

Abstract
Lymph nodes (LNs) are important sentinal organs, populated by circulating lymphocytes and antigen-bearing cells exiting the tissue beds. Although cellular and humoral immune responses are induced in LNs by antigenic challenge, it is not known if LNs are essential for acquired immunity. We examined immune responses in mice that lack LNs due to genetic deletion of lymphotoxin ligands or in utero blockade of membrane lymphotoxin. We report that LNs are absolutely required for generating contact hypersensitivity, a T cell–dependent cellular immune response induced by epicutaneous hapten. We show that the homing of epidermal Langerhans cells in response to hapten application is specifically directed to LNs, providing a cellular basis for this unique LN function. In contrast, the spleen cannot mediate contact hypersensitivity because antigen-bearing epidermal Langerhans cells do not access splenic white pulp. Finally, we formally demonstrate that LNs provide a unique environment essential for generating this acquired immune response by reversing the LN defect in lymphotoxin-α−/− mice, thereby restoring the capacity for contact hypersensitivity.
Keywords: haptens, antigens, Langerhans cells, lymphoid tissue, spleen
Introduction
Secondary lymphoid organs are sites of immunological coordination, where diverse cell types, including T cells, B cells, APCs, and follicular dendritic cells (FDCs), are brought together within a highly structured, organized environment. This organization has evolved to maximize antigen presentation to the rare T cells capable of specific TCR-mediated antigen recognition and clonal expansion, and is considered necessary for the efficient development of immune responses 1 2 3. Examples of secondary lymphoid organs include the spleen, LNs, Peyer's patches (PPs), mucosa-associated lymphoid tissues (MALTs), and lymphocyte aggregates such as the follicles and cryptopatches of the intestines and colon. These lymphoid compartments share striking similarities in cellular organization, such as discrete T and B cell areas, yet each also has unique, specialized portals for cell or antigen entry, e.g., the afferent lymphatics and high endothelial venules (HEVs) of LNs, the marginal zone (MZ) of the spleen, and the M cell–rich dome of PPs. LNs are found throughout the body, yet despite intensive investigation of the role of LNs in cellular and humoral immune responses, it has not been demonstrated whether these organs are absolutely required for any specific immune function.
In this study, we investigate whether LNs are required to generate a T cell–dependent cellular immune response, contact hypersensitivity (CH). It has been recognized for many years that efficient epidermal immune responses depend on Langerhans cell (LC) uptake of antigen and traffic to LNs 4 5. However, studies directly addressing the role of LNs in CH responses have used either physical disruption of LN lymphatic connections 6 7, or agents capable of blocking LCs or T cell access to LNs 8 9, thereby isolating antigen and responding cells. These studies showed that the CH response is not primed in situ, i.e., in the skin itself, and, along with the observation that LCs arrest in draining LNs 10, suggested that LNs are sufficient to mediate CH responses. Although these data demonstrate a correlation between LC traffic to LNs, intact lymphatic vasculature, and CH reactions, it remains unclear if LNs are the sole, and therefore essential, site where T cell priming to epidermal antigens occurs. Therefore, we have developed novel model systems to test whether LNs are absolutely required for CH responses.
Here we utilize lymphotoxin (LT)α−/− and LTβ−/− mice, which have developmental defects in LN genesis, to study whether LNs are essential to generate a CH response. Interaction of the membrane-bound, heteromeric ligand LTαβ with its receptor (LTβ-R) is a critical signal for the genesis of LNs and PPs during embryonic development 3 11 12. The LTα−/− mouse is essentially devoid of LNs and PPs 13 14. The LTβ−/− mouse has a similar phenotype; however, these mice can retain a subset of mucosa-associated LNs 15 16. LT gene–deficient mice thus provide potentially useful models for investigating the role of LNs in various immune responses. However, the phenotypes of these knockout mice include additional alterations in the immune system (e.g., loss of the splenic MZ and of FDC networks in spleen and LNs [3, 17 18 19 20 21]) which lead to the disruption of splenic germinal center formation and Ab responses 3. Therefore, we have employed in utero manipulation to induce LN formation in LTα−/− mice and to block LN formation in wild-type mice 12 22 23, to create model systems which allow us to determine if the absence of LNs, or the loss of LT ligand expression, impacts the CH response. Our data clearly demonstrate that epidermal CH responses are not generated in the absence of LNs, illustrating an essential role for LNs in this immune response. The specific trafficking of LCs to LNs is shown to provide a cellular basis for this unique LN function. These results have important implications for our understanding of LN function in local immunity and the trafficking of LCs to different lymphoid compartments.
Materials and Methods
CH and LC Trafficking Studies.
