

Abstract
Leukocyte migration is the hallmark of inflammation, and integrin αMβ2 and its ligand fibrinogen (Fg) are key participants in this cellular response. Cells expressing wild-type or mutant αMβ2 and Fg or its derivatives have been used to dissect the molecular requirements for this receptor–ligand pair to mediate cell migration. The major conclusions are that (a) Fg, its D fragment, and its P1 and P2 αMβ2 recognition peptides support a chemotactic response; (b) when the I domain of αL was replaced with the I domain of αM, the chimeric receptor supported cell migration to Fg; however, the αM subunit, containing the I domain but lacking the β2 subunit, supported migration poorly, thus, the αMI domain is necessary but not sufficient to support chemotaxis, and efficient migration requires the β2 subunit and αMI domain; and (c) in addition to supporting cell migration, P2 enhanced αMβ2-mediated chemotaxis to Fg and the P1 peptide. This activation was associated with exposure of the activation-dependent epitope recognized by monoclonal antibody 7E3 and was observed also with human neutrophils. Taken together, these data define specific molecular requirements for αMβ2 to mediate cell migration to Fg derivatives and assign a novel proinflammatory activity to the P2 peptide.
Keywords: adhesion molecules, fibrinogen, integrin, CD11b/CD18, inflammation
Introduction
The leukocyte integrin αMβ2 (CD11b/CD18, Mac-1, CR3) and fibrinogen (Fg) are key molecular participants in the immune/inflammatory response 1 2 3 4. Within the currently accepted scenario for leukocyte trafficking during the inflammatory reaction, αMβ2 is thought to play a pivotal role in the firm adhesion of leukocytes to the endothelium and in the subsequent transmigration of the adherent cells to sites of inflammation. Evidence for these functions of αMβ2 has been developed in studies with PMNs 5, monocytes 6, lymphocytes 7, and eosinophils 8. The characterization of αMβ2-deficient mice has confirmed these findings by showing that a variety of leukocyte-dependent responses are diminished in these animals 9 10 11. Also, Fg is believed to play a multifaceted role in the immune and inflammatory response. For example, afibrinogenemic patients fail to develop the induration associated with an inflammatory response 12, depletion of mice of Fg blunts their inflammatory response to biomaterial implants 4, and Fg-deficient mice exhibit greatly reduced joint inflammation in an antigen-induced arthritis model 13. Linking these two inflammatory/immune participants, Fg is a ligand for αMβ2 14 15. Deposits of Fg/fibrin are adhesive for leukocytes via αMβ2, an interaction demonstrable in numerous in vitro and in vivo studies 4 16. Fg also has been shown to bridge αMβ2-bearing leukocytes to intercellular adhesion molecule (ICAM)-1 on endothelial cells 17 18. Engagement of αMβ2 by Fg induces a series of intracellular signaling events and cellular responses, which are relevant to the inflammatory response including cytokine secretion and nuclear factor κB activation 19 20. Most studies of Fg–αMβ2 interactions have centered on the engagement of the molecule with the receptor leading to cell adhesion 17 21. However, little is known regarding the role of Fg in initiating the diapedesis and migration of leukocytes 18 22.
αMβ2 is a member of the integrin family of α/β heterodimeric adhesion receptors and shares the same β2 subunit as αLβ2 (CD11a/CD18, LFA-1), αXβ2 (CD11c/CD18, p150,95), and αDβ2 (αD/CD18) 23 24 25. High affinity binding of many protein ligands, including Fg, to αMβ2 involves a segment of ∼200 amino acids in the αM subunit, termed the I (or A) domain 23 26 27. Other I domain ligands include ICAM-1, C3bi, Candida albicans, and the helminth protein neutrophil inhibitory factor (NIF; references 24, 25, 28). Critical to the ligand-binding functions of the αM I domain is the metal ion–dependent adhesion site (MIDAS). In this MIDAS motif, five noncontiguous amino acids, including the D(140), S(142), S(144), T(209), and D(242) (numbering refers to the amino acid positions in the mature αM subunit sequence), provide coordination sites for a divalent cation and ligands bind in close proximity to this motif 29 30. Optimal recognition of many of the αM I domain ligands can be influenced by the activation of the αMβ2-bearing cell, which alters receptor avidity/affinity 23 24 31. αMβ2 recognizes mannose and β-glucan carbohydrate structures via a lectin-like ligand-binding function. This lectin binding site is also located in the αM subunit 32. Ligands of the lectin domain include zymosan, Saccharomyces cerevisiae, and nonspecific activators of cell-mediated immunity such as lentinan 32.
Fg is a dimeric molecule composed of three pairs of nonidentical peptide chains. It is organized into a central E and two peripheral D domains, which give rise to E and D fragments when Fg is degraded by most proteolytic enzymes, including plasmin 33. Two specific sequences within the γ chain moiety of fragment D are recognized by αMβ2. The peptides corresponding to these sequences are designated P1 (γ190–202) 34 and P2 (γ377–395) 21. The P1 and P2 peptides not only inhibit αMβ2-mediated adhesion to Fg derivatives but also support adhesion of αMβ2-bearing cells. On a molar basis, P2 is a more potent inhibitor of αMβ2 adhesion to Fg than P1 21. The binding sites for both the P1 and P2 peptides in αMβ2 have been mapped to the αM I domain, and cells expressing the αM subunit in the absence of the β2 subunit adhere well to Fg and these recognition peptides 21.
We have previously used HEK 293 cells transfected with wild-type (WT) and mutant forms of αMβ2 to demonstrate the importance of the αMI domain for high affinity binding of NIF 35, C3bi 36, C. albicans 37, and Fg 21 38. The experiments in this study describe the utilization of these transfectants to investigate αMβ2-dependent cell migration to Fg and its derivatives. We demonstrate that αMβ2 can, indeed, mediate directed cell migration to Fg and that the recognition peptides influence this response. Furthermore, the structural components of αMβ2 that are required for cell migration are distinct from those for cell adhesion. Also, receptor activation is shown to play a major role in regulating αMβ2-mediated cell migration to Fg and its derivatives. Indeed, one of the recognition peptides of Fg is capable of inducing such activation not only in the αMβ2 transfectants but also in neutrophils.
Materials and Methods
Fg and Fibronectin Peptides.
Plasminogen-depleted human Fg was purified as described previously 39 or purchased from Enzyme Research Laboratories. Fragment D100 (M r 100,000) was prepared by digestion of human Fg with plasmin and purified by ion-exchange chromatography on CM-Sephadex followed by gel filtration on Sephacryl S-200 40. The Fg P1 (γ190–202), P2 (γ377–395), and γ400–411 peptides were synthesized using an Applied Biosystems model 430 peptide synthesizer and purified by HPLC as described previously 21.
NIF, mAbs, and Reagents.
NIF was a gift from Corvas International. mAbs used were as follows: OKM1 (anti-CD11b, IgG2b), 44a (anti-CD11b, IgG1), LM2/1 (anti-CD11b, IgG1), M1/70 (anti-CD11b), IB4 (anti-CD18, IgG2a), and W6/32 (anti-MHC class I, IgG1), TS2/18 (anti-αLβ2, IgG1), and TS1/18 (anti-β2, IgG1). The hybridoma cell lines producing these mAbs were obtained from the American Type Culture Collection and purified from their conditioned media using recombinant protein G columns as described by the manufacturer (Zymed Laboratories). The anti-β1 mAb F4611 and anti-αVβ3 mAb LM609 were purchased from Chemicon. All of these mAbs were of mouse origin, and the secondary Ab used for immunofluorescence analyses was FITC goat anti–mouse IgG (Zymed Laboratories). c7E3, the humanized chimeric Fab fragment, was provided by Drs. M. Nakada and R. Jordan of Centocor (Malvern, PA) and was labeled with FITC (Sigma-Aldrich) according to the manufacturer's instructions. β-Glucan was purchased from Molecular Probes.
