

Abstract
Human, but not murine, adenosine deaminase (ADA) forms a complex with the cell membrane protein CD26/dipeptidyl peptidase IV. CD26-bound ADA has been postulated to regulate extracellular adenosine levels and to modulate the costimulatory function of CD26 on T lymphocytes. Absence of ADA–CD26 binding has been implicated in causing severe combined immunodeficiency due to ADA deficiency. Using human–mouse ADA hybrids and ADA point mutants, we have localized the amino acids critical for CD26 binding to the helical segment 126–143. Arg142 in human ADA and Gln142 in mouse ADA largely determine the capacity to bind CD26. Recombinant human ADA bearing the R142Q mutation had normal catalytic activity per molecule, but markedly impaired binding to a CD26+ ADA-deficient human T cell line. Reduced CD26 binding was also found with ADA from red cells and T cells of a healthy individual whose only expressed ADA has the R142Q mutation. Conversely, ADA with the E217K active site mutation, the only ADA expressed by a severely immunodeficient patient, showed normal CD26 binding. These findings argue that ADA binding to CD26 is not essential for immune function in humans.
Keywords: adenosine deaminase deficiency, severe combined immunodeficiency, T lymphocyte, protein–protein interaction, adenosine deaminase complexing protein
Introduction
Adenosine deaminase (ADA) deficiency in humans causes profound lymphopenia, which is evident by midgestation and results in SCID during infancy 1 2. Various effects of adenosine (Ado) and 2′-deoxyadenosine (dAdo) have been implicated in pathogenesis 3. Skeletal, neurologic, and hepatic abnormalities that occur in some patients may also be due to the metabolic disorder, but these are of less clinical relevance than the immunodeficiency. In contrast to the phenotype in humans, ADA knockout mice have normal lymphoid development at birth and die perinatally of hepatic and pulmonary injury 4 5. Lymphopenia develops postnatally in strains genetically engineered to express only placental ADA, but these animals die at a few weeks of age from lung injury 4 5 6. The greater lymphoid selectivity in humans than mice may reflect differences in purine metabolism, tissue sensitivity to ADA substrates, or timing of development. In this context it is intriguing that human ADA, but not murine ADA, can form a complex with a multifunctional membrane protein that has a role in regulating thymocyte proliferation and T cell activation.
The ADA of human erythrocytes behaves as a soluble monomer of ∼41 kD. Larger forms (>200 kD) found in extracts of other tissues of humans, rabbits, and cattle are due to the binding of ADA to a homodimeric membrane glycoprotein with subunit M r ∼ 110 kD 7 8 9. This so-called “ADA complexing protein” (ADA-CP) was shown to bind two ADA monomers with KA of 4–20 nM 8 10. ADA-CP occurs on secretory or absorptive surfaces of epithelia of liver, gut, kidney, and exocrine glands 11. Mice lack “large” forms of ADA 12, but were found to possess a membrane protein with the tissue distribution and size of ADA-CP, which cross-reacted with Ab to human ADA-CP, but bound neither murine nor human ADA 13. Unlike human and bovine ADA, murine ADA did not bind to rabbit ADA-CP 10.
ADA-CP has been identified as a protein known both as CD26 and dipeptidyl peptidase IV (DPPIV; 14, 15). CD26 was first defined as an antigen on activated human T lymphocytes 16, and DPPIV as a widely distributed ectoenzyme that cleaves peptides with Pro or Ala at position 2, including several hormones, neuropeptides, and cytokines 17 18. CD26 and DPPIV cDNAs from human T cells and intestine predict the same 766-residue, 88-kD polypeptide 19 20. This type II membrane protein has 6 NH2-terminal cytoplasmic residues, a 34-residue membrane anchor, a large extracellular domain consisting of a glycosylated “stalk”, a cysteine-rich segment (residues 290–552), and a COOH-terminal region that bears the serine protease (DPPIV) active site (residues 628–632 21). Residues 294 and 340–343 of the cysteine-rich segment are essential for binding ADA 22 23. In addition to ADA, CD26 has been reported to bind collagen, fibronectin, the CD45 tyrosine phosphatase, and the HIV-1 Tat protein (for reviews, see references 21 and 24).
Among lymphoid cells, CD26 is expressed on medullary thymocytes, blood T cells with a helper/memory phenotype, activated B cells, and NK cells. On T cells, CD26 acts as a “co-stimulator” of antigen receptor–mediated activation 24. CD26 ligation also stimulates the proliferation of thymocytes and other hematopoietic cells in the mouse and rat 25 26. It has been variously proposed that in humans, binding of ADA is important to the costimulatory function of CD26, that such binding protects lymphocytes from effects of extracellular Ado, and that immunodeficiency in patients with ADA deficiency is due to the absence of CD26-associated ADA 15 23 24 27. We have defined the amino acid residues of human ADA that are essential for CD26 binding, and investigated the effects on CD26 binding of ADA mutations from individuals with ADA deficiency. Our findings are not consistent with some predictions about the nature of the CD26 binding site; neither do they support the hypothesis that ADA–CD26 binding is essential for immune function in humans.
Materials and Methods
Materials
DEAE-Sepharose fast flow, Superose 12, and CNBr-activated Sepharose 4B were obtained from Amersham Pharmacia Biotech. ADA-Sepharose was prepared by coupling calf mucosal ADA (Sigma-Aldrich and Boehringer) to CNBr-activated Sepharose 4B according to the manufacturer's instructions. Plasmid pZC11 and Escherichia coli SØ3834 were provided by Dr. Rod Kellems (University of Texas, Houston, TX). Goat anti–human ADA antiserum was provided by Dr. Dan Wiginton (University of Cincinnati, Cincinnati, OH). The mouse mAb 1C5 was prepared against ADA purified from human T cell leukemia cells (Hershfield, M.S., unpublished details).
Lymphoid Cells and Cell Lines
Activated T lymphocytes were prepared by culturing blood mononuclear cells with phytohemagglutinin and IL-2 as described 28, except that the cells were grown in AIM-V serum-free medium (Life Technologies). The HTLV-1–transformed AlNe cell line derived from an ADA-deficient patient (provided by Dr. Ken Weinberg, Los Angeles Children's Hospital, Los Angeles, CA) was also maintained in AIM-V medium.
Enzyme Assays
DPPIV activity was determined using Gly-Pro-p-nitroanilide tosylate (Sigma-Aldrich) as substrate, monitoring A405 at 37°C 29. ADA activity was determined by monitoring the decrease in A263 at 37°C using as substrate 150 μM Ado in 50 mM Tris-HCl, pH 7.4. Both the CD26 and ADA assays were performed in 96-well microtiter plates (Costar UV plate; Corning Inc.) in reaction volumes of 0.2 ml, using a SpectraMax plus spectrophotometer (Molecular Devices). ADA was also assayed by a radiochemical-TLC method 28. Protein was determined using the bicinchoninic acid (BCA) method (Pierce Chemical Co.) with BSA as standard.
Rabbit Kidney CD26/DPPIV
CD26/DPPIV was purified from New Zealand white rabbit kidney homogenates by a published procedure involving DEAE-Sepharose and ADA-Sepharose 4B chromatography 30. In some preparations, the ADA-Sepharose column was eluted with 6 M urea in 0.01 M KPO4, pH 7.4, 0.1% Triton X-100, as described 31. Fractions with DPPIV activity were pooled, dialyzed against 0.01 M KPO4, pH 7.4, 0.1% Triton X-100, and stored at −20°C. The purified preparation had a DPPIV-specific activity of 7,883 μmol min−1mg−1. A partially purified preparation used for screening ADA–CD26 binding had DPPIV activity of 6.5–7.2 μmol min−1mg−1. ADA activity of these preparations was <0.003 μmol min−1mg−1.
Bovine ADA cDNA
Standard methods were used to amplify, clone, and sequence cDNA 32 33. Degenerate reverse transcription PCR primers were used to clone the ADA coding region from bovine thymus RNA: (+)5′ATCGAAGCTTCCATGGCCCAGACRCCCGC- MTTC, (−)5′GGCCATGGAGAGGTAGCCACGACACCTT- CACAGACA. Both strands were sequenced by the dideoxy method (sequence data are available from EMBL/GenBank/DDBJ under accession no. AF280603). The expressed product of the bovine ADA cDNA was enzymatically active (Kelly, S.J., and M.S. Hershfield, unpublished data).
Construction of Human–Mouse ADA cDNA Hybrids and Point Mutants, and Expression of ADA for CD26 Binding Studies
cDNAs consisting of segments from both the human 34 and mouse 35 ADA coding regions were made by overlap extension PCR (36; Table and Table ). The P126Q, R142Q, R149Q, A215T, and E217K ADA mutants have been identified in human subjects 37 38 39 40; their expression in E. coli strain SØ3834 has been reported 41. Other mutations (see Results) were introduced into the wild-type human or mouse ADA cDNAs by PCR mutagenesis essentially as described 37 41. All final ADA cDNA PCR products were cloned into pBluescript II KS or pBluescript SK and fully sequenced using the ABI 377 PRISM DNA Sequencing Instrument (Applied Biosystems).
Table 1.
Human–Mouse ADA Hybrids
| Construct | Templates(ADA cDNAs) | Primers |
|---|---|---|
| M1–81/H82–363 | I: M wild-type | 1, 2 |
| II: H wild-type | 3, 4 | |
| III: Products of I and II | 1, 4 | |
| H1–81/M 82–125/H126–363 | I: H 1–81/ M 82–352 | 5, 6 |
| II: H wild-type | 7, 4 | |
| III: Products of I and II | 5, 4 | |
| H1–125/M126–143/H144–363 | I: H wild-type | 5, 8 |
| II: H wild-type | 9, 4 | |
| III: Products of I and II | 5, 4 | |
| H1–143/M144–247/H248–363 | I: H wild-type | 5, 10 |
| II: M 1–247/H 248–363 | 11, 4 | |
| III: Products of I and II | 5, 4 | |
| H1–247/M248–352 | I: H wild-type | 5, 12 |
| II: M wild-type | 13, 14 | |
| III: Products of I and II | 5, 14 | |
| H1–81/M82–352 | I: H wild-type | 5, 15 |
| II: M wild-type | 16, 14 | |
| III: Products of I and II | 5, 14 | |
| M1–247/ 248–363 | I: M wild-type | 1, 17 |
| II: H wild-type | 18, 4 | |
| III: Products of I and II | 1, 4 | |
| M1–125/H126–143/M144–352 | I: M wild-type | 1, 6 |
| II: M wild-type | 11, 14 | |
| III: Products of I and II | 1, 14 |
Table 2.
