

Abstract
Serum antibody (Ab) can play several roles during B cell immune responses. Among these is to promote the deposition of immune complexes (ICs) on follicular dendritic cells (FDCs). ICs on FDCs are generally thought to be critical for normal germinal center (GC) formation and the development and selection of memory B cells. However, it has been very difficult to test these ideas. To determine directly whether FDC-bound complexes do indeed function in these roles, we have developed a transgenic (Tg) mouse in which all B lymphocytes produce only the membrane-bound form of immunoglobulin M. Immune Tg mice have 10,000-fold less specific Ab than wild-type mice and lack detectable ICs on FDCs. Nonetheless, primary immune responses and the GC reaction in these mice are robust, suggesting that ICs on FDCs do not play critical roles in immune response initiation and GC formation. Moreover, as indicated by the presence and pattern of somatic mutations, memory cell formation and selection appear normal in these IC-deficient GCs.
Keywords: B lymphocyte, transgenic mouse, surface immunoglobulin, lambda light chain, immunological memory
Introduction
Immune responses in the spleen begin with proliferating B and T cells at the interface of the T and B cell zones 1 2. Within a few days, early Ab-forming cells (AFCs) are seen, which provide the first sources of serum Ab. The B cells that participate in the early extrafollicular reaction are generally of low affinity 3. By day 6 after immunization, the first signs of the germinal center (GC) reaction are evident. Over the next 10 d, the GC reaction peaks, culminating in the selection of higher affinity mutants and the differentiation of both memory cells and plasma cells.
Follicular dendritic cells (FDCs) are thought to play a key role in the formation of GCs, as well as in selection, differentiation, and maintenance of memory B cells. FDCs are dendritiform, nonphagocytic cells found in the follicles of secondary lymphoid organs 4. A striking characteristic of FDCs is their ability to retain native immune complexes (ICs) on their surface for long periods of time 5. The half-life of radiolabeled Ags on FDCs is months, if not years 6. Complement receptors (CRs) provide the main mechanism for ICs trapping on FDCs. C3 fragments bound to ICs bind CR1 (CD35) and CR2 (CD21), which are highly expressed on FDCs 7 8. Depletion of C3 by cobra venom factor inhibits Ag localization to FDCs 9, as does treatment with anti-C3 10 or anti-CR1/2 11. ICs also bind Fc receptors on FDCs, particularly during the GC response 11. Hence, except for certain Ags that might fix sufficient complement autonomously, both mechanisms for Ag retention by FDCs require that Ag be complexed with Ab.
Several important functions have been postulated for the ICs that deposit on FDCs. It has been hypothesized that the initial localization of ICs on FDCs nucleates the GC response and provides the Ag to which GC B cells respond 6 12. This idea provides an explanation for the delayed appearance of GCs, since several days would be needed to provide the Ab required to allow ICs to form and deposit. Not all earlier data agree with this role of IC deposition on FDCs. Kroese et al. reported that rats which are reconstituted with mature lymphocytes after irradiation can make early GC responses to SRBC immunization even though their FDCs are transiently impaired in IC trapping at the time GCs are first observed 13. ICs on FDCs have also been suggested to provide the stimulus that rescues centrocytes from apoptosis 14, thus leading to the formation of memory B cells. The initiation of somatic mutation, which normally occurs in the GC, may also depend on encounter with Ag on the FDCs. Competition with previously formed Ab for Ag sequestered in ICs on FDCs is thought to drive affinity maturation of B cells 15. Thus, ICs are thought to drive the initiation and normal progression of GC and memory cell development.
Although these proposed roles for ICs retained on FDCs fit with various correlative data, they have been difficult to evaluate experimentally. Mice that lack TNFR family receptors or ligands lack FDCs and also generally lack GCs. The study of immune responses in these animals has been used to draw conclusions about the roles of FDCs 16 17. Although there are clearly some memory formation defects in these mice, the interpretation of these data is complicated. It is difficult to attribute the mechanism of immune response defects because these mice have markedly disorganized lymphoid architecture, as well as carry mutations in key cytokine or chemokine pathways 18 19 20 that may have direct effects on lymphocyte responses. Moreover, studies in these mutant strains do not distinguish between the role of FDC-bound ICs and other essential functions of the FDCs since neither are present.
An additional informative approach has been the study of mice that lack CR1/2, in which the FDC's ability to capture Ag is reduced. These mice make very weak Ab responses to T cell–dependent Ags, as well as reduced GC responses 21 22. However, at least a substantial part of the deficiency was due to disruption of CR1/2 expression on B cells, and not primarily due to FDCs lacking the ability to capture ICs via CR1/2 21 23. In any case, Ag may deposit on FDCs via Fc receptors even in the absence of the CR1/2 receptor 11.
To better define the roles of ICs on FDCs, it would be advantageous to study B cell immune responses in the absence of secreted Ab and hence in the absence of IC deposition on FDCs. To accomplish this, we have developed a transgenic (Tg) mouse in which B cells are functional but cannot secrete Ab. The membrane Ig (mIg) Tg consists of a rearranged VDJ joined to an IgM H chain from which the secreted exon and polyadenylation site had been excised 24. We chose to use Vh186.2 so that pairing of the Tg H chain with endogenous λ light chain would result in a small, readily detectable population of cells specific for the hapten (4-hydroxy -3-nitrophenyl)acetyl (NP). As mIg Tg mice have markedly reduced Ab and undetectable IC deposition on FDCs, we have used them to investigate the role of Abs in the development and maintenance of primary and secondary immune responses.
Materials and Methods
mIg Construct and Tg Mouse Production.
The mIg H chain construct was as described 24. In brief, a rearranged VDJ segment containing Vh186.2 was ligated to an IgMa constant region from which the secreted exon and polyadenylation site had been excised, resulting in production of only membrane-bound Ig. The construct was tested by transfection into CH1, a λ/IgMb-expressing cell line 25. CH1-mIg transfectants expressed surface IgMa anti-NP, yet did not secrete any detectable IgMa into culture supernatant (data not shown). A control strain, the membrane plus secreted Ig ([m+s]Ig) Tg, was produced using a similar construct with an intact secreted exon.
Tg-positive founders were identified by Southern blot. mIg Tg mice were first backcrossed with CB.17 mice (IgMb congenics of BALB/c). Levels of Tg-derived surface IgMa expression on B cells from the founder lines were compared with the (m+s)Ig control strain by FACS®; two that matched the (m+s)Ig Tg mice closely were chosen for study. To eliminate endogenous H chain production, the mIg Tg × CB.17 mice were backcrossed onto the Jh knockout strain (JhD/CB.17). The resulting strain, referred to as mIg Tg, has >99% CB.17 background genes. mIg Tg mice are housed under specific pathogen-free conditions and receive Sulfatrim in drinking water 4 d/wk.
Ags and Immunizations.
Chicken gamma globulin (CGG; Sigma-Aldrich) and human serum albumin (HSA; Amour Pharmaceutical) were haptenated with NP-hydroxysuccinimide ester (Cambridge Research Biochemicals/Genosys). 7–12-wk-old mice were immunized intraperitoneally with 50 μg alum-precipitated NP24CGG or NP15HSA. For secondary immunizations, 50 μg soluble Ag was administered in PBS.
ELISA.
Anti-NP was measured as λ+ Ig that binds to (4-hydroxy-3-nitro-2-iodo-phenyl)acetyl (NIP)-BSA–coated plates. In the context of the Vh186.2 Tg, this should account for virtually all anti-NP, and includes all Ig H chain isotypes. Immulon 2 plates (Dynex Technologies) were coated overnight with NIP26-BSA. Plates were blocked with 1% BSA, followed by incubation with serial dilutions of serum at 37°C for 1 h. After washing and incubation with polyclonal anti-λ–alkaline phosphatase (Southern Biotechnology Associates, Inc.) plates were developed with p-nitrophenyl phosphate (Sigma-Aldrich). For anti-IgM ELISA, plates were coated overnight with b7-6 (anti-IgM) and blocked with 1% BSA; anti-IgM–alkaline phosphatase (Southern Biotechnology Associates, Inc.) was used as the secondary Ab.