Cells and Cell Lines.
The HEK 293 cell lines were maintained as described previously 35 in DMEM-F12 plus 10% FBS, 100 U/ml penicillin, 100 mg/ml streptomycin, and 2 mM l-glutamine (all from BioWhittaker). Adherent cells were removed for passage and experiments using enzyme-free cell dissociation buffer (GIBCO BRL). Construction of 293 cells stably expressing WT or mutant forms of αMβ2 and αLβ2 has been described previously 35 41. Before use in adhesion or cell migration assays, receptor expression levels were verified to be similar by flow cytometry (FACS®) using a FACStar™ instrument (Becton Dickinson). The mean fluorescent intensities for the WT and mutants used were ∼300 when stained with an anti-αM mAb (OKM1, 44a), the β2 mAb TS1/18 or TS2/18 for the αLβ2 cells compared with <25 for mock-transfected cells. U937 monocytoid cells were obtained from the American Type Culture Collection and maintained in RPMI 1640 with the same supplements as for the HEK 293 cells. Human neutrophils were isolated from the peripheral blood of healthy adult volunteers using Ficoll-Paque as per the manufacturer's instructions (Amersham Pharmacia Biotech) followed by dextran sedimentation and hypotonic lysis of residual erythrocytes as described previously 42. The purity of the neutrophils used was routinely >96%.
Cell Migration Assays.
Cell migration assays were performed under sterile conditions using Costar 24-well transwell plates with 8-μm pore size uncoated polycarbonate filters (Corning, Inc.). Experiments were carried out in DMEM-F12 (BioWhittaker) or, for selected experiments, in hybridoma serum-free medium (GIBCO BRL). Lower wells contained 600 μl medium, whereas upper wells contained a final volume of 200 μl after the addition of cells. To begin the assay, 50 μl of cells (5 × 105 cells) in medium was added to upper wells, and the transwells were placed in a humidified incubator at 37°C/5% CO2. For inhibition/stimulation experiments, cells were preincubated for 30 min at 22°C with the test agents before addition to the transwells. Assays were stopped after 22 h by removing the upper wells and wiping the inside of upper wells twice with a cotton swab to remove nonmigrated cells. Quantitation of the migrated cells depended on the cells under analysis. For the HEK 293 cell lines, which remained adherent to the undersurface of the membrane, the upper transwell assembly was immersed in 10% formalin for 1 h at 22°C. Cells were then stained for at least 1 h with Mayers hematoxylin (Richard-Allan Scientific). Migrating cells were counted using an inverted microscope with an eyepiece counting grid at 100× magnification. Data are presented as the mean cell number per high power field (HPF), a 0.1-mm2 area of duplicate wells from three or more experiments with at least five random HPFs counted per well. Neutrophils did not adhere to the membrane between the chambers but rather accumulated as a suspension in the medium in the lower chamber. These cells were quantitated using the CyQuant Cell proliferation kit (Molecular Probes) according to the manufacturer's protocol. In brief, neutrophils within the medium of the lower chamber were recovered by centrifugation, and the cells were frozen for at least 3 h at −70°C. Upon thawing, the cells were resuspended in the CyQuant reagent, and the fluorescence was measured with a CytoFluor II fluorescence multiwell plate reader (PerSeptive Biosystems, Inc.) using a 485-nm excitation and a 530-nm emission filter. The data from these experiments are presented as the total number of migrated cells, determined from a standard curve developed with a known amount of CyQuant-labeled cells. Data obtained in these assays were consistent with those obtained by microscopic counting of the neutrophils. Statistical analyses of data from cell migration assays were performed using the SigmaPlot software program (Jandel) and the Student's t test.
Results
αMβ2-mediated Cell Migration to Fg.
To investigate the relationship between αMβ2, Fg, and cell migration, we compared the ability of transfected cells expressing αMβ2 or mock-transfected cells to migrate toward Fg in a transwell system. Migration was allowed to proceed for 22 h at 37°C, at which time the cells adherent to the underside of the filter were fixed, stained, and counted. Cell viability, as judged by trypan blue exclusion, remained high (>95%) during the course of the assays. When the cells were placed in the upper chamber and a Fg concentration of 50 μg/ml in the lower chamber, a dramatic difference in the migration of the αMβ2 and mock-transfected cells was observed (Fig. 1 A). With the mock-transfected cells, 15 ± 3 cells migrated per HPF, whereas 396 ± 58 of the αMβ2-transfected cells migrated at the same time point. Background migration for each cell type was measured using medium alone in lower wells. Although the number of αMβ2-expressing cells recovered on the lower surface of the filter with only buffer present in the lower chamber was slightly higher (59 ± 9 cells/HPF) than for mock-transfected cells (15 ± 3 cells/HPF), the migration of the αMβ2 cells toward Fg was nearly sevenfold more than this background migration. Over the course of five experiments, the increase in migration of the αMβ2-transfected cells to Fg versus buffer was 671 ± 15%. Nevertheless, the mock-transfected cells were able to migrate as demonstrated when fibronectin (Fn) was placed in the lower wells. This migration of the mock-transfected cells to Fn was similar in extent to that of the αMβ2-transfected cells to Fg and was inhibited by an mAb (F4611) to the integrin β1 subunit (20 μg/ml), which had no effect on αMβ2 migration to Fg (see Fig. 1 A). These experiments were conducted in protein-free DMEM-F12, but similar results also were obtained in hybridoma serum-free medium.
Figure 1.


Migration of αMβ2 WT transfectants but not mock transfectants to Fg. Human 293 cells (5 × 105/well) expressing αMβ2 (WT, black bars) or mock-transfected cells containing only the neomycin resistance plasmid (white bars) were assessed for their ability to migrate to Fg (50 μg/ml, 150 nM), Fn (50 μg/ml), or medium alone placed in the lower wells of transwell plates. Some cells were pretreated with 20 μg/ml anti-β1 blocking mAb F4611 (A) or 100 nM NIF; M1/70 and 44a, 20 μg/ml αM-blocking mAb; IB4, 20 μg/ml β2-blocking mAb; or LM2/1, 20 μg/ml αM-nonblocking mAb (B) for 30 min before addition to the upper wells. Migration was assessed for 22 h at 37°C and migrated cells were fixed, stained, and counted. Migration data are expressed as mean cells/HPF ± SD for five random fields per well (A) or as a percentage of the migration of the WT cells to Fg (B) with duplicate wells in each experiment from three or more experiments. The x-axis indicates the addition to the upper wells containing cells over the addition to the lower wells. ***Medium alone.
Direct evidence that the migration of the transfected cells to Fg was dependent on αMβ2 was sought. Several specific inhibitors of αMβ2 function were tested for their ability to inhibit the migration of the αMβ2 transfectants to Fg (Fig. 1 B). The test inhibitors were preincubated with the αMβ2 transfectants for 30 min before their addition to the upper chamber of the transwell system with 50 μg/ml Fg in the lower chamber. NIF, a high affinity ligand for the αM I domain, eliminated migration of the αMβ2 cells to Fg. The αM I domain–specific mAb 44a also eliminated migration of the αMβ2 transfectants to Fg, while a second αM I–domain specific mAb M1/70 inhibited migration by 80%. These results are consistent with the effects of these I domain mAbs on αMβ2 -mediated adhesion to a variety of ligands including Fg 26. In contrast, a nonblocking αM I domain–specific mAb LM2/1 26 had no effect on migration of the αMβ2 transfectants. The β2-specific mAb IB4 also eliminated migration of the αMβ2 transfectants to Fg. Taken together, these results demonstrate that migration of the transfected cells to Fg is αMβ2 dependent, and agents that block αMβ2-mediated cell adhesion to Fg also block its capacity to mediate cell migration to this ligand.