PCR Primers Used in Constructing Human–Mouse ADA Hybrids
| Primer sequence | Location in ADA cDNA | |
|---|---|---|
| 1 | (+)CGCGCGAATTCGGGCACCATGGCCCAGACACCCGCATTC | M1–21 (EcoRI/NcoI) |
| 2 | (−)CATCTCTACAAACTCATAGGCGATCCTCTTGATGGCCTCTCTGCA | Reverse of primer 3 |
| 3 | (+)TGCAGAGAGGCCATCAAGAGGATCGCCTATGAGTTTGTAGAGATG | M223–243/H244–267 |
| 4 | (−)GCGCAAGCTTCGGGCCATGGTCTTCAGAGGTTCTGCCCTGCAG | H1073–1092 (HindIII/NcoI) |
| 5 | (+)CGCGCGAATTCGGGCACCATGGCCCAGACGCCCGCCTTCGAC | H1–24 (EcoRI/NcoI) |
| 6 | (−)GTCTCGCTCCCCCTCCTGCAGGCCCTGGCCCACTAGGGCTACCACCT | Reverse of primer 7 |
| CGTCTGGGGTGACGTCCCCTTCAGTCTGGTTCCAGGG | ||
| 7 | (+)CCCTGGAACCAGACTGAAGGGGACGTCACCCCAGACGAGGTGGTAGC | M346–375/H376–429 |
| CCTAGTGGGCCAGGGCCTGCAGGAGGGGGAGCGAGAC | ||
| 8 | (−)TGCTTGCTCTCCCTCCTGCAGGCCCTGGTTCACAAGATCCACAACGT | H346–375/M376–429 |
| CATCAGGGGTGAGGTCCCCTTCAGCCTGGTTCCAGGG | ||
| 9 | (+)CCTGATGACGTTGTGGATCTTGTGAACCAGGGCCTGCAGGAGGGAGA | M376–429/H430–459 |
| GCAAGCATTCGGGGTCAAGGCTCGGTCCATCCTGTGC | ||
| 10 | (−)GCACAGAATGGACCGGACCTTGATGCCAAAGTCTCGCTCCCCCTCCT | Reverse of primer 11 |
| GCAGGCCCTGGCCCACTAGGGCTACCACCTCGTCTGG | ||
| 11 | (+)CCAGACGAGGTGGTAGCCCTAGTGGGCCAGGGCCTGCAGGAGGGGGA | H376–429/M430–459 |
| GCGAGACTTTGGCATCAAGGTCCGGTCCATTCTGTGC | ||
| 12 | (−)TTCTTTCAGTAGTCTGTTGTAGAGGGCCTGGTCTTCCAGGGTGTG | Reverse of primer 13 |
| 13 | (+)CACACCCTGGAAGACCAGGCCCTCTACAACAGACTACTGAAAGAA | H721–741/M742–765 |
| 14 | (−)GCGCGATATCCGGGCCATGGTCTCTATTGGTATTCTCTGTAGAGCC | M1037–1059 (EcoRV/NcoI) |
| 15 | (−)CATCTCCACAAACTCGTAGGCGATCCTTTTGATAGCCTCCCGGCA | Reverse of primer 16 |
| 16 | (+)TGCCGGGAGGCTATCAAAAGGATCGCCTACGAGTTTGTGGAGATG | H223–243/M 244–267 |
| 17 | (−)TTCCTGCCGCAGCCTGTTATAAAGAGCTTCATCCTCGATGGTGTG | Reverse of primer 18 |
| 18 | (+)CACACCATCGAGGATGAAGCTCTTTATAACAGGCTGCGGCAGGAA | M721–741/H742–765 |
ADA cDNAs were ligated into the NcoI site of pZ (derived from pZC11 from which wild-type human ADA cDNA had been excised [42]). pZ/ADA plasmids were used to transform E. coli SØ3834, which has a deletion of the bacterial ADA gene 42. Single transformant colonies were grown at 37°C in Luria broth/carb/tet medium to constitutively express ADA, as described 41. Under these conditions, the yield of wild-type mouse ADA was four- to sixfold higher than for human ADA, determined both by activity assay (Table ) and by Western blotting with cross-reacting goat anti–human ADA antiserum (not shown). Studies with human–mouse ADA hybrids (Table ) and results not presented indicate that this difference in expression is related to the presence in human ADA of NH2-terminal codons used infrequently in E. coli.
Table 3.
ADA Activity of Human–Mouse Hybrids Expressed in E. coli SØ3834
| ADA cDNA | ADA activity(mean ± SD) | Wild-typehuman ADA |
|---|---|---|
| μmol/min per mg protein | % | |
| Human wt | 3.3 ± 0.7 | 100.0 |
| Mouse wt | 12.4 ± 1.3 | 375.7 |
| M1–81/H82–363 | 13.5 ± 2.2 | 409.0 |
| H1–81/M82–125/H126–363 | 4.3 ± 0.4 | 130.3 |
| H1–125/M126–143/H144–363 (“m126–143”) | 1.2 ± 0.1 | 36.3 |
| H1–143/M144–247/H248–363 | 2.9 ± 0.3 | 87.8 |
| H1–247/M248–352 | 2.0 ± 0.2 | 60.6 |
| H1–81/M82–352 | 1.8 ± 0.7 | 54.5 |
| M1–247/H248–363 | 12.3 ± 3.8 | 372.7 |
| M1–125/H126–143/M144–352 (“h126–143”) | 13.1 ± 3.6 | 396.9 |
wt, wild-type; H, human; M, mouse.
To prepare ADA for CD26 binding studies, the cells from 100-ml overnight cultures were sonicated in 3 ml of lysis buffer 41. In some experiments the cells from 1-liter cultures were disrupted in 20 ml of lysis buffer using a Microfluidizer Model M-110L (Microfluidics, Inc.). After centrifugation (20,190 g, 15 min, 4°C), supernatants were passed through 0.2 μm filters and dialyzed at 4°C against 0.01 M KPO4, pH 7.4. In addition to assaying ADA activity, aliquots of these lysates (20-μg protein) were analyzed by Western blotting using goat anti–human ADA antiserum, as described 41. Western blotting was also performed with the 1C5 mouse mAb to human ADA (1:2,000 dilution of ammonium sulfate–concentrated mouse ascites); a goat anti–mouse IgG–horseradish peroxidase conjugate (Santa Cruz Biotechnology, Inc.) was used as a second Ab, and was detected with the Enhanced Chemiluminescence System (Amersham Pharmacia Biotech).
ADA-CD26 Binding
FPLC Screening.
In the standard assay, dialyzed (0.01 M KPO4, pH 7.4) lysates of pZ/ADA cDNA-transformed E. coli SØ3834 cells containing 100–250 nmol/min of ADA activity (diluted as necessary with dialyzed lysate of untransformed SØ3834 cells) were combined with 10–30 nmol/min (DPPIV activity) of rabbit CD26 in 0.01 M KPO4, pH 7.4, 0.1% Triton X-100, total volume 150 μl. After incubating for 2 h at 37°C, the entire mixture was injected onto a 1.5 × 30 cm Superose 12 column equilibrated with 50 mM Tris-HCl, pH 8.4 (fast protein liquid chromatography [FPLC] system; Amersham Pharmacia Biotech). The column was eluted with this buffer at room temperature, flow rate 0.5 ml/min. Fractions (0.5 ml) were assayed for ADA and DPPIV activities, and the percentage of total ADA activity associated with the peak of DPPIV activity (CD26) was calculated. In some experiments the column was equilibrated and eluted with 50 mM Tris-HCl, pH 8.4, containing 150 mM NaCl. This shifted the peak of free (41 kD) ADA activity from fraction 25 to fraction 27, but neither the DPPIV activity peak nor the amount of ADA activity associated with the DPPIV peak were altered.
Gel Mobility Shift Assay.
[35S]Met-labeled ADA was generated in vitro from ADA cDNA constructs in pBluescript, using the TNT Coupled Wheat Germ Extract System (Promega) according to the manufacturer's instructions (this system was used in preference to rabbit reticulocyte extracts because it possessed very low endogenous ADA-like activity). Translation products were analyzed by SDS-PAGE and fluorography, and were also electrophoresed on cellulose acetate and stained for ADA activity in situ, as described 28. Aliquots of the translation mixtures containing equal amounts of 35S-labeled products were then incubated (2 h, 37°C) with 0.7–2.2 nmol/min of rabbit CD26/DPPIV in 0.01 M KPO4, pH 7.4, 0.1% Triton X-100, total volume 20 μl. These reaction mixtures were then electrophoresed on a 3–15% gradient nondenaturing PAGE gel (pH 8.8, 30% acrylamide, 0.8% bisacrylamide; running conditions 60 V/cm, 18 h, 4°C 43). [35S]ADA was located by fluorography.
Recombinant ADA Binding to AlNe T Cells
Aliquots (25 μl) of pZ/ADA cDNA-transformed SØ3834 lysates containing 400 nmol/min of ADA activity were added to 2 × 107 AlNe T cells in 1 ml of serum-free growth medium and incubated 2 h at 37°C. The T cells were then washed three times with PBS, pH 7.4, and lysed by freezing and thawing (or by sonication, which gave identical results) in 150 μl of 25 mM Tris-HCl, pH 7.5, 1 mM EDTA, 1 mM DTT, 0.1% Triton X-100. After centrifugation, the supernatant was analyzed as described above for the FPLC CD26 binding assay. ADA binding to intact AlNe cells was determined by flow cytometry using the 1C5 anti-ADA mAb (see below). For these studies, 106 AlNe cells were incubated for 60 min at 37°C in 1 ml of medium containing aliquots of untransformed or ADA-expressing SØ3834 lysates. The cells were then washed with cold PBS containing 1% BSA and 0.1% sodium azide (PBSW) and processed for flow cytometry.
Flow Cytometry
Surface antigens and bound ADA on AlNe T cells were detected with the following reagents: unconjugated anti-ADA mAb 1C5 and control mAb P3; and fluorochrome-conjugated Ta1-RD1 (CD26), B1-RD1 (CD20), T11-RD1 (CD2), mouse IgG1-RD1, and either fluorescein- or PE-conjugated goat anti–mouse IgG1 provided by Beckman Coulter (Fullerton, CA). All Ab incubations were performed in PBSW at 4°C. After washing with PBSW, cells were fixed with 0.4% paraformaldehyde, and analyzed using an Epics Elite XL flow cytometer (Beckman Coulter). Data were processed using the FACSConvert™ and CellQuest™ software programs (Becton Dickinson).
Results
Binding of Recombinant ADAs to Rabbit CD26.