Flow Cytometry.
FACS® was performed as described 26. PE (Molecular Probes) haptenated with NIP-hydroxysuccinimide ester (NIP-PE; Cambridge Research Biochemicals/Genosys) was used to identify NP-specific B cells, as Vh186.2/λ recognizes NIP with high affinity. In addition to NIP-PE, splenocytes were stained with anti-λ–fluorescein (Southern Biotechnology Associates, Inc.), and GL-1–biotin (anti–B7-2) followed by streptavidin–Spectral red (Southern Biotechnology Associates, Inc.).
Histology.
5 μm frozen spleen sections were thaw mounted onto poly-l-lysine (Sigma-Aldrich)–coated slides, fixed in cold acetone for 10 min, and stored at −80°C. Tissue was blocked with 1% BSA (Gemini Bioproducts) in PBS/0.1% Tween 20, or with 1% ovalbumin (ICN Biomedicals) containing 10% rat serum (Gemini Bioproducts) when staining with mouse anti-HSA. Sections were stained with peanut agglutinin–horseradish peroxidase (PNA-HRP; EY Labs), anti-λ–alkaline phosphatase (Southern Biotechnology Associates, Inc.), anti-CD35–biotin (BD PharMingen), anti-CGG–biotin (Accurate), or anti-HSA–biotin. Anti-HSA was affinity purified from serum of HSA-immune BALB/c mice; serum was complement inactivated at 56°C for 30 min, diluted 1:1 with PBS containing 0.05% sodium azide, and run over a column of HSA-coupled Affi-Gel 15 (Bio-Rad Laboratories). Eluted anti-HSA was then conjugated to biotin-XX (Molecular Probes).
In Vitro Deposition of ICs.
Goat anti-CGG (Southern Biotechnology Associates, Inc.) and CGG protein were mixed together at a weight ratio of 2:1 with fresh 10% mouse serum in PBS on ice for 30 min to form ICs at the dilutions indicated. ICs were incubated at 37°C for 1 h on acetone-fixed sections of spleen from an mIg Tg mouse that had received 50 μg NP-CGG in alum 12 d earlier. The deposited complexes were detected with a biotinylated donkey F(ab′)2 anti-CGG Ab (Accurate) and developed with streptavidin–alkaline phosphatase reagent. The in vivo–localized Ag was developed at the same time in both the mIg Tg mouse (same mouse as used for the in vitro localization) and the day 9 immune (m+s)Ig Tg mouse for negative and positive controls, respectively, using the same detection Ab and substrate.
Sequencing.
λ+ day 16 GCs were microdissected from stained spleen sections. Cell clusters were digested overnight at 37°C in 20 μl 25% PBS containing 10 μg proteinase K. Vλ1 sequences were amplified by nested PCR using Pfu (Stratagene): external primers 5′-GCACCTCAAGTCTTGGAGAG-3′ and 5′-ACTCTCTCTCCTGGCTCTCA-3′, and internal primers 5′-CTACACTGCAGTGGGTATGCAACAATGCG-3′ and 5′-GTTCTCTAGACCTAGGACAGTCAGTTTGG-3′. Amplified DNA was isolated by phenol/chloroform/isoamyl alcohol (25:24:1) extraction followed by gel purification, digestion of gel blocks with GELase (Epicentre), and ethanol precipitation. Vλ1 DNA was ligated via T4 DNA ligase (New England Biolabs, Inc.) to pBluescript II KS (Stratagene) cut with PstI and XbaI (New England Biolabs, Inc.). Ligation products were transformed into DH5α. Vλ1 DNA was further amplified by placing colonies directly into PCR reactions containing the following primers: 5′-AATTAACCCTCACTAAAGGG-3′ and 5′-GTAATACGACTCACTATAGGGC-3′. DNA was purified from the PCR reaction mixture with GFX DNA and Gel Band Purification kit (Amersham Pharmacia Biotech), mixed with sequencing primer 5′-TCGAGGTCGACGGTATC-3′, and sequenced by the Keck Biotechnology Resource Laboratory at Yale University School of Medicine using Applied Biosystems 377 DNA sequencers.
Statistics.
The unpaired t test was performed using StatView v4.5 (Abacus Software). P < 0.05 was considered significant.
Results
The mIg Tg Reconstitutes NP Binding by λ1 B Cells but without Detectable Anti-NP Ab.
The mIg Tg has been backcrossed onto the Jh knockout strain (JhD/JhD), and thus only the Tg-encoded H chain V region is expressed. As predicted, expression of Vh186.2 in all B cells yields 2–4% of splenocytes in an unimmunized mIg Tg mouse that are NP specific and λ+ by FACS® analysis (Fig. 1). The control (m+s)Ig Tg strain, made from an otherwise identical construct that retains the intact secreted exon, has a similar NP-specific population. To confirm that mIg Tg B cells were not capable of secreting IgM upon stimulation in vitro, mIg Tg splenocytes were cultured for 5 d with 50 μg/ml LPS (Fig. 2). As expected, mIg Tg splenocytes did not produce any detectable IgM. Splenocytes from mIg Tg mice with intact endogenous Ig Jh alleles did produce IgM, presumably emanating from a small number of B cells that expressed endogenous H chains.
Figure 1.

The mIgM Tg rescues B cell development and confers specificity for NIP on λ+ B cells when expressed on the JhD background. FACS® profiles of splenocytes from CB.17 (left) or mIg Tg mice (JhD background) are shown. The top row demonstrates sole expression of IgMa in the mIg Tg mice. The bottom row demonstrates that λ+ B cells in the mIg Tg mice bind to NIP (as NIP-PE) in proportion to the amount of surface λ. There are practically no detectable λ+ NP-binding cells in the CB.17 mice (bottom left).
Figure 2.

Lack of secretion of IgM from mIg Tg mice after stimulation by LPS in vitro. Splenocytes from mIg Tg mice (i.e., on the JhD background) and the original mIg × CB.17 mice (which expressed some endogenous H chain) were cultured with 50 μg/ml LPS for 5 d to determine their capacity to secrete IgM in vitro. Supernatants from multiple wells were pooled and total IgM was quantitated by ELISA.
No NP-specific Ig could be detected in sera from naive mIg Tg mice, even with an assay sensitivity of 20 ng/ml; indeed, mIg Tg sera were indistinguishable from JhD/JhD control sera (data not shown). Anti-NP concentrations in unimmunized (m+s)Ig Tg and CB.17 mice were in the range of 1 μg/ml.
mIg Tg Mice Mount Primary Responses.
Having eliminated preexisting Ab, we asked whether mIg Tg mice would develop a normal primary response, since ICs might be important in the initial stimulation of B cells 27 28. To determine if primary responses in mice with similar B cell repertoires would be diminished by lack of Ab, mIg Tg and (m+s)Ig Tg mice were immunized with NP-HSA, NP-CGG, or CGG alone, and splenocytes were analyzed by FACS® between days 0 and 28 (Fig. 3). Ag-specific B cells bound NIP-PE and expressed λ light chain (Fig. 1). In NP-immunized mIg Tg and (m+s)Ig Tg mice, Ag-specific B cells comprised an increasingly large percentage of the splenocytes (Fig. 3 A), peaking between days 12 and 16. Similar effects were seen on the total number of NP-specific cells (data not shown). There is a similar proportional increase of NP-specific cells in both strains. By day 28, the percentage of NP-specific cells in both Tg strains had dropped, although not yet back to baseline levels. NP-specific B cells in carrier-immunized controls remained near baseline levels.