All animal studies were performed in accordance with Institutional Animal Care and Use Committee standards. For LC trafficking studies, LTα−/−, LTβ−/−, LTα1/+, or wild-type mice were painted on the shaved skin of the upper back with 100 μl of 5 mg/ml FITC (Sigma-Aldrich) dissolved in 50% acetone/50% dibutylphthalate (Sigma-Aldrich). LNs were removed the next day for analysis of LC content by FACS® analysis and immunohistochemistry.
For CH experiments, dose response data were generated for each hapten using wild-type Balb/c and C57Bl/6 mice. The amount of hapten applied at both the sensitization phase and elicitation phase was titered to arrive at a dose that induced maximal response without excessive hapten. This was done to reduce the possibility that excess vehicle application could cause penetration beyond the epidermis, or induce other unanticipated responses, such as hapten tolerance 5 24 25. Mice were sensitized, rested for 7 d, then rechallenged on the ear. Caliper measurements of the ear were taken just before rechallenge and 24 h afterward, and the data calculated as the percent increase in thickness. For the hapten FITC, a dose of 100 μl of 5 mg/ml at sensitization and 5 μl of 5 mg/ml at elicitation gave a maximal response. Using the hapten 4-ethoxymethylene-2-phenyl-2-oxazolin-5-one (oxazalone) dissolved in ethanol, a dose of 100 μl of 0.3% at sensitization and 10 μl 0.3% at elicitation was sufficient to elicit a maximal response. For the dinitrofluorobenzene (DNFB) studies, mice were painted on the shaved belly using 10 μl 1% solution in vehicle (4:1 acetone/olive oil) followed by 10 μl on the footpad 5 d later, a protocol similar to those successfully used in several recent studies 8 9. Swelling of the footpad was measured after 24 h (challenged site − control site, in mm). Croton oil (ICN Biomedicals) was diluted to 0.8% in acetone. 10 μl was applied to each ear, and the swelling response was measured after 16 h. Phenyltrimethylammoaniline (TMA) was obtained from Bachem Inc. For experiments using TMA, mice were sensitized on the shaved abdomen either by subcutaneous injection or skin painting with 50 μl of a 10 mM solution in saline (subcutaneous) or DMSO (painting). Elicitation of the footpad was with 20 μl TMA presented either subcutaneously or by painting. Footpad swelling is measured as thickness at site of challenge − thickness of contralateral control.
Adoptive transfer experiments were performed essentially as described 9. In brief, Balb/c mice treated in utero with LTβ-R-Ig (see below) to ablate peripheral LN (pLN) and PP development were sensitized with oxazalone. 4 d later the mesenteric LN (mLN) and spleen were removed, and cells were isolated by mechanical disruption and red blood cell lysis. mLN cells isolated from untreated Balb/c mice were used as a negative control. 2.5 × 107 isolated cells were injected intravenously into normal, naive, syngeneic recipients, which were immediately challenged by ear painting with oxazalone. 24 h later the percent increase in ear swelling was measured. The results were compared with the CH response induced using the normal challenge/sensitization regimen.
In all experiments, mice were housed individually to prevent grooming of the sensitized area of the skin. All mice were coded and were measured in random order. Statistical significance of the change in ear thickness was determined using tests of the equivalence of the means.
Irradiation and Bone Marrow Transfer.
LTα1/+ and LTα−/− mice were lethally irradiated using 1,100 rad (Gamma cell; Nordion International Inc.) and reconstituted within 4 h with 2 × 106 bone marrow (BM) cells derived from the donor mice. The BM cell suspension was subjected to depletion using anti-Thy1.2 and rabbit complement (Accurate Chemical & Scientific Corp.) before use to remove mature T cells. After reconstitution, <5% of hemopoietic cells were recipient derived.
LN Induction and Ablation Protocols.
To generate peripheral and mucosal LNs in the progeny, pregnant female LTα−/− mice were injected with 250 μg of agonist anti–LTβ-R mAb AF.H6 via the tail vein beginning on gestation day 11 or 12, followed by additional injections intravenously every 48 h, as described 12. Other pregnant LTα−/− mice were treated with control hamster mAb Ha4/8 (anti-KLH). To ablate LN development, pregnant female Balb/c or C57Bl/6 mice were injected intravenously multiple times with 100 μg of purified murine LTβ-R-Ig, 100 μg TNF-R55-Ig, or both together, as described 12. The presence or absence of LNs was carefully determined by dissection of representative progeny of each litter, and confirmed in each individual animal at necropsy.
Immunohistochemistry and FACS® Analysis.
Mouse spleen and LNs were harvested, frozen, sectioned, and stained for B220 and CD11c as described previously 22 23. Trafficking of LCs to LNs was analyzed by UV induced fluorescence of the captured FITC, in combination with PE-coupled anti-B220 mAb (BD PharMingen). All sections were viewed under 50× or 100× optics and captured as digitized image files using either single or dual filters. The images shown are representative of those obtained from a minimum of four animals per treatment group from a minimum of four independent experiments. The identity of FITC-laden LCs in LNs was confirmed using FACS® analysis and CELLQuest™ software (Becton Dickinson).
