

Abstract
The firm adhesion and transplatelet migration of leukocytes on vascular thrombus are both dependent on the interaction of the leukocyte integrin, Mac-1, and a heretofore unknown platelet counterreceptor. Here, we identify the platelet counterreceptor as glycoprotein (GP) Ibα, a component of the GP Ib-IX-V complex, the platelet von Willebrand factor (vWf) receptor. THP-1 monocytic cells and transfected cells that express Mac-1 adhered to GP Ibα–coated wells. Inhibition studies with monoclonal antibodies or receptor ligands showed that the interaction involves the Mac-1 I domain (homologous to the vWf A1 domain), and the GP Ibα leucine-rich repeat and COOH-terminal flanking regions. The specificity of the interaction was confirmed by the finding that neutrophils from wild-type mice, but not from Mac-1–deficient mice, bound to purified GP Ibα and to adherent platelets, the latter adhesion being inhibited by pretreatment of the platelets with mocarhagin, a protease that specifically cleaves GP Ibα. Finally, immobilized GP Ibα supported the rolling and firm adhesion of THP-1 cells under conditions of flow. These observations provide a molecular target for disrupting leukocyte–platelet complexes that promote vascular inflammation in thrombosis, atherosclerosis, and angioplasty-related restenosis.
Keywords: inflammation, leukocytes, platelets, adhesion, receptors
Introduction
Adhesive interactions between vascular cells play important roles in orchestrating the inflammatory response. Recruitment of circulating leukocytes to vascular endothelium requires multistep adhesive and signaling events including selectin-mediated attachment and rolling, leukocyte activation, and integrin-mediated firm adhesion and diapedesis that result in the infiltration of inflammatory cells into the blood vessel wall 1. Firm attachment is mediated by members of the β2 integrin family, LFA-1 (αLβ2, CD11a/CD18), Mac-1 (αMβ2, CD11b/CD18), and p150,95 (αXβ2, CD11c/CD18), which bind to endothelial counterligands (e.g., intercellular adhesion molecule 1 [ICAM-1]) 2, to endothelial-associated extracellular matrix proteins (e.g., fibrinogen) 3, or to glycosaminoglycans 4.
Leukocyte recruitment and infiltration also occur at sites of vascular injury where the lining endothelial cells have been denuded and platelets and fibrin have been deposited. In vivo studies show that leukocytes and platelets colocalize at sites of hemorrhage, within atherosclerotic and postangioplasty restenotic lesions, and in areas of ischemia-reperfusion injury 5 6 7 8. This heterotypic interaction between platelets and leukocytes links the hemostatic/thrombotic and inflammatory responses 5. Although less well characterized, a similar sequential adhesion model of leukocyte attachment to and transmigration across surface-adherent platelets has been proposed 9 10 11 12 13 14. The initial tethering and rolling of leukocytes on platelet P-selectin 9 10 15 are followed by their firm adhesion and transplatelet migration, processes that are dependent on the leukocyte integrin, Mac-1 12 13 14. In addition to promoting the accumulation of leukocytes at sites of platelet coverage within the vasculature, the binding of platelets to neutrophils influences key cellular effector responses by inducing neutrophil activation, upregulating expression of cell adhesion molecules 16, and generating signals that promote integrin activation 13, chemokine synthesis 17 18, and the respiratory burst 16. Interestingly, both neutrophil–platelet and monocyte–platelet aggregates have been identified in the peripheral blood of patients with coronary artery disease 16 19 and may be markers of disease activity 16.
Leukocyte Mac-1 binds endothelial cell adhesion molecules of the immunoglobulin superfamily, most prominently ICAM-1. This receptor is not found on platelets, although platelets express a related receptor, ICAM-2 20. Nevertheless, Diacovo et al. 12 have shown that ICAM-2 blockade has no effect on the firm adhesion of neutrophils on monolayers of activated platelets under flow. Because activated Mac-1 binds fibrinogen, one possibility is that firm adhesion is mediated by Mac-1 binding to fibrinogen that has been immobilized on platelet glycoprotein (GP) IIb-IIIa (αIIbβ3). Indeed, Weber and Springer 21 found that neutrophil adhesion to activated platelets under flow was partially blocked by an antibody against GP IIb-IIIa and that platelets from a patient with Glanzmann thrombasthenia (which lack GP IIb-IIIa) did not support neutrophil accumulation nearly as well as did wild-type platelets. Nevertheless, neither of these manipulations was successful in completely blocking neutrophil accumulation, even in conjunction with ICAM-2–blocking antibodies. Against a role for the Mac-1–fibrinogen–GP IIb-IIIa axis in neutrophil arrest, Ostrovsky et al. 22 found that neither arginine-glycine-aspartate-serine (RGDS) peptides nor the replacement of normal platelets with thrombasthenic platelets affected the accumulation of the leukocytes on platelets. Despite the differences in the two studies, both groups proposed the existence of another Mac-1 receptor on the platelet surface.
Evaluation of the structural features of integrins has provided us with insights into a candidate platelet counterreceptor for Mac-1. Integrins are heterodimeric proteins composed of one α and one β subunit 23. A subset of integrin α subunits, including CD11b of Mac-1, contains an inserted domain (I domain) of ∼200 amino acids that is implicated in ligand binding 24 25 26 27 28 and is strikingly similar to the A domains of von Willebrand factor (vWf) 29 30 31, one of which, A1, mediates the interaction of vWf with its platelet receptor, the GP Ib-IX-V complex. Through this interaction, platelets are able to adhere to regions of vascular injury in a process entirely analogous to the interaction between leukocytes and activated endothelium. Platelets recognize vWf in the subendothelium, and roll along the region until they become activated and adhere firmly through GP IIb-IIIa 32.
One interesting aspect of the GP Ib-IX-V–vWf interaction is that both platelets and vWf circulate freely in blood, but do not interact unless vWf is immobilized on the subendothelium or in the presence of very high shear stresses (such as might be found at sites of arterial stenosis). Under static conditions in vitro, the interaction requires the presence of modulators: botrocetin, a snake venom protein, or ristocetin, a peptide antibiotic from the soil bacterium Nocardia lurida 33. The modulators induce conformational changes in vWf (and possibly also in GP Ibα, in the case of ristocetin) that enable the interaction.
The GP Ib-IX-V complex comprises four phylogenetically related polypeptides, GP Ibα, GP Ibβ, GP IX, and GP V, of which only GP Ibα has been shown to bind ligands 34. The ligand-binding domain of this polypeptide resides within the NH2-terminal 300 amino acids, a region containing seven 24–amino acid leucine-rich repeats, which assign this polypeptide to a large protein superfamily with similar motifs. This region is held above the platelet membrane by a heavily O-glycosylated mucin-like region called the macroglycopeptide. Following a single transmembrane domain, a cytoplasmic domain of ∼100 amino acids mediates association of the entire complex with the cytoskeleton and with signaling proteins 33.
Because of the similarity of the vWf A1 domain and the αM I domain, we hypothesized that GP Ibα might also be able to bind Mac-1. Here, we report that GP Ibα is a constitutively expressed counterreceptor for Mac-1.
Materials and Methods
Materials.
Human fibrinogen depleted of plasminogen, vWf, and fibronectin were purchased from Enzyme Research Laboratories. Porcine heparin (10,000 U/ml) was obtained from Elkins-Sinn, Inc. TGF-β1 was from Collaborative Research, Inc., and 1,25-(OH)2 vitamin D3 was from Calbiochem. The snake venom metalloprotease, mocarhagin, was purified as described previously 35. Glycocalicin was purified by a minor modification of the method of Canfield et al. 36 by successive chromatography on wheat germ lectin Sepharose 6MB (Amersham Pharmacia Biotech) and jacalin agarose (Pierce Chemical Co.) (Fig. 1). A 39/34-kD dispase fragment of vWf encompassing the A1 domain (Leu480–Gly718) was purified as described previously 37. Peptide P2, corresponding to amino acid residues 377–395 in the γ chain of fibrinogen, which binds to the I domain of Mac-1 and blocks fibrinogen binding 38, was obtained from the W.M. Keck Biotechnology Resource Center (Yale University, New Haven, CT). 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein, acetoxymethyl ester (BCECF AM) was purchased from Molecular Probes.
Figure 1.
Purified glycocalicin. Purified glycocalicin from human platelets was subjected to SDS and 7.5% PAGE and stained with Coomassie brilliant blue.
CD11/CD18 mAbs used included the following: LPM19c, directed to the αM subunit of human Mac-1 (CD11b) and capable of blocking fibrinogen, ICAM-1, and C3bi binding (a gift of Dr. Karen Pulford, John Radcliffe Hospital, Oxford, UK) 24; M1/70, directed to the αM subunit of mouse Mac-1 (CD11b), with broad ligand blocking properties (39; American Type Culture Collection); TS1/22, directed to the αL subunit of LFA-1 (CD11a) and capable of blocking ICAM binding (provided by Dr. Lloyd Klickstein, Brigham and Women's Hospital); and IB4, directed to the common β2 subunit (CD18; provided by Dr. Lloyd Klickstein). The stimulating CD18 mAb KIM 127 was a gift of Dr. Martyn Robinson (Celltech Ltd., Slough, England) 40.