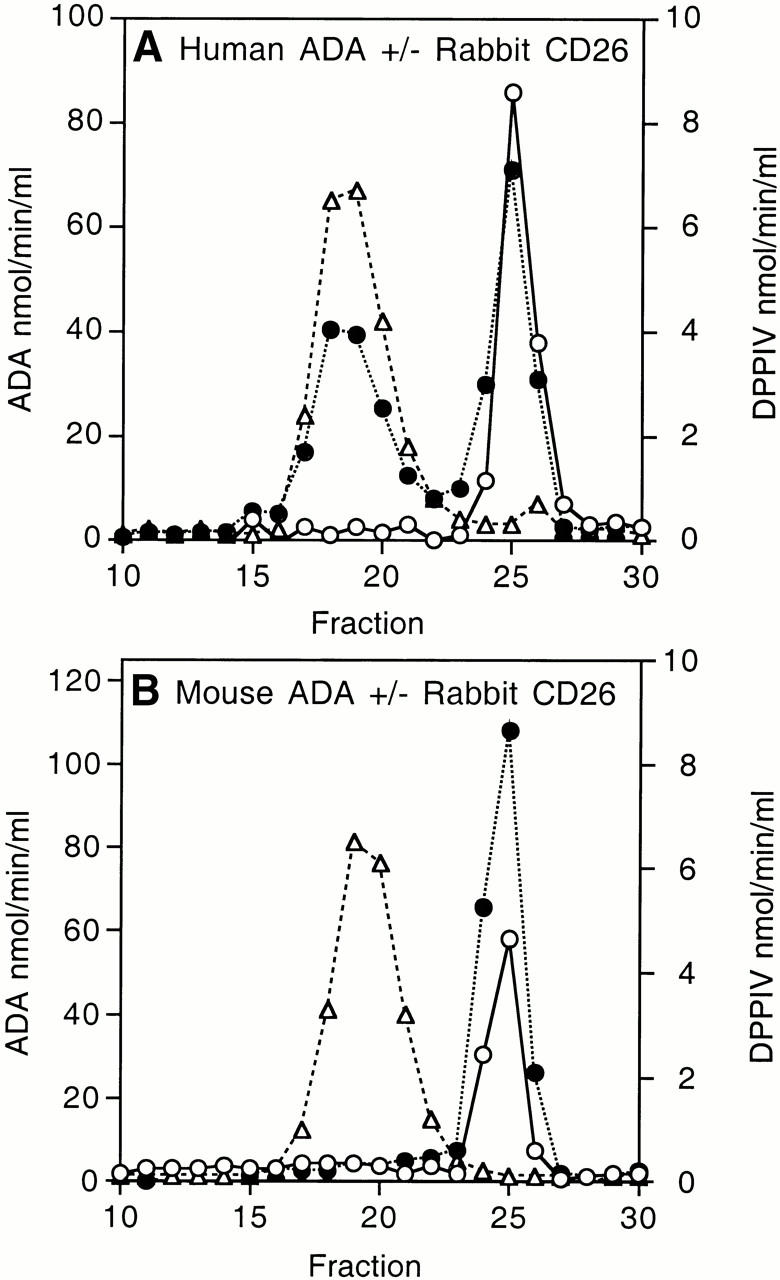
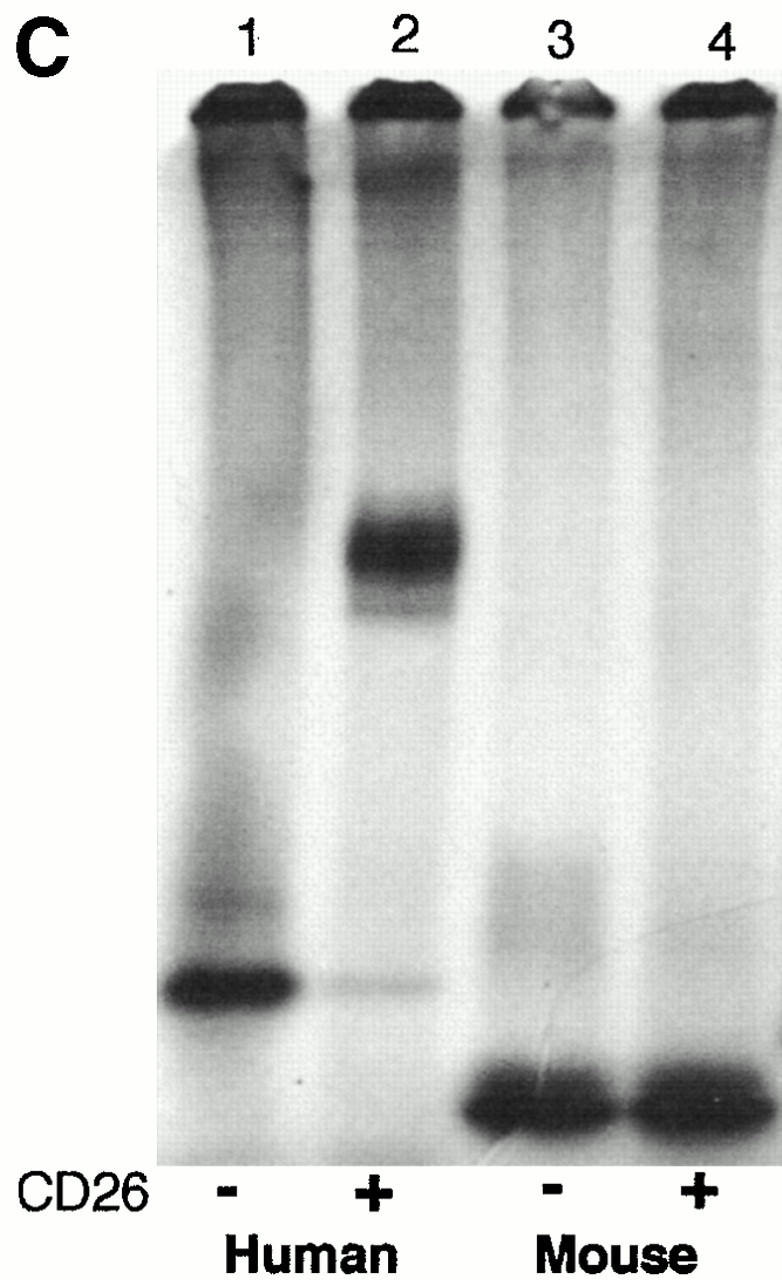
Like the enzymes isolated from tissues 10, human and mouse ADA expressed in E. coli SØ3834 differ markedly in ability to bind rabbit kidney CD26/DPPIV. When equal amounts of ADA activity were compared, ∼50% of human and <1% of mouse ADA activity coeluted with DPPIV activity from a Superose 12 FPLC column (Fig. 1A and Fig. B). The difference in CD26 binding could also be demonstrated using nondenaturing PAGE to assess the effect of rabbit CD26 on the migration of 35S-labeled human and mouse ADA in vitro translation products (Fig. 1 C). Addition of unlabeled human ADA expressed in E. coli SØ3834 (100 nmol/min) blocked the CD26-induced shift in migration of the human [35S]ADA translation product, whereas the same amount of recombinant mouse ADA had no effect (not shown).
Figure 1.
Binding of recombinant wild-type human and mouse ADA to rabbit CD26. (A and B) FPLC assay. Lysates of E. coli SØ3834 cells containing equal units of wild-type human ADA (A) or wild-type mouse ADA (B) were incubated in the absence (open circles) or presence (filled circles) of rabbit CD26, then fractionated on Superose 12. Circles, ADA activity; triangles, DPPIV activity. (C) Nondenaturing PAGE assay. 35S-labeled ADA in vitro translation products were incubated in the presence (+) or absence (−) of rabbit CD26 and then subjected to nondenaturing PAGE, followed by fluorography. Lane 1, wild-type human ADA; lane 2, wild-type human ADA plus rabbit CD26; lane 3, wild-type mouse ADA; lane 4, wild-type mouse ADA plus rabbit CD26.
Based on these results, we constructed human–mouse ADA cDNA hybrids (Table Table Table ) and used the above assays to locate residues necessary for CD26 binding (analogous to human–rat CD26 “swap mutants” used to identify CD26 residues necessary for ADA binding [22]). To refine our search, we also used point mutants found in ADA-deficient individuals, which we have previously expressed in SØ3834 41. ADA mutants with sufficient catalytic activity were screened by FPLC, although in some cases using less ADA activity than in the “standard” assay (see Materials and Methods). The PAGE assay was used for ADA mutants with very low catalytic activity. Some mutants could not be tested because their activity was too low for the FPLC assay, and their in vitro translation products did not enter the nondenaturing gel (results not shown).
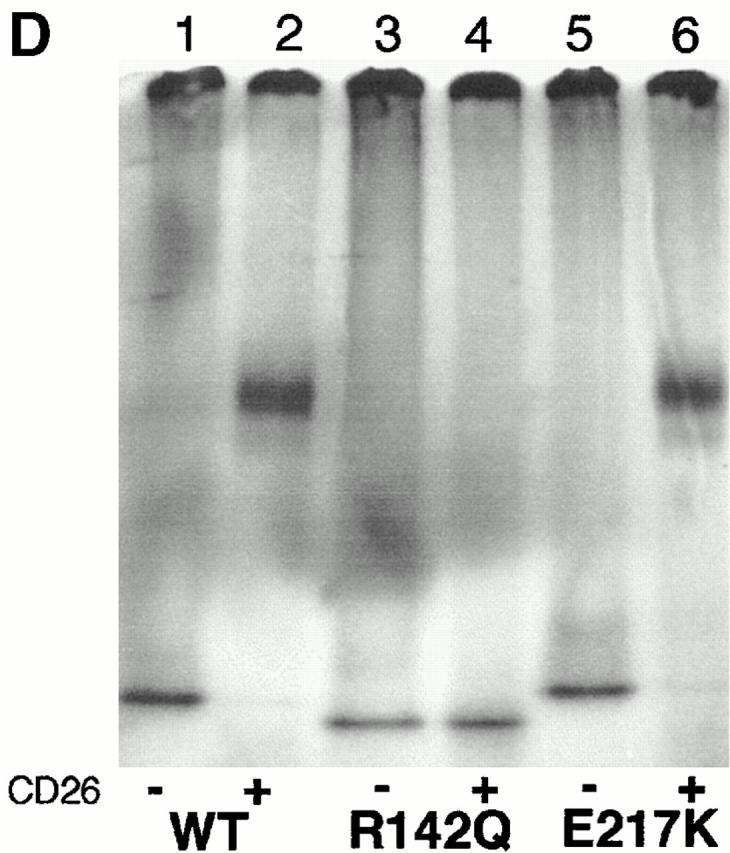
Initial hybrid screening placed the CD26 binding site between residues 82 and 248 (Fig. 2A and Fig. B). This interval was narrowed by results with the R142Q point mutant, which expressed 28% of wild-type human ADA activity in SØ3834 41: in four standard FPLC binding experiments, 10.4 ± 2.9% (mean ± SD) of R142Q ADA activity coeluted with DPPIV activity (Fig. 2 C), resulting in 0.9 ± 0.5 ADA units bound per unit DPPIV. By comparison, in five standard experiments with wild-type human ADA, 48.7 ± 5.1% of ADA activity coeluted with DPPIV activity, resulting in the binding of 5.9 ± 1.4 units of ADA per unit of DPPIV (Fig. 2A and Fig. B). We also tested the P126Q, R149Q, and A215T mutants, which expressed 0.3, 6.2, and 4.8% of wild-type ADA activity, respectively 41. Experiments with these mutants were performed with less ADA activity than under standard conditions. However, 75% to >90% of the added ADA activity coeluted with DPPIV activity (Fig. 2 C), suggesting that these mutations did not impair CD26 binding. Association of 35S-labeled R142Q in vitro translation product with CD26 was undetectable by PAGE, whereas 35S-labeled E217K, an active site mutant with <0.005% of wild-type ADA activity 41 44, showed normal binding (Fig. 2 D). Reduced CD26 binding by the R142Q mutant was of particular interest because (a) glutamine is the “wild-type” amino acid at position 142 in mouse ADA, and (b) R142Q was identified in a healthy adult whose other ADA allele was not expressed 38.
Figure 2.