Figure 3.

Time course analysis of primary response to NP immunization. mIg and (m+s)Ig Tg splenocytes were analyzed by FACS® at various time points after immunization with NP-HSA, NP-CGG, or CGG alone. (A) Mean percentages of NP-specific splenocytes; (B) the mean percentage of NP-specific splenocytes that are B7-2high. Error bars indicate SEM. Numbers of mice: d0, n = 4; d28, n = 3; d5–16, NP-HSA, n = 3; CGG mIg Tg, n = 4–5; NP-CGG mIg Tg, n = 5–6; CGG (m+s)Ig Tg, n = 3; NP-CGG (m+s)Ig Tg, n = 4–7. Data are combined from three experiments.
To provide evidence of Ag-specific activation of mIg Tg B cells, we determined the percentage of NP-specific splenocytes expressing high levels of B7-2 after immunization with NP or carrier (Fig. 3 B). The percentage of B7-2high NP-specific cells peaks at day 12 in both mIg Tg and (m+s)Ig Tg mice, whereas levels in carrier-immunized mice remain low. These results indicate that mIg Tg B cells become activated upon immunization, with kinetics similar to those of (m+s)Ig Tg controls.
Ab Levels in mIg Tg Serum Are Markedly Reduced.
In spite of robust proliferative responses that resulted in 15–20% of total splenocytes carrying the NP specificity, serum Ab responses to immunization in mIg Tg mice were severely blunted and required an ultrasensitive assay for detection. At day 12 after immunization with NP-HSA, there was a mean of 90 ng/ml anti-NP in mIg Tg sera; five of six mice had ≤60 ng/ml (Fig. 4). This quantity of NP Ab is 100-fold less than in immunized (m+s)Ig Tg mice and 10,000-fold less than in immunized CB.17 mice, despite the much higher precursor frequency of NP-specific B cells in mIg Tg mice than in CB.17 mice.
Figure 4.

Ab secretion is markedly reduced in mIg Tg mice. NP Ab levels from mIg Tg, (m+s)Ig Tg, and CB.17 mice bled on day 12 after receiving 50 μg NP-HSA. Error bars indicate SEM. Note that mIg Tg mice produce ∼10,000-fold less NP Ab compared with wild-type CB.17 mice.
It was surprising to find any serum Ab in mIg Tg mice since endogenous Jh loci in these mice are inactivated. Shedding of mIgM is unlikely to account for this, given our inability to detect any secreted IgM in vitro after LPS stimulation. Some of the Ab is IgG (data not shown), and thus we believe that these minute quantities of Ab arise via rare interchromosomal isotype switch events between the Tg locus and the endogenous IgH loci 29. Regardless of the mechanism by which this Ab emerges, the quantity of anti-NP at day 12 after immunization in mIg Tg mice is three orders of magnitude less than the amount of immunizing Ag, whereas in CB.17 mice, Ab is far in excess of Ag. This indicates that if any ICs form in the mIg Tg model, the amount must be radically reduced. In particular, the likelihood that two Ab molecules would bind Ag in close proximity, as required for C′ activation, is remote given the extreme Ag excess.
mIg Tg Mice Produce NP-specific GCs.
The second question addressed in these studies was whether GCs would form in the absence of IC deposition. This question was raised by the suggestion that early and/or preexisting Ab was required in order to promote the shift to the GC reaction 12. If so, GC formation in mIg Tg mice—with no preexisting anti-NP and a 4-log reduction in Ab at a time when the GC response peaks—should be impaired. To investigate this, spleen sections from mIg Tg, (m+s)Ig Tg, and CB.17 mice, removed on day 12 after immunization, were double-stained with PNA and anti-λ to identify λ+ (NP-specific) GCs. mIg Tg mice produced NP-specific GCs upon immunization with either NP-CGG or NP-HSA; carrier-immunized mIg Tg mice produced only λ− (presumably carrier-specific) GCs (Fig. 5 and Fig. 6). The number of NP-specific GCs present in the mIg Tg spleens was equal to or greater than that in the (m+s)Ig Tg or CB.17 spleens (Fig. 6), indicating that the mIg Tg GC response is not diminished by lack of Ab or ICs. Indeed, as is evident from the substantially greater size of the GCs in both types of Tg mice compared with those in CB.17 mice (Fig. 6), the Tg mice make extremely vigorous GCs. Moreover, the kinetics of the GC response is similar in the two types of Tg mice (data not shown). The normal GC response in mIg Tg mice is not solely attributable to the enriched anti-NP repertoire, as carrier Ags also induce GCs despite the presumably below normal precursor frequencies for the carrier epitopes (Fig. 5 B, Fig. 6, and data not shown).
Figure 5.
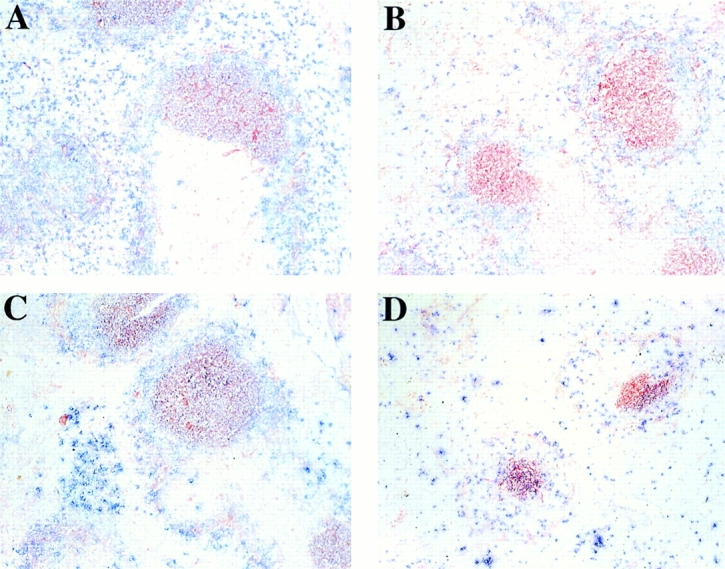
mIg Tg mice produce GCs. Spleens from mIg Tg, (m+s)Ig Tg, and CB.17 mice were removed on day 12 after immunization with NP-CGG or CGG. Spleen sections were stained with PNA-HRP (red) to detect GCs and anti-λ light chain (blue) to detect λ-expressing B cells. Immunization with NP-CGG produces λ+ NP-specific GCs: (A) mIg Tg, (C) (m+s)Ig Tg, (D) CB.17. Immunization with CGG produces only λ− GCs (B; mIg Tg). Note the much larger size of the GCs from mIg Tg and (m+s)Ig Tg mice (A–C) compared with wild-type mice (D).
Figure 6.

mIg Tg mice make a normal number of GCs in response to immunization. The mean number of NP-specific GCs per random 40× view of spleen sections; error bars indicate SEM. Means represent counts from three to eight mice per treatment group. Bar heights show total number of GCs. The number of λ+ GCs is indicated by the hatched bars and the number of λ− GCs by the gray bars. unim, unimmunized.
Lack of Detectable Ag Localization in mIg Tg Mice.