The GP Ibα mAbs used were: AK2, AP1, VM16d, SZ2, and WM23. All but WM23 bind within the GP Ibα ligand-binding region, within the first 282 amino acids at the GP Ibα NH2 terminus. AK2 blocks vWf binding induced by both ristocetin and botrocetin and binds within the first leucine-rich repeat (amino acid residues 36–59). AP1 (provided by Dr. Dermot Kenny, Beaumont Hospital, The Royal College of Surgeons, Dublin, Ireland) also blocks both ristocetin- and botrocetin-induced vWf binding (epitope 201–268); VM16d (gift of Dr. Alexey Mazurov, Russian Ministry of Health, Moscow, Russia) only blocks botrocetin-induced vWf binding (epitope 201–268), as does SZ2 (epitope 269–282) 41. WM23 binds within the GP Ibα macroglycopeptide and does not interfere with the binding of any GP Ibα ligands 42. The polyclonal GP Ibα antibody was prepared in rabbits immunized with glycocalicin (the soluble extracellular region of GP Ibα) and affinity-purified over a glycocalicin Affigel 10/15 column 43.
7E3 and 10E5 44, murine mAbs to GP IIb and/or IIIa that block platelet fibrinogen binding, were provided by Dr. Barry S. Coller (Mt. Sinai Medical Center, New York, NY). Y2/51 (Dako) is directed against CD61 (GP IIIa) and was purchased conjugated to FITC. TUK4 (Dako) is directed against CD14 on the monocyte surface and was purchased conjugated to PE. Y2/51 and TUK4 are IgG murine antibodies. Murine IgG-FITC (Dako) was used as a nonspecific isotype control. All mAbs were purified unless otherwise indicated.
Cell Lines and Culture Conditions.
THP-1 monocytic cells (American Type Culture Collection) were maintained in RPMI 1640 supplemented with 10% fetal bovine serum, 2 mM glutamine, 25 mM Hepes, penicillin, and streptomycin. Differentiation of THP-1 cells (106/ml), which is accompanied by increased expression of Mac-1, was induced by treatment with 1 ng/ml TGF-β1 and 50 nM 1,25-(OH)2 vitamin D3 (TGF-β/vit D3) for 24 h 45. Nontransfected 293 cells (American Type Culture Collection) were maintained in DMEM/F12 supplemented with 10% fetal bovine serum, 25 mM Hepes, penicillin, and streptomycin; 293 cells expressing human LFA-1, Mac-1, or a chimeric LFA-1 receptor that contained the I domain of Mac-1 (i.e., LFA-1 [IαM]-transfected 293 cells), established as described previously 46 47 48, were maintained in similar media containing G418.
Preparation of Neutrophils and Platelets.
Mac-1–deficient (Mac-1−/−) mice, generated as described previously 49, were backcrossed for >12 generations onto the C57BL/J6 strain. Murine neutrophils were harvested from the peritoneal cavity of wild-type C57BL/J6 (Mac-1+/+) or Mac-1−/− mice by lavage with 10 ml RPMI 4 h after the intraperitoneal injection of 1 ml sterile 3% thioglycollate broth. Isolated neutrophils were resuspended in RPMI containing low-endotoxin BSA (0.5%). Cytospin preparations stained with Giemsa revealed that >90% of the cells were neutrophils. Cell viability (>95%) was assessed in all cases using trypan blue exclusion.
Venous blood was obtained from volunteers who had not consumed aspirin or other nonsteroidal antiinflammatory drugs for at least 10 d, and was anticoagulated with 13 mM trisodium citrate. Platelet-rich plasma was prepared by centrifugation at 150 g for 10 min. Gel-filtered platelets were obtained by passage of platelet-rich plasma over a Sepharose 2B column in calcium-free Tyrode's Hepes buffer, as described previously 50. Platelet counts were measured using a Coulter counter (model ZM) and adjusted to 150,000/μl by the addition of buffer.
Adhesion Assays.
Adherent cells were assayed by colorimetry 45 51 or by loading THP-1 or 293 cells and thioglycollate-elicited murine neutrophils with BCECF AM (1 μM) according to the manufacturer's protocol. Cells (105/well) were placed in 96-well microtiter plates coated with purified GP Ibα (10 μg/ml) or fibrinogen (10 μg/ml) and blocked with gelatin (0.2%). Adhesion was stimulated with PMA (17 ng/ml) or the β2-stimulating mAb KIM 127 (5 μg/ml). Plates were washed with 0.9% NaCl (three to five times), adherent cells were fixed in methanol for 15 min and stained with Giemsa, and adhesion was quantified by measuring absorbance at 540 nm. Alternatively, adhesion was quantified by measuring the fluorescence of BCECF AM–loaded cells using a Cytofluor II fluorescence multiwell microplate reader (PerSeptive Biosystems). The effect of anti-CD11/CD18 mAbs or soluble Mac-1 ligands (i.e, fibrinogen, heparin) on adhesion was assessed by preincubating cells with the indicated mAb (10 μg/ml) or ligand for 15 min at 37°C; the effect of anti-GP Ibα mAbs on adhesion was investigated by incubating the indicated mAb (10 μg/ml) with GP Ibα–coated wells for 30 min at 37°C before the addition of cells. Data are expressed as percent inhibition of maximum adherent responses of respective sets of treatment.
In the case of 293 cell adhesion experiments, low passage (1 to 3) human saphenous vein endothelial cells (provided by Dr. Peter Libby, Brigham and Women's Hospital) were grown to confluence in 96-well microtiter wells and stimulated with TNF-α (10 ng/ml) for 4 h to upregulate ICAM-1 expression 52. 293 cells were loaded with BCECF AM for 45 min at 37°C, washed, and stimulated with KIM 127 (5 μg/ml) before adding to endothelial cell monolayers.
Purified I Domain Binding Experiments.
High-binding microtiter plates (MaxiSorp; Nunc) were coated with purified I domain (10 μg/ml), obtained as described previously 38, in Tris-buffered saline (TBS), pH 7.4, and then blocked with buffer containing 0.5% gelatin. Biotinylated glycocalicin (0–50 μg/ml) was added to each well in TBS containing 1 mM CaCl2 and MgCl2 and 0.5% gelatin, and plates were incubated for 60 min at 25°C. After washing, bound glycocalicin was quantified with avidin peroxidase. Specific binding was determined by subtracting binding to wells coated with gelatin alone and accounted for up to 40% of the total binding.
Neutrophil Adhesion to Surface-adherent Platelets.
Neutrophil adhesion to surface-adherent platelets was investigated as described previously 12. Gel-filtered human platelets (∼1.5 × 107) were added to 96-well microtiter plates coated overnight with 0.2% gelatin. After 45 min at 37°C, unbound platelets were removed by washing. Neutrophils (1.5 × 105) were loaded with 1 μM BCECF AM, washed twice, and then added to each well for 60 min at 37°C in 5% CO2. After washing, neutrophil adhesion was quantified as the percentage of total cells adherent by measuring the fluorescence of BCECF AM–loaded cells using a Cytofluor II fluorescence multiwell microplate reader (PerSeptive Biosystems). Fluorescence of input neutrophils before washing served as a measure of total cell number. The effect of mAbs on neutrophil adhesion to platelets was assessed as described above for purified GP Ibα; the effect of the snake venom metalloprotease, mocarhagin, which cleaves GP Ibα at peptide bond 282–283 35, on leukocyte adhesion to platelets was examined by preincubating surface-adherent platelets with mocarhagin for 30 min at 37°C. Data are expressed as percent inhibition of maximum adherent responses of respective sets of treatment.
Whole Blood Detection of Platelet–Leukocyte Aggregates.
Leukocyte–platelet aggregates were measured by two-color flow cytometry in a FACSCalibur™ flow cytometer (Becton Dickinson) by slight modifications of methods described previously 19. Peripheral blood was drawn from a healthy volunteer or, as indicated, from a patient with Bernard-Soulier syndrome (BSS) 53 who had not ingested aspirin or other antiplatelet drugs during the previous 10 d. The first 2 ml of drawn blood was discarded. Blood was then drawn into a 3.2% sodium citrate Vacutainer (Becton Dickinson). The sample was diluted 1:1 with modified Tyrode's Hepes buffer, pH 7.4, and then immediately incubated at 22°C for 10 min with either (a) buffer alone; (b) 0.5 μM ADP (Biodata); or (c) 5 μM thrombin receptor activating peptide (TRAP; 14-mer; Calbiochem) and saturating concentrations of Y2/51-FITC and TUK4-PE. The samples were then fixed at 22°C for 10 min to a final concentration of 1% formalin (Polysciences, Inc.) and 1.5× HBSS concentrate (GIBCO BRL). The samples were then diluted with 500 μl of distilled H2O, vortexed, and incubated at 22°C for 10 min to lyse the red blood cells. Monocytes and neutrophils were identified by CD14-PE positivity and their characteristic light scatter.
THP-1 Adhesion to Glycocalicin under Flow.