Fg as a Migratory Stimulus.
In the above analyses, the single concentration of 50 μg/ml Fg was used as a migratory stimulus. As shown in Fig. 2, the extent of migration of the αMβ2-transfected cells was dependent on the available Fg concentration. Concentrations of Fg as low as 1 μg/ml induced a measurable increase in cell migration. Maximum migration was observed at a concentration of 50 μg/ml. At a still higher dose of Fg, migration of the αMβ2 transfectants decreased markedly, a pattern often encountered in such assays 43.
Figure 2.

Migration of αMβ2 transfectants to Fg. 293 cells (5 × 105/well) expressing αMβ2 (WT cells) were assessed for their ability to migrate to various concentrations of Fg or medium alone (0 μg/ml). Migration data are expressed as mean cells/HPF ± SD for five random fields per well with duplicate wells in each experiment from three or more experiments.
To investigate whether the observed αMβ2 cell migration to Fg was due to chemokinesis or chemotaxis, a checkerboard analysis was conducted. Varying concentrations of Fg were placed either in the upper wells alone, the lower wells alone, or both wells simultaneously (Table ). Data are presented as the percentage of migration of the αMβ2 transfectants to the optimal (50 μg/ml) concentration of Fg in the lower well (% WT). With equal concentrations of Fg in the upper and lower wells, migration of the αMβ2-transfected cells was only 24%, a value not significantly different (P > 0.05) from the 15% migration with no Fg present. With 50 μg/ml in the upper well with the cells and none in the lower chamber, 32% of the maximal migration was observed, suggesting that Fg may cause a modest increase in cell motility. The chemotactic influence of Fg is supported by the inhibition of migration to a constant concentration of 50 μg/ml Fg in the lower well as increasing concentrations of Fg were placed in the upper well: 1 μg/ml (0% inhibition), 10 μg/ml (35% inhibition), 50 μg/ml (76% inhibition). These analyses indicate that the migration of the αMβ2 transfectants to Fg is largely directional and, therefore, chemotactic.
Table 1.
Fg Checkerboard Analysis
| Fn concentration (upper well) | ||||
|---|---|---|---|---|
| Fg concentration(lower well) | 50 μg/ml | 10 μg/ml | 1 μg/ml | Medium only |
| Medium | 32 | 18 | 12 | 16 |
| 1 μg/ml | 25 | 18 | 15 | 29 |
| 10 μg/ml | 26 | 29 | 26 | 58 |
| 50 μg/ml | 24 | 65 | 101 | 100 |
Migration of αMβ2 WT 293 transfected cells was assessed as in the legend to Fig. 1 A to different concentrations of Fg placed either in the upper or lower transwells. Data are means of cells per HPF for duplicate wells from three or more experiments expressed as percentage of WT (migration to 50 μg/ml Fg, lower well).
Structural Requirements within αMβ2 for Cell Migration to Fg.
To investigate the role of the various domains of αMβ2 in Fg-induced cell migration, three different cell lines expressing various forms of the receptors were examined (Fig. 3 A). The three transfectants tested were HEK 293 cells expressing αLβ2 (LFA-1), only the αM and not the β2 subunit, and the L/M mutant, an αLβ2 heterodimer in which the αL I domain was switched to the αM I domain. All three cell lines have been characterized previously for their ability to support adhesion to αMβ2 ligands 21 35. All three were determined to express similar levels of receptor or the αM subunit as the WT αMβ2-expressing cells by FACS® analyses. As shown in Fig. 3 A, the αLβ2-expressing cells demonstrated no specific migration to Fg relative to their background migration to medium alone. The αM alone cells exhibited a weak migratory response to Fg relative to its medium control although this level of migration was not significantly different from the background migration of the WT transfectants to medium. These data demonstrate a requirement for the β2 chain of the intact heterodimer for efficient migration to Fg. Of particular note, the L/M cells exhibited the same extent of migration to Fg as the WT αMβ2 cells, indicating the αM I domain is sufficient to confer the migratory phenotype to the αLβ2 heterodimer.
Figure 3.


Migration of αMβ2 WT and mutant cell lines to Fg. In A, the cell lines are either mock-transfected or transfected cells expressing either αMβ2 WT, αLβ2, αM only (expressing the αM but not β2 subunit), or L/M chimeric (the αM I domain switched into αLβ2) receptors. In B, the cell lines are αM(P147-R152)β2, in which residues P(147)-R(152) of the αM I domain have been switched to the corresponding residues of the αL I domain; αM(Q190)-S(197)β2, in which residues Q(190)-S(197) have been switched to the corresponding residues of the αL I domain; and point mutations of D(134) and S(136) to A in the β2 subunit. Details of these mutations are described in reference 35. Migration of each cell type is to 50 μg/ml Fg. Data are means of cells per HPF with duplicate wells in each experiment from three or more experiments ND expressed as the percentage of WT ± SD.
We have described previously a series of homologue scanning mutants in which the crystal structures of I domains were used as a guide to replace individual small secondary structural elements in the αM I domain with the corresponding segment of the αL I domain in the context of the αMβ2 heterodimer expressed in the same HEK 293 cells 35. Because of the extensive sequence and structural similarity between the two I domains, such swaps should not perturb overall conformation. These mutant cell lines have been used to map the binding sites for several αMβ2 ligands 35 36 37 38. To further dissect the role of individual segments of the I domain in αMβ2-mediated cell migration to Fg, two representative mutant receptors were tested for their ability to support the migratory response. Mutant P(147)-R(152), a mutant in which the segment between these residues on the MIDAS face of the I domain was switched to the corresponding residues in αL, failed to migrate to Fg (Fig. 3 B). This mutant also fails to recognize NIF, iC3b, and C. albicans 35 36 37, indicative of the key role of this segment in ligand recognition by αMβ2, including a role in directed cell migration. At the other extreme, mutant Q(190)-S(197) was tested. This mutant was previously shown to exhibit enhanced adhesion to Fg relative to the WT αMβ2 cells. However, as shown in Fig. 3 B, the migration of these cells was similar to that of WT cells, again providing evidence that the structural requirements for adhesion and migration are distinct. In view of the importance of the β2 subunit in cell migration to Fg, we also tested the effect of single-point mutations of either D134 and S136 to alanines in the β2 subunit. These mutations reside at residues which are likely to serve as cation coordination sites in a MIDAS motif in integrin β subunits 44 45, and this MIDAS motif may be formed by an I domain–like fold within the β subunits 46 47. Regardless of the validity of these structural speculations, these mutations are known to abolish ligand binding to multiple integrins 48 49. As shown in Fig. 3 B, these mutations also abolished the migration of the cells to Fg.
Structural Requirements within Fg for αMβ2-mediated Migration.
Two peptide sequences, designated P1 (γ191–202) and P2 (γ373–395), in the γ chain of the D domain of Fg have been identified as αMβ2 recognition sites 21 34; these peptides inhibit αMβ2-mediated adhesion to Fg and directly support cell adhesion via αMβ2 by interacting with the I domain of the receptor 21. Accordingly, we tested the capacity of the D100 fragment produced by plasmin cleavage of Fg and synthetic P1 and P2 peptides to support αMβ2-mediated cell migration. As shown in Fig. 4, the D100 fragment of Fg supported cell migration at very low concentrations; 1 and 10 nM concentrations of D100 were more effective than 150 nM (50 μg/ml) Fg. The P1 and P2 peptides also supported cell migration although higher concentrations of the recognition peptides were required. With P2, 3–6 μM peptide was as effective as Fg in supporting migration of the αMβ2 transfectants. P1 was less potent but did support a migratory response in a concentration-dependent fashion in the 1.5–6 μM range.