To confirm that the marked reduction in specific Ab levels in mIg Tg mice prevented normal localization of Ag to the FDCs, adjacent spleen sections were stained with a polyclonal antiserum specific for the protein Ag carrier or with monoclonal anti-CD35, along with PNA. This strategy identifies Ag deposited on FDCs in GCs. A rigorous test of Ab-mediated localization of Ag is secondary immunization, in which case any leakiness in the system should be revealed. 5 wk after receiving 50 μg NP-CGG, mIg and (m+s)Ig Tg mice were given 50 μg NP-CGG intravenously in PBS. Spleens were removed at day 3 after reimmunization, and serial spleen sections were stained. There was no detectable localization of Ag in mIg Tg spleens, whereas numerous FDC networks in (m+s)Ig Tg spleens stained darkly with anti-CGG (Fig. 7A–D). Similar results were achieved when mIg and (m+s)Ig Tg mice were reimmunized with NP-HSA 12 wk after primary immunization and spleen sections were stained with anti-HSA (Fig. 7E–H). Note from the serial sections stained with anti-CR1/2 to reveal FDCs that the pattern of Ag deposition matched the distribution of FDCs. We also investigated primary responses: as expected from the secondary response data, at day 12 after NP immunization, no Ag was detectable in mIg Tg mice, whereas Ag was clearly visible in many (m+s)Ig Tg GCs (Fig. 8 and data not shown).
Figure 7.
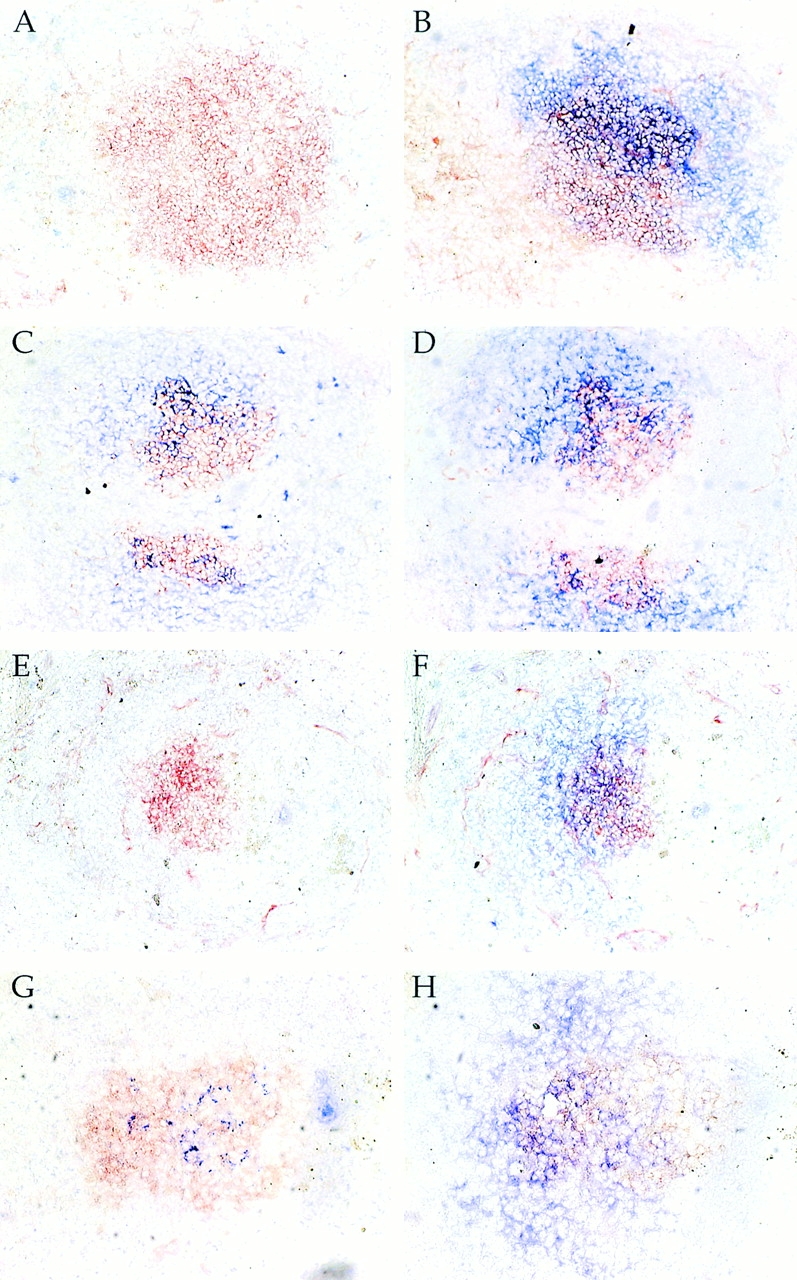
Lack of Ag localization in mIg Tg mice. Spleens were removed on day 3 or 5 after secondary immunization with NP-CGG (A–D) or NP-HSA (E–H), respectively, from either mIg Tg (A, B, E, and F) or (m+s)Ig Tg (C, D, G, and H) mice. Serial spleen sections were stained with PNA-HRP to show GCs (red), plus anti-CGG (A and C) or anti-HSA (E and G) to detect localized Ag (blue), and anti-CD35 (B, D, F, and H) to visualize FDCs (blue). FDCs are present in mIg Tg mice; however, no Ag localization was detected in any mIg Tg GCs. Original magnification: ×40 (A–F) and ×100 (G and H).
Figure 8.

Estimation of the sensitivity of IC detection on FDCs in vivo. ICs that had been naturally deposited in vivo or artificially in vitro were detected using the same reagents at the same time, as described in detail in Materials and Methods. Since mIg Tg mice are negative in the in vivo deposition assay (B and see Fig. 7), we undertook this experiment to estimate the dynamic range of the assay and to establish the minimum amount of reduction needed in the mIg Tg in order to have a negative result. Thus, a dilution series was constructed for the in vitro ICs, which were then assayed on serial sections of mIg Tg spleen. All staining and development were performed simultaneously and stopped at the same time. A shows typical in vivo localization in (m+s)Ig Tg mice. B shows the lack of such in a mIg Tg mouse. D–F show various dilutions of the IC titrations: (C) 20 μg/ml; (D) 2.5 μg/ml; (E) 0.15 μg/ml; and (F) 0.05 μg/ml. The intensity of staining in C was comparable to the in vivo positive in A. At 400-fold less ICs (F), staining is barely detectable in nearly all GCs (F; and other similar GCs not shown), indicating an endpoint for the titration. Thus, there is an ∼400-fold dynamic range of the assay, and lack of staining roughly corresponds to a 400-fold or greater reduction in deposited ICs.
The sensitivity of detection was investigated using an in vitro IC-localization assay to construct a standard curve for comparison with simultaneous staining of in vivo samples (see Materials and Methods). In vitro–prepared ICs using CGG and a polyclonal anti-CGG were incubated in the presence of fresh mouse serum with spleen sections from day 12 immune mIg Tg mice. Since essentially all GCs in mIg Tg mice are λ+ and thus NP specific (see Fig. 6), there should be few if any B cells in GCs of these immune mice that could bind the carrier Ag CGG. Binding to FDCs was detected using labeled anti-CGG, as performed for in vivo Ag localization studies in Fig. 7. The quantities of ICs were then titrated and an endpoint dilution was established (Fig. 8). The pattern of this distribution resembles that of FDCs, as determined by staining with anti-CR1/2 on adjacent sections (data not shown). Moreover, no such localization was seen in the absence of a source of fresh C′ (data not shown), again strongly indicating that deposition on FDCs, rather than binding to B cells, is being observed. The dilution that gave similar intensity of staining to the in vivo localization that was detected simultaneously on sections from (m+s)IgM Tg mice was identified. By comparing this to the endpoint, a minimum estimate of the sensitivity was obtained. We estimate this to be 400-fold less than the intensity routinely seen in the (m+s)IgM Tg mice, thus attributing an order of magnitude to the meaning of lack of detectable ICs in the mIg mice.
mIg Tg GC B Cells Undergo Somatic Mutation of the Vλ Region.