The interaction of TGF-β1/vit D3–stimulated THP-1 cells with immobilized glycocalicin was examined using a parallel-plate flow chamber system, which has been described previously 54. Glass coverslips, which form the bottom of the chamber, were coated with purified glycocalicin (100 μg/ml) by immersing them in the glycocalicin solution for 3 h at 37°C. Residual nonspecific binding sites were then blocked with 3% BSA for 30 min. The chamber was then assembled and mounted onto an inverted-stage microscope (DIAPHOT-TMD; Nikon) equipped with a silicon-intensified target video camera (model C2400; Hammatus) connected to a video cassette recorder. Cells were suspended in culture medium and the suspension was brought to room temperature. 1 ml of the cell suspension (106 cells/ml) was then injected into the chamber and incubated for 1 min. The cells were allowed to settle onto the glycocalicin substrate for 1 min; the chamber was then perfused with the PBS at a flow rate calculated to generate fluid shear stress of 2 dyn/cm2. The attachment and rolling of cells in a single-view field were recorded in real time for 3 min, and the video data were then analyzed using imaging software (IC-300 Modular Image Processing Workstation; Inovision Corp.) to calculate the rolling velocities. The rolling events scored were ligand specific as confirmed in parallel determinations on control substrates coated with heat-stable antigen (HSA). In the study to inhibit glycocalicin, the coverslip was incubated with the GP Ibα mAb VM16d (25 μg/ml) for 30 min at room temperature and washed before assembly of the chamber. To test the effect of Mac-1 inhibition, the THP-1 cells were incubated with LMP19c (25 μg/ml) for 30 min at room temperature before injection into the chamber. Rolling cells were those observed to be moving in the direction of flow while maintaining constant contact with the glycocalicin substrate.
Statistics.
All data are presented as the mean ± SD. Groups were compared using the nonpaired t test. P values < 0.05 were considered significant.
Results
Mac-1–expressing Cells Bind to GP Ibα.
Given the homology between the receptor-binding vWf A1 domain and the Mac-1 I domain, we hypothesized that Mac-1 would bind the platelet GP Ib-IX-V complex. To assess this potential interaction, we assayed the adhesion of Mac-1–bearing cells to purified glycocalicin, the soluble extracellular domain of GP Ibα. We have previously shown that stimulation of THP-1 monocytic cells (which constitutively express Mac-1) with TGF-β1 and 1,25-(OH)2 vitamin D3 increases Mac-1 surface expression ∼2.0-fold 45. When cytokine-treated THP-1 cells were stimulated with either phorbol ester (PMA) or KIM 127, an mAb to CD18 that induces a change in the conformation of CD18 and promotes both LFA-1– and Mac-1–dependent adhesion 40, they adhered robustly to wells coated with GP Ibα and blocked with gelatin, but not to wells coated with gelatin alone (Fig. 2). Adhesion of the cells to GP Ibα was inhibited by IB4, an anti-CD18 mAb that blocks both LFA-1– and Mac-1–dependent functions, indicating that adhesion is CD18 dependent (Fig. 2, and Table ). THP-1 cell adhesion to GP Ibα was also completely blocked by LPM19c, an mAb that binds to the I domain of the αM subunit of Mac-1 (CD11b) and blocks the binding of fibrinogen, C3bi, and ICAM 24. In contrast, the anti–LFA-1 mAb TS1/22 had no effect on adhesion. Similar results were obtained with integrin mAbs whether THP-1 cell adhesion was stimulated with KIM 127 or PMA, ruling out the possibility that the mAbs inhibited adhesion by interfering with the binding of KIM 127 to CD18.
Figure 2.

THP-1 cell adhesion to GP Ibα is Mac-1 dependent. Cytokine-treated THP-1 cells were added to glycocalicin (GP Ibα)–coated and gelatin-blocked wells. Adhesion was promoted by the addition of the stimulating mAb KIM 127 (5 μg/ml). The effect of anti-CD18 (IB4), anti-CD11a (TS1/22), and anti-CD11b (LPM19c) mAbs on adhesion was assessed as described in Materials and Methods. After washing, (A) adherent cells were stained with Giemsa and (B) adhesion was quantified by measuring the fluorescence of BCECF AM–loaded THP-1 cells. Triplicate determination (mean ± SD) representative of three to five separate experiments.
Table 1.
Summary of Inhibition of KIM 127–stimulated THP-1 Adhesion to GP Ibα by Antibodies or Soluble Ligands
| Antibody or ligand | Receptor (epitope) | Percent inhibition |
|---|---|---|
| IB4 | Anti-CD18 | 99 ± 1 |
| TS1/22 | Anti-CD11a (I domain) | 10 ± 13 |
| LPM19c | Anti-CD11b (I domain) | 92 ± 12 |
| Polyclonal | Anti-GP Ibα | 80 ± 17 |
| Polyclonal | Rabbit IgG control | 16 ± 20 |
| VM16d | Anti-GP Ibα (aa residues 201–268) | 83 ± 16 |
| AP1 | Anti-GP Ibα (aa residues 201–268) | 86 ± 10 |
| AK2 | Anti-GP Ibα (aa residues 36–59) | 16 ± 6 |
| SZ2 | Anti-GP Ibα (sulfated tyrosine residues 269–282) | 8 ± 6 |
| WM23 | Anti-GP Ibα (macroglycopeptide) | 11 ± 5 |
| 7E3 | Anti-GP IIb-IIIa, -αvβ3, –Mac-1 | 16 ± 8 |
| 10E5 | Anti-GP IIb-IIIa | 18 ± 6 |
| Fibrinogen (2 μM) | Mac-1 | 99 ± 1 |
| P2γ377–395 (10 μM) | Mac-1 | 0 |
| Heparin (200 U/ml) | Mac-1 | 93 ± 9 |
| vWF A1 (20 μg/ml) | GP Ibα | 83 ± 13 |
The adhesion of cytokine-treated THP-1 cells to glycocalicin (GP Ibα)–coated microtiter wells was stimulated by the addition of KIM 127 (5 μg/ml) in the presence and absence of antibodies directed to or soluble ligands of Mac-1 and GP Ibα. Polyclonal antibodies were added at 20 μg/ml and purified mAbs at 10 μg/ml as described in Materials and Methods. Soluble ligand concentrations are indicated. CD11/CD18 mAbs included IB4, TS1/22, and LPM19c; GP Ibα mAbs used were AK2, AP1, VM16d, SZ2, and WM23. Data are expressed as percent inhibition of maximal KIM-stimulated adhesion by the antibodies or soluble ligands (mean ± SD, n = 3–5). aa, amino acids.
To confirm that Mac-1 mediates the adhesion of THP-1 cells to GP Ibα and to eliminate the possibility that mAbs to Mac-1 inhibit THP-1 cell adhesion indirectly, we assessed the adhesion of 293 cells transfected with either LFA-1 or Mac-1 to GP Ibα. Flow cytometry confirmed similar expression levels of LFA-1 and Mac-1 (Fig. 3 A). Both LFA-1–transfected and Mac-1–transfected 293 cells adhered robustly to human endothelial cells expressing ICAM-1 (Fig. 3 B), indicating that these transfected 293 cells are functional. Mac-1–expressing 293 cells adhered to GP Ibα, and this adhesion was enhanced by KIM 127 (Fig. 3 C). KIM 127–stimulated adhesion was blocked by mAbs directed to both CD11b and CD18, and to GP Ibα (VM16d). Neither untransfected 293 cells nor LFA-1–transfected 293 cells bound to GP Ibα.
Figure 3.

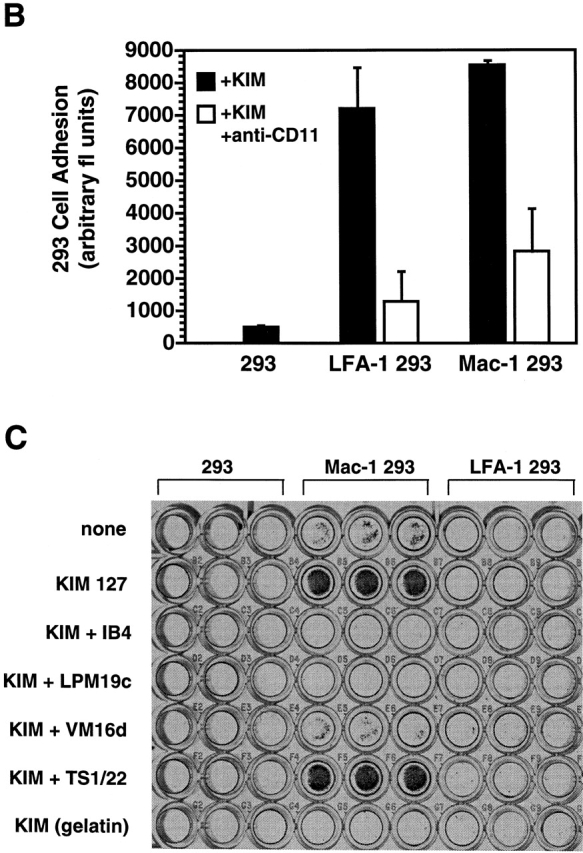
Mac-1 expression is required for adhesion to GP Ibα. (A) Expression of CD11/CD18 integrin in 293 cells was confirmed by flow cytometry using the anti-CD18 mAb IB4. Equivalent expression of CD18 is observed in 293 cells transfected with human LFA-1, Mac-1, or a chimeric LFA-1 containing the I domain of Mac-1, designated LFA-1 (IαM). (B) Adhesion of untransfected 293 cells and 293 cells transfected with human Mac-1 or LFA-1 to human endothelial cells stimulated with TNF-α to express ICAM-1. Adhesion was promoted by the addition of the stimulating mAb KIM 127 (5 μg/ml) and quantified by measuring the fluorescence of BCECF AM–loaded 293 cells; triplicate determination (mean ± SD) representative of two separate experiments. (C) 293 cells were added to glycocalicin-coated and gelatin-blocked wells. Adhesion was promoted by the addition of KIM 127 (5 μg/ml). After washing, adherent cells were stained with Giemsa. Triplicate determination representative of three separate experiments.
