

The essence of the immune system is built on two separate foundation pillars: one is specific or adaptive immunity which is characterized by relatively slow response kinetics and the ability to remember. The other is nonspecific or innate immunity exhibiting rapid response kinetics but lacking memory. Lymphocytes are the key players of the adaptive immune system. Each lymphocyte expresses antigen receptors of unique specificity. Upon recognizing an antigen via the receptor, lymphocytes proliferate and develop effector function. Few lymphocytes exhibit specificity for a given antigen or pathogen and massive proliferation is usually required before an effector response can be measured. Hence, the slow kinetics of the adaptive immune system. Because a significant proportion of the expanded lymphocytes survive and may maintain some effector function after elimination of the antigen, the adaptive immune system reacts faster when encountering the antigen a second time. This is the basis of its ability to remember. In contrast to the situation with lymphocytes, where specificity for a pathogen is confined to few cells that must expand to gain function, the cells and molecules of the innate immune system are usually present in massive numbers and recognize a limited number of invariant features associated with pathogens 1. Examples of such patterns include LPS, nonmethylated CG-rich DNA (CpG), or double-stranded RNA, which are specific for bacterial and viral infections, respectively. Most research in immunology has focused on the adaptive immune system and only recently has the innate immune system entered the focus of interest. Historically, the adaptive and innate immune system were treated and analyzed as two separate entities that had little in common. Such was the disparity that few researchers wondered why antigens were much more immunogenic for the specific immune system when applied with adjuvants that stimulated innate immunity 2 3 4. However, the answer posed by this question is critical to our understanding of the immune system and for comprehending the balance between protective immunity and autoimmunity. This commentary tries to highlight a few important links between the innate and adaptive immune systems and how those may impinge on the course of autoimmune diseases.
Association of Autoimmunity with Infection: Molecular Mimicry versus Aberrant Presentation of Self-Antigens.
It has long been suspected that infections may be responsible for the induction or precipitation of autoimmune diseases. Prominent examples include an association of chlamydial infection with heart disease or Coxsackie virus infection with type I diabetes. A favored explanation for the potential of pathogens to cause autoimmunity is known as molecular mimicry. It is suspected that the pathogen shares T or B cell epitopes with the host which may result in the induction of a self-specific immune response upon infection (Fig. 1 a; reference 5). This view is supported by the observation that pathogen-specific T cell clones readily cross-react with defined self-peptides 6 and sometimes cause disease in animal models. Further support for the notion of molecular mimicry comes from the fact that when self-epitopes are displayed in a highly repetitive manner on viral surfaces, B cell tolerance can be broken 7 8. Nevertheless, it has recently become evident that cross-reactivity between pathogen-derived and self-derived antigens may not always be the cause of infection-induced autoimmunity. Bystander T cell activation may in fact be as important a factor as molecular mimicry (Fig. 1 a; references 9 and 10). Cell damage is an inevitable side effect of any infection, be it through direct pathogen-mediated cell destruction or through the action of CTLs. Self-antigens are released during this process and taken up and processed by macrophages and dendritic cells (DCs). Simultaneously, these APCs become activated by the above mentioned pathogen-associated “nonspecific” factors such as LPS, CpGs, double-stranded RNA, or possibly even cell debris. This would serve to increase their antigen-processing ability, upregulate expression of costimulatory molecules, and trigger their migration to secondary lymphoid organs where they efficiently prime T cell responses 11 12. Surprisingly, it is not only pathogen-derived structures that are able to nonspecifically trigger the maturation program in DCs under these conditions. Factors secreted by T cells also induce maturation of virtually all DCs in lymphoid organs during viral infections 13. This brings adaptive and innate immunity closer together and further blurs their differences. Thus, generalized activation of APCs may often be the cause for triggering self-specific lymphocytes and autoimmunity. This view is compatible with a past observation that administration of LPS together with thyroid extracts is able to overcome tolerance and trigger autoimmune thyroiditis 14. Moreover, in a transgenic mouse model, it was recently shown that administration of self-peptide alone failed to cause autoimmunity unless APCs were activated by a separate pathway 15. The link between innate immunity and autoimmune disease is further underscored by the observation that LPS, viral infections, or generalized activation of APCs delays or prevents the establishment of peripheral tolerance 16 17 18. In this way, innate immunity not only enhances the activation of self-specific lymphocytes but also inhibits their subsequent elimination.