Figure 4.

Migration of αMβ2 WT cells to Fg D100 fragment and Fg recognition peptides P1 and P2. Migration of WT cells was assessed to various concentrations of either the Fg D100 fragment, P1 peptide, or P2 peptide placed in the lower transwells. Data are means of cells per HPF with duplicate wells for each experiment from three or more experiments and expressed as percentage WT ± SD.
Stimulation of αMβ2-mediated Cell Migration by the P2 Peptide of Fg.
To further examine the migratory activity of these Fg peptides, their ability to inhibit migration to Fg and each other was assessed (Fig. 5). When placed only in the upper well together with the cells, neither of the two peptides or the D100 fragment stimulated spontaneous migration of the αMβ2 cells, suggesting that the induction of cell migration to these derivatives was a chemotactic response. Furthermore, when placed in equal amounts in both the upper and lower wells, P1, P2, and the D100 fragment inhibited migration to itself, consistent with a chemotactic activity (data not shown). When testing the effects of the Fg derivatives in influencing cell migration to each other, some unexpected results were encountered. As anticipated, when the P1 peptide was added to the αMβ2-transfected cells in the upper chamber, it did inhibit migration to P2 and Fg in the lower chamber. In contrast, when P2 was added to the cells in the upper well, it stimulated rather than inhibited migration to P1 in the lower well (Fig. 5). To further explore the stimulatory effect of P2, its influence on αMβ2-mediated cell migration to Fg was assessed. As shown in Fig. 5, when added to the cells, P2 also stimulated migration to Fg. At a 6 μM concentration, P2 increased migration to Fg by 50% (relative to the migration of the cells to Fg alone, 100%). In contrast, both D100 (10 nM, 82% inhibition) and P1 (6 μM, 77% inhibition) inhibited migration of the αMβ2 cells under the same conditions. The Fg γ400–411 peptide, reported by other investigators to inhibit αMβ2 adhesion 50 had no effect on WT migration to Fg in concentrations as high as 50 μM (data not shown). This stimulatory effect of P2 was similar to that induced by two known activators of αMβ2. As shown in Fig. 6, 10 nM PMA and 2 μg/ml β-glucan were found to stimulate migration by 25 and 70%, respectively. The increases in migration induced by all three activators were statistically significant (P < 0.05) relative to the migration to Fg in their absence. Collectively, these data demonstrate a novel role for the P2 peptide in stimulating αMβ2-mediated migration to Fg.
Figure 5.

Migration of αMβ2 WT cells to Fg is inhibited by P1 but stimulated by P2. Migration of αMβ2 WT cells was assessed to various combinations of optimal concentrations of 50 μg/ml Fg, 6 μM P1 peptide, 6 μM P2 peptide, or 10 nM D100 fragment in the upper or lower transwells as indicated. Fg or peptides added to the upper well were preincubated with cells for 30 min before addition of the entire mixture to the transwell. Data are means of cells per HPF with duplicate wells for each experiment from three or more experiments and expressed as percentage of WT ± SD.
Figure 6.

Stimulation of αMβ2 WT cell migration to Fg. Migration of αMβ2 WT cells to 50 μg/ml Fg was assessed in the presence of various reagents after preincubation for 30 min. Reagents are Fg recognition peptides P1 (6 μM) or P2 (6 μM), 2 μg/ml β-glucan, and 10 nM PMA. Data are means of cells per HPF with duplicate wells for each experiment from three or more experiments and expressed as percentage of WT ± SD.
We sought to determine if the activating activity of P2 on αMβ2 occurs with cells other than the HEK 293 transfectants. For this purpose, we tested the effects of P2 on migration of peripheral blood neutrophils to Fg. As shown in Fig. 7, neutrophils exhibited a substantial migratory response to Fg, which was αMβ2 dependent as indicated by the inhibitory effect of mAb 44a. The neutrophils also migrated to P2. When added to the lower wells at a 10 μM concentration, the extent of cell migration to P2 was similar to that induced by 50 μg/ml Fg. Addition of P2 at this concentration (or 5 μM) to neutrophils in the upper chamber enhanced their migration to Fg by >50%. This increment was also αMβ2 dependent as the migration was fully inhibited by mAb 44a. Under these conditions, addition of the same concentration P1 to the upper chamber inhibited the migration of the neutrophils to Fg by ∼50% (data not shown). Thus, P2 exerted its activating effect on a cell which expresses αMβ2 naturally.
Figure 7.

Stimulation of neutrophil migration to Fg by P2. Isolated human neutrophils were placed in the upper wells and 20 μg/ml Fg or 10 μM P2 in the lower wells, and migration was measured after 24 h. In the experiments in which P2 or mAb 44a were used in the upper chambers, these were preincubated with the neutrophils for 30 min before addition of the cells to the upper wells. Migration data are expressed as mean cells per HPF ± SD for five random fields per well with duplicate wells in each experiment from three or more experiments.
P2 Activation of αMβ2 Is Associated with Exposure of the 7E3 Epitope.
To further investigate the mechanism of P2 stimulation of αMβ2-mediated migration to Fg, we conducted blocking studies with the chimeric Fab fragment (abciximab) of mAb 7E3. This mAb was originally developed against integrin αIIbβ3 51 but has subsequently been shown to interact with αVβ3 and αMβ2 (for a review, see reference 52). The epitope in αMβ2 resides in the I domain and appears to require activation of the receptor for expression 53 54. Although this mAb blocks Fg binding to the activated αMβ2 on leukocytes, it does not react with our nonstimulated WT αMβ2 transfectants as assessed by FACS® analyses (data not shown). Based on these characteristics of the 7E3 epitope, we hypothesized that c7E3 might exhibit a differential effect on nonstimulated and P2-stimulated migration of the αMβ2 transfectants to Fg. To maximize the stimulatory effect of P2, a lower concentration of 10 μg/ml Fg was employed in the transwell migration assay. As shown in Fig. 8 A, 40 μg/ml c7E3 had no inhibitory effect on αMβ2-mediated migration to Fg alone. When 6 μM P2 was added to the cells, migration was stimulated ∼2.5-fold. However, when c7E3 and P2 were added to the cells in the upper well, the increment in cell migration was inhibited. A similar inhibitory effect of c7E3 on the migration of the αMβ2-transfected cells stimulated by β-glucan was also observed (90% inhibition; Fig. 8 A). To rule out possible recognition of αVβ3 as the basis of the observed effects of c7E3, a potent αVβ3-blocking mAb, 20 μg/ml LM609, was substituted for c7E3 and found to have no effect on P2-stimulated (or baseline, data not shown), αMβ2-mediated cell migration to Fg. The data in Fig. 8 A indicate that the P2 induces, not suppresses, expression of the c7E3 epitope. We also considered whether P2 influences c7E3 reactivity with its epitope. This question was addressed with platelets. 10 μM P2 did not inhibit the binding of c7E3 to platelets as assessed by FACS®.
Figure 8.