Neutrophil Binding to GP Ibα Requires Mac-1.
To investigate further the role of Mac-1 in leukocyte adhesion to GP Ibα, we examined the binding of neutrophils from wild-type (Mac-1+/+) and Mac-1–deficient (Mac-1−/−) mice to GP Ibα. Mac-1+/+ neutrophils bound to GP Ibα, adhesion that was enhanced by PMA pretreatment (Fig. 4). In contrast, Mac-1−/− neutrophils showed significantly reduced adhesion to GP Ibα, whether in the absence or presence of PMA. Taken together, these observations indicate that Mac-1 mediates adhesion to purified GP Ibα.
Figure 4.

Neutrophil binding to GP Ibα requires Mac-1. Thioglycollate-elicited neutrophils from wild-type (Mac-1+/+) or Mac-1–deficient (Mac-1−/−) mice were added to glycocalicin-coated and gelatin-blocked wells in the absence (black bars) and presence (white bars) of PMA (17 ng/ml). Adhesion was promoted by the addition of PMA (17 ng/ml). Adhesion was quantified by measuring the fluorescence of BCECF AM–loaded neutrophils. Triplicate determination (mean ± SD) representative of three separate experiments.
The Mac-1 I Domain Serves as a Recognition Site for GP Ibα.
The adhesion of Mac-1–bearing THP-1 cells to GP Ibα was inhibited by LPM19c (Fig. 2), an mAb that binds to the I domain of the αM subunit of Mac-1 (CD11b). To establish definitively that the Mac-1 I domain serves as a recognition site for GP Ibα, we transfected 293 cells with a chimeric LFA-1 receptor that contained the I domain of Mac-1 (i.e., LFA-1 [IαM]–transfected 293 cells). Flow cytometry showed comparable expression of LFA-1 (IαM), LFA-1, and Mac-1 in transfected 293 cells (Fig. 3 A). LFA-1–transfected 293 cells did not adhere to GP Ibα (Fig. 3 C); in contrast, LFA-1 (IαM)–transfected 293 cells adhered robustly to GP Ibα (Fig. 5 A), indicating that the I domain of Mac-1 is required for adhesion to GP Ibα. We next investigated whether GP Ibα would directly bind to purified I domain of Mac-1. Biotinylated glycocalicin bound to immobilized I domain in a concentration-dependent manner over the range 0–200 nM (Fig. 5 B).
Figure 5.

Mac-1 I domain serves as a recognition site for GP Ibα. (A) 293 cells transfected with human Mac-1, LFA-1, or a chimeric LFA-1 containing the I domain of Mac-1 (LFA-1 [IαM]) were added to glycocalicin-coated and gelatin-blocked wells. Adhesion was promoted by the addition of the stimulating mAb KIM 127 (5 μg/ml) and quantified by measuring the fluorescence of BCECF AM–loaded 293 cells. Triplicate determination (mean ± SD) representative of three separate experiments. (B) Interaction of purified glycocalicin and Mac-1 I domain. Specific binding of biotinylated glycocalicin (0–200 nM) to purified I domain–coated and gelatin-blocked wells as described in Materials and Methods (B). Triplicate determination (mean ± SD), n = 2.
To evaluate further the I domain as a recognition site on Mac-1 for GP Ibα, we examined the ability of Mac-1 I domain ligands (e.g., fibrinogen [24] and heparin [4]) to inhibit THP-1 adhesion to GP Ibα. Soluble fibrinogen (50% inhibitory concentration [IC50] ∼ 1 μM) and heparin (IC50 ∼ 75 U/ml) both inhibited adhesion in a dose-dependent manner (Fig. 6A–C). Inhibition by fibrinogen suggested to us the possibility that the GP Ibα and fibrinogen binding sites in Mac-1 may be partially overlapping. To exclude potential nonspecific steric hindrance of fibrinogen, we examined the effect of peptide P2, corresponding to amino acid residues 377–395 in the γ chain of fibrinogen, that binds to the I domain of Mac-1 and blocks fibrinogen binding (IC50 ∼ 1 μM) 38. P2 had no effect on THP-1 adhesion to GP Ibα, suggesting that the GP Ibα and fibrinogen binding sites are probably distinct.
Figure 6.
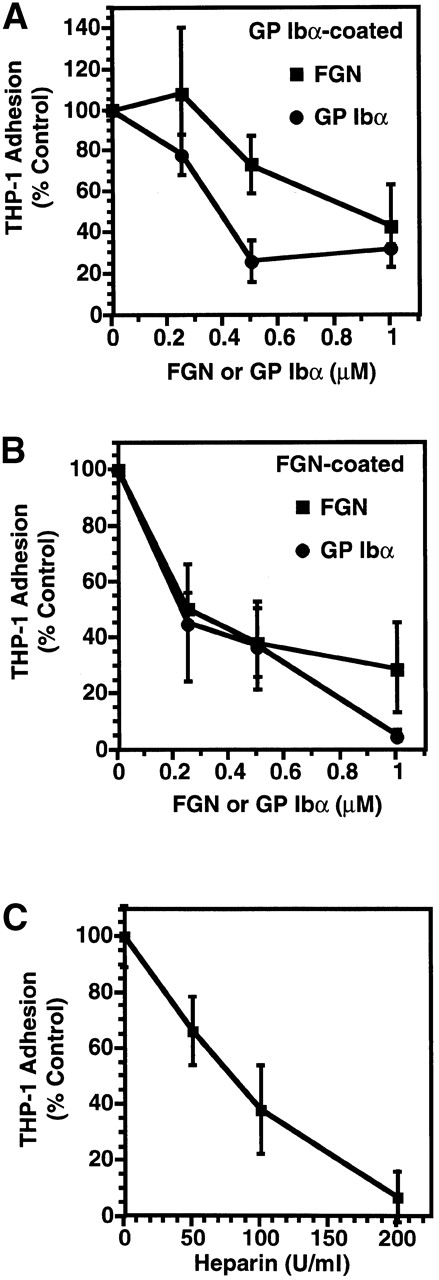
Mac-1–GP Ibα binding and Mac-1 I domain ligands. Cytokine-treated THP-1 cells were added to (A) glycocalicin-coated or (B) fibrinogen (FGN)-coated wells. Adhesion was promoted by the addition of KIM 127 (5 μg/ml) in the presence of increasing concentrations (0–2 μM) of soluble GP Ibα (○) or the soluble Mac-1 I domain ligand fibrinogen (▪). (C) The adhesion of THP-1 cells to glycocalicin (GP Ibα)–coated wells in the presence of increasing concentrations of a second Mac-1 I domain ligand, heparin (0–100 U/ml), was also examined. Adhesion was quantified by measuring the fluorescence of BCECF AM–loaded cells and expressed relative to maximal adhesion stimulated with KIM 127. Mean ± SD, n = 3.
To evaluate the interaction between Mac-1 and GP Ibα more quantitatively and to confirm that Mac-1 did not recognize a binding site present solely on immobilized GP Ibα, we tested soluble GP Ibα for its ability to inhibit Mac-1–dependent THP-1 cell adhesion to immobilized GP Ibα (Fig. 6 B) or fibrinogen (Fig. 6 C). Soluble GP Ibα inhibited THP-1 adhesion to wells coated with either GP Ibα (IC50 = 0.5 μM) or fibrinogen (IC50 = 0.25 μM). Conversely, the vWf A1 domain (20 μg/ml), a ligand for GP Ibα, blocked binding of THP-1 cells to immobilized GP Ibα (percent inhibition = 83 ± 13) (Table ).
Identifying the Mac-1 Binding Site within GP Ibα.