Figure 1.
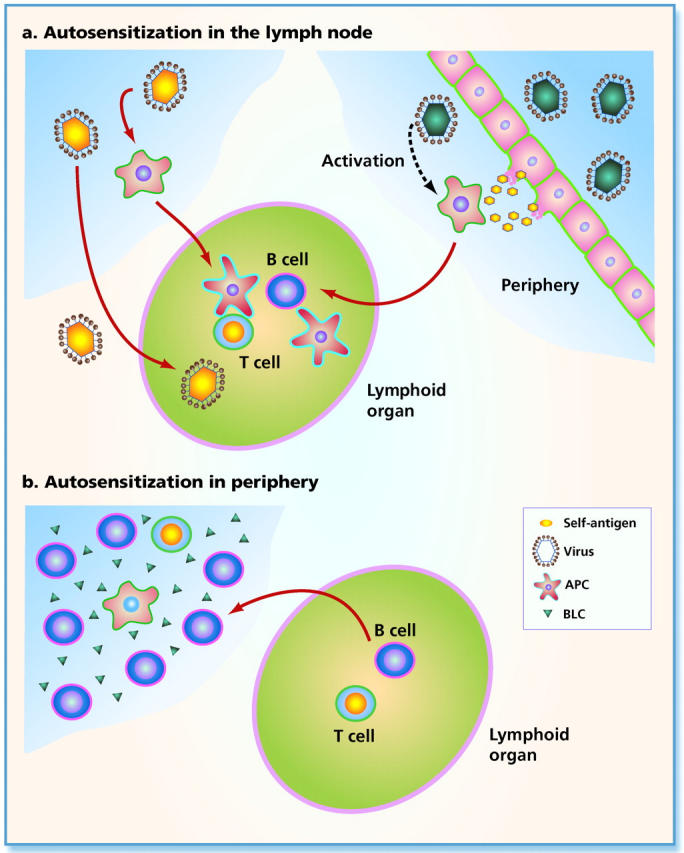
Models for autosensitization of lymphocytes. (a) Self-antigens are transported to lymphoid organs where activation of self-specific lymphocytes occurs. On the left side is indicated that pathogens carrying B or T cell epitopes shared with the host may directly reach lymphoid organs for induction of an immune response. Alternatively, pathogens may infect DCs or are processed by them. Activated DCs subsequently migrate to lymphoid organs and induce autoimmunity. On the right side is illustrated that pathogens may induce cell lysis, either directly or by activation of cytotoxic T cells. Self-antigens (shown in yellow) are released during this process and are taken up and processed by local DCs. In addition, the DCs are stimulated by pathogen-associated patterns triggering their migration to lymphoid organs where autosensitization occurs. (b) Lymphocytes are recruited to the periphery, where activation of self-specific lymphocytes occurs. Chemokines, such as BLC (triangles), are secreted by DCs which attracts B cells from lymphoid organs and probably the peritoneum. B1 cells may be attracted preferentially under these conditions. These B cells together with additionally recruited T cells and the BLC-producing DCs generate an environment ideal for activation of lymphocytes and autosensitization may occur locally.
Induction of Controlled Autoimmunity: A Challenge for Tumor Therapy.
Rationalized manipulation of the innate immune system to deliberately induce a self-specific T cell response provides a means for T cell–based tumor therapy. Hence, the focus of most current therapies is on the use of activated DCs as antigen carriers for the induction of sustained T cell responses 19. Similarly, in vivo activators of the innate immune system, such as CpGs or anti-CD40 antibodies, are being incorporated into the vaccines in order to enhance their immunogenicity 20 21. Considerable success has been achieved by using these approaches. Nevertheless, concommitant induction of autoimmunity is a common side effect of tumor immunotherapy. Melanoma therapy with tyrosinase-derived peptides is an example, where normal melanocytes are sometimes destroyed by CTLs leading to vitiligo (white patches in defined regions of the skin). As long as the affected cell types are dispensable, this may not cause severe complications. However, prolonged treatment of transgenic mice with DCs presenting the neo–self-antigen rapidly induces de novo formation of lymph node–like organs in peripheral organs and lethal autoimmunity, illustrating the fine line one has to walk for CTL-based immunotherapy 22.