Binding of rabbit CD26 by human–mouse ADA hybrids and ADA point mutants. (A and B) FPLC assay of human–mouse ADA hybrids. Hybrid structures are shown as bars in which white indicates human (h) and black indicates mouse (m) ADA amino acid residues (numbered beside bars). Gray bars in A and B show the percentage of input ADA activity associated with DPPIV activity (A), or the units of ADA bound per unit of DPPIV (B). Error bars (SD) shown for wild-type human ADA (A–C) and the R142Q mutant (C) are based on five and four standard binding experiments (see Materials and Methods), respectively. (C) FPLC assay of human (Hum) and mouse (Mus) ADA point mutants. Black bars are mutants found in ADA-deficient individuals; gray bars are mutants prepared for this study from human or mouse wild-type cDNA, as indicated. Standard FPLC assay conditions were used for screening the hybrids and point mutants, except for the R149Q, A215T, P126Q mutants from ADA-deficient individuals. In those cases, a lower amount of ADA activity was used, accounting for a greater percentage of input ADA activity becoming associated with CD26/DPPIV than was found with wild-type human ADA. (D) Nondenaturing PAGE assay. Lane 1, wild-type (WT) human ADA; lane 2, wild-type human ADA plus rabbit CD26; lane 3, R142Q human ADA mutant; lane 4, R142Q plus rabbit CD26; lane 5, E217K human ADA mutant; lane 6, E217K plus rabbit CD26.
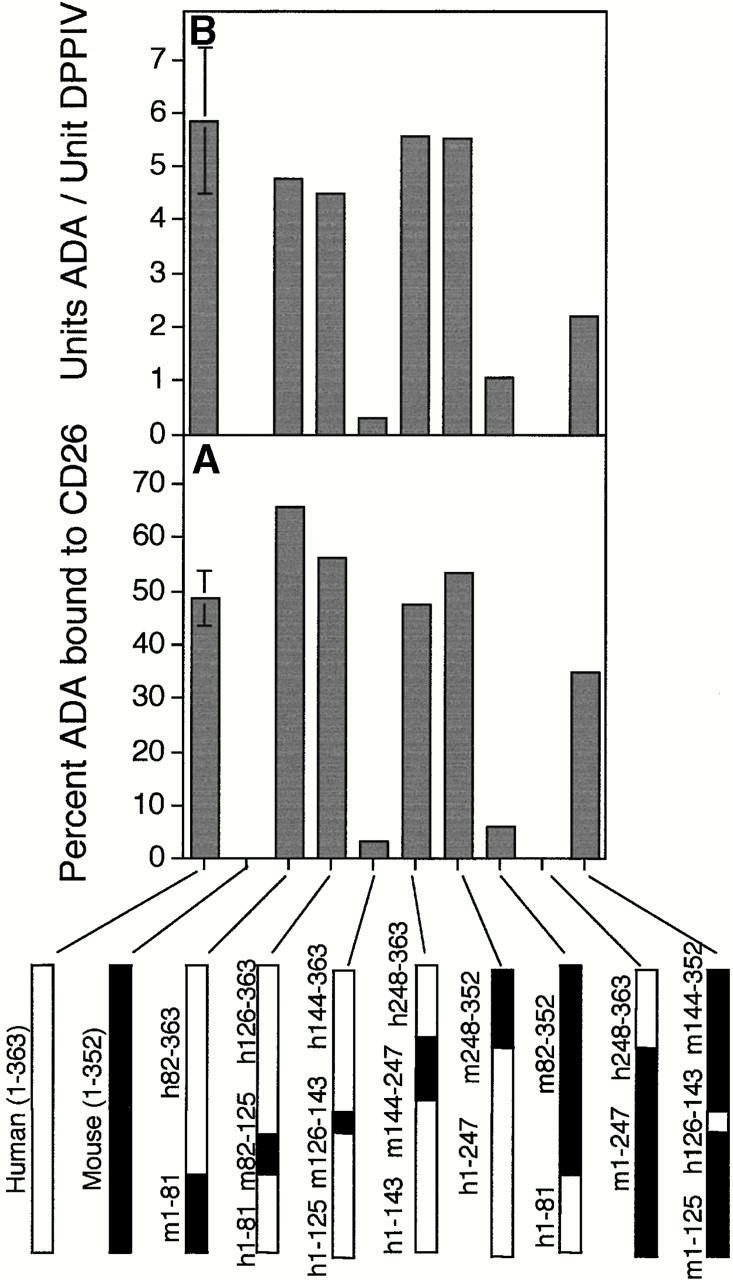

In the crystal structure of murine ADA, Gln142 lies in the 18-residue segment 126–143, which forms the peripheral α2 helix (44; Fig. 3). A hybrid consisting of human ADA substituted with mouse residues 126–143 (referred to as “m126–143”) expressed 36% of wild-type human ADA activity in SØ3834 (Table ), but it showed minimal binding to CD26 by FPLC (Fig. 2A and Fig. B) and none by PAGE (not shown). Binding was also reduced markedly in other hybrids that possessed the murine α2 helix residues (Fig. 2A and Fig. B). Hybrids in which all other regions of human ADA, except for residues 126–143, were replaced with their murine counterparts had substantial ADA activity (Table ) and showed essentially normal CD26 binding (Fig. 2A and Fig. B). Modifying mouse ADA by substituting human residues 126–143 for the corresponding mouse residues (“h126–143”), or by making the Q142R point mutation, conferred some ability to bind CD26, which could be appreciated by FPLC (Fig. 2A Fig. c). Together, these results show that the amino acids critical for CD26 binding lie within segment 126–143 of human ADA.
Figure 3.
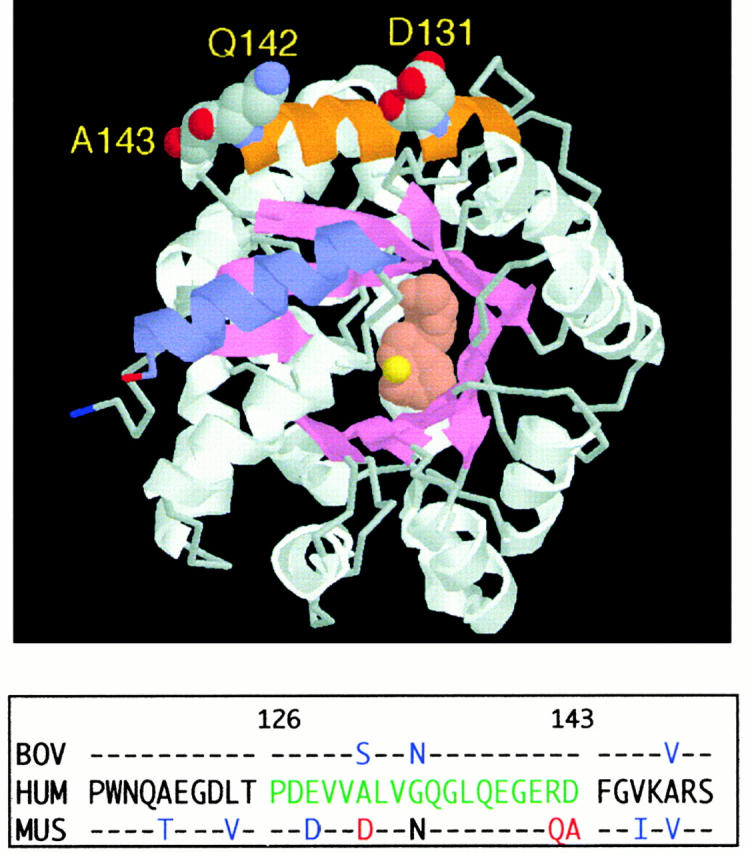
Location of the α2 helix (CD26 binding site) in the murine ADA crystal structure. The RasMol model is based on coordinates, as reported (reference 44). The α2 helix (residues 126–143) is shown in orange, with space-filling display of residues D131, Q142, and A143. For orientation, the COOH-terminal helix (residues 337–351) is shown in blue. Bound inhibitor (rose) and the zinc ion (yellow) are space-filling structures at the active site. The panel below the model compares the partial amino acid sequences (residues 115–150) of human (HUM), murine (MUS), and bovine (BOV) ADA. Amino acids 126–143 of human ADA, green letters; amino acids D131, Q142, and A143 of murine ADA, red letters; other amino acids of bovine and murine ADA that differ from the human sequence, blue letters.
As bovine ADA also binds CD26, we amplified cDNA carrying the ADA coding region from bovine thymus RNA. The predicted amino acid sequences of residues 115–150 of human, bovine, and murine ADA are compared in Fig. 3. Within segment 126–143, human and bovine ADA differ at only 2 of 18 positions, whereas human and murine ADA differ at 5, and murine and bovine ADA at 4 positions. The most notable differences are (a) residues 141–143 of human and bovine ADA are all charged, Glu-Arg-Asp, whereas only Glu141 is charged in the mouse sequence, Glu-Gln-Ala; and (b) residue 131 in mouse ADA is Asp, whereas Ala and Ser are found in human and bovine ADA, respectively. We prepared mutants of human ADA in which α2 helix residues from murine ADA replaced the corresponding human residues. All were active when expressed in E. coli SØ3834 (data not shown), but they fell into two groups with respect to CD26 binding. The single mutants E128D, A131D, G134N, and D143A all showed CD26 binding in the FPLC assay (Fig. 2 C). Binding by the double mutants E128D/R142Q and R142Q/D143A, the triple mutant E128D/R142Q/D143A, and the quadruple mutant E128D/A131D/R142Q/D143A was in each case reduced, but comparable to that of R142Q alone (Fig. 2 C).
Binding of Recombinant ADAs to Human T Cell–associated CD26.
The AlNe HTLV-1 transformed T cell line is derived from an immunodeficient patient homozygous for a mutation in ADA intron 10 that activates a cryptic 3′ splice site 45. ADA activity was unmeasurable in extracts of AlNe cells when cultured in serum-free (ADA-free) medium, and ADA protein could not be detected by immunoblotting with polyclonal Ab to human ADA (not shown). AlNe cells express a substantial level of CD26 (see below). We compared the binding of several recombinant ADAs to CD26 on AlNe cells (see Fig. 4 Fig. 5 Fig. 6).
Figure 4.
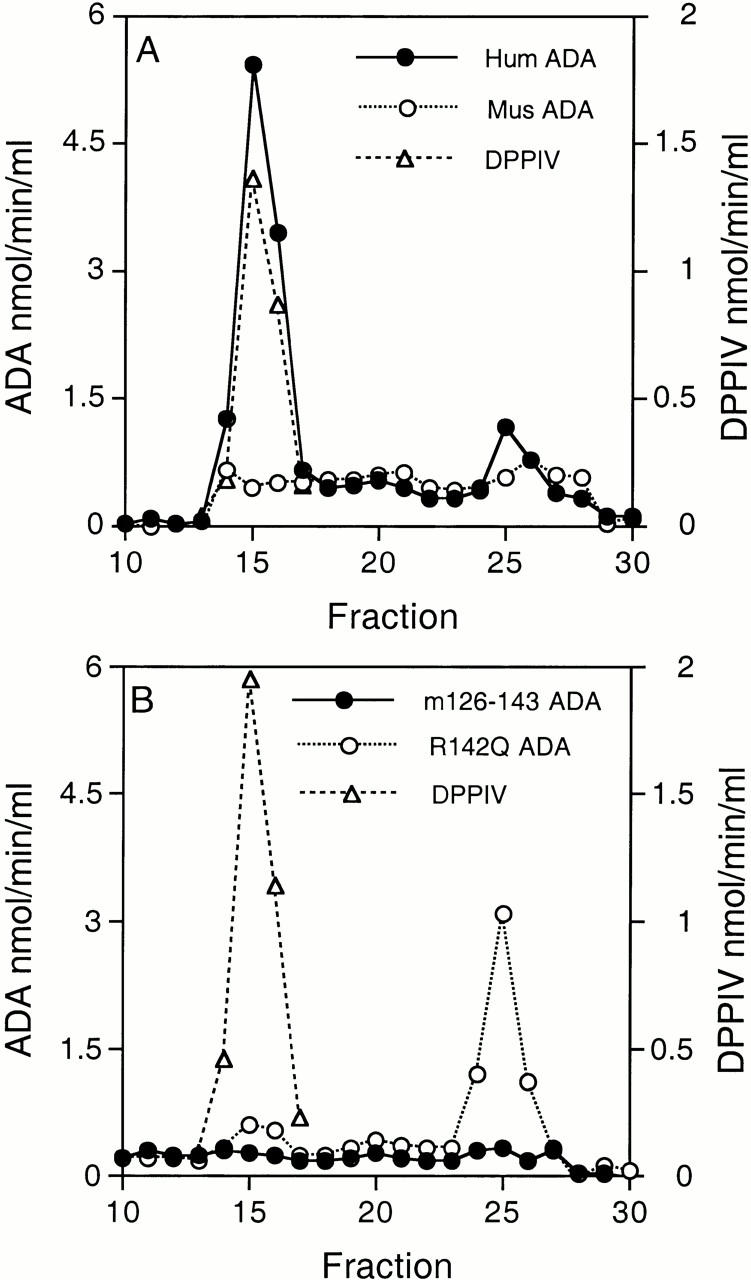
Association of recombinant ADAs with CD26/DPPIV on the AlNe human ADA-deficient T cell line. (A) Binding of wild-type human ADA (hum; filled circles) and wild-type mouse ADA (mus; open circles). (B) Binding of the m126–143 human–mouse ADA hybrid (filled circles) and the R142Q human ADA mutant (open circles). Circles, ADA activity; triangles, DPPIV activity.