We next turned our attention to identifying the Mac-1 interaction site within GP Ibα by assessing the effect of monoclonal and polyclonal antibodies to GP Ibα on Mac-1–dependent THP-1 cell adhesion to glycocalicin (Fig. 7, and Table ). Polyclonal anti–GP Ibα, but not control rabbit IgG, significantly reduced THP-1 cell adhesion. VM16d and AP1, which map to the leucine-rich COOH-terminal flanking region of GP Ibα (amino acids 201–268), inhibited KIM 127–stimulated adhesion of THP-1 cells to GP Ibα (percent inhibition: VM16d = 83 ± 16; AP1 = 86 ± 10). A similar inhibitory effect of these mAbs was noted when THP-1 cell adhesion to GP Ibα was stimulated by PMA (percent inhibition: VM16d = 90 ± 4; AP1 = 84 ± 2). In contrast, neither AK2, directed to the first leucine-rich repeat (amino acids 36–59), nor WM23, directed to the macroglycopeptide mucin core region of GP Ibα, had any significant effect on THP-1 cell adhesion. An irrelevant mAb against GP IIb-IIIa (10E5) also had no significant effect on THP-1 cell adhesion.
Figure 7.

Epitope map of mAbs to GP Ibα. Schematic representation of the extracellular domain of GP Ibα. The GP Ibα mAbs employed were: AK2, AP1, VM16d, SZ2, and WM23. AK2 binds within the first leucine-rich repeat (amino acid residues 36–58). AP1 and VM16d bind to the COOH-terminal flanking and leucine-rich repeat region (201–268). SZ2 maps to the sulfated tyrosine residues encompassing amino acids 268–282 (reference 41). WM23 binds within the macroglycopeptide region of GP Ibα (reference 42).
Our observation that heparin inhibited Mac-1–dependent adhesion to GP Ibα led us to consider the possibility that the region within GP Ibα containing sulfated tyrosine residues (an anionic stretch between residues Asp269 and Asp289 which contains three sulfated tyrosines, Tyr276, Tyr278, and Tyr279) might be involved in the interaction with Mac-1. We tested this possibility by assessing the effect of SZ2, an anti-GP Ibα mAb that maps within the region containing the sulfated tyrosines and requires sulfation for its epitope. SZ2 did not affect THP-1 cell adhesion to GP Ibα, suggesting that heparin likely inhibits adhesion by binding the Mac-1 I domain and interfering with GP Ibα binding rather than by directly mimicking a binding site on GP Ibα 4.
Glycocalicin Supports the Rolling and Firm Adhesion of THP-1 Cells under Flow.
To evaluate the potential for the GP Ibα–Mac-1 interaction to support the adhesion of blood cells under flow, we perfused THP-1 cells over coverslips coated with a glycocalicin matrix using a parallel-plate flow chamber system. The cells were either kept in their native state or treated with TGF-β1 and 1,25-(OH)2 vitamin D3 to increase Mac-1 expression. THP-1 cells were able to adhere to the glycocalicin, whether or not they were induced to differentiate, but did not adhere to control, BSA-coated coverslips (Fig. 8). However, undifferentiated cells that adhered to the glycocalicin exhibited mainly rolling behavior, whereas differentiated cells adhered more firmly. The antibodies LMP19c and VM16d both greatly decreased the number of attached cells, confirming the involvement of both Mac-1 and GP Ibα, respectively, in the adhesion of THP-1 cells under flow.
Figure 8.


Adhesion of THP-1 cells to glycocalicin under flow. (A) Video images of THP-1 cells rolling on or firmly adherent to immobilized glycocalicin at a wall shear stress of 2 dyn/cm2 in a parallel-plate flow chamber. Images were created using a digital image processing system to snap frames of previously recorded experiments. The cells were either untreated (Undifferentiated) or induced to differentiate with TGF-β1 and 1,25-(OH)2 vitamin D3 then injected into the chamber and allowed to settle on the matrix for 1 min. The chamber was then perfused with buffer at a velocity calculated to generate the desired shear stress. Images were created by overlapping 30 frames taken over 1 s. The effect of mAbs VM16d (anti-GP Ibα) and LMP19c (anti-CD11b) were assessed by preincubating, respectively, either the coverslip or the cells with saturating concentrations of antibody. BSA-coated coverslips were used as a control matrix. (B) Quantitation of rolling and firmly adherent cells from experiments such as those represented in A (mean ± SEM, n = 4).
Mac-1 and GP Ibα Facilitate the Interaction between Leukocytes and Platelets.
Finally, to establish that Mac-1 and GP Ibα facilitate the heterotypic interaction between leukocytes and platelets, we assayed the adhesion of wild-type and Mac-1–deficient neutrophils to platelets. Thioglycollate-elicited Mac-1+/+ neutrophils bound to adherent platelets, and this adhesion was promoted by PMA (Fig. 9 A). In contrast, Mac-1−/− neutrophils demonstrated markedly reduced adhesion to platelets. Adhesion of Mac-1+/+ neutrophils was also blocked by the rat anti–mouse Mac-1 mAb M1/70 (percent inhibition = 95 ± 5) and the anti-GP Ibα mAb VM16d (percent inhibition = 55 ± 10). Furthermore, Mac-1+/+ leukocyte adhesion to platelets was inhibited dose-dependently by soluble glycocalicin (percent inhibition = 72 ± 18) and by pretreatment of adherent platelets with the snake venom metalloprotease, mocarhagin (percent inhibition = 89 ± 8), which cleaves GP Ibα at peptide bond 282–283 as the only detectable proteolytic event on the platelet surface 35 (Fig. 9 B). Taken together, these observations indicate that neutrophil adhesion to platelets is primarily mediated by Mac-1 and GP Ibα.
Figure 9.

Neutrophil binding to platelets requires Mac-1 and GP Ibα. (A) Thioglycollate-elicited neutrophils from wild-type (Mac-1+/+, black bars) or Mac-1–deficient (Mac-1−/−, white bars) mice were added to surface-adherent platelets. Adhesion was promoted by the addition of PMA (17 ng/ml). The contribution of Mac-1 or GP Ibα to wild-type neutrophil adhesion to platelets was assayed by the addition of the rat anti–mouse CD11b mAb, M1/70 (10 μg/ml), or the anti-GP Ibα mAb, VM16d (10 μg/ml), respectively. (B) The effect of soluble glycocalicin (0–20 μg/ml) and the snake venom metalloprotease, mocarhagin, which cleaves GP Ibα at peptide bond 282–283, on neutrophil adhesion was also examined. Adhesion was quantified by measuring the fluorescence of BCECF AM–loaded neutrophils. Triplicate determination (mean ± SD) representative of three separate experiments.
Finally, to provide additional evidence supporting a role for GP Ibα in platelet binding to leukocytes, we assessed the presence of leukocyte–platelet aggregates in whole blood obtained from a normal volunteer or a patient with BSS. We have previously shown that spontaneous or agonist-induced leukocyte–platelet aggregate formation require an interaction between P-selectin glycoprotein ligand 1 (PSGL-1) and P-selectin that is strengthened by integrins 19. Leukocyte–platelet aggregates were decreased in the circulation of a patient with BSS compared with a normal control (2 vs. 7% platelet-positive neutrophils; Fig. 10). Moreover, these leukocyte–platelet aggregates were less likely to form in the BSS patient after agonist stimulation with either 0.5 μM ADP (4 vs. 21% platelet-positive neutrophils) or 5 μM TRAP (13 vs. 86% platelet-positive neutrophils). Mac-1 dependence in leukocyte–platelet aggregate formation in this assay was confirmed by the fact that the anti–Mac-1 mAb LPM19c completely inhibited agonist-induced platelet–neutrophil complex formation.
Figure 10.

Absence of GP Ibα reduces neutrophil–platelet aggregates in whole blood. Leukocyte–platelet aggregates in peripheral blood obtained from a normal volunteer (black bars) or a patient with BSS (white bars) were measured by two-color flow cytometry as described in Materials and Methods. Formation of leukocyte–platelet aggregates was stimulated by incubating blood with ADP (0.5 μM) or TRAP (5 μM). Data shown are mean values of duplicate determinations.
Discussion
In this study, we have identified a direct interaction between the leukocyte integrin Mac-1 and platelet GP Ibα. The following evidence was obtained for this interaction: (a) mAbs to both Mac-1 and GP Ibα inhibited THP-1 cell adhesion to purified GP Ibα; (b) 293 cells that express Mac-1, but not LFA-1, bound strongly to GP Ibα, and this adhesion was inhibited specifically by mAbs; (c) wild-type, but not Mac-1–deficient, neutrophils adhered to platelets and to purified GP Ibα; (d) neutrophil adhesion to platelets was inhibited by mAbs to Mac-1 and GP Ibα and by pretreatment of the platelets with the snake venom metalloprotease, mocarhagin, whose major platelet substrate is GP Ibα 35; and (e) basal and agonist-stimulated leukocyte–platelet aggregates were decreased in whole blood of a patient with BSS compared with a normal control.
By virtue of binding diverse ligands including, among others, fibrin(ogen) 55 56, ICAM-1 57, factor X 58, C3bi 55, high molecular weight kininogen 59, and heparin 4, Mac-1 regulates important leukocyte functions including adhesion, migration, coagulation, proteolysis, phagocytosis, oxidative burst, and signaling 49 60 61 62. However, these ligands do not account for all of Mac-1's adhesive interactions. Although previous studies have shown that Mac-1, the primary fibrin(ogen) receptor on leukocytes, directly facilitates the recruitment of leukocytes at sites of platelet and fibrin deposition 12 13 14, the precise platelet counterreceptor was unidentified.