A Link between Chemokine Expression and Autoimmunity.
In both mechanisms of autoimmunity discussed so far, the self-antigen or mimic thereof is brought to B and T cells in lymphoid organs, leading to their activation (Fig. 1 a). The paper by Ishikawa et al. in this issue 23 now shows that the reverse may also occur, namely that lymphocytes are attracted to self-antigens in the periphery, causing autoimmunity (Fig. 1 b). The molecule responsible for this pathological migration of lymphocytes was B lymphocyte chemokine (BLC; CXCL13, B cell-attracting chemokine 1 [BCA-1]), a member of the chemokine family. The physiological role of BLC is to orchestrate the generation and maintenance of B cell follicles in lymph nodes and spleen, by attracting CXCR5 receptor–expressing B cells 24. Upon stimulation by BLC, B cells produce lymphotoxin β (LT-β), which is essential for the generation of follicular DCs and formation of B cell follicles 25. Also, expression of secondary lymphoid tissue chemokine (SLC) is induced which attracts T cells (in addition to BLC) and leads to the formation of T cell regions adjacent to the B cell follicles 26. Thus, BLC acts as a Spemann's organizer and its presence alone is sufficient for the induction of lymphoid organs in any part of the body. Indeed, overexpression of BLC in the pancreas of transgenic mice has previously been shown to induce local lymph node–like organs, and insulitis, giving first indications that ectopic expression of chemokines may be linked to autoimmunity 26. The paper by Ishikawa et al. 23 now presents compelling evidence for a role of BLC in inducing lupus-like symptoms in (NZB × NZW)F1 (BWF1) mice. In these mice, BLC expression was increased up to 10,000-fold in organs such as thymus and kidney, which are typically infiltrated by lymphocytes. Surprisingly, the authors found that myeloid DCs rather than follicular DCs were the major producers of BLC in these afflicted organs, again indicating a pivotal role for the DCs in regulating the balance between protective immunity and autoimmune disease. The molecular mechanism for this disregulated expression and localization of BLC so far remains unknown. Similarly as observed previously in the transgenic mouse expressing BLC in the pancreas 26, the authors here report large B cell–dominated infiltrates in the thymus and kidney. Interestingly, B1 cells were particularly frequent within these infiltrates, an observation that is consistent with an old hypothesis that B1 cells are critical for disease in BWF1 mice. Also, the authors demonstrate that B1 cells migrate more efficiently to BLC than conventional B2 cells, which seems explained by the fact that peritoneal B1 cells express higher levels of CXCR5 than splenic B2 cells. Therefore, enhanced expression of BLC in DCs of BWF1 mice may attract B1 cells and later also T cells, thus creating a lymphoid environment in a nonlymphoid organ — a situation tailored to cause autoimmunity. B1 cells are discussed as the major source of natural antibodies and are prone to produce autoantibodies. This is in part because they originate in the peritoneum, which is secluded from many self-antigens 27, and also because they may be selected rather than deleted by self-antigen 28. It is therefore conceivable that the presence of elevated numbers of B1 cells in the thymus or kidney of mice leads to the production of self-specific antibodies involved in disease progression. It will be interesting to see whether lymphoid-like organs observed in patients with Hashimoto's thyroiditis or type I diabetes may have a similar origin. Moreover, these observations offer potential therapeutic opportunities for the treatment of some autoimmune diseases. Instead of treating autoimmunity by generalized immunosuppression, it may be sufficient to block the action of BLC, or its downstream effector molecule LT-β. This could serve to inhibit chronic inflammation of target organs and thereby prevent the vicious circle of attracting lymphocytes to peripheral organs, which in turn leads to autoimmunity and consequently enhanced target organ inflammation. As blocking LT-β virtually eliminates lymph node–like structures in the BLC transgenic mouse model 26, this may indeed be an avenue to follow.