Figure 5.
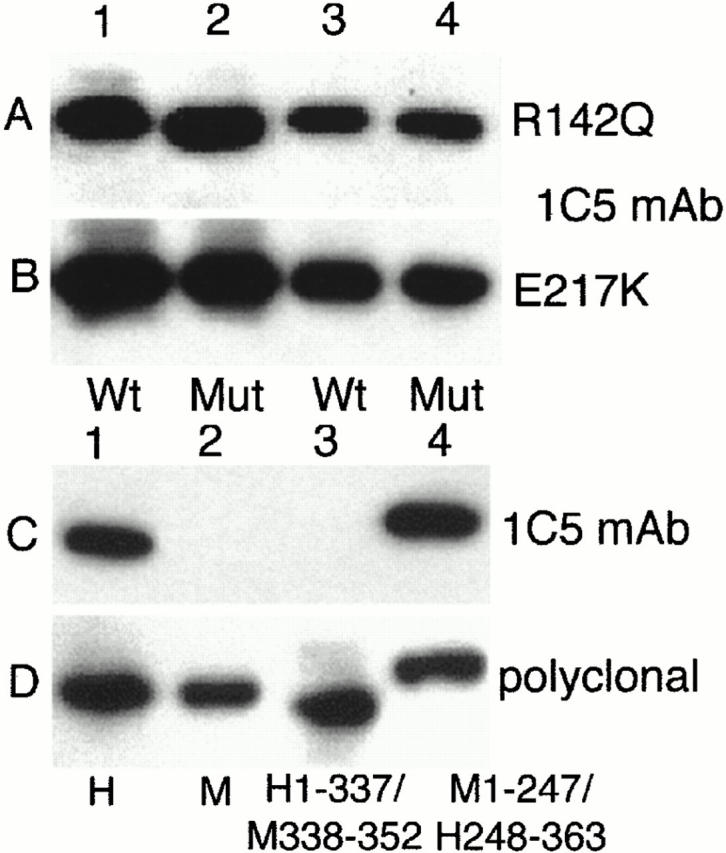
Western blot of recombinant ADAs with mAb 1C5 (A–C) or polyclonal antiserum (D). (A) Lanes 1 and 3, wild-type human ADA; lanes 2 and 4, the R142Q mutant. Equal amounts of wild-type and R142Q ADA activity were loaded: 209 nmol/min in lanes 1 and 2, and 42 nmol/min in lanes 3 and 4. (B) Lanes 1 and 3, wild-type human ADA; lanes 2 and 4, the E217K mutant. Equal amounts of wild-type and E217K extract protein were loaded: 15 μg in lanes 1 and 2; 3 μg in lanes 3 and 4 (for wild-type ADA, this represented 209 and 42 nmol/min in lanes 1 and 3, respectively). (C) All lanes were loaded with 12.5 nmol/min of ADA activity. Lane 1, wild-type human ADA; lane 2, wild-type mouse ADA; lane 3, hybrid h1–337/m338–352 (amino acids 1–337 from human and 338–352 from mouse ADA sequences; for comparison with results shown in Table , this hybrid expressed 2.8 μmol/min of ADA activity, or 85% of wild-type human ADA); lane 4, hybrid m1–247/h248–363 (see Table ). (D) As described for C, except that the blot was probed with a goat anti-ADA antiserum as described previously 41. WT, wild-type; Mut, mutant; H, human, M, mouse.
Figure 6.

Flow cytometry analysis of CD26 and ADA on the surface of AlNe cells. (A) CD26 expression. Shown are reactivity of AlNe cells with PE-conjugated anti-CD26 mAb Ta1 (shaded histogram) and control mAb IgG1-PE (open histogram). (B and C) ADA. Anti-ADA mAb 1C5 binding to AlNe cells that had been washed after incubation with lysate of untransformed SØ3834 (B) or with lysate of SØ3834 expressing wild-type human ADA (400 nmol/min per ml of medium) (C). (D) Binding of wild-type (wt) human ADA (circles), R142Q (triangles), and E217K (squares) ADA mutants. AlNe cells were incubated with 0.3, 1.7, 8.3, 42, 209, and 400 nmol/min per ml of recombinant wild-type and R142Q ADAs. The amounts of E217K-expressing SØ3834 lysate protein used (0.024–30 μg) were made equal to wild-type ADA lysate protein. After washing, cell surface–associated ADA was determined by reactivity of mAb 1C5 and flow cytometry. For clarity, the horizontal axis shows only units of ADA activity. Data shown are from one of two experiments.
Virtually no ADA activity was found in Superose 12–fractionated extracts of AlNe cells that had been incubated with a high concentration (400 nmol/min) of recombinant murine ADA (Fig. 4 A), or with the m126–143 human–mouse hybrid (Fig. 4 B). After incubation with wild-type human ADA, 91% of cell-associated ADA activity coeluted with DPPIV, resulting in a ratio of ADA to DPPIV activity in this peak of 3.7 (Fig. 4 A). With the R142Q mutant, ∼40% as much total ADA activity became cell associated as was found with wild-type human ADA. However, only 9% of the bound R142Q ADA activity coeluted with DPPIV, resulting in a ratio of R142Q ADA to DPPIV activity of 0.1 (Fig. 4 B). Virtually identical results were obtained for wild-type human ADA and the R142Q mutant in a second experiment using the same protocol. The R142Q ADA activity eluting as a 41-kD monomer may have resulted from unstable binding of mutant ADA to CD26 on AlNe cells, with subsequent dissociation in the extract or during FPLC. This transient binding probably reflects the high concentration of exogenous ADA activity used in these experiments (see below).
We next examined binding of recombinant ADAs to the surface of AlNe cells by flow cytometry, using mouse mAb 1C5 to human ADA. Western blot experiments indicated that 1C5 recognizes a COOH-terminal epitope present in human but not in mouse ADA, and it reacts approximately as well with the R142Q and E217K mutants as with wild-type human ADA (Fig. 5). In these experiments (and in flow cytometry studies shown below in Fig. 6 D), equal units of R142Q and wild-type ADA activity were compared (Fig. 5 A), whereas equal amounts of total SØ3834 lysate protein were used to compare E217K and wild-type ADA (Fig. 5 B). Taken together with previous studies of expression 38 41, these results indicate that the R142Q protein has the same activity per molecule as wild-type ADA, but is somewhat less stable in cells, whereas the E217K protein is about as stable as the wild-type enzyme, but is catalytically inactive. The stability of E217K is unusual because most ADA missense mutations identified in SCID patients result in unstable proteins that are difficult to detect with antisera to ADA, both in patient-derived cell lines and when expressed in E. coli SØ3834 41 46.
In flow cytometry experiments, AlNe cells expressed high levels of CD26 (Fig. 6 A). They did not bind anti-ADA mAb 1C5 (Fig. 6 B). Incubating AlNe cells with recombinant wild-type human ADA (followed by washing) induced 1C5 binding (Fig. 6C and Fig. D), as did the R142Q and E217K mutants (Fig. 6 D). 1C5 binding increased with the amount of ADA to which the cells were exposed (Fig. 6 D). Based on mean fluorescence values, at the lowest concentrations tested, E217K induced about half the 1C5 binding induced by wild-type ADA, but equivalent binding occurred at the higher end of the range (because expression in SØ3834 is somewhat variable [41], the amounts of wild-type and E217K ADA protein may not have been identical). 1C5 binding was detected at all concentrations of wild-type and E217K proteins tested, but was not measurable with R142Q ADA at 0.3 and 1.7 nmol/min per ml medium. Wild-type ADA induced 7.5-fold more 1C5 binding than R142Q at ADA activities of 8.3 and 42 nmol/min, and fourfold greater binding at 209 nmol/min (very similar results were found in a second experiment not shown). ADA bound to CD26 is derived from an extracellular source 47. Serum ADA1 activity (the isozyme derived from the ADA gene) for 320 normal humans was reported to be 4.2 ± 1.5 nmol/min per ml 48. Thus, under physiological conditions, binding of circulating R142Q ADA to T cells via CD26 may be 10-fold lower than normal.
Binding of Naturally Occurring R142Q ADA to Human CD26.
Human ADA expressed in E. coli might interact differently with CD26 than would ADA produced in human cells. To address this possibility, we studied the binding of ADA in extracts of erythrocytes and cultured T cells from the individual in whom the R142Q allele was identified, whose second ADA allele carries a nonsense mutation in codon 3 38. A limited quantity of R142Q cells was available for these studies, but the results support those obtained with the recombinant enzyme.
As a control for the erythrocyte studies, we used red cells from an ADA heterozygote whose nonwild-type allele also has a nonsense mutation. ADA activities of the control and R142Q hemolysates was 18.8 and 5.5 nmol/h per mg protein (normal: 63 ± 41 nmol/h per mg), respectively. As erythrocytes lack CD26, we examined CD26 binding by incubating aliquots of hemolyzed red cells with AlNe T cells (ADA-deficient). After washing, Triton X-100 extracts of the AlNe cells were analyzed by FPLC (Fig. 7 A). About 2.6-fold more control (wild-type) than R142Q ADA activity eluted as monomer, proportional to the amounts of ADA activity in the incubations. However, ∼14-fold more control (wild-type) than R142Q ADA activity coeluted with DPPIV activity.
Figure 7.