I or A domains are regions of ∼200 amino acids that are present in 1 or more copies in many proteins involved in cell–cell, cell–matrix, and matrix–matrix interactions 25 31. This superfamily motif is present in integrin α subunits, including CD11a, CD11b, CD11c, CD11d, CD49a, CD49b, and αE 25, the complement proteins Factors B and C2 63 64, collagens 65 66, and vWf 29. For CD11b, experimental evidence supports the notion that the I domain is responsible for the binding of all Mac-1 ligands except for factor X 24 67. vWf has three similar domains, in this case termed A domains. The first and third of these, A1 and A3, mediate binding to GP Ibα and collagen, respectively 68. High-resolution crystal structures of the CD11b I domain and the vWf A1 domain show that both of these domains adopt a classic α/β “Rossman” fold 25 69. The Mac-1 I domain also contains a metal ion–dependent adhesion site (MIDAS) for binding protein ligands, a motif, however, not present in the vWf A1 domain due to the presence of an arginine and an alanine instead of a serine and an aspartate, respectively, at two of the critical amino acids forming the MIDAS motif. The observation that mutations of the αM I domain that correspond to gain-of-function mutations of the vWf A1 domain also alter the binding activity of Mac-1 48 supports the notion that Mac-1 and vWf may be functionally similar with respect to GP Ib-IX-V binding.
The binding of GP Ibα to vWf and Mac-1 has several similarities and some interesting differences. First, as expected, the binding involves the homologous I or A domains. In addition, in both cases binding requires a conformational change of the A or I domain, in the case of vWf requiring ristocetin, botrocetin, or shear stress, and in the case of Mac-1 requiring activation of the integrin to its ligand-competent form. Distinguishing the two interactions is the distinct pattern of inhibition by GP Ibα antibodies. AK2, for example, is a potent inhibitor of vWf binding to GP Ibα, whether induced by ristocetin, botrocetin, or shear stress. This antibody failed to inhibit the interaction of Mac-1 with GP Ibα. VM16d, on the other hand, does not inhibit vWf binding to GP Ibα, except as induced by botrocetin, but is a potent inhibitor of Mac-1 binding. However, the sites are not completely distinct, as indicated by the observation that the isolated vWf A1 domain blocks the interaction of Mac-1–expressing cells with GP Ibα and by the ability of the mAb AP1, which also blocks ristocetin- and botrocetin-induced vWf binding, to inhibit the interaction.
The identification of the interaction between GP Ibα and Mac-1 provides a tantalizing lead into the nature of leukocyte–platelet adhesion, helping to clarify the sequential adhesion model of neutrophil attachment to surface-adherent platelets proposed by Diacovo et al. 12. Nevertheless, our data do not rule out the possibility of additional platelet surface receptors for Mac-1. Other potential Mac-1 ligands present on the platelet membrane include fibrinogen (bound to GP IIb-IIIa) 55 56, ICAM-2 70, high molecular weight kininogen 59, and glycosaminoglycans 4. A leukocyte–platelet interaction mediated by fibrinogen bridging between Mac-1 and GP IIb-IIIa has been discounted by Ostrovsky et al. 22, who found that neither RGDS peptides nor the replacement of normal platelets with thrombasthenic platelets (i.e., lacking GP IIb-IIIa) affected the accumulation of the leukocytes on platelets, and recently by Furman et al. 71, who found that GP IIb-IIIa antagonists and RGDS peptides did not reduce leukocyte–platelet aggregate formation in whole blood.
Other interactions contributing to Mac-1–independent leukocyte–platelet complex formation include thrombospondin bridging between GP IV receptors on platelets and monocytes 72, and P-selectin on activated platelets binding with leukocyte PSGL-1 73 74. Nevertheless, under the experimental conditions employed in the present study, which assayed the adhesion of activated neutrophils (i.e., thioglycollate-elicited peritoneal neutrophils) to surface-adherent platelets after vigorous washing, the predominant interaction between neutrophils and platelets appeared to be between Mac-1 and GP Ibα.
These present observations also suggest a possible target for therapeutic intervention. In particular, the distinct difference in the inhibitory patterns of GP Ibα antibodies suggests that it might be possible to prevent leukocyte attachment to platelets by targeting GP Ibα without inhibiting platelet adhesion to the vessel wall. Our recent observations have identified Mac-1 as a molecular determinant of neointimal thickening after experimental arterial injury that produces endothelial denudation and platelet and/or fibrin deposition. We found that antibody-mediated blockade 7 or selective absence 75 of Mac-1 impaired transplatelet leukocyte migration into the vessel wall, diminishing medial leukocyte accumulation and neointimal thickening after experimental angioplasty or endovascular stent implantation. Therefore, future studies aimed at identifying the precise binding site(s) responsible for Mac-1–GP Ibα binding might provide a molecular strategy for disrupting leukocyte–platelet complexes that promote vascular inflammation in thrombosis, atherosclerosis, and angioplasty-related restenosis.
Acknowledgments
The authors would like to thank Paula McColgan for manuscript preparation.
This work was supported in part by grants from the National Institutes of Health (HL57506 to D.I. Simon and HL54218 to J.A. López), from the American Heart Association (96002750 and 96012670 to J.A. López), and from the National Health and Medical Research Council of Australia. J.A. López is an Established Investigator of the American Heart Association.
Footnotes
Abbreviations used in this paper: BSS, Bernard-Soulier syndrome; GP, glycoprotein; IC50, 50% inhibitory concentration; ICAM-1, intercellular adhesion molecule 1; RGDS, arginine-glycine-aspartate-serine; TRAP, thrombin receptor activating peptide; vWf, von Willebrand factor.
References
- Springer T.A. Traffic signals for lymphocyte recirculation and leukocyte emigrationa multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- Smith C.W., Marlin S.D., Rothlein R., Toman C., Anderson D.C. Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J. Clin. Invest. 1989;83:2008–2017. doi: 10.1172/JCI114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Languino L.R., Duperray A., Joganic K.J., Fornaro M., Thornton G.B., Altieri D.C. Regulation of leukocyte-endothelial interactions and leukocyte transendothelial migration by intercellular adhesion molecule 1-fibrinogen recognition. Proc. Natl. Acad. Sci. USA. 1995;92:7734–7738. doi: 10.1073/pnas.92.5.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond M.S., Alon R., Parkos C.A., Quinn M.T., Springer T.A. Heparin is an adhesive ligand for the leukocyte integrin Mac-1 (CD11b/CD18) J. Cell Biol. 1995;130:1473–1482. doi: 10.1083/jcb.130.6.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus A.J. Thrombosis and inflammation as multicellular processessignificance of cell-cell interactions. Semin. Hematol. 1994;31:261–269. [PubMed] [Google Scholar]
- Ross R. Mechanisms of diseaseatherosclerosis—an inflammatory disease. N. Engl. J. Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- Rogers C., Edelman E.R., Simon D.I. A monoclonal antibody to the β2-leukocyte integrin Mac-1 (CD11b/CD18) reduces intimal thickening after angioplasty or stent implantation in rabbits. Proc. Natl. Acad. Sci. USA. 1998;95:10134–10139. doi: 10.1073/pnas.95.17.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinder C.S., Bonan J.L., Rinder H.M., Matthew J., Hines R., Smith B.R. Cardiopulmonary bypass induces leukocyte-platelet adhesion. Blood. 1992;79:1201–1205. [PubMed] [Google Scholar]
- Larsen E., Celi A., Gilbert G.E., Furie B.C., Erban J.K., Bonfanti R., Wagner D.D., Furie B. PADGEM proteina receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell. 1989;59:305–312. doi: 10.1016/0092-8674(89)90292-4. [DOI] [PubMed] [Google Scholar]
- Hamburger S.A., McEver R.P. GMP-140 mediates adhesion of stimulated platelets to neutrophils. Blood. 1990;75:550–554. [PubMed] [Google Scholar]
- Yeo E.L., Sheppard J.-A.I., Feuerstein I.A. Role of P-selectin and leukocyte activation in polymorphonuclear cell adhesion to surface adherent activated platelets under physiologic shear conditions (an injured vessel wall model) Blood. 1994;83:2498–2507. [PubMed] [Google Scholar]