In conclusion, the innate immune system is able to facilitate autosensitization in various ways (Fig. 1). Immune responses specific for self-antigens may be enhanced by innate immunity mostly through APC activation. Thus, APCs that carry self-antigen or a mimic thereof are stimulated by components of pathogens, resulting in enhanced presentation of self-antigens in lymphoid organs. On the other hand, as shown in the present paper by Ishikawa et al. 23, chemokines aberrantly produced in nonlymphoid organs may be able to attract lymphocytes into the periphery. Due to the capacity of chemokines to orchestrate together with lymphocytes de novo formation of lymph node–like organs, these lymphocytes may convert the usually poorly immunogenic peripheral environment into an organ prone to induce autoimmunity.
Acknowledgments
We would like to thank Gary Jennings and Christiane Ruedl for critically reading the manuscript and helpful discussions, and Pia Wildhaber for excellent secretarial assistance.
References
- Medzhitov R., Janeway C.A., Jr. Innate immunitythe virtues of a nonclonal system of recognition. Cell. 1997;91:295–298. doi: 10.1016/s0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- Janeway C. Approaching the Asymptote? Evolution and Revolution in Immunology 1989. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: pp. 13 [DOI] [PubMed] [Google Scholar]
- Zinkernagel R.M. Immunology taught by viruses. Science. 1996;271:173–178. doi: 10.1126/science.271.5246.173. [DOI] [PubMed] [Google Scholar]
- Matzinger P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- Oldstone M. Molecular mimicry and autoimmune disease. Cell. 1987;50:819–820. doi: 10.1016/0092-8674(87)90507-1. [DOI] [PubMed] [Google Scholar]
- Hausmann S., Wucherpfennig K.W. Activation of autoreactive T cells by peptides from human pathogens. Curr. Opin. Immunol. 1997;9:831–1838. doi: 10.1016/S0952-7915(97)80186-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann M.F., Hoffmann R.U., Kündig T.M., Bürki K., Hengartner H., Zinkernagel R.M. The influence of antigen organisation on B cell responsiveness. Science. 1993;262:1448–1451. doi: 10.1126/science.8248784. [DOI] [PubMed] [Google Scholar]
- Chackerian B., Lowy D.R., Schiller J.T. Induction of autoantibodies to mouse CCR5 with recombinant papillomavirus particles. Proc. Natl. Acad. Sci. USA. 1999;96:2373–2378. doi: 10.1073/pnas.96.5.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande S.P., Lee S., Zheng M., Song B., Knipe D., Kapp J.A., Rouse B.T. Herpes simplex virus-induced keratitisevaluation of the role of molecular mimicry in lesion pathogenesis. J. Virol. 2001;75:3077–3088. doi: 10.1128/JVI.75.7.3077-3088.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz M.S., Bradley L.M., Harbertson J., Krahl T., Lee J., Sarvetnick N. Diabetes induced by Coxsackie virusinitiation by bystander damage and not molecular mimicry. Nat. Med. 1998;4:781–785. doi: 10.1038/nm0798-781. [DOI] [PubMed] [Google Scholar]
- Cella M., Engering A., Pinet V., Pieters J., Lanzavecchia A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature. 1997;388:782–787. doi: 10.1038/42030. [DOI] [PubMed] [Google Scholar]
- Reis e Sousa C., Germain R.N. Analysis of adjuvant function by direct visualization of antigen presentation in vivoendotoxin promotes accumulation of antigen-bearing dendritic cells in the T cell areas of lymphoid tissue. J. Immunol. 1999;162:6552–6561. [PubMed] [Google Scholar]