Somatic mutation of Ig genes is a hallmark of memory B cells 30. Selection of mutations, presumably based on higher affinity, occurs concurrent with memory B cell development 31. In TNFR family knockout mice, memory B cell development was abnormal. To determine whether lack of ICs on FDCs affected memory B cell development by altering mutation or selection in the mIg Tg mice, we microdissected NP-specific GCs from spleen sections of mIg Tg and (m+s)Ig Tg mice at day 16 after immunization and sequenced the endogenous Vλ1 genes.
Clearly, mutation is occurring in mIg Tg mice, with the average Vλ having 1.4 mutations in ∼300 basepairs sequenced. This frequency is comparable to the number of Vh chain mutations in λ+ GCs of normal mice 32. In both mIg Tg and (m+s)Ig Tg mice, the numbers of replacement mutations in λ CDRs are significantly higher than would be predicted at random, suggesting that selection of mutants by Ag also takes place in mIg Tg GCs (Table ). We compiled a list of partial sequences showing all the Vλ codons that contained replacement mutations in any of the sequences from mIg Tg and (m+s)Ig Tg mice, plus those reported in NP-CGG–immunized C57BL/6 mice (33; Fig. 9). Several mutations seen in mIg Tg CDRs are also seen in (m+s)Ig Tg and normal mice. These recurrent amino acid replacements are found almost exclusively in the CDRs, again suggesting that Ag-driven selection is occurring and that it is qualitatively comparable to that in Ab-secreting mice.
Table 1.
Number of Replacement (R) and Silent (S) Mutations in Vλ1 Sequences from GC B Cells
| FR | CDR | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Strain | Ag | n | R | S | All | R | S | All | P value |
| mIg Tg | NP-CGG | 30 | 9 | 8 | 17 | 18 | 1 | 19 | 3.6 × 10−2 |
| mIg Tg | NP-HSA | 23 | 12 | 4 | 16 | 20 | 4 | 24 | 6.6 × 10−6 |
| (m+s)Ig Tg | NP-CGG | 16 | 16 | 1 | 17 | 23 | 2 | 25 | <10−6 |
P values are shown for the difference in CDR replacement mutations from expected frequencies. P values and the expected number of replacement mutations for the Vλ1 sequence were calculated using programs by Chang and Casali (reference 51).
Figure 9.

Vλ1 sequences from mIg Tg mice contain replacement (R) mutations common to (m+s)Ig Tg and nontransgenic mice. GC cells from mIg Tg and (m+s)Ig Tg mice were microdissected on day 16 after immunization with NP-CGG or NP-HSA, and Vλ1 sequenced. All mIg Tg and (m+s)Ig Tg Vλ1 codons containing replacement mutations in any of the sequences are listed, in addition to those reported in C57BL/6 mice after NP-CGG immunization by Taketani et al. (reference 50). The number of sequences represented is shown in parentheses; see Table for additional details. Shaded columns indicate codons at which mIg Tg sequences share mutations with (m+s)Ig Tg and/or published non-Tg sequences.
Discussion
These studies test several long-held ideas about the role of preexisting Ab and ICs in the normal development of B cell immune responses. By providing a scenario in which B cells exist in the near absence of Ab, the mIg Tg mouse model allows us to distinguish between processes in which ICs actually participate and those that simply coincide with the presence of Ag on FDCs. The ability to make these distinctions without resorting to gene deletions that result in pleiomorphic defects suggests a reevaluation of some of the current concepts for the role of FDC-bound ICs in primary B cell responses.
A 4-log reduction in specific Ab was achieved in the mIg Tg mice. Consistent with this reduction, there was no detectable deposition (>400-fold reduction) of Ag on FDCs in vivo in mIg Tg mice. The lack of IC deposition confirmed the widely held concept that such deposits are Ab dependent, with the ICs binding FcRs and CR1/2 11 on FDCs. Since Ag is deposited on FDCs as ICs, it seems likely that for most Ags the process is dependent on the classical pathway, an idea confirmed by our findings that observable deposition required the presence of Ab both in vivo and in vitro. In addition, immune responses are grossly normal in Factor B–deficient mice 34, unlike the situation for C4-deficient mice 35.
The proposed model we are testing is that the visible immune deposits on FDCs are the limiting factor for GC development, division, maturation, and memory B cell selection. Any process for which FDC-retained Ag is essential should display inhibition at least proportional to the lowered Ab level. Were they not the limiting factor, the functions attributed to these deposits in controlling the onset and size of the GC and in providing limiting amounts of Ag to promote affinity selection would not be possible. We have eliminated the main if not the only mechanism of IC deposition. According to the prevailing hypothesis, this should have resulted in a detectable decrease in some or all of these functions.
ICs were not required for a normal proliferative or GC response, since GC reactions of normal size, frequency, and kinetics were observed in mIg Tg mice. This is not simply an effect of high anti-NP precursor frequency, as carrier Ags alone also elicit a good GC response. Thus, our data are consistent with the interpretations of Kroese et al., who inferred that IC localization may not be necessary for the early GC response by studying irradiated rats, which transiently demonstrate poor IC trapping by FDCs yet initiate GC responses a few days before detectable IC localization returns after irradiation 13. Here we further show that IC deposition is not required for normal kinetics and size of the GC reaction, or for later events such as mutation and selection.
Recently, Ehrenstein et al. 27 concluded that natural IgM accelerates the primary Ab response, an interpretation based on delayed IgG production in mutant mice that lacked secreted IgM but that contained secreted IgG. Similarly, Boes et al. 28 inferred that IgM-containing ICs stimulate GC reactions, since the GC response was moderately impaired in a similar mutant model. However, the fact that the GC response in mIg Tg mice is not diminished compared with (m+s)Ig Tg or CB.17 mice argues against this interpretation. The muted immune responses may reflect dominant signaling through the inhibitory FcγRIIB 36 in mice that contain only IgG and no IgM. No such imbalance would have existed in mIg Tg mice.
Time course analysis of the relative size and activation state of NP-specific B cell populations revealed similar kinetics for primary responses in mIg Tg and (m+s)Ig Tg mice. Based on in vitro data, Liu et al. suggested that B cell receptor (BCR) signaling, as a consequence of binding Ag on FDCs, rescues centrocytes from apoptosis 14. If such interactions were truly critical, then we should have observed massive loss of centrocytes and thus much smaller GCs. Instead, we see large GCs with normal kinetics and well-developed light zones. Therefore, although interaction with Ag may well be critical for the preservation of centrocytes in the GC, the source of Ag need not be ICs bound to FDCs.
The normal kinetics of the proliferative and GC responses in mIg mice also has implications for the mechanisms for turning off primary responses. The termination of the primary response has been attributed both to Ab-mediated Ag clearance and to inhibitory signals mediated by FcγR coligation with the BCR, as would occur when B cells bind IgG-containing ICs 36. Since the kinetics of GC appearance are normal in mIg Tg mice, other intrinsic, non–Ab-dependent mechanisms must also be important in dampening the immune response.
Once the GC reaction is established, GC B cells undergo somatic mutation of Ig genes 37. In the normal spleen, mutation appears restricted to GCs 32, although the stimulus for onset of mutation remains unclear. Direct sequencing of Vλ genes microdissected from GCs shows that mutation occurs normally in mIg Tg GC B cells, indicating that ICs on FDCs are not responsible for the initiation of mutation, although other signals from FDCs may still play a role. Affinity maturation is thought to involve preferential expansion and/or survival of those GC B cells that acquire affinity-enhancing mutations. It is thought that higher-affinity mutants are selected by their ability to compete with Ab already bound to Ag on FDCs. Analysis of mutations in mIg Tg mice does not support this concept. The number of replacement mutations in λ1 sequences is higher than would be predicted for a nonselected population. Further, comparison of patterns of replacement mutations in mIg Tg mice with (m+s)Ig Tg and non-Tg mice reveals mutations common to the three strains at several sites in CDRs. Both of these features indicate that Ag-driven selection is occurring in mIg Tg mice.