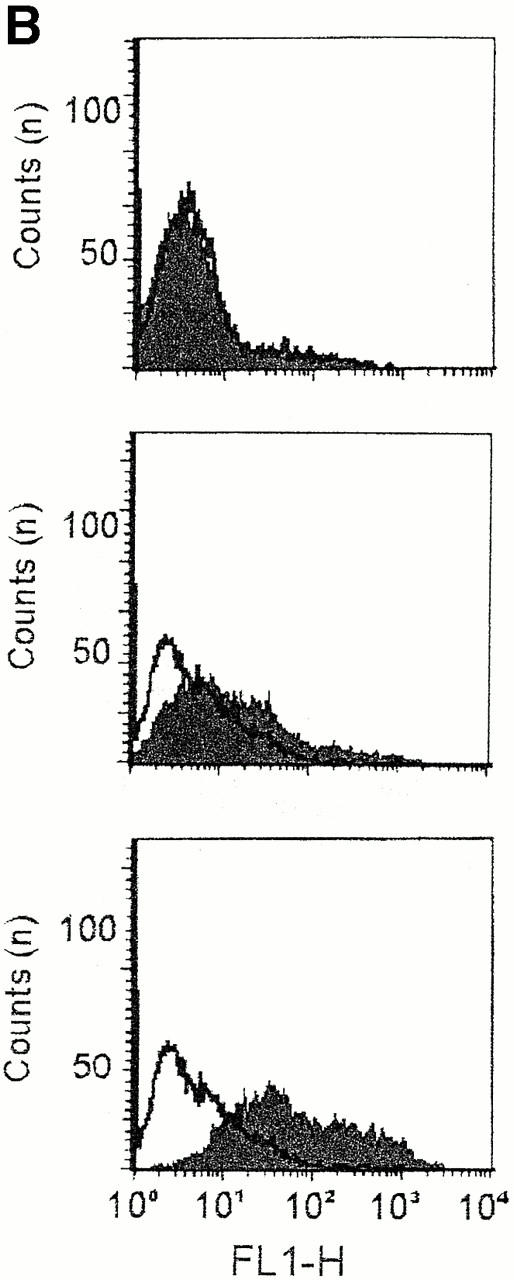
Role of the c7E3 epitope in αMβ2-mediated cell migration to Fg. In A, migration of αMβ2 WT cells was assessed to a concentration of 10 μg/ml Fg in the lower transwell. Cells were pretreated for 30 min with peptides or reagents before addition to the upper transwell. Reagents are 2 μg/ml β-glucan, 6 μM Fg P2 peptide, 40 μg/ml αIIbβ3 mAb c7E3, 20 μg/ml αM mAb 44a, and 20 μg/ml αVβ3 mAb LM609. Data are means of cells per HPF with duplicate wells for each experiment from three or more experiments and expressed as percentage of WT (migration to 10 μg/ml Fg, lower well) ± SD. In B, αMβ2 WT cells, treated with (gray scans) or without (white scans) P2, were recovered from the upper wells of migration chambers at 0 (upper), 1 (middle), or 12 h (lower), stained with fluor-escein-labeled c7E3 and analyzed by flow cytometry.
These data support a model in which an activated state of αMβ2, defined by expression of the 7E3 epitope, is induced by P2 and supports enhanced migration to Fg. To assess this possibility, the reactivity of the αMβ2 transfectants with 7E3 was assessed by FACS® at various time points during their migration to Fg with or without P2 present in the upper chamber (Fig. 8 B). As noted above, the αMβ2 transfectants did not express the 7E3 epitope, and this lack of reactivity was not changed by addition of P2 to the cells. On the other hand, the cells developed c7E3 reactivity over time in the presence of P2. The reactivity was noted at 1 h and increased still further by 12 h (Fig. 8 B). This reactivity with c7E3 was enhanced substantially in the presence of P2.
Discussion
Leukocyte migration is the hallmark of inflammation in vivo, and αMβ2 and Fg have been shown to contribute to leukocyte migration in multiple systems 23 24. This study has used αMβ2 transfectants and selected mutants to dissect the molecular requirements for αMβ2-mediated cell migration to Fg and its derivatives. The major conclusions of our study are the following. (a) Fg supports a chemotactic cell migration mediated by αMβ2. This response is dependent on Fg concentration and occurs at low (1–50 μg/ml) Fg levels. (b) The αM I domain is necessary but not sufficient to support cell migration to Fg. In contrast to cell adhesion to Fg, efficient migration requires the β2 subunit. (c) The P1 and P2 peptides, as well as the D100 fragment, support cell migration. Thus, the same Fg derivatives that mediate αMβ2-dependent cell adhesion also support cell migration. (d) The P2 peptide stimulates αMβ2-mediated cell migration to Fg and the P1 peptide, in a manner similar to other αMβ2 activators, β-glucan, and PMA. In addition, the activation-dependent epitope of c7E3 is induced by P2, c7E3 inhibits P2, and β-glucan stimulated αMβ2-mediated migration to Fg. Many of these findings, including the activating activity of P2, are also observed in the migration of human neutrophils to Fg. Thus, these data reveal new information regarding the molecular interactions between αMβ2 and Fg that are necessary for cell migration and define a novel mechanism whereby the P2 peptide of Fg stimulates αMβ2-mediated chemotaxis.
More than a decade ago, Fg was identified as a ligand for αMβ2 14, and numerous subsequent studies have examined the structure function requirements for αMβ2-mediated cell adhesion to Fg 38 41. The capacity of this ligand–receptor interaction to mediate chemotaxis has received less attention but, in view of the propensity of Fg/fibrin to accumulate at sites of inflammation, it is of clear physiological relevance. Indeed, Fg and the P1 peptide have been shown to mediate an αMβ2-dependent inflammatory response in vivo 53 55. Two notable in vitro studies have also shown that Fg, as well as the D100 fragment and P1 peptide, induce leukocyte transmigration and chemotaxis which is αMβ2 dependent 56 57. Thus, the demonstration that αMβ2 transfectants migrate to Fg and its recognition peptides is consistent with published data on leukocytes and supports the relevance of these cells to dissect the molecular requirements for the chemotactic response. In this regard, we found that the I domain of the receptor was not sufficient in the αM alone cells to mediate efficient migration to Fg. However, when the αM I domain was placed in the context of another β2 integrin, αLβ2, that does not mediate migration to Fg, chemotaxis of these L/M cells was observed. Thus, within the context of an intact heterodimer, the αM I domain is necessary and sufficient to confer migration to Fg. Furthermore, within the αM I domain, at least some of the sequences which are key for recognition of Fg and other αMβ2 ligands also influence αMβ2-mediated cell migration. Thus, the P147-152 αMβ2 mutant that fails to adhere to NIF 35, iC3b 36, C. albicans 37, and the D100 fragment of Fg 38 also fails to migrate to Fg. These data are consistent with results demonstrating that Fg is a ligand for αMβ2 but not αLβ2, adhesion to Fg is mediated principally via the I domain of αMβ2, and sequences on the MIDAS face of the αM I domain are involved in the migratory and adhesive response of the receptor. However, the β2 subunit is more influential in cell migration than in cell adhesion 21. The role of the β2 subunit may depend on the interaction of its cytoplasmic tail with cytoskeletal elements, such as talin and paxillin, which must undergo rearrangements in order for cells to migrate 24. Such requirements for the β subunit cytoplasmic segment for migration were demonstrated with Chinese hamster ovary cells expressing integrin αIIbβ3 and migrating on Fg 58.