- Diacovo T.G., Roth S.J., Buccola J.M., Bainton D.F., Springer T.A. Neutrophil rolling, arrest, and transmigration across activated, surface-adherent platelets via sequential action of P-selectin and the beta 2-integrin CD11b/CD18. Blood. 1996;88:146–157. [PubMed] [Google Scholar]
- Evangelista V., Manarini S., Rontondo S., Martelli N., Polischuk R., McGregor J.L., de Gaetano G., Cerletti C. Platelet/polymorphonuclear leukocyte interaction in dynamic conditionsevidence of adhesion cascade and cross talk between P-selectin and the β2 integrin CD11b/CD18. Blood. 1996;88:4183–4194. [PubMed] [Google Scholar]
- Kuijper P.H.M., Gallardo Torres H.I., Lammers J.-W.J., Sixma J.J., Koenderman L., Zwaginga J.J. Platelet and fibrin deposition at the damaged vessel wallcooperative substrates for neutrophil adhesion under flow conditions. Blood. 1997;89:166–175. [PubMed] [Google Scholar]
- McEver R.P., Cummings R.D. Role of PSGL-1 binding to selectins in leukocyte recruitment. J. Clin. Invest. 1997;100:S97–S103. [PubMed] [Google Scholar]
- Ott I., Neumann F.-J., Gawaz M., Schmitt M., Schomig A. Increased neutrophil-platelet adhesion in patients with unstable angina. Circulation. 1996;94:1239–1246. doi: 10.1161/01.cir.94.6.1239. [DOI] [PubMed] [Google Scholar]
- Weyrich A.S., McIntyre T.M., McEver R.P., Prescott S.M., Zimmerman G.A. Monocyte tethering by P-selectin regulates monocyte chemotactic protein-1 and tumor necrosis factor-α secretion. J. Clin. Invest. 1995;95:2297–2303. doi: 10.1172/JCI117921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyrich A.S., Elstad M.R., McEver R.P., McIntyre T.M., Moore K.L., Morrissey J.H., Prescott S.M., Zimmerman G.A. Activated platelets signal chemokine synthesis by human monocytes. J. Clin. Invest. 1996;97:1525–1534. doi: 10.1172/JCI118575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman M.I., Benoit S.E., Barnard M.R., Valeri C.R., Borbone M.L., Becker R.C., Hechtman H.B., Michelson A.D. Increased platelet reactivity and circulating monocyte-platelet aggregates in patients with stable coronary artery disease. J. Am. Coll. Cardiol. 1998;31:352–358. doi: 10.1016/s0735-1097(97)00510-x. [DOI] [PubMed] [Google Scholar]
- Diacovo T.G., de Fougerolles A.R., Bainton D.F., Springer T.A. A functional integrin ligand on the surface of plateletsintercellular adhesion molecule-2. J. Clin. Invest. 1994;94:1243–1251. doi: 10.1172/JCI117442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber C., Springer T.A. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to αIIbβ3 and stimulated by platelet-activating factor. J. Clin. Invest. 1997;100:2085–2093. doi: 10.1172/JCI119742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrovsky L., King A.J., Bond S., Mitchell D., Lorant D.E., Zimmerman G.A., Larsen R., Niu X.F., Kubes P. A juxtacrine mechanism for neutrophil adhesion on platelets involves platelet-activating factor and a selectin-dependent activation process. Blood. 1998;91:3028–3036. [PubMed] [Google Scholar]
- Diamond M.S., Springer T.A. The dynamic regulation of integrin adhesiveness. Curr. Biol. 1994;4:506–517. doi: 10.1016/s0960-9822(00)00111-1. [DOI] [PubMed] [Google Scholar]
- Diamond M., Garcia-Aguiliar J., Bickford J., Corbi A., Springer T. The I domain is a major recognition site on the leukocyte integrin Mac-1 (CD11b/CD18) for four distinct ligands. J. Cell Biol. 1993;120:1031–1043. doi: 10.1083/jcb.120.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.O., Rieu P., Arnaout M.A., Liddington R. Crystal structure of the A domain from the alpha subunit of integrin CR3 (CD11b/CD18) Cell. 1995;80:631–638. doi: 10.1016/0092-8674(95)90517-0. [DOI] [PubMed] [Google Scholar]
- Qu A., Leahy D.J. Crystal structure of the I-domain from the CD11a/CD18 (LFA-1, alpha L beta 2) integrin. Proc. Natl. Acad. Sci. USA. 1995;92:10277–10281. doi: 10.1073/pnas.92.22.10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.O., Bankston L.A., Arnaout M.A., Liddington R.C. Two conformations of the integrin A-domain (I-domain)a pathway for activation? Structure. 1995;3:1333–1340. doi: 10.1016/s0969-2126(01)00271-4. [DOI] [PubMed] [Google Scholar]
- Li R., Rieu P., Griffith D.L., Scott D., Arnaout M.A. Two functional states of the CD11b A-domaincorrelations with key features of two Mn2+-complexed crystal structures. J. Cell Biol. 1998;143:1523–1534. doi: 10.1083/jcb.143.6.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler J.E., Shelton-Inloes B.B., Sorace J.M., Harlan J.M., Titanti K., Davie E.W. Cloning and characterization of two cDNAs coding for human von Willebrand factor. Proc. Natl. Acad. Sci. USA. 1985;82:6394–6398. doi: 10.1073/pnas.82.19.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson R.S., Corbi A.L., Berman L., Springer T.A. Primary structure of the leukocyte function-associated molecule-1 alpha subunitan integrin with an embedded domain defining a protein superfamily. J. Cell Biol. 1989;108:703–712. doi: 10.1083/jcb.108.2.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombatti A., Bonaldo P. The superfamily of proteins with von Willebrand factor type A-like domainsone theme common to components of extracellular matrix, hemostasis, cellular adhesion, and defense mechanisms. Blood. 1991;77:2305–2315. [PubMed] [Google Scholar]
- Ruggeri Z.M., Dent J.A., Saldivar E. Contribution of distinct adhesive interactions to platelet aggregation in flowing blood. Blood. 1999;94:172–178. [PubMed] [Google Scholar]
- Andrews R.K., Shen Y., Gardiner E.E., Dong J., Lopez J.A., Berndt M.C. The glycoprotein Ib-IX-V complex in platelet adhesion and signaling. Thromb. Haemost. 1999;82:357–364. [PubMed] [Google Scholar]
- Lopez J.A., Dong J.F. Structure and function of the glycoprotein Ib-Ix-V complex. Curr. Opin. Hematol. 1997;4:323–329. doi: 10.1097/00062752-199704050-00005. [DOI] [PubMed] [Google Scholar]
- Ward C.M., Andrews R.K., Smith A.I., Berndt M.C. Mocarhagin, a novel cobra venom metalloproteinase, cleaves the platelet von Willebrand factor receptor glycoprotein Ibα. Identification of the sulfated tyrosine/anionic sequence Tyr-276-Glu-282 of glycoprotein Ibα as a binding site for von Willebrand factor and α-thrombin. Biochemistry. 1996;35:4929–4938. doi: 10.1021/bi952456c. [DOI] [PubMed] [Google Scholar]
- Canfield V.A., Ozols J., Nugent D., Roth G.J. Isolation and characterization of the alpha and beta chains of human platelet glycoprotein Ib. Biochem. Biophys. Res. Commun. 1987;147:526–534. doi: 10.1016/0006-291x(87)90963-6. [DOI] [PubMed] [Google Scholar]
- Andrews R.K., Gorman J.J., Booth W.J., Corino G.L., Castaldi P.A., Berndt M.C. Cross-linking of a monomeric 39/34-kDa dispase fragment of von Willebrand factor (Leu-480/Val-481-Gly-718) to the N-terminal region of the α-chain of membrane glycoprotein Ib on intact platelets with bis(sulfosuccinimidyl) suberate. Biochemistry. 1989;28:8326–8336. doi: 10.1021/bi00447a010. [DOI] [PubMed] [Google Scholar]
- Ugarova T.P., Solovjov D.A., Zhang L., Loukinov D.I., Yee V.C., Medved L.V., Plow E.F. Identification of a novel recognition sequence for integrin αβ within the αβ chain of fibrinogen. J. Biol. Chem. 1998;273:22519–22527. doi: 10.1074/jbc.273.35.22519. [DOI] [PubMed] [Google Scholar]
- Ault K.A., Springer T.A. Cross-reaction of a rat-anti-mouse phagocyte-specific monoclonal antibody (anti-Mac-1) with human monocytes and natural killer cells. J. Immunol. 1981;120:359–364. [PubMed] [Google Scholar]
- Robinson M.K., Andrew D., Rosen H., Brown D., Ortlepp S., Stephens P., Butcher E.C. Antibody directed against the Leu-CAM beta-chain (CD18) promotes both LFA-1- and CR3-dependent adhesion events. J. Immunol. 1982;148:1080–1085. [PubMed] [Google Scholar]
- Shen Y., Romo G., Dong J.-F., Schade A., McIntire L.V., Kenny D., Whisstock J.C., Berndt M.C., Lopez J.A., Andrews R.K. Requirement of leucine-rich repeats of glycoprotein (GP) Ibα for shear-dependent and static binding of von Willebrand factor to the platelet membrane GP Ib-IX-V complex. Blood. 2000;95:903–910. [PubMed] [Google Scholar]
- Berndt M.C., Du X., Booth W.J. Ristocetin-dependent reconstitution of binding of von Willebrand factor to purified human platelet membrane glycoprotein Ib-IX complex. Biochemistry. 1988;27:633–640. doi: 10.1021/bi00402a021. [DOI] [PubMed] [Google Scholar]