It is important to compare our findings with those of either CR1/2 knockout or TNF/TNFR family knockout mice. GCs developed normally in CR1/2−/− mice that were provided with wild-type bone marrow, in which only the FDCs lacked CR1/2 21. However, a firm conclusion about IC localization was not drawn, since IC deposition could still occur via FcRs 11. Moreover, a second group found reduced GC formation when FDCs lacked CR1/2, suggesting a role of CR1/2 on FDCs 38. Thus, previous results in this area were conflicting with regard to the role of IC localization and GC initiation. The finding in mIg Tg mice that vigorous GC formation does occur in the absence of deposition of ICs on FDCs establishes that this event is not essential for GC formation.
The situation is also complex in TNF/TNFR family knockouts. Affinity maturation and somatic hypermutation have been reported in lymphotoxin (LT)-α−/− mice, which lack FDCs, have abnormal splenic architecture, and do not form GCs 19. However, this response is suboptimal, requires high dose Ag or multiple immunizations, and for some Ags such as SRBCs, IgG responses and memory formation are dramatically reduced 16 19. A similar picture was seen in the LT-β −/− mice 20. TNFR1−/− mice can form long-lived memory cells if infected with replicating virus, but respond poorly and lack long-lived IgG or memory if immunized with killed virus 17. However, given the pleiotropic defects including lymphoid architecture disruption consequent to lack of LT-α or TNFR1 expression throughout the body, it is difficult to draw specific conclusions about the physiologic role of IC deposition on FDCs from the LT-α or TNFR1 knockouts. The lack of defects in follicular architecture and other functions of FDCs in mIg Tg mice contrasts with the situation in LT-deficient or TNFR1−/− mice, thus implicating disruptions in these aspects, rather than lack of FDC-bound Ag, in the immune response defects. Recent work from Chaplin and colleagues highlights this point, demonstrating that the normal lymphoid architecture is required to allow wild-type memory B cells to function 16. mIg Tg mice have apparently normal mutation and selection; mutation and possibly some selection were also observed in the LT-deficient mice, suggesting that GCs were not required for this function. Thus GCs, but not FDC-bound ICs, are needed for optimal mutation and selection, though the GCs are not required for some degree of mutation.
It has been suggested that B cell CR1/2 cross-linking by C3 fragments fixed on ICs was essential for B cell activation 35 39 40. If this were true, the normal activation and expansion of NP-specific B cells in mIg Tg mice would be surprising. There are at least two possible explanations for this paradox: complement-complexed Ag is believed to augment B cell stimulation at particularly low doses of Ag; the dose of the Ag (50 μg) used in our case, though comparable to that used in the above studies, may not have been limiting. However, even at 1/4 of this dose per mouse we saw normal responses (data not shown). This suggests that CR1/2 must by itself be present on a responding B cell for it to participate effectively in a GC reaction, even if its BCR is strongly cross-linked. An additional possibility is that mIgM that is aggregated on the cell surface by BCR ligation can fix complement 41 42, and that C3b deposition occurs locally at the surface of the B cell. If this were the case, then the immune responses in mIg Tg mice suggest that such a mechanism is sufficient to drive B cell activation via CR1/2 and sIg cocross-linking.
The results reported here raise two general questions. First, if Ag bound to FDCs is not required to promote the primary GC reaction, what is the source of Ag to drive the GC response? Second, what is the function of retained Ag on FDCs if GC formation, mutation, and selection and memory B cell development can occur in its absence? As for the source of Ag, we do believe there must be a continuous source to maintain the response. Normally, this would be in the form of a proliferating pathogen that would supply a fresh source until the pathogen was eliminated. In the case of protein Ags, it is noteworthy that optimal immunization requires a depot preparation as in alum or Freund's adjuvant. These depots would release a continuous supply of Ags in a way that simulates the pathogen situation. In either case, there would be free Ag in the animal, which we believe is the normal source of Ag for B cells. This should not be surprising, as free Ag is almost certainly the source that initiates the extrafollicular as well as T cell–independent responses. An additional interesting possibility is marginal zone (MZ) B cells, which have been shown to transport Ag into follicles 13 43. However, in mIg Tg mice FDCs are not evidently receptors of Ag transported via MZ B cells. Perhaps Ig secretion by MZ B cells is an important aspect of their ability to transfer ICs to FDCs. On the other hand, MZ B cells could well serve to transport Ag directly to GC B cells in mIg Tg and possibly normal mice. Given the results of Kroese et al. 13, this is unlikely to be important for initiation of GCs but it could be critical for the maintenance of GCs. Indeed, MacLennan and colleagues have recently shown that there can be a GC response that is T cell independent but not sustained, suggesting that requirements for initiation and maintenance of GCs are likely to be different 44. Finally, we cannot formally exclude the possibility that alterations in Ag metabolism in Ab-free mice could affect Ag distribution and at least partially compensate for the lack of ICs bound to FDCs. However, the unaltered kinetics of the GC response in mIg Tg mice suggests that Ag availability is not markedly altered. Nor did we detect an increase in B cell–bound Ag (which was not readily observed in any situation, Fig. 5 and Fig. 8) in these mice.
Regarding the functions of FDC-bound Ag, there are several possibilities. Although it is apparent from mutation analysis that some selection does occur, we have not ruled out impairment in mIg Tg mice. Long-term maintenance of memory B cells remains an additional possible function of retained Ag. Quantitation of memory B cells would help resolve this, as would very long-term functional memory experiments. FDCs could act as an IC “sink” for the GC 45. By trapping any ICs present in the GC, FDCs would reduce potential FcγR-mediated downregulation of activated GC B cells. The mIg Tg mouse is neutral in this regard, as there are no ICs to be removed or to mediate downregulation. An additional possibility is that retained Ag could play a role in the regulation of Ab production, for example by helping to promote plasma cell longevity. Recent studies have revealed a population of long-lived plasma cells in the bone marrow and spleen that continue to undergo affinity maturation after the GC reaction has waned 46 47. Addition of IgG-complexed neutralized Ag after primary immunization increased long-term specific Ab titers, suggesting a connection between the presence of retained Ag and continuing Ab production 48. Interestingly, AFC survival in TNFR−/− mice is much more strongly impaired than memory cell survival 17. These data are also consistent with the idea that, as Ab levels wane, ICs on FDCs could dissociate, releasing some bound Ag and thereby stimulating plasma cells to renew Ab production.
The FDC network is a prominent and dynamic feature of the B cell follicle and GC. The deposition and retention of ICs on FDCs are striking and no doubt important aspects of FDC function. Our results suggest that the prevailing concepts of how ICs contribute to the development, selection, and possibly maintenance of memory B cells should be reconsidered. More detailed evaluation will be needed to further clarify the roles of IC capture and FDCs in general. The system we describe here, along with others such as the CR1/2−/− 21 22 and FcR−/− 49, should be instrumental in this effort.
Acknowledgments
We thank Cynthia Chi and Hong Zhou for technical assistance; Garnett Kelsoe and Joe Dal Porto for instruction on GC microdissection; and Alfred Bothwell, Michael Cancro, and Martin Weigert for critical review of the manuscript.
This work was supported by National Institutes of Health grant AI43603 (M.J. Shlomchik). A.M. Haberman was supported by a grant from the Donaghue Foundation, and L.G. Hannum was supported by National Institutes of Health Training Grant AI07019 and a Richard K. Gershon Predoctoral Fellowship.