- Ruedl C., Kopf M., Bachmann M.F. CD8(+) T cells mediate CD40-independent maturation of dendritic cells in vivo. J. Exp. Med. 1999;189:1875–1994. doi: 10.1084/jem.189.12.1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigle W.O. Analysis of autoimmunity through experimental models of thyroiditis and allergic encephalomyelitis. Adv. Immunol. 1980;30:159–273. doi: 10.1016/s0065-2776(08)60196-0. [DOI] [PubMed] [Google Scholar]
- Garza K.M., Chan S.M., Suri R., Nguyen L.T., Odermatt B., Schoenberger S.P., Ohashi P.S. Role of antigen-presenting cells in mediating tolerance and autoimmunity. J. Exp. Med. 2000;191:2021–2027. doi: 10.1084/jem.191.11.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella A.T., McCormack J.E., Linsley P.S., Kappler J.W., Marrack P. Lipopolysaccharide interferes with the induction of peripheral T cell death. Immunity. 1995;2:261–270. doi: 10.1016/1074-7613(95)90050-0. [DOI] [PubMed] [Google Scholar]
- Ehl S., Hombach J., Aichele P., Rulicke T., Odermatt B., Hengartner H., Zinkernagel R., Pircher H. Viral and bacterial infections interfere with peripheral tolerance induction and activate CD8+ T cells to cause immunopathology. J. Exp. Med. 1998;187:763–774. doi: 10.1084/jem.187.5.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell J.R., Campbell J.D., Kim C.H., Vella A.T. CD40 activation boosts T cell immunity in vivo by enhancing T cell clonal expansion and delaying peripheral T cell deletion. J. Immunol. 1999;162:2024–2034. [PubMed] [Google Scholar]
- Fong L., Engleman E.G. Dendritic cells in cancer immunotherapy. Annu. Rev. Immunol. 2000;18:245–273. doi: 10.1146/annurev.immunol.18.1.245. [DOI] [PubMed] [Google Scholar]
- Sotomayor E.M., Borrello I., Tubb E., Rattis F.M., Bien H., Lu Z., Fein S., Schoenberger S., Levitsky H.I. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat. Med. 1999;5:780–787. doi: 10.1038/10503. [DOI] [PubMed] [Google Scholar]
- Diehl L., den Boer A.T., Schoenberger S.P., van der Voort E.I., Schumacher T.N., Melief C.J., Offringa R., Toes R.E. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat. Med. 1999;5:774–779. doi: 10.1038/10495. [DOI] [PubMed] [Google Scholar]
- Ludewig B., Odermatt B., Ochsenbein A.F., Zinkernagel R.M., Hengartner H. Role of dendritic cells in the induction and maintenance of autoimmune diseases. Immunol. Rev. 1999;169:45–54. doi: 10.1111/j.1600-065x.1999.tb01305.x. [DOI] [PubMed] [Google Scholar]
- Ishikawa S., Sato T., Abe M., Nagai S., Onai N., Yoneyama H., Zhang Y., Suzuki T., Hashimoto S., Shirai T., Lipp M., Matsushima K. Aberrant high expression of B lymphocyte chemokine (BLC/CXCL13) by C11b+ CD11c+ dendritic cells in murine lupus and preferential chemotaxis of B1 cells towards BLC. J. Exp. Med. 2001;193:1393–1402. doi: 10.1084/jem.193.12.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser B., Loetscher P. Lymphocyte traffic control by chemokines. Nat. Immunol. 2001;2:123–128. doi: 10.1038/84219. [DOI] [PubMed] [Google Scholar]
- Ansel K.M., Ngo V.N., Hyman P.L., Luther S.A., Forster R., Sedgwick J.D., Browning J.L., Lipp M., Cyster J.G. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature. 2000;406:309–314. doi: 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- Luther S.A., Lopez T., Bai W., Hanahan D., Cyster J.G. BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis. Immunity. 2000;12:471–481. doi: 10.1016/s1074-7613(00)80199-5. [DOI] [PubMed] [Google Scholar]
- Murakami M., Tsubata T., Okamoto M., Shimizu A., Kumagai S., Imura H., Honjo T. Antigen-induced apoptotic death of Ly-1 B cells responsible for autoimmune disease in transgenic mice. Nature. 1992;357:77–80. doi: 10.1038/357077a0. [DOI] [PubMed] [Google Scholar]
- Wortis H.H., Berland R. Cutting edge commentaryorigins of B-1 cells. J. Immunol. 2001;166:2163–2166. doi: 10.4049/jimmunol.166.4.2163. [DOI] [PubMed] [Google Scholar]