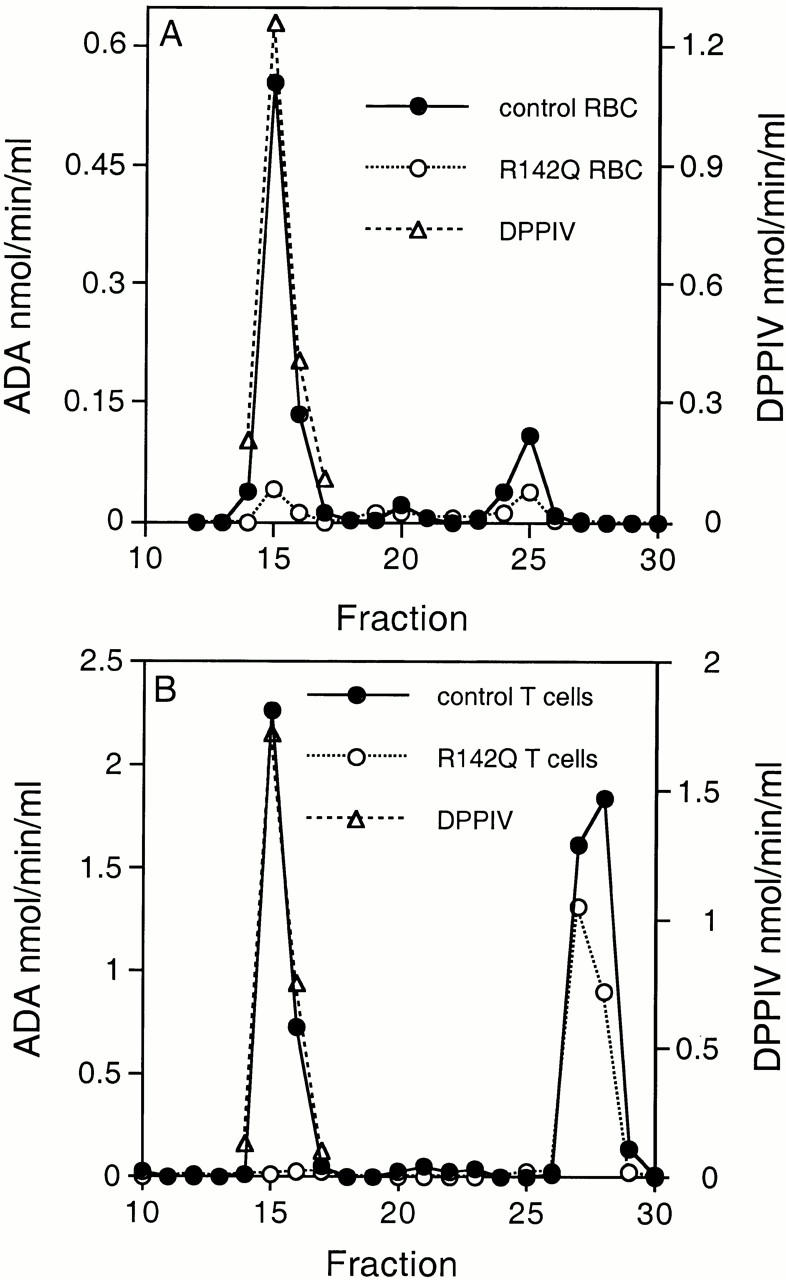
Binding of naturally occurring R142Q ADA to human CD26. (A) Binding of ADA from erythrocyte lysates (RBC) to CD26 from AlNe T cells. 107 AlNe T cells in 1 ml medium were incubated 2 h at 37°C with 0.2 ml aliquots of control (filled circles) and R142Q (open circles) hemolysates. ADA activities present in the control and R142Q binding assays were 10.9 and 4.7 nmol/min, respectively. The AlNe cells were then washed, lysed, fractionated on Superose 12, and assayed as described in Materials and Methods. Circles, ADA activity; triangles, DPPIV activity. (B) Binding of endogenous T cell ADA to endogenous T cell CD26. 2 × 107 control (filled circles) and R142Q (open circles) T cells were lysed, fractionated on Superose 12, and assayed as in A, except that the Superose 12 column was equilibrated and eluted with 50 mM Tris-HCl, pH 8.4, 150 mM NaCl (see Materials and Methods). Circles, ADA activity; triangles, DPPIV activity.
For the T cell studies we assessed the interaction of endogenous ADA with endogenous CD26 (Fig. 7 B). The control T cells were obtained from an individual presumed to be homozygous for wild-type ADA. ADA activity of the control and R142Q T cell extracts were 1,750 and 592 nmol/h per mg protein, respectively; they expressed approximately equal levels of CD26 by flow cytometry (not shown). After FPLC fractionation of lysates of these cells, ∼1.6-fold more control (wild-type) than R142Q ADA activity eluted as monomer, whereas 45-fold more control than R142Q ADA activity coeluted with DPPIV activity, resulting in ratios of ADA units bound per unit CD26 of 1.8 for the control and 0.03 for the R142Q T cells (Fig. 7 B).
Discussion
Murine ADA has a parallel α/β architecture with an eight-stranded central β barrel and eight peripheral α helixes; the active site containing an essential Zn2+ ion lies at the COOH-terminal end of the β barrel (44; Fig. 3). Human ADA is 83% identical in sequence and presumably has the same overall structure. Using a panel of recombinant human–mouse hybrids that retain catalytic activity, we have identified the COOH end of the peripheral α2 helix as the primary determinant of the difference in the ability of mouse and human ADA to bind CD26/DPPIV. Replacing the entire 18-residue segment in human ADA with murine residues 126–143 modestly reduced the expression of ADA activity in E. coli, but abolished stable binding to rabbit and human CD26. Both effects could largely be achieved by replacing Arg142 of human ADA with Gln, the residue found at this position in murine ADA. At the resolution of our screening assays, human to mouse substitutions at the other four nonidentical positions within this segment did not reduce CD26 binding significantly, and in various combinations with R142Q they had no greater effect than the R142Q substitution alone. Making the converse Q142R substitution in mouse ADA, or introducing the entire human 126–143 segment, conferred some ability to bind rabbit CD26. No other regions of human ADA appear to be involved in CD26 binding, including the 11 COOH-terminal residues, which are absent in murine ADA.
Residues 294 (Leu) and 340–343 (Leu-Val-Ala-Arg) of human CD26 have been identified as essential for binding ADA 22 23. It was postulated that stable ADA–CD26 binding is due to contacts between these CD26 residues and a nonconserved hydrophobic surface of human ADA, possibly involving residues Leu346, Ala350, and Gly352, which are respectively Arg, Glu, and Gln in mouse ADA 23. This cannot be correct because the human–mouse ADA hybrid h1–247/m248–352 binds CD26 (Fig. 2). Moreover, residues 141–143 are Glu-Arg-Asp in human and bovine ADA, each of which binds CD26, and Glu-Gln-Ala in mouse ADA, which does not, suggesting that charged residues of ADA are critical for binding CD26. This type of contact between proteins, involving mainly hydrophobic residues of one partner and charged residues of the other, is not unique, and has recently been observed in the complex of hen egg white lysozyme with the antilysozyme mAb HyHEL-63 49. Stable ADA–CD26 binding may also arise from interactions of the critical hydrophobic CD26 and charged ADA residues, not with each other, but with other partners that will be identified when the 3D structures of human ADA and a CD26–ADA complex have been determined.
Significance of the R142Q Mutation.
Most patients with ADA deficiency have SCID, but 15–20% have a milder immune deficit with a later clinical onset 37 45 50. Screening of populations and studies of the relatives of patients with SCID has identified some healthy individuals, most of African ancestry, with low or absent ADA activity in red cells, but 5–70% of normal activity in nucleated cells; this “partial ADA deficiency” has been shown to result from several different missense mutations 37 39 51. Red cells of subjects with partial ADA deficiency show no (or minimal) accumulation of dAdo nucleotides (dAXP), whereas a massive increase occurs in patients with SCID, and a more modest elevation in those with milder immune deficiency 3 41 45 52. This correlation, and evidence that dATP is toxic to lymphoid cells and can induce apoptosis by p53- and caspase-dependent mechanism(s) 53 54 55 56 57 58 59, has implicated dAdo-induced dATP pool expansion as the principal cause of lymphopenia in ADA deficiency.
With this background, the finding of <20% of normal red cell ADA activity in the healthy Somalian father of a child with SCID led to our discovery that he had transmitted an ADA allele with a codon 3 nonsense mutation to his affected daughter, and that his second ADA allele carried the R142Q mutation 38. We postulated that the R142Q mutation, which is distant from the active site, reduced cellular ADA activity by impairing enzyme folding or stability, but that residual activity due to the R142Q allele was sufficient to catabolize dAdo, preventing dATP accumulation and immune deficiency. Our present studies with the 1C5 anti-ADA mAb show that the R142Q protein has a specific activity per molecule close to that of wild-type human ADA. In our original report of the R142Q allele, we also identified SCID patients who were homozygous for a codon 142 nonsense mutation 38. Both the R142Q and R142X mutations arose from a CGA (Arg) codon. We speculated that during murine evolution there might have been some advantage in replacing this Arg codon, which contains a mutagenic CpG dinucleotide, with a CAA Gln codon 38. Our finding that Arg 142 is the “keystone” of the bridge between ADA and CD26 permits an additional speculation: that at the protein level, a Gln142 residue might have conferred some evolutionary advantage on mice by reducing interaction between ADA and CD26.
Our studies of CD26 binding by the R142Q and E217K mutants, derived from ADA-deficient individuals with distinctive phenotypes, are relevant to theories implicating CD26 in the pathogenesis of the immune defect in ADA deficiency. These have postulated either: (a) that ADA binding per se (unrelated to catalytic activity) is crucial to an essential function of CD26 in lymphocyte development or activation 27 60; or (b) that CD26-bound “ecto-ADA”, by controlling the level of extracellular Ado, regulates signaling through cell surface Ado receptors (assumed to be critical for lymphocyte development or function [15, 24, 27, 47]). The first hypothesis ignores evidence, discussed above, of the correlation between mutant ADA catalytic activity and the metabolic and clinical phenotypes of ADA-deficient individuals 3 41 61. The ADA active site mutation E217K was identified in a patient with early onset SCID, whose other ADA allele is not expressed 40. We have shown that the catalytically inactive E217K protein is stable and binds to CD26 on the surface of T cells in a manner similar to wild-type human ADA, suggesting that ADA–CD26 binding is not sufficient to preserve immune function.
The second hypothesis, that CD26-bound “ecto-ADA” regulates levels of extracellular Ado, has been supported by in vitro studies showing lesser effects of exogenous Ado (0.1–10 mM) on proliferation and IL-2 production by CD26-transfected compared with untransfected Jurkat cells 47. Flow cytometry with anti-ADA antiserum indicated that only the former possessed cell surface–associated ADA (although CD26 on these cells was not saturated with ADA). Whole cell lysates of the transfected and untransfected cells had the same total ADA, as estimated by Western blotting. The physiologic relevance and interpretation of these observations may be questioned. The Ado concentrations used were at least 100-fold higher than occurs in normal plasma and at least 10-fold higher than has been found in plasma of patients with ADA deficiency 3. The differences in proliferation and IL-2 production observed were attributed to different rates of Ado deamination (despite identical total ADA levels), but this was not measured. This result, if true, would be surprising because ADA substrates (at physiologic levels) can equilibrate efficiently across the cell membrane via a ubiquitous, nonconcentrative nucleoside transporter 62. As a result of this transport, transfused ADA+ erythrocytes 63 and polyethylene glycol–modified ADA acting in plasma 64 can correct metabolic abnormalities in cells of ADA-deficient individuals.