Footnotes
L.G. Hannum's present address is University of Southern Maine/Lewiston-Auburn College, 51 Westminster St., Lewiston, ME 04240.
Abbreviations used in this paper: AFC, Ab-forming cell; BCR, B cell receptor; CGG, chicken gamma globulin; CR, complement receptor; FDC, follicular dendritic cell; GC, germinal center; HSA, human serum albumin; IC, immune complex; JhD, Jh knockout strain; LT, lymphotoxin; mIg, membrane Ig; MZ, marginal zone; PNA-HRP, peanut agglutinin–horseradish peroxidase; Tg, transgenic or transgene.
References
- Jacob J., Kassir R., Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I. the architecture and dynamics of responding cell populations. J. Exp. Med. 1991;173:1165–1175. doi: 10.1084/jem.173.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.J., Zhang J., Lane P.J., Chan E.Y., MacLennan I.C. Sites of specific B cell activation in primary and secondary responses to T cell-dependent and T cell-independent antigens. Eur. J. Immunol. 1991;21:2951–2962. doi: 10.1002/eji.1830211209. [DOI] [PubMed] [Google Scholar]
- Dal Porto J.M., Haberman A.M., Shlomchik M.J., Kelsoe G. Antigen drives very low affinity B cells to become plasmacytes and enter germinal centers. J. Immunol. 1998;161:5373–5381. [PubMed] [Google Scholar]
- Tew J.G., Kosco-Vilbois M.H., Burton G.H., Szakal A.K. Follicular dendritic cells as accessory cells. Immunol. Rev. 1990;117:185–211. doi: 10.1111/j.1600-065x.1990.tb00573.x. [DOI] [PubMed] [Google Scholar]
- Tew J., Mandel T.E., Burgess A.W. Retention of intact HSA for prolonged periods in the popliteal lymph nodes of specifically immunized mice. Cell. Immunol. 1979;45:207–212. doi: 10.1016/0008-8749(79)90378-2. [DOI] [PubMed] [Google Scholar]
- Nossal G.J.V., Abbot A., Mitchell J., Lummus Z. Antigens in immunity. XV. Ultrastructural features of antigen capture in primary and secondary lymphoid follicles. J. Exp. Med. 1968;127:277–290. doi: 10.1084/jem.127.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosco M.H., Tew T.G., Szakal A.K. Antigenic phenotyping of isolated and in situ rodent follicular dendritic cells (FDC) with emphasis on the ultrastructural demonstration of Ia antigens. Anat. Rec. 1986;215:201–213. doi: 10.1002/ar.1092150303. [DOI] [PubMed] [Google Scholar]
- Reynes M., Aubert J.P., Cohen J.H.M., Audouin J., Tricottet V., Diebold J., Kazatchkine M.D. Human follicular dendritic cells express CR1, CR2, and CR3 complement receptor antigens. J. Immunol. 1985;135:2687–2694. [PubMed] [Google Scholar]
- Papamichail M., Gutierrez G., Embling P., Johnson P., Holborow E.J., Pepys M.B. Complement dependence of localization of aggregated IgG in germinal centers. Scand. J. Immunol. 1975;4:343–347. doi: 10.1111/j.1365-3083.1975.tb02635.x. [DOI] [PubMed] [Google Scholar]
- van den Berg T.K., Dopp E.A., Daha M.R., Kraal G., Dijkstra C.D. Selective inhibition of immune complex trapping by follicular dendritic cells with monoclonal antibodies against rat C3. Eur. J. Immunol. 1992;22:957–962. doi: 10.1002/eji.1830220412. [DOI] [PubMed] [Google Scholar]
- Yoshida K., Van Den Berg T.K., Dijkstra C.D. Two functionally different follicular dendritic cells in secondary lymphoid follicles of mouse spleen, as revealed by CR1/2 and FcRγII-mediated immune-complex trapping. Immunology. 1993;80:34–39. [PMC free article] [PubMed] [Google Scholar]
- Klaus G.G.B. The generation of memory cells II. Generation of B memory cells with preformed antigen-antibody complexes. Immunology. 1978;34:643–652. [PMC free article] [PubMed] [Google Scholar]
- Kroese F.G.M., Wubbena A.S., Nieuwenhuis P. Germinal centre formation and follicular antigen trapping in the spleen of lethally X-irradiated and reconstituted rats. Immunology. 1986;57:99–104. [PMC free article] [PubMed] [Google Scholar]
- Liu Y.-J., Joshua D.E., Williams G.T., Smith C.A., Gordon J., MacLennan I.C.M. Mechanism of antigen-driven selection in germinal centres. Nature. 1989;342:929–931. doi: 10.1038/342929a0. [DOI] [PubMed] [Google Scholar]
- MacLennan I.C.M., Gray D. Antigen-driven selection of virgin and memory B cells. Immunol. Rev. 1986;91:61–85. doi: 10.1111/j.1600-065x.1986.tb01484.x. [DOI] [PubMed] [Google Scholar]
- Fu Y.X., Huang G., Wang Y., Chaplin D.D. Lymphotoxin-alpha-dependent spleen microenvironment supports the generation of memory B cells and is required for their subsequent antigen-induced activation. J. Immunol. 2000;164:2508–2514. doi: 10.4049/jimmunol.164.5.2508. [DOI] [PubMed] [Google Scholar]
- Karrer U., Lopez-Macias C., Oxenius A., Odermatt B., Bachmann M.F., Kalinke U., Bluethmann H., Hengartner H., Zinkernagel R.M. Antiviral B cell memory in the absence of mature follicular dendritic cell networks and classical germinal centers in TNFR1−/− mice. J. Immunol. 2000;164:768–778. doi: 10.4049/jimmunol.164.2.768. [DOI] [PubMed] [Google Scholar]
- Mariathasan S., Matsumoto M., Baranyay F., Nahm M.H., Kanagawa O., Chaplin D.D. Absence of lymph nodes in lymphotoxin-α(LTα)-deficient mice is due to abnormal organ development, not defective lymphocyte migration. J. Inflamm. 1995;45:72–78. [PubMed] [Google Scholar]
- Matsumoto M., Lo S.F., Carruthers C.J.L., Min J., Mariathasan S., Huang G., Plas D.R., Martin S.M., Geha R.S., Nahm M.H., Chaplin D.D. Affinity maturation without germinal centres in lymphotoxin-α-deficient mice. Nature. 1996;382:462–466. doi: 10.1038/382462a0. [DOI] [PubMed] [Google Scholar]
- Koni P.A., Sacca R., Lawton P., Browning J.L., Ruddle N.H., Flavell R.A. Distinct roles in lymphoid organogenesis for lymphotoxins alpha and beta revealed in lymphotoxin beta-deficient mice. Immunity. 1997;6:491–500. doi: 10.1016/s1074-7613(00)80292-7. [DOI] [PubMed] [Google Scholar]
- Ahearn J.M., Fischer M.B., Croix D., Goerg S., Ma M., Xia J., Zhou X., Howard R.G., Rothstein T.L., Carroll M.C. Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired B cell response to T-dependent antigen. Immunity. 1996;4:251–262. doi: 10.1016/s1074-7613(00)80433-1. [DOI] [PubMed] [Google Scholar]
- Molina H., Holers V.M., Li B., Fang Y.-F., Mariathasan S., Goellner J., Strauss-Schoenberger J., Karr R.W., Chaplin D.D. Markedly impaired humoral immune response in mice deficient in complement receptors 1 and 2. Proc. Natl. Acad. Sci. USA. 1996;93:3357–3361. doi: 10.1073/pnas.93.8.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croix D.A., Ahearn J.M., Rosengard A.M., Han S., Kelsoe G., Ma M., Carroll M.C. Antibody response to a T-dependent antigen requires B cell expression of complement receptors. J. Exp. Med. 1996;183:1857–1864. doi: 10.1084/jem.183.4.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan O.T., Hannum L.G., Haberman A.M., Madaio M.P., Shlomchik M.J. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J. Exp. Med. 1999;189:1639–1648. doi: 10.1084/jem.189.10.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynes M.A., Lanier L.L., Babcock G.F., Wettstein P.J., Haughton G. Antigen-induced murine B cell lymphomas. I. Induction and characterization of CH1 and CH2. J. Immunol. 1978;121:2352–2357. [PubMed] [Google Scholar]
