 |
| +/+ | OVA | 65.3 | 0.68 | 0.47 | 8.09 |
| BMCMC→W/WV | OVA | 75.8 | 0.68 | 0.50 | 8.24 |
Mean serum levels of OVA-specific antibodies and total IgE and IgG1 were determined by ELISA.
Conclusions.
We have found that the extent to which mast cells can contribute to the airway hyperreactivity and chronic inflammation associated with models of allergic asthma in mice is highly dependent on the specific experimental model investigated. We used two different “asthma models” to assess three characteristics of the airway response to allergen challenge in OVA-sensitized mice: airway reactivity to aerosolized methacholine, infiltration of airway tissues with eosinophils, and numbers of BrdU+ cells in the airway epithelium. For each of these features, responses to OVA in antigen-challenged mast cell–deficient W/Wv mice that had been sensitized with OVA in the absence of alum were substantially and significantly lower than those in the corresponding normal mice, and responses in mast cell–reconstituted W/Wvmice were statistically indistinguishable from those in the wild-type animals. Notably, of six prior studies that employed W/Wv and +/+ mice to investigate allergen-induced eosinophil recruitment to the airways 14 15 16 17 18 29, only one detected significantly reduced responses (by ∼50%) in W/Wv versus +/+ mice 14. In that study, as in ours, the mice had been sensitized without an adjuvant 14.
By contrast, we detected no statistically significant effects of mast cells on either airway reactivity to methacholine or airway eosinophil infiltration in mice that were sensitized to OVA with alum. These findings are thus in accord with those reported by Takeda et al. 15, who investigated the same asthma model. Using a different (“weaker”) model of OVA/alum sensitization and OVA challenge, Kobayashi et al. 29 detected a mast cell–dependent contribution to airway hyperreactivity to cholinergic stimulation but not to eosinophil recruitment.
It will be important to identify the specific mechanisms by which mast cells can augment airway hyperreactivity, eosinophil infiltration, and numbers of BrdU+ cells in the airways, and to characterize the specific epithelial and nonepithelial cell populations that are affected. Indeed, the striking increase in BrdU+ cells after allergen challenge represents a previously unrecognized feature of these asthma models in mice, and one which may have a mast cell–dependent component whether the mice have been sensitized to antigen either in the presence or absence of alum. We suspect that multiple mediators and cytokines, and both direct and indirect mechanisms, may link mast cell activation to the specific aspects of the airway responses that we have studied. For example, some “mast cell–dependent” effects on AHR or numbers of BrdU+ cells could be mediated, at least in part, by eosinophils. Nevertheless, our data strongly support the hypothesis that the mast cell can represent an important local amplifier of antigen-dependent chronic inflammation in asthma, and that this cell can contribute significantly to multiple features of this disorder.
Acknowledgments
We thank Z.-S. Wang for excellent technical assistance, and Hans C. Oettgen for helpful advice.
This work was supported by United States Public Health Science Grants CA/AI72074, AI/GM23990, and 5U19 AI41995 (Project 1) (to S.J. Galli).