Ado receptor–mediated signaling is functional in mice despite the lack of ADA–CD26 binding. In addition, a strain of Fischer 344 rats is homozygous for a CD26/DPPIV missense mutation that abolishes both peptidase catalytic activity and cell surface expression of the protein 65 66. Studies of these rats have indicated a role for DPPIV in renal and intestinal hydrolysis and absorption of prolyl dipeptides 67 68. Neither CD26/DPPIV-deficient rats nor normal mice manifest the purine metabolic abnormalities and profound immunodeficiency (or nonlymphoid pathology) associated with ADA deficiency in humans and mice.
Advocates of ADA–CD26 hypotheses may consider rodents to be poor models for ADA deficiency in humans 24. In this regard our observations have special relevance. There is good evidence that ADA bound to CD26 on human T cells is not derived from within these cells, but is acquired from the extracellular space (possibly after release from senescent cells [47]). In the range of ADA activity found in normal human plasma 48, we observed ∼10-fold less binding of recombinant R142Q than wild-type human ADA to CD26+ ADA− AlNe T cells. The defect in CD26 binding was even greater with ADA from erythrocytes and T cells obtained from the R142Q-expressing subject (Fig. 7). It is reasonable to expect that his lymphocytes and other tissues also have a markedly reduced level of ADA-ligated CD26 in vivo. The existence of a healthy adult with defective ADA–CD26 binding suggests that interaction of these proteins is not essential for the development or maintenance of immune function in humans, as is certainly the case in mice and perhaps some other species.
Acknowledgments
We wish to acknowledge Dr. Ann Mary Achyuthan for assaying ADA-CP expression on AlNe T cells using Ab to human ADA-CP that was kindly provided by Dr. William P. Schrader. Dr. Pawan Bali assisted in the development of assays for ADA and DPPIV, and Ed Geisinger with preparing mutant ADA cDNAs. We wish to thank the carrier of the R142Q mutation and Dr. Chaim Roifman for providing material for study.
Supported by grants RO1 DK20902 (M.S. Hershfield) and RO1 AI47604 (D.D. Patel) from the National Institutes of Health, and by a grant from Enzon, Inc. (M.S. Hershfield). E. Richard was supported by Fellowship 98/9329 from Fondo de Investigación Sanitaria, Instituto de Salud Carlos III, Ministerio de Sanidad y Consumo, Spain.
Footnotes
Abbreviations used in this paper: ADA, adenosine deaminase; ADA-CP, ADA complexing protein; Ado, adenosine; dAdo, 2′-deoxyadenosine; DPPIV, dipeptidyl peptidase IV; FPLC, fast protein liquid chromatography.
References
- Giblett E.R., Anderson J.E., Cohen F., Pollara B., Meuwissen H.J. Adenosine deaminase deficiency in two patients with severely impaired cellular immunity. Lancet. 1972;2:1067–1069. doi: 10.1016/s0140-6736(72)92345-8. [DOI] [PubMed] [Google Scholar]
- Hershfield M.S. Immunodeficiency caused by deficiency of adenosine deaminase. Immunol. Allergy Clin. North Am. 2000;20:161–175. [Google Scholar]
- Hershfield M.S., Mitchell B.S. Immunodeficiency diseases caused by adenosine deaminase deficiency and purine nucleoside phosphorylase deficiency. 7th ed. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill Inc; New York: 1995. pp. 1725–1768. [Google Scholar]
- Wakamiya M., Blackburn M.R., Jurecic R., McArthur M.J., Geske R.S., Cartwright J., Jr., Mitani K., Vaishnav S., Belmont J.W., Kellems R.E. Disruption of the adenosine deaminase gene causes hepatocellular impairment and perinatal lethality in mice. Proc. Natl. Acad. Sci. USA. 1995;92:3673–3677. doi: 10.1073/pnas.92.9.3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migchielsen A.A.J., Breuer M.L., van Roon M.A., te Riele H., Zurcher C., Ossendorp F., Toutain S., Hershfield M.S., Berns A., Valerio D. Adenosine deaminase-deficient mice die perinatally and exhibit liver-cell degeneration, atelectasis and small intestinal cell death. Nat. Genet. 1995;10:279–287. doi: 10.1038/ng0795-279. [DOI] [PubMed] [Google Scholar]
- Blackburn M.R., Datta S.K., Kellems R.E. Adenosine deaminase-deficient mice generated using a two-stage genetic engineering strategy exhibit a combined immunodeficiency. J. Biol. Chem. 1998;273:5093–5100. doi: 10.1074/jbc.273.9.5093. [DOI] [PubMed] [Google Scholar]
- Nishihara H., Ishikawa S., Shinkai K., Akedo H. Multiple forms of human adenosine deaminase. II. Isolation and properties of a conversion factor from human lung. Biochim. Biophys. Acta. 1973;302:429–442. doi: 10.1016/0005-2744(73)90172-1. [DOI] [PubMed] [Google Scholar]
- Daddona P.E., Kelley W.N. Human adenosine deaminase binding protein. Assay, purification, and properties. J. Biol. Chem. 1978;253:4617–4623. [PubMed] [Google Scholar]
- Andy R.J., Kornfeld R. The adenosine deaminase binding protein of human skin fibroblasts is located on the cell surface. J. Biol. Chem. 1982;257:7922–7925. [PubMed] [Google Scholar]
- Schrader W.P., West C.A., Miczek A.D., Norton E.K. Characterization of the adenosine deaminase-adenosine deaminase complexing protein binding reaction. J. Biol. Chem. 1990;265:19312–19318. [PubMed] [Google Scholar]
- Dinjens W.N., ten Kate J., van der Linden E., Wijnen J.T., Khan P.M., Bosman F.T. Distribution of adenosine deaminase complexing protein (ADCP) in human tissues. J. Histochem. Cytochem. 1989;37:1869–1875. doi: 10.1177/37.12.2573631. [DOI] [PubMed] [Google Scholar]
- Trotta P.P., Ahland M.P., Brown G.F., Balis M.E. Studies on the effects of infusion of enzyme inhibitors on mouse adenosine deaminase. Mol. Pharmacol. 1978;14:199–209. [PubMed] [Google Scholar]
- Dinjens W.N., ten Kate J., Wijnen J.T., van der Linden E.P., Beek C.J., Lenders M.H., Khan P.M., Bosman F.T. Distribution of adenosine deaminase-complexing protein in murine tissues. J. Biol. Chem. 1989;264:19215–19220. [PubMed] [Google Scholar]
- Morrison M.E., Vijayasaradhi S., Engelstein D., Albino A.P., Houghton A.N. A marker for neoplastic progression of human melanocytes is a cell surface ectopeptidase. J. Exp. Med. 1993;177:1135–1143. doi: 10.1084/jem.177.4.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameoka J., Tanaka T., Nojima Y., Schlossman S., Morimoto C. Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science. 1993;261:466–469. doi: 10.1126/science.8101391. [DOI] [PubMed] [Google Scholar]
- Fox D.A., Hussey R.E., Fitzgerald K.A., Acuto O., Poole C., Palley L., Daley J.F., Schlossman S.F., Reinhertz E.L. TA1, a novel 105 KD human T cell activation antigen defined by a monoclonal antibody. J. Immunol. 1984;133:1250–1256. [PubMed] [Google Scholar]
- Hegen M., Niedobitek G., Klein C.E., Stein H., Fleischer B. The T cell triggering molecule Tp103 is associated with dipeptidyl peptidase IV activity. J. Immunol. 1990;144:2908–2914. [PubMed] [Google Scholar]
- Ulmer A.J., Mattern T., Feller A.C., Heymann E., Flad H.D. CD26 antigen is a surface dipeptidyl peptidase IV (DPPIV) as characterized by monoclonal antibodies clone TII-19-4-7 and 4EL1C7. Scand. J. Immunol. 1990;31:429–435. doi: 10.1111/j.1365-3083.1990.tb02789.x. [DOI] [PubMed] [Google Scholar]
- Darmoul D., Lacasa M., Baricault L., Marguet D., Sapin C., Trotot P., Barbat A., Trugnan G. Dipeptidyl peptidase IV (CD26) gene expression in enterocyte-like colon cancer cell lines HT-29 and Caco-2. Cloning of the complete human coding sequence and changes of dipeptidyl peptidase IV mRNA levels during cell differentiation. J. Biol. Chem. 1992;267:4824–4833. [PubMed] [Google Scholar]
- Tanaka T., Camerini D., Seed B., Torimoto Y., Dang N.H., Kameoka J., Dahlberg H.N., Schlossman S.F., Morimoto C. Cloning and functional expression of the T cell activation antigen CD26. J. Immunol. 1992;149:481–486. [PubMed] [Google Scholar]
- Fleischer B. CD26a surface protease involved in T-cell activation. Immunol. Today. 1994;15:180–184. doi: 10.1016/0167-5699(94)90316-6. [DOI] [PubMed] [Google Scholar]
- Dong R.P., Tachibana K., Hegen M., Munakata Y., Cho D., Schlossman S.F., Morimoto C. Determination of adenosine deaminase binding domain on CD26 and its immunoregulatory effect on T cell activation. J. Immunol. 1997;159:6070–6076. [PubMed] [Google Scholar]
- Abbott C.A., McCaughan G.W., Levy M.T., Church W.B., Gorell M.D. Binding to human dipeptidyl peptidase IV by adenosine deaminase and antibodies that inhibit ligand binding involves overlapping, discontinuous sites on a predicted B propeller domain. Eur. J. Biochem. 1999;266:798–810. doi: 10.1046/j.1432-1327.1999.00902.x. [DOI] [PubMed] [Google Scholar]
- Morimoto C., Schlossman S.F. The structure and function of CD26 in the T-cell immune response. Immunol. Rev. 1998;161:55–70. doi: 10.1111/j.1600-065x.1998.tb01571.x. [DOI] [PubMed] [Google Scholar]
- Vivier I., Marguet D., Naquet P., Bonicel J., Black D., Li C.X., Bernard A.M., Gorvel J.P., Pierres M. Evidence that thymocyte activating molecule is mouse CD26 (dipeptidyl peptidase IV) J. Immunol. 1991;147:447–454. [PubMed] [Google Scholar]
- Bristol L.A., Sakaguchi K., Appella E., Doyle D., Takacs L. Thymocyte costimulating antigen is CD26 (dipeptidyl peptidase IV). Costimulation of granulocyte, macrophage, and T cell lineage cell proliferation via CD26. J. Immunol. 1992;149:367–372. [PubMed] [Google Scholar]
- Franco R., Valenzuela A., Lluis C., Blanco J. Enzymatic and extraenzymatic role of ecto-adenosine deaminase in lymphocytes. Immunol. Rev. 1998;161:27–42. doi: 10.1111/j.1600-065x.1998.tb01569.x. [DOI] [PubMed] [Google Scholar]
- Arredondo-Vega F.X., Kurtzberg J., Chaffee S., Santisteban I., Reisner E., Povey M.S., Hershfield M.S. Paradoxical expression of adenosine deaminase in T cells cultured from a patient with adenosine deaminase deficiency and combined immunodeficiency. J. Clin. Invest. 1990;86:444–452. doi: 10.1172/JCI114730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatsu T., Hino M., Fuyamada H., Hayakawa T., Sakakibara S., Nakagawa Y., Takemoto T. New chromogenic substrates for X-prolyl dipeptidyl-aminopeptidase. Anal. Biochem. 1976;74:466–476. doi: 10.1016/0003-2697(76)90227-x. [DOI] [PubMed] [Google Scholar]