Integrin αMβ2-mediated migration was triggered not only by intact Fg but also by its D100 fragment and by peptides duplicating its γ191–202 and γ373–395 sequences. While Fg was a potent chemoattractant, the plasmin-derived D100 fragment was active at 150-fold lower concentrations. The activity of the D100 fragment and other degradation products may be important in the recruitment of leukocytes to inflammatory sites, where proteolysis is a major mechanism for resolution of Fg/fibrin deposits. In addition, fibrin(ogen) degradation products in blood could potentially suppress leukocyte emigration. Both the P1 and P2 peptide sequences reside in the D100 fragment. We have found that these sequences are poorly exposed in soluble Fg but become exposed upon its deposit or degradation (unpublished results). The combined activity and exposure of these sequences may account for the potency of the D100 fragment as a chemoattractant. The P2 peptide was more potent than P1 in supporting and inhibiting αMβ2-dependent cell migration. This difference in activity is consistent with the greater apparent affinity of P2 for αMβ2 21.
When P2 was added to the αMβ2 transfectants, it enhanced cell migration. This increase appears to reflect an activation of αMβ2 by the peptide. This interpretation is supported by the observation that the epitope for c7E3, which is expressed by activated αMβ2 53, was induced by P2, and c7E3 inhibited the increase in cell migration evoked by P2. The activation of integrins by peptide ligands was originally demonstrated with αIIbβ3 59. Particularly relevant to the present observation, an αMβ2 ligand peptide derived from ICAM-2 activates αMβ2-mediated cell migration 60. The fact that P2, but not P1, induced such activation implies that, even though both peptides bind to the αM I domain, they must interact in a fundamentally different way to initiate a differential response in the receptor. Activation of αMβ2 has been shown to be important for optimal recognition of Fg to mediate cell adhesion 15 and migration 57. Such activation can arise from interactions within 60 or outside of the αM I domain 61, or outside of αMβ2 altogether (PMA stimulation). β-Glucan is a ligand for the lectin-binding domain of αMβ2 61, and its activation of the receptor has been studied extensively 62. β-Glucan activation of αMβ2 also is known to result in expression of the αM I domain activation neoepitope 61 recognized by mAb CBRM1/5, and this mAb eliminates αMβ2 adhesion to Fg 63. Nevertheless, mAbs CBRM1/5 and c7E3 do not compete with each other for binding to activated αMβ2 54. Our data support a model in which β-glucan and P2 stimulation results in an activated αMβ2 conformation, which expresses the c7E3 epitope. The greater inhibition by c7E3 of β-glucan– versus P2-stimulated migration suggests that the mechanism and/or extent of αMβ2 activation may be different. Also, P2 was shown to enhance neutrophil migration to Fg, emphasizing that modulation of αMβ2 function may be of physiological significance. The γ chain of Fg in which P2 resides is capable of undergoing conformational modulations, including upon ligation by integrins 64. Whether activation of the receptor via a P2-dependent mechanism is in itself of physiological importance can only be the subject of speculation at this time.
In summary, we have demonstrated that αMβ2 can mediate a chemotactic cell migration to Fg and Fg derivatives. Both the P1 and P2 recognition peptides can support this migratory response. Such migration depends on the αM I domain and is influenced by other domains of αMβ2. While our earlier studies identified a negative role for the β2 subunit in modulating adhesion 37, this study identifies a positive role for the β2 subunit in influencing cell migration. Thus, the structural requirements for αMβ2-mediated cell adhesion and migration to the same ligand, Fg, are distinct. In addition, we identify a novel proinflammatory function for the P2 sequence, as well as β-glucan, to activate αMβ2 and stimulate cell migration to Fg.
Acknowledgments
C.B. Forsyth was supported by an American Heart Association and a National Institutes of Health (NIH) Postdoctoral Fellowship. This work was supported in part by NIH grants HL54924 and HL63199 and by a grant-in-aid from the American Heart Association.
Footnotes
Abbreviations used in this paper: Fg, fibrinogen; Fn, fibronectin; HPF, high power field; ICAM, intercellular adhesion molecule; MIDAS, metal ion–dependent adhesion site; NIF, neutrophil inhibitory factor; WT, wild-type.
References
- Jaeschke H., Farhood A., Bautista A.P., Spolarics Z., Spitzer J.J., Smith C.W. Functional inactivation of neutrophils with a Mac-1 (CD11b/CD18) monoclonal antibody protects against ischemia-reperfusion injury in rat liver. Hepatology. 1993;17:915–923. [PubMed] [Google Scholar]
- Taylor P.C., Chur C.Q., Plater-Zyberk C., Maini R.N. Transfer of type II collagen-induced arthritis from DBA/1 to severe combined immunodeficiency mice can be prevented by blockade of Mac-1. Immunology. 1996;88:315–321. doi: 10.1111/j.1365-2567.1996.tb00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers C., Edelman E.R., Simon D.I. A mAb to the β2-leukocyte integrin Mac-1 (CD11b/CD18) reduces intimal thickening after angioplasty or stent implantation in rabbits. Proc. Natl. Acad. Sci. USA. 1998;95:10134–10139. doi: 10.1073/pnas.95.17.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L., Eaton J.W. Fibrin(ogen) mediates acute inflammatory responses to biomaterials. J. Exp. Med. 1993;178:2147–2156. doi: 10.1084/jem.178.6.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J.-X., Issekutz A.C. Mac-1 (CD11b/CD18) is the predominant β2 (CD18) integrin mediating human neutrophil migration through synovial and dermal fibroblast barriers. Immunology. 1996;88:463–470. doi: 10.1046/j.1365-2567.1996.d01-662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang X.Z., Issekutz A.C. Contribution of CD11a/CD18, CD11b/CD18, ICAM-1 (CD54) and -2 (CD102) to human monocyte migration through endothelium and connective tissue fibroblast barriers. Eur. J. Immunol. 1998;28:1970–1979. doi: 10.1002/(SICI)1521-4141(199806)28:06<1970::AID-IMMU1970>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Kavanaugh A.F., Lightfoot E., Lipsky P.E., Oppenheimer-Marks N. Role of CD11/CD18 in adhesion and transendothelial migration of T cells. J. Immunol. 1991;146:4149–4150. [PubMed] [Google Scholar]
- Jia G.Q., Gonzalo J.A., Hidalgo A., Wagner D., Cybulsky M., Gutierrez-Ramos J.C. Selective eosinophil transendothelial migration triggered by eotaxin via modulation of Mac-1/ICAM-1 and VLA-4/VCAM-1 interactions. Int. Immunol. 1999;11:1–10. doi: 10.1093/intimm/11.1.1. [DOI] [PubMed] [Google Scholar]