References
- Worldwide variation in prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and atopic eczema: ISAAC The International Study of Asthma and Allergies in Childhood (ISAAC) Steering Committee. Lancet. 1998;351:1225–1232. [PubMed] [Google Scholar]
- Barnes P.J. Therapeutic strategies for allergic diseases Nature. 402Suppl1999. B31 B38 [DOI] [PubMed] [Google Scholar]
- Holt P.G., Macaubas C., Stumbles P.A., Sly P.D. The role of allergy in the development of asthma Nature. 402Suppl1999. B12 B17 [DOI] [PubMed] [Google Scholar]
- Corry D.B., Kheradmand F. Induction and regulation of the IgE response Nature. 402Suppl1999. B18 B23 [DOI] [PubMed] [Google Scholar]
- Turner H., Kinet J-P. Signaling through the high-affinity IgE receptor FcεRI Nature. 402Suppl1999. B24 B30 [DOI] [PubMed] [Google Scholar]
- Wills-Karp M. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu. Rev. Immunol. 1999;17:255–281. doi: 10.1146/annurev.immunol.17.1.255. [DOI] [PubMed] [Google Scholar]
- Galli S.J. Complexity and redundancy in the pathogenesis of asthmareassessing the roles of mast cells and T cells. J. Exp. Med. 1997;186:343–347. doi: 10.1084/jem.186.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlhop P.D., van de Rijn M., Goldberg A.B., Brewer J.P., Kurup V.P., Martin T.R., Oettgen H.C. Allergen-induced bronchial hyperreactivity and eosinophilic inflammation occur in the absence of IgE in a mouse model of asthma. Proc. Natl. Acad Sci. USA. 1997;94:1344–1349. doi: 10.1073/pnas.94.4.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corry D.B., Grunig G., Hadeiba H., Kurup V.P., Warnock M.L., Sheppard D., Rennick D.M., Locksley R.M. Requirements for allergen-induced airway hyperreactivity in T and B cell deficient mice. Mol. Med. 1998;4:344–355. [PMC free article] [PubMed] [Google Scholar]
- Korsgren M., Erjefalt J.S., Korsgren O., Sundler F., Persson C.G. Allergic eosinophil-rich inflammation develops in lungs and airways of B cell–deficient mice. J. Exp. Med. 1997;185:885–892. doi: 10.1084/jem.185.5.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean J.A., Sauty A., Luster A.D., Drazen J.M., De Sanctis G.T. Antigen-induced airway hyperresponsiveness, pulmonary eosinophilia, and chemokine expression in B cell-deficient mice. Am. J. Respir. Cell Mol. Biol. 1999;20:379–387. doi: 10.1165/ajrcmb.20.3.3291. [DOI] [PubMed] [Google Scholar]
- Hamelmann E., Cieslewicz G., Schwarze J., Ishizuka T., Joetham A., Heusser C., Gelfand E.W. Anti-interleukin 5 but not anti-IgE prevents airway inflammation and airway hyperresponsiveness. Am. J. Respir. Crit. Care Med. 1999;160:934–941. doi: 10.1164/ajrccm.160.3.9806029. [DOI] [PubMed] [Google Scholar]
- Hamelmann E., Tadeda K., Oshiba A., Gelfand E.W. Role of IgE in the development of allergic airway inflammation and airway hyperresponsiveness—a murine model. Allergy. 1999;54:297–305. doi: 10.1034/j.1398-9995.1999.00085.x. [DOI] [PubMed] [Google Scholar]
- Kung T.T., Stelts D., Zurcher J.A., Jones H., Umland S.P., Kreutner W., Egan R.W., Chapman R.W. Mast cells modulate allergic pulmonary eosinophilia in mice. Am. J. Respir. Cell Mol. Biol. 1995;12:404–409. doi: 10.1165/ajrcmb.12.4.7695919. [DOI] [PubMed] [Google Scholar]
- Takeda K., Hamelmann E., Joetham A., Shultz L.D., Larsen G.L., Irvin C.G., Gelfand E.W. Development of eosinophilic airway inflammation and airway hyperresponsiveness in mast cell–deficient mice. J. Exp. Med. 1997;186:449–454. doi: 10.1084/jem.186.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogami M., Suko M., Okudaira H., Miyamoto T., Shiga J., Ito M., Kasuya S. Experimental pulmonary eosinophilia in mice by Ascaris suum extract. Am. Rev. Respir. Dis. 1990;141:1289–1295. doi: 10.1164/ajrccm/141.5_Pt_1.1289. [DOI] [PubMed] [Google Scholar]
- Brusselle G.G, Kips J.C., Tavernier J.H., van der Heyden J.G., Cuvelier C.A., Pauwels R.A., Bluethmann H. Attenuation of allergic airway inflammation in IL-4-deficient mice. Clin. Exp. Allergy. 1994;24:73–80. doi: 10.1111/j.1365-2222.1994.tb00920.x. [DOI] [PubMed] [Google Scholar]