- De Meester I., Vanhoof G., Lambier A.M., Scharpé S. Use of immobilized adenosine deaminase (EC 3.5.4.4) for the rapid purification of native human CD26/dipeptidyl peptidase IV (EC 3.4.14.5) J. Immunol. Methods. 1996;189:99–105. doi: 10.1016/0022-1759(95)00239-1. [DOI] [PubMed] [Google Scholar]
- Schrader W.P., Harder C.M., Schrader D.K. Adenosine deaminase complexing proteins of the rabbit. Comp. Biochem. Physiol. B. 1983;75:119–126. doi: 10.1016/0305-0491(83)90048-2. [DOI] [PubMed] [Google Scholar]
- Erlich H.A. PCR Technology 1989. Principles and Applications for DNA Amplificiation. Stockton Press; New York: pp. 246 pp [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. Molecular Cloning. A Laboratory Manual. 2nd ed. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- Wiginton D.A., Adrian G.S., Hutton J.J. Sequence of human adenosine deaminase cDNA including the coding region and a small intron. Nucleic Acids Res. 1984;12:2439–2446. doi: 10.1093/nar/12.5.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung C.-Y., Ignolia D.E., Roth D.B., Shoemaker C., Al-Ubaidi A.U., Yen J.-Y., Ching C., Bobonis C., Kaufman R.J., Kellems R.E. Identification of functional murine adenosine deaminase cDNA clones by complementation in Escherichia coli . J. Biol. Chem. 1985;260:10299–10307. [PubMed] [Google Scholar]
- Ho S.N., Hunt H.D., Horton R.M., Pullen J.K., Pease L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Ozsahin H., Arredondo-Vega F.X., Santisteban I., Fuhrer H., Tuchschmid P., Jochum W., Aguzzi A., Lederman H.M., Fleischman A., Winkelstein J.A. Adenosine deaminase deficiency in adults. Blood. 1997;89:2849–2855. [PubMed] [Google Scholar]
- Santisteban I., Arredondo-Vega F.X., Kelly S., Loubser M., Meydan N., Roifman C., Howell P.L., Bowen T., Weinberg K.I., Schroeder M.L., Hershfield M.S. Three new adenosine deaminase mutations that define a splicing enhancer and cause severe and partial phenotypesimplications for evolution of a CpG hotspot and expression of a transduced ADA cDNA. Hum. Mol. Genet. 1995;4:2081–2087. doi: 10.1093/hmg/4.11.2081. [DOI] [PubMed] [Google Scholar]
- Hirschhorn R., Tzall S., Ellenbogen A. Hot spot mutations in adenosine deaminase deficiency. Proc. Natl. Acad. Sci. USA. 1990;87:6171–6175. doi: 10.1073/pnas.87.16.6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn R., Nicknam M.N., Eng F., Yang D.R., Borkowsky W. Novel deletion and a new missense mutation (Glu 217 Lys) at the catalytic site in two adenosine deaminase alleles of a patient with neonatal onset adenosine deaminase—severe combined immunodeficiency. J. Immunol. 1992;149:3107–3112. [PubMed] [Google Scholar]
- Arredondo-Vega F.X., Santisteban I., Daniels S., Toutain S., Hershfield M.S. Adenosine deaminase deficiencygenotype-phenotype correlations based on expressed activity of 29 mutant alleles. Am. J. Hum. Genet. 1998;63:1049–1059. doi: 10.1086/302054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Z.Y., Nygaard P., Chinault A.C., Kellems R.E. Deduced amino acid sequence of Escherichia coli adenosine deaminase reveals evolutionarily conserved amino acid residuesimplications for catalytic function. Biochemistry. 1991;30:2273–2280. doi: 10.1021/bi00222a033. [DOI] [PubMed] [Google Scholar]
- Bollag D.M., Rozycki M.D., Edelstein S.J. Protein Methods 2nd ed 1996. Wiley-Liss; New York: pp. 107–172 [Google Scholar]
- Wilson D.K., Rudolph F.B., Quiocho F.A. Atomic structure of adenosine deaminase complexed with a transition-state analogunderstanding catalysis and immunodeficiency mutations. Science. 1991;252:1278–1284. doi: 10.1126/science.1925539. [DOI] [PubMed] [Google Scholar]
- Santisteban I., Arredondo-Vega F.X., Kelly S., Mary A., Fischer A., Hummell D.S., Lawton A., Sorensen R.U., Stiehm E.R., Uribe L., Weinberg K., Hershfield M.S. Novel splicing, missense, and deletion mutations in 7 adenosine deaminase-deficient patients with late/delayed onset of combined immunodeficiency disease. Contribution of genotype to phenotype. J. Clin. Invest. 1993;92:2291–2302. doi: 10.1172/JCI116833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiginton D.A., Hutton J.J. Immunoreactive protein in adenosine deaminase deficient human lymphoblast cell lines. J. Biol. Chem. 1982;257:3211–3217. [PubMed] [Google Scholar]
- Dong R.P., Kameoka J., Hegen M., Tanaka T., Xu Y., Schlossman S.F., Morimoto C. Characterization of adenosine deaminase binding to human CD26 on T cells and its biologic role in immune response. J. Immunol. 1996;156:1349–1355. [PubMed] [Google Scholar]
- Muraoka T., Katsuramaki T., Shiraishi H., Yokoyama M.M. Automated enzymatic measurement of adenosine deaminase isoenzyme activities in serum. Anal. Biochem. 1990;187:268–272. doi: 10.1016/0003-2697(90)90455-i. [DOI] [PubMed] [Google Scholar]
- Li Y., Li H., Smith-Gill S.J., Mariuzza R.A. Three-dimensional structures of the free and antigen-bound Fab from monoclonal antilysozyme antibody HyHEL-63. Biochemistry. 2000;39:6296–6309. doi: 10.1021/bi000054l. [DOI] [PubMed] [Google Scholar]
- Shovlin C.L., Simmonds H.A., Fairbanks L.D., Deacock S.J., Hughes J.M., Lechler R.I., Webster A.D., Sun X.M., Webb J.C., Soutar A.K. Adult onset immunodeficiency caused by inherited adenosine deaminase deficiency. J. Immunol. 1994;153:2331–2339. [PubMed] [Google Scholar]
- Jenkins T., Rabson A.R., Nurse G.T., Lane A.B. Deficiency of adenosine deaminase not associated with severe combined immunodeficiency. J. Pediatr. 1976;89:732–736. doi: 10.1016/s0022-3476(76)80792-5. [DOI] [PubMed] [Google Scholar]
- Hirschhorn R. Overview of biochemical abnormalities and molecular genetics of adenosine deaminase deficiency Pediatr. Res. 33Suppl1993. S35 S41 [DOI] [PubMed] [Google Scholar]
- Ullman B., Gudas L.J., Cohen A., Martin D.W., Jr. Deoxyadenosine metabolism and cytotoxicity in cultured mouse T lymphoma cellsa model for immunodeficiency disease. Cell. 1978;14:365–375. doi: 10.1016/0092-8674(78)90122-8. [DOI] [PubMed] [Google Scholar]
- Seto S., Carrera C.J., Kubota M., Wasson D.B., Carson D.A. Mechanism of deoxyadenosine and 2-chlorodeoxyadenosine toxicity to nondividing human lymphocytes. J. Clin. Invest. 1985;75:377–383. doi: 10.1172/JCI111710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizaki H., Shimada H., Ohsaka F., Sakurada T. Adenosine, deoxyadenosine, and deoxyguanosine induce DNA cleavage in mouse thymocytes. J. Immunol. 1988;141:1652–1657. [PubMed] [Google Scholar]
- Benveniste P., Cohen A. p53 expression is required for thymocyte apoptosis induced by adenosine deaminase deficiency. Proc. Natl. Acad. Sci. USA. 1995;92:8373–8377. doi: 10.1073/pnas.92.18.8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X., Knudsen T.B., Ibrahim M.M., Haldar S. Bcl-2 relieves deoxyadenylate stress and suppresses apoptosis in pre-B leukemia cells. Cell Death Differ. 1995;2:69–78. [PubMed] [Google Scholar]
- Liu X., Kim C.N., Yang J., Jemmerson R., Wang X. Induction of apoptotic program in cell-free extractsrequirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Li P., Nijhawan D., Budihardjo I., Srinivasula S.M., Ahmad M., Alnemri E.S., Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Martin M., Huguet J., Centelles J.J., Franco R. Expression of ecto-adenosine deaminase and CD26 in human T cells triggered by the TCR-CD3 complex. J. Immunol. 1995;155:4630–4643. [PubMed] [Google Scholar]
- Hirschhorn R. Adenosine deaminase deficiencymolecular basis and recent developments. Clin. Immunol. Immunopathol. 1995;76:S219–S227. doi: 10.1016/s0090-1229(95)90288-0. [DOI] [PubMed] [Google Scholar]
- Griffith D.A., Jarvis S.M. Nucleoside and nucleobase transport systems of mammalian cells. Biochim. Biophys. Acta. 1996;1286:153–181. doi: 10.1016/s0304-4157(96)00008-1. [DOI] [PubMed] [Google Scholar]
- Cohen A., Hirschhorn R., Horowitz S.D., Rubinstein A., Polmar S.H., Hong R., Martin D.W., Jr. Deoxyadenosine triphosphate as a potentially toxic metabolite in adenosine deaminase deficiency. Proc. Natl. Acad. Sci. USA. 1978;75:472–475. doi: 10.1073/pnas.75.1.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershfield M.S., Buckley R.H., Greenberg M.L., Melton A.L., Schiff R., Hatem C., Kurtzberg J., Markert M.L., Kobayashi R.H., Kobayashi A.L., Abuchowski A. Treatment of adenosine deaminase deficiency with polyethylene glycol-modified adenosine deaminase. N. Engl. J. Med. 1987;316:589–596. doi: 10.1056/NEJM198703053161005. [DOI] [PubMed] [Google Scholar]
- Tsuji E., Misumi Y., Fujiwara T., Takami N., Ogata S., Ikehara Y. An active-site mutation (Gly633→Arg) of dipeptidyl peptidase IV causes its retention and rapid degradation in the endoplasmic reticulum. Biochemistry. 1992;31:11921–11927. doi: 10.1021/bi00162a035. [DOI] [PubMed] [Google Scholar]
- Erickson R.H., Suzuki Y., Sedlmayer A., Kim Y.S. Biosynthesis and degradation of altered immature forms of intestinal dipeptidyl peptidase IV in a rat strain lacking the enzyme. J. Biol. Chem. 1992;267:21623–21629. [PubMed] [Google Scholar]
- Tiruppathi C., Miyamoto Y., Ganapathy V., Roesel R.A., Whitford G.M., Leibach F.H. Hydrolysis and transport of proline-containing peptides in renal brush-border membrane vesicles from dipeptidyl peptidase IV-positive and dipeptidyl peptidase IV-negative rat strains. J. Biol. Chem. 1990;265:1476–1483. [PubMed] [Google Scholar]
- Tiruppathi C., Miyamoto Y., Ganapathy V., Leibach F.H. Genetic evidence for the role of DPPIV in intestinal hydrolysis and assimilation of prolyl peptides. Am. J. Physiol. 1993;265:G81–G89. doi: 10.1152/ajpgi.1993.265.1.G81. [DOI] [PubMed] [Google Scholar]