- Shlomchik M.J., Zharhary D., Camper S., Saunders T., Weigert M. A rheumatoid factor transgenic mouse model of autoantibody regulation. Int. Immunol. 1993;5:1329–1341. doi: 10.1093/intimm/5.10.1329. [DOI] [PubMed] [Google Scholar]
- Ehrenstein M.R., O'Keefe T.L., Davies S.L., Neuberger M.S. Targeted gene disruption reveals a role for natural secretory IgM in the maturation of the primary immune response. Proc. Natl. Acad. Sci. USA. 1998;95:10089–10093. doi: 10.1073/pnas.95.17.10089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boes M., Esau C., Fischer M.B., Schmidt T., Carroll M., Chen J. Enhanced B-1 cell development, but impaired IgG antibody responses in mice deficient in secreted IgM. J. Immunol. 1998;160:4776–4787. [PubMed] [Google Scholar]
- Durdik J., Gerstein R.M., Rath S., Robbins P.F., Nisonoff A., Selsing E. Isotype switching by a microinjected μ immunoglobulin heavy chain gene in transgenic mice. Proc. Natl. Acad. Sci. USA. 1989;86:2346–2350. doi: 10.1073/pnas.86.7.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKean D., Huppi K., Bell M., Straudt L., Gerhard W., Weigert M. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. Proc. Natl. Acad. Sci. USA. 1984;81:3180–3184. doi: 10.1073/pnas.81.10.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke S.H., Staudt L.M., Kavaler J., Schwartz D., Gerhard W.U., Weigert M.G. V region gene usage and somatic mutation in the primary and secondary responses to influenza virus hemagglutinin. J. Immunol. 1990;144:2795–2801. [PubMed] [Google Scholar]
- Jacob J., Przylepa J., Miller C., Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. III. The kinetics of V region mutation and selection in germinal center B cells. J. Exp. Med. 1993;178:1293–1307. doi: 10.1084/jem.178.4.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimoto H., Nagaoka H., Adachi Y., Mizuochi T., Azuma T., Yagi T., Sata T., Yonehara S., Tsunetsugu-Yokota Y., Taniguchi M., Takemori T. Accumulation of somatic hypermutation and antigen-driven selection of rapidly cycling surface Ig+ germinal center (GC) B cells which occupy GC at a high frequency during the primary anti-hapten response in mice. Eur. J. Immunol. 1997;27:268–279. doi: 10.1002/eji.1830270140. [DOI] [PubMed] [Google Scholar]
- Matsumoto M., Fukuda W., Circolo A., Goellner J., Strauss-Schoenberger J., Wang X., Fujita S., Hidvegi T., Chaplin D.D., Colten H.R. Abrogation of the alternative complement pathway by targeted deletion of murine factor B. Proc. Natl. Acad. Sci. USA. 1997;94:8720–8725. doi: 10.1073/pnas.94.16.8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M.B., Ma M., Goerg S., Zhou X., Xia J., Finco O., Han S., Kelsoe G., Howard R.G., Rothstein T.L. Regulation of the B cell response to T-dependent antigens by classical pathway complement. J. Immunol. 1996;157:549–556. [PubMed] [Google Scholar]
- Phillips N.E., Parker D.C. Cross-linking of B lymphocyte Fcγ receptors and membrane immunoglobulin inhibits anti-immunoglobulin-induced blastogenesis. J. Immunol. 1984;132:627–632. [PubMed] [Google Scholar]
- Jacob J., Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. II. A common clonal origin for periarteriolar lymphoid sheath–associated foci and germinal centers. J. Exp. Med. 1992;176:679–687. doi: 10.1084/jem.176.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y., Xu C., Fu Y.-X., Holers V.M., Molina H. Expression of complement receptors 1 and 2 on follicular dendritic cells is necessary for the generation of a strong antigen-specific IgG response. J. Immunol. 1998;160:5273–5279. [PubMed] [Google Scholar]
- Pepys M.B. Studies in vivo of cobra factor and murine C3. Immunology. 1975;28:369–377. [PMC free article] [PubMed] [Google Scholar]
- Heyman B., Wiersma E.J., Kinoshita T. In vivo inhibition of the antigen response by a complement receptor–specific monoclonal antibody. J. Exp. Med. 1990;172:665–668. doi: 10.1084/jem.172.2.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohishi K., Kanoh M., Shinomiya H., Hitsumoto Y., Utsumi S. Complement activation by cross-linked B cell-membrane IgM. J. Immunol. 1995;154:3173–3179. [PubMed] [Google Scholar]
- Olesen E.H., Johnson A.A., Damgaard G., Leslie R.G.Q. The requirement of localized, CR2-mediated, alternative pathway activation of complement for covalent deposition of C3 fragments on normal B cells. Immunology. 1998;93:177–183. doi: 10.1046/j.1365-2567.1998.00429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray D., Kumararatne D.S., Lortan J., Khan M., MacLennan I.C.M. Relation of intra-splenic migration of marginal zone B cells to antigen localization on follicular dendritic cells. Immunology. 1984;52:659–669. [PMC free article] [PubMed] [Google Scholar]
- de Vinuesa C.G., Cook M.C., Ball J., Drew M., Sunners Y., Cascalho M., Wabl M., Klaus G.G., MacLennan I.C. Germinal centers without T cells. J. Exp. Med. 2000;191:485–494. doi: 10.1084/jem.191.3.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tew J.G., Wu J., Qin D., Helm S., Burton G.F., Szakal A.K. Follicular dendritic cells and presentation of antigen and costimulatory signals to B cells. Immunol. Rev. 1997;156:39–52. doi: 10.1111/j.1600-065x.1997.tb00957.x. [DOI] [PubMed] [Google Scholar]
- Slifka M.K., Antia R., Whitmire J.K., Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8:363–372. doi: 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- Takahashi Y., Dutta P.R., Cerasoli D.M., Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. V. Affinity maturation develops in two stages of clonal selection. J. Exp. Med. 1998;187:885–895. doi: 10.1084/jem.187.6.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann M.F., Kundig T.M., Hengartner H., Zinkernagel R.M. Regulation of IgG antibody titers by the amount persisting of immune-complexed antigen. Eur. J. Immunol. 1994;24:2567–2570. doi: 10.1002/eji.1830241046. [DOI] [PubMed] [Google Scholar]
- Takai T., Ono M., Hikida M., Ohmori H., Ravetch J.V. Augmented humoral and anaphylactic responses in FcγRII. Nature. 1996;379:346–349. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- Taketani M., Naitoh A., Motoyama N., Azuma T. Role of conserved amino acid residues in the complementarity determining regions on hapten-antibody interaction of anti-(4-hydroxy-3-nitrophenyl) acetyl antibodies. Mol. Immunol. 1995;32:983–990. doi: 10.1016/0161-5890(95)00057-l. [DOI] [PubMed] [Google Scholar]
- Chang B., Casali P. The CDR1 sequences of a major proportion of human germline Ig VH genes are inherently susceptible to amino acid replacement. Immunol. Today. 1994;15:367–373. doi: 10.1016/0167-5699(94)90175-9. [DOI] [PMC free article] [PubMed] [Google Scholar]