- Lu H., Smith C.W., Perrard J., Bullard D., Tang L., Entman M.L., Beaudet A.L., Ballantyne C.M. LFA-1 is sufficient in mediating neutrophil emigration in Mac-1 deficient mice. J. Clin. Invest. 1997;99:1340–1350. doi: 10.1172/JCI119293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz A.R., Coxon A., Maurer M., Gurish M.F., Austen K.F., Friend D.S., Galli S.J., Mayadas T.N. Impaired mast cell development and innate immunity in Mac-1. J. Immunol. 1998;161:6463–6467. [PubMed] [Google Scholar]
- Soriano S.G., Coxon A., Wang Y.F., Frosch M.P., Lipton S.A., Hickey P.R., Mayadas T.N. Mice deficient in Mac-1 (CD11b/CD18) are less susceptible to cerebral ischemia/reperfusion injury. Stroke. 1999;30:134–139. doi: 10.1161/01.str.30.1.134. [DOI] [PubMed] [Google Scholar]
- Colvin R.B., Mosesson M.W., Dvorak H.F. Delayed-type hypersensitivity skin reactions in congenital afibrinogenemia lack fibrin deposition and induration. J. Clin. Invest. 1979;63:1302–1306. doi: 10.1172/JCI109425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busso N., Peclat V., Van Ness K., Kolodziesczyk E., Degen J., Bugge T., So A. Exacerbation of antigen-induced arthritis in urokinase-deficient mice. J. Clin. Invest. 1998;102:41–50. doi: 10.1172/JCI2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altieri D.C., Mannucci P.M., Capitanio A.M. Binding of fibrinogen to human monocytes. J. Clin. Invest. 1986;78:968–976. doi: 10.1172/JCI112687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altieri D.C., Bader R., Mannucci P.M., Edgington T.S. Oligospecificity of the cellular adhesion receptor MAC-1 encompasses an inducible recognition specificity for fibrinogen. J. Cell Biol. 1988;107:1893–1900. doi: 10.1083/jcb.107.5.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijper P.H.M., Torres G., Lammers J.-W.J., Sixma J.J., Koenderman L., Zwaginga J.J. Platelet and fibrin deposition at the damaged vessel wallcooperative substrates for neutrophil adhesion under flow conditions. Blood. 1997;89:166–175. [PubMed] [Google Scholar]
- Languino L.R., Plescia J., Duperray A., Brian A.A., Plow E.F., Geltosky J.E., Altieri D.C. Fibrinogen mediates leukocyte adhesion to vascular endothelium through an ICAM-1-dependent pathway. Cell. 1993;73:1423–1434. doi: 10.1016/0092-8674(93)90367-y. [DOI] [PubMed] [Google Scholar]
- Sriramarao P., Languino L.R., Altieri D.C. Fibrinogen mediates leukocyte-endothelium bridging in vivo at low shear forces. Blood. 1996;88:3416–3423. [PubMed] [Google Scholar]
- Perez R.L., Roman J. Fibrin enhances the expression of IL-1β by human peripheral blood mononuclear cells. Implications in pulmonary inflammation. J. Immunol. 1995;154:1879–1887. [PubMed] [Google Scholar]
- Sitrin R.G., Pan P.M., Srikanth S., Todd R.F., III. Fibrinogen activates NF-κB transcription factors in mononuclear phagocytes. J. Immunol. 1998;161:1462–1470. [PubMed] [Google Scholar]
- Ugarova T.P., Solovjov D.A., Zhang L., Loukinov D.I., Yee V.C., Medved L.V., Plow E.F. Identification of a novel recognition sequence for integrin αMβ2 within the γ-chain of fibrinogen. J. Biol. Chem. 1998;273:22519–22527. doi: 10.1074/jbc.273.35.22519. [DOI] [PubMed] [Google Scholar]
- Diacovo T.G., Roth S.J., Buccola J.M., Bainton D.F., Springer T.A. Neutrophil rolling, arrest, and transmigration across activated, surface-adherent platelets via sequential action of P-selectin and the β2 integrin CD11b/CD18. Blood. 1996;88:146–157. [PubMed] [Google Scholar]
- Arnaout M.A. Structure and function of the leukocyte adhesion molecules CD11/CD18. Blood. 1990;75:1037–1050. [PubMed] [Google Scholar]
- Gahmberg C.G., Tolvanen M., Kotovuori P. Leukocyte adhesion-structure and function of human leukocyte β2-integrins and their cellular ligands. Eur. J. Biochem. 1997;245:215–232. doi: 10.1111/j.1432-1033.1997.00215.x. [DOI] [PubMed] [Google Scholar]
- Harris E.S., McIntyre T.M., Prescott S.M., Zimmerman G.A. The leukocyte integrins. J. Biol. Chem. 2000;275:23409–23412. doi: 10.1074/jbc.R000004200. [DOI] [PubMed] [Google Scholar]
- Diamond M.S., Garcia-Aguilar J., Bickford J.K., Corbí A.L., Springer T.A. The I domain is a major recognition site on the leukocyte integrin MAC-1 (CD11b/CD18) for four distinct adhesion ligands. J. Cell Biol. 1993;120:1031–1043. doi: 10.1083/jcb.120.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitinger B., Hogg N. Integrin I domains and their function. Biochem. Soc. Trans. 1999;27:826–832. doi: 10.1042/bst0270826. [DOI] [PubMed] [Google Scholar]
- Plow E.F., Haas T.A., Zhang L., Loftus J., Smith J.W. Ligand binding to integrins. J. Biol. Chem. 2000;275:21785–21788. doi: 10.1074/jbc.R000003200. [DOI] [PubMed] [Google Scholar]
- Lee J.-O., Rieu P., Arnaout M.A., Liddington R. Crystal structure of the A domain from the α subunit of integrin CR3 (CD11b/CD18) Cell. 1995;80:631–638. doi: 10.1016/0092-8674(95)90517-0. [DOI] [PubMed] [Google Scholar]
- Lee J.-O., Bankston L.A., Arnaout M.A., Liddington R.C. Two conformations of the integrin A-domain (I-domain)a pathway for activation? Structure. 1995;3:1333–1340. doi: 10.1016/s0969-2126(01)00271-4. [DOI] [PubMed] [Google Scholar]
- Shimaoka M., Shifman J.M., Jing H., Takagi L., Mayo S.L., Springer T.A. Computational design of an integrin I domain stabilized in the open high affinity conformation. Nat. Struct. Biol. 2000;7:674–678. doi: 10.1038/77978. [DOI] [PubMed] [Google Scholar]
- Thornton B.P., Vetvicka V., Pitman M., Goldman R.C., Ross G.D. Analysis of the sugar specificity and molecular location of the β-glucan-binding lectin site of complement receptor type 3. J. Immunol. 1996;156:1235–1246. [PubMed] [Google Scholar]
- Doolittle R.F. Fibrinogen and fibrin. Annu. Rev. Biochem. 1984;53:195–229. doi: 10.1146/annurev.bi.53.070184.001211. [DOI] [PubMed] [Google Scholar]
- Altieri D.C., Plescia J., Plow E.F. The structural motif glycine 190-valine 202 of the fibrinogen γ chain interacts with CD11b/CD18 integrin (αMβ2, Mac-1) and promotes leukocyte adhesion. J. Biol. Chem. 1993;268:1847–1853. [PubMed] [Google Scholar]
- Zhang L., Plow E.F. Identification and reconstruction of the binding pocket within αMβ2 for a specific and high affinity ligand, NIF. J. Biol. Chem. 1997;272:17558–17564. doi: 10.1074/jbc.272.28.17558. [DOI] [PubMed] [Google Scholar]
- Zhang L., Plow E.F. Amino acid sequences within the α subunit of integrin αMβ2 (Mac-1) critical for specific recognition of C3bi. Biochemistry. 1999;38:8064–8071. doi: 10.1021/bi990141h. [DOI] [PubMed] [Google Scholar]
- Forsyth C.B., Plow E.F., Zhang L. Interaction of the fungal pathogen Candida albicans with integrin CD11b/CE18recognition by the I domain is modulated by the lectin-like domain and the CD18 subunit. J. Immunol. 1998;161:6198–6205. [PubMed] [Google Scholar]
- Yakubenko V.P., Solovjov D.A., Zhang L., Yee V.C., Plow E.F., Ugarova T.P. Identification of the binding site for fibrinogen recognition peptide γ383-395 within the αM I-domain of integrin αMβ2 . J. Biol. Chem. 2001;276:13995–14003. doi: 10.1074/jbc.M010174200. [DOI] [PubMed] [Google Scholar]