- Okudaira H., Nogami M., Matsuzaki G., Dohi M., Suko M., Kasuya S., Takatsu K. T-cell-dependent accumulation of eosinophils in the lung and its inhibition by monoclonal anti-interleukin-5. Int. Arch. Allergy. Appl. Immunol. 1991;94:171–173. doi: 10.1159/000235354. [DOI] [PubMed] [Google Scholar]
- Martin T.R., Takeishi T., Katz H.R., Austen K.F., Drazen J.M., Galli S.J. Mast cell activation enhances airway responsiveness to methacholine in the mouse. J. Clin. Invest. 1993;91:1176–1182. doi: 10.1172/JCI116277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIntyre D., Boyd G. Factors influencing the occurrence of a late reaction to allergen challenge in atopic asthmatics. Clin. Allergy. 1984;14:311–317. doi: 10.1111/j.1365-2222.1984.tb02211.x. [DOI] [PubMed] [Google Scholar]
- Sulakvelidze I., Inman M.D., Rerecich T., O'Byrne P.M. Increases in airway eosinophils and interleukin-5 with minimal bronchoconstriction during repeated low-dose allergen challenge in atopic asthmatics. Eur. Respir. J. 1998;11:821–827. doi: 10.1183/09031936.98.11040821. [DOI] [PubMed] [Google Scholar]
- Kitamura Y., Go S., Hatanaka S. Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood. 1978;52:447–452. [PubMed] [Google Scholar]
- Nakano T., Sonoda T., Hayashi C., Yamatodani A., Kanayama Y., Yamamura T., Asai H., Yonezawa T., Kitamura Y., Galli S.J. Fate of bone marrow–derived cultured mast cells after intracutaneous, intraperitoneal, and intravenous transfer into genetically mast cell–deficient W/Wvmice. Evidence that cultured mast cells can give rise to both connective tissue type and mucosal mast cells. J. Exp. Med. 1985;162:1025–1043. doi: 10.1084/jem.162.3.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli S.J., Zsebo K.M., Geissler E.N. The kit ligand, stem cell factor. Adv. Immunol. 1994;55:1–96. doi: 10.1016/s0065-2776(08)60508-8. [DOI] [PubMed] [Google Scholar]
- Hamelmann E., Schwarze J., Takeda K., Oshiba A., Larsen G.L., Irvin C.G., Gelfand E.W. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am. J. Respir. Crit. Care Med. 1997;156:766–775. doi: 10.1164/ajrccm.156.3.9606031. [DOI] [PubMed] [Google Scholar]
- Grouls V., Helpap B. Selective staining of eosinophils and their immature precursors in tissue sections and autoradiographs with Congo red. Stain Technol. 1981;56:323–325. doi: 10.3109/10520298109067335. [DOI] [PubMed] [Google Scholar]
- Arizono N., Shiota T., Yamada M., Matsumoto Y., Yoshikawa H., Matsuda S., Tegoshi T. Bromodeoxyuridine labeling studies on the proliferation of intestinal mucosal mast cells in normal and athymic rats. APMIS. 1990;98:369–376. doi: 10.1111/j.1699-0463.1990.tb01046.x. [DOI] [PubMed] [Google Scholar]
- Spergel J.M., Mizoguchi E., Brewer J.P., Martin T.R., Bahn A.K., Geha R.S. Epicutaneous sensitization with protein antigen induces localized allergic dermatitis and hyperresponsiveness to methacholine after single exposure to aerosolized antigen in mice. J. Clin. Invest. 1998;101:1614–1622. doi: 10.1172/JCI1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Miura T., Haba T., Sato M., Serizawa I., Nagai H., Ishizaka K. An essential role of mast cells in the development of airway hyperresponsiveness in a murine asthma model. J. Immunol. 2000;164:3855–3861. doi: 10.4049/jimmunol.164.7.3855. [DOI] [PubMed] [Google Scholar]
- Busse W., Elias J., Sheppard D., Banks-Schlegel S. Airway remodeling and repair. Am. J. Respir. Crit. Care Med. 1999;160:1035–1042. doi: 10.1164/ajrccm.160.3.9902064. [DOI] [PubMed] [Google Scholar]
- Holgate S.T., Lackie P.M., Davies D.E., Roche W.R., Walls A.F. The bronchial epithelium as a key regulator of airway inflammation and remodelling in asthma Clin. Exp. Allergy. 29Suppl. 21999. 90 95 [DOI] [PubMed] [Google Scholar]
- Holgate S.T. The inflammation-repair cycle in asthmathe pivotal role of the airway epithelium Clin. Exp. Allergy. 28Suppl. 51998. 97 103 [DOI] [PubMed] [Google Scholar]