- Cramer E.M., Lu H., Caen J.P., Soria C., Berndt M.C., Tenza D. Differential redistribution of platelet glycoproteins Ib and IIb-IIIa after plasmin stimulation. Blood. 1991;77:694–699. [PubMed] [Google Scholar]
- Coller B.S., Peerschke E.I., Scudder L.E., Sullivan C.A. A murine monoclonal antibody that completely blocks the binding of fibrinogen to platelets produces a thrombasthenic-like state in normal platelets and binds to glycoproteins IIb and/or IIIa. J. Clin. Invest. 1983;72:325–338. doi: 10.1172/JCI110973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon D.I., Rao N.K., Xu H., Wei Y., Majdic O., Ronne E., Kobzik L., Chapman H.A. Mac-1 (CD11b/CD18) and the urokinase receptor (CD87) form a functional unit on monocytic cells. Blood. 1996;88:3185–3194. [PubMed] [Google Scholar]
- Zhang L., Plow E.F. Identification and reconstruction of the binding site within αMβ2 for a specific and high affinity ligand, NIF. J. Biol. Chem. 1997;272:17558–17564. doi: 10.1074/jbc.272.28.17558. [DOI] [PubMed] [Google Scholar]
- Zhang L., Plow E.F. Overlapping, but not identical, sites are involved in the recognition of C3bi, neutrophil inhibitory factor, and adhesive ligands by the αMβ2 integrin. J. Biol. Chem. 1996;271:18211–18216. doi: 10.1074/jbc.271.30.18211. [DOI] [PubMed] [Google Scholar]
- Zhang L., Plow E.F. A discrete site modulates activation of I domains. J. Biol. Chem. 1996;271:29953–29957. doi: 10.1074/jbc.271.47.29953. [DOI] [PubMed] [Google Scholar]
- Lu H., Smith C.W., Perrard J., Bullard D., Tang L., Shappell S.B., Entman M.L., Beaudet A.L., Ballantyne C.M. LFA-1 is sufficient in mediating neutrophil emigration in Mac-1-deficient mice. J. Clin. Invest. 1997;99:1340–1350. doi: 10.1172/JCI119293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawiger J., Parkinson S., Timmons S. Prostacyclin inhibits mobilization of fibrinogen-binding sites on human ADP- and thrombin-treated platelets. Nature. 1980;283:195–198. doi: 10.1038/283195a0. [DOI] [PubMed] [Google Scholar]
- Wei Y., Waltz D.A., Rai N., Drummond R.J., Rosenberg S., Doyle M.V., Chapman H.A. Identification of the urokinase receptor as an adhesion receptor for vitronectin. J. Biol. Chem. 1994;269:32380–32388. [PubMed] [Google Scholar]
- Bevilacqua M.P., Pober J.S., Wheeler M.E., Cotran R.S., Gimbrone M.A.J. Interleukin 1 acts on cultured human vascular endothelium to increase the adhesion of polymorphonuclear leukocytes, monocytes, and related leukocyte cell lines. J. Clin. Invest. 1985;76:2003–2011. doi: 10.1172/JCI112200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRosa C.A., Rohrer M.J., Benoit S.E., Barnard M.R., Michelson A.D. Neutrophil cathepsin G modulates the platelet surface expression of the glycoprotein (GP) Ib-IX complex by proteolysis of the von Willebrand factor binding site on GPIbα and by a cytoskeletal-mediated redistribution of the remainder of the complex. Blood. 1994;84:158–168. [PubMed] [Google Scholar]
- Romo G.M., Dong J.F., Schade A.J., Gardiner E.E., Kansas G.S., Li C.Q., McIntire L.V., Berndt M.C., Lopez J.A. The glycoprotein Ib-IX-V complex is a platelet counterreceptor for P-selectin. J. Exp. Med. 1999;190:803–814. doi: 10.1084/jem.190.6.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S.D., Weitz J.S., Huang A.J., Levin S.M., Silverstein S.C., Loike J.D. Complement receptor type three (CD11b/CD18) of human polymorphonuclear leukocytes recognizes fibrinogen. Proc. Natl. Acad. Sci. USA. 1988;85:7734–7738. doi: 10.1073/pnas.85.20.7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altieri D.C., Bader R., Mannucci P.M., Edgington T.S. Oligospecificity of the cellular adhesion receptor Mac-1 encompasses an inducible recognition specificity for fibrinogen. J. Cell Biol. 1988;107:1893–1900. doi: 10.1083/jcb.107.5.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond M.S., Staunton D.E., Marlin S.D., Springer T.A. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell. 1991;65:961–971. doi: 10.1016/0092-8674(91)90548-d. [DOI] [PubMed] [Google Scholar]
- Altieri D.C., Morrissey J.H., Edgington T.S. Adhesive receptor Mac-1 coordinates the activation of factor X on stimulated cells of monocytic and myeloid differentiationan alternative initiation of the coagulation cascade. Proc. Natl. Acad. Sci. USA. 1988;85:7462–7466. doi: 10.1073/pnas.85.20.7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachtfogel Y.T., DeLa Cadena R.A., Kunapuli S.P., Rick L., Miller M., Schultz R.L., Altieri D.C., Edgington T.S., Colman R.W. High molecular weight kininogen binds to Mac-1 on neutrophils by its heavy chain (domain 3) and its light chain (domain 5) J. Biol. Chem. 1994;269:19307–19312. [PubMed] [Google Scholar]
- Arnaout M.A. Structure and function of the leukocyte adhesion molecules CD11/CD18. Blood. 1990;75:1037–1050. [PubMed] [Google Scholar]
- Plow E.F., Zhang L. A MAC-1 attackintegrin functions directly challenged in knockout mice. J. Clin. Invest. 1997;99:1145–1146. doi: 10.1172/JCI119267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coxon A., Rieu P., Barkalow F.J., Askari S., Sharpe A.H., von Andrian U.H., Arnaout M.A., Mayadas T.N. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosisa homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- Campbell R.D., Porter R.R. Molecular cloning and characterization of the gene coding for human complement protein factor B. Proc. Natl. Acad. Sci. USA. 1983;80:4464–4468. doi: 10.1073/pnas.80.14.4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley D.R. Primary structure of human complement component C2. Biochem. J. 1986;239:339–345. doi: 10.1042/bj2390339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu M.-L., Zhang R.-Z., Pan T.-C., Stokes D., Conway D., Kuo H.-J., Glanville R., Mayer U., Mann K., Deutzmann R., Timple R. Mosaic structure of globular domains in the human type VI collagen α3 chainsimilarity to von Willebrand factor, fibronectin, actin, salivary proteins and aprotinin type protease inhibitors. EMBO (Eur. Mol. Biol. Organ.) J. 1990;9:385–393. doi: 10.1002/j.1460-2075.1990.tb08122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombati A., Bonaldo P., Doliana R. Type A modulesinteracting domains found in several non-fibrillar collagens and in other matrix proteins. Matrix. 1993;13:297–306. doi: 10.1016/s0934-8832(11)80025-9. [DOI] [PubMed] [Google Scholar]
- Zhou L., Lee D.H., Plescia J., Lau C.Y., Altieri D.C. Differential ligand binding specificities of recombinant CD11b/CD18 integrin I-domain. J. Biol. Chem. 1994;269:17075–17079. [PubMed] [Google Scholar]
- Ruggeri Z.M. Structure and function of von Willebrand factor. Thromb. Haemost. 1999;82:576–584. [PubMed] [Google Scholar]
- Emsley J., Cruz M., Handin R., Liddington R. Crystal structure of the von Willebrand Factor A1 domain and implications for the binding of platelet glycoprotein Ib. J. Biol. Chem. 1998;273:10396–10401. doi: 10.1074/jbc.273.17.10396. [DOI] [PubMed] [Google Scholar]
- Xie J., Li R., Kotovuori P., Vermont-Desroches C., Wijdenes J., Arnaout M.A., Nortamo P., Gahmberg C.G. Intercellular adhesion molecule-2 (CD102) binds to the leukocyte integrin CD11b/CD18 through the A domain. J. Immunol. 1995;155:3619–3628. [PubMed] [Google Scholar]
- Furman M.I., Krueger L.A., Frelinger A.L., III, Barnard M.R., Mascelli M.A., Nakada M.T., Michelson A.D. Tirofiban and eptifibatide, but not abciximab, induce leukocyte-platelet aggregation Circulation 100 1999. 1 681(Abstr.) [Google Scholar]
- Silverstein R., Asch A.S., Nachman R.L. Glycoprotein IV mediates thrombospondin-dependent platelet-monocyte and platelet-U937 cell adhesion. J. Clin. Invest. 1989;84:546–552. doi: 10.1172/JCI114197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M.P., Lucas C.M., Burns G.F., Chesterman C.N., Berndt M.C. GMP-140 binding to neutrophils is inhibited by sulfated glycans. J. Biol. Chem. 1991;266:5371–5374. [PubMed] [Google Scholar]
- Moore K.L., Varki A., McEver R.P. GMP-140 binds to a glycoprotein receptor on human neutrophilsevidence for a lectin-like interaction. J. Cell Biol. 1991;112:491–499. doi: 10.1083/jcb.112.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon D.I., Chen Z., Seifert P., Edelman E.R., Ballantyne C.M., Rogers C. Decreased neointimal formation in Mac-1−/− mice reveals a role for inflammation in vascular repair after angioplasty. J. Clin. Invest. 2000;105:293–300. doi: 10.1172/JCI7811. [DOI] [PMC free article] [PubMed] [Google Scholar]