- Doolittle R.F., Schubert D., Schwartz S.A. Amino acid sequence studies on artiodactyl fibrinopeptides I. Dromedary camel, mule deer, and cape buffalo. Arch. Biochem. Biophys. 1967;118:456–467. doi: 10.1016/0003-9861(67)90374-8. [DOI] [PubMed] [Google Scholar]
- Ugarova T.P., Budzynski A.Z. Interaction between complementary polymerization sites in the structural D and E domains of human fibrin. J. Biol. Chem. 1992;267:13687–13693. [PubMed] [Google Scholar]
- Zhang L., Plow E.F. Overlapping, but not identical sites, are involved in the recognition of C3bi, NIF, and adhesive ligands by the αMβ2 integrins. J. Biol. Chem. 1996;271:18211–18216. doi: 10.1074/jbc.271.30.18211. [DOI] [PubMed] [Google Scholar]
- Miles L.A., Plow E.F. Receptor mediated binding of the fibrinolytic components, plasminogen and urokinase, to peripheral blood cells. Thromb. Haemost. 1987;58:936–942. [PubMed] [Google Scholar]
- Palecek S.P., Loftus J.C., Ginsberg M.H., Lauffenburger D.A., Horwitz A.F. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature. 1997;385:537–540. doi: 10.1038/385537a0. [DOI] [PubMed] [Google Scholar]
- Tozer E.C., Liddington R.C., Sutcliffe M.J., Smeeton A.H., Loftus J.C. Ligand binding to integrin αIIbβ3 is dependent on a MIDAS-like domain in the β3 subunit. J. Biol. Chem. 1996;271:21978–21984. doi: 10.1074/jbc.271.36.21978. [DOI] [PubMed] [Google Scholar]
- Loftus J.C., Liddington R.C. Perspectives seriescell adhesion in vascular biology. New insights into integrin-ligand interaction. J. Clin. Invest. 1997;99:2302–2306. doi: 10.1172/JCI119408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzon-McLaughlin W., Takada Y. Critical residues for ligand binding in an I domain-like structure of the integrin β1 subunit. J. Biol. Chem. 1996;271:20438–20443. doi: 10.1074/jbc.271.34.20438. [DOI] [PubMed] [Google Scholar]
- Huang C.C., Zang Q., Takagi J., Springer T.A. Structural and functional studies with antibodies to the integrin β2 subunita model for the I-like domain. J. Biol. Chem. 2000;275:21514–21524. doi: 10.1074/jbc.M002286200. [DOI] [PubMed] [Google Scholar]
- Bajt M.L., Loftus J.C. Mutation of a ligand binding domain of β3 integrinintegral role of oxygenated residues in αIIbβ3 receptor function. J. Biol. Chem. 1994;269:20913–20919. [PubMed] [Google Scholar]
- Goodman T.G., Bajt M.L. Identifying the putative metal ion-dependent adhesion site in the β2 (CD18) subunit required for αLβ2 and αMβ2 ligand interactions. J. Biol. Chem. 1996;271:23729–23736. doi: 10.1074/jbc.271.39.23729. [DOI] [PubMed] [Google Scholar]
- Wright S.D., Weitz J.I., Huang A.J., Levin S.M., Silverstein S.C., Loike J.D. Complement receptor type three (CD11b/CD18) of human polymorphonuclear leukocytes recognizes fibrinogen. Proc. Natl. Acad. Sci. USA. 1988;85:7734–7738. doi: 10.1073/pnas.85.20.7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller B.S. A new murine monoclonal antibody reports an activation-dependent change in the conformation and/or microenvironment of the platelet glycoprotein IIb/IIIa complex. J. Clin. Invest. 1985;76:101–108. doi: 10.1172/JCI111931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller B.S. Binding of abciximab to αVβ3 and activated αMβ2 receptorswith a review of platelet-leukocyte interactions. Thromb. Haemost. 1999;82:326–336. [PubMed] [Google Scholar]
- Plescia J., Conte M.S., VanMeter G., Ambrosini G., Altieri D.C. Molecular identification of the cross-reacting epitope on αMβ2 integrin I domain recognized by anti-αIIbβ3 monoclonal antibody 7E3 and its involvement in leukocyte adherence. J. Biol. Chem. 1998;273:20372–20377. doi: 10.1074/jbc.273.32.20372. [DOI] [PubMed] [Google Scholar]
- Simon D.I., Xu H., Ortlepp S., Rogers C., Rao N.K. 7E3 monoclonal antibody directed against the platelet glycoprotein IIb/IIIa cross-reacts with the leukocyte integrin Mac-1 and blocks adhesion to fibrinogen and ICAM-1. Arterioscler. Thromb. Vasc. Biol. 1997;17:528–535. doi: 10.1161/01.atv.17.3.528. [DOI] [PubMed] [Google Scholar]
- Tang L., Ugarova T.P., Plow E.F., Eaton J.W. Molecular determinants of acute inflammatory responses to biomaterials. J. Clin. Invest. 1996;97:1329–1334. doi: 10.1172/JCI118549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Languino L.R., Duperray A., Joganic K.J., Fornaro M., Thornton G.B., Altieri D.C. Regulation of leukocyte-endothelium interaction and leukocyte transendothelial migration by intercellular adhesion molecule 1-fibrinogen recognition. Proc. Natl. Acad. Sci. USA. 1995;92:1505–1509. doi: 10.1073/pnas.92.5.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross T.J., Leavell K.J., Peterson M.W. CD11b/CD18 mediates the neutrophil chemotactic activity of fibrin degradation product D domain. Thromb. Haemost. 1997;77:894–900. [PubMed] [Google Scholar]
- Huttenlocher A., Palecek S.P., Lu Q., Zhang W., Mellgren R.L., Lauffenburger D.A., Ginsberg M.H., Horwitz A.F. Regulation of cell migration by the calcium-dependent protease calpain. J. Biol. Chem. 1997;272:32719–32722. doi: 10.1074/jbc.272.52.32719. [DOI] [PubMed] [Google Scholar]
- Du X., Plow E.F., Frelinger A.L., III, O'Toole T.E., Loftus J.C., Ginsberg M.H. Ligands “activate” integrin αIIbβ3 (platelet GPIIb-IIIa) Cell. 1991;65:409–416. doi: 10.1016/0092-8674(91)90458-b. [DOI] [PubMed] [Google Scholar]
- Li R., Xie J., Kantor C., Koistinen V., Altieri D.C., Nortamo P., Gahmberg C.G. A peptide derived from the intercellular adhesion molecule-2 regulates the avidity of the leukocyte integrins CD11b/CD18 and CD11c/CD18. J. Cell Biol. 1995;129:1143–1153. doi: 10.1083/jcb.129.4.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetvicka V., Thornton B.P., Ross G.D. Soluble β-glucan polysaccharide binding to the lectin site of neutrophil or natural killer cell complement receptor type 3 (CD11b/CD18) generates a primed state of the receptor capable of mediating cytotoxicity of iC3b-opsonized target cells. J. Clin. Invest. 1996;98:50–61. doi: 10.1172/JCI118777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeish K.R., Klein J.B., Coxon P.Y., Head K.Z., Ward R.A. Bacterial phagocytosis activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase cascades in human neutrophils. J. Leukoc. Biol. 1998;64:835–844. [PubMed] [Google Scholar]
- Oxvig C., Lu C., Springer T.A. Conformational changes in tertiary structure near the ligand binding site of an integrin I domain. Proc. Natl. Acad. Sci. USA. 1999;96:2215–2220. doi: 10.1073/pnas.96.5.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugarova T.P., Budzynski A.Z., Shattil S.J., Ruggeri Z.M., Ginsberg M.H., Plow E.F. Conformational changes in fibrinogen elicited by its interaction with platelet membrane glycoprotein GPIIb-IIIa. J. Biol. Chem. 1993;268:21080–21087. [PubMed] [Google Scholar]