Results
LTα−/− Mice Cannot Mount a CH Response to Haptens.
Immunohistochemical staining revealed that the distribution of LCs in the skin of LTα−/− mice was essentially indistinguishable from wild-type mice (unpublished data). Therefore, we tested if these LCs were capable of supporting CH responses in the absence of LNs. We first established dose response curves for the response of wild-type mice to the haptens FITC and oxazalone (Table ) and confirmed that our established doses were within the linear portion of the response. Then, we initiated our studies of CH responses by comparing the ability of LTα−/− mice and LT+/+ mice to respond to hapten challenge. In LTα−/− mice, the CH response to FITC was greatly reduced, approaching background levels (Table ). This result was independent of the hapten used, as LTα−/− mice were also unable to respond to oxazalone (Table ) and DNFB (Table ).
Table 1.
Titration of the Hapten Doses Used in CH Experiments
| Hapten | Sensitization dose | Elicitation dose | CH response |
|---|---|---|---|
| FITC-isothiocyanate | Fixed concentration (μl) | ||
| 200 | 10 | 65 ± 17 | |
| 100 | 10 | 100 ± 26 | |
| 50 | 10 | 39 ± 21 | |
| 25 | 10 | 20 ± 15 | |
| 0 | 10 | 0 ± 6 | |
| Oxazalone | Fixed volume (%) | ||
| 1.0 | 0.3 | 109 ± 7 | |
| 0.3 | 0.3 | 107 ± 31 | |
| 0.3 | 0.1 | 93 ± 27 | |
| 0.1 | 0.1 | 79 ± 23 | |
| 0 | 0.3 | 12 ± 12 | |
Mice were shaved and painted on the upper trunk with the indicated doses of hapten.
Table 2.
LTα− /− Mice Cannot Mount a CH Response to Secondary Hapten Challenge but Retain a Sensitivity Response to Hapten Presented Subcutaneously
| CH responses | Subcutaneous and CH responses to TMA | ||||||
|---|---|---|---|---|---|---|---|
| Strain | Sensitization | Elicitation | Swelling (%) | Strain | n | Priming dose and route | Swelling (mm) |
| LT+/+ | FITC | Vehicle | 9 ± 3 | LTα+/− | 5 | 10 mm, 50 μl s.c. | 0.90 ± 0.23 |
| LT+/+ | FITC | FITC | 61 ± 8 | LTα+/− | 6 | 10 mm, 50 μl s.c. | 0.70 ± 0.12 |
| LTα−/− | FITC | FITC | 16 ± 8 | Control | 3 | 0 | 0.18 ± 0.02 |
| LT+/+ | Vehicle | 0.3% oxazalone | 5 ± 8.5 | LTα−/− | 5 | 10 mm, 50 μl e.c. | 0.68 ± 0.16 |
| LT+/+ | 0.3% oxazalone | 0.3% oxazalone | 31.6 ± 13.6 | LTα−/− | 6 | 10 mm, 50 μl e.c. | 0.28 ± 0.11 |
| LTα−/− | 0.3% oxazalone | 0.3% oxazalone | 5.1 ± 4.3 | Control | 3 | 0 | 0.10 ± 0.03 |
| LT+/+ | 2% oxazalone | 0.3% oxazalone | 40 ± 22 | ||||
| LTα−/− | 2% oxazalone | 0.3% oxazalone | 4.7 ± 6.1 | ||||
| LT+/+ | None | 0.8% croton oil | 23.5 ± 2.8 | ||||
| LTα−/− | None | 0.8% croton oil | 31.5 ± 9.7 | ||||
For skin sensitization, mice were painted with 100 μl hapten. 10 d later, CH was elicited by painting the ears with 10 μl hapten. Ear thickness was measured and the average percent increase in thickness (n = 6, FITC; n = 8, oxazalone; n = 4, croton oil) was determined for each set of mice. FITC and oxazalone data are representative of four independent experiments. Croton oil responses were not statistically different from each other. For experiments using TMA, mice were sensitized on the shaved abdomen with 50 μl either by subcutaneous (s.c.) injection or skin painting (e.c.). Elicitation of the footpad was with 20 μl TMA presented either subcutaneously or by painting. Footpad swelling is thickness at site of challenge − thickness of contralateral control. The TMA experiments were performed three times with similar results.
Table 3.
BM-derived Cells Are Not Responsible for the Defect in CH Response in LTα−/− Mice
| Experiment | Donor | Recipient | Priming dose | Footpad swelling |
|---|---|---|---|---|
| % | mm | |||
| 1 | +/+ | +/+ | 0.5 | 0.97 ± 0.09 |
| +/+ | LTα−/− | 0.5 | 0.13 ± 0.06 | |
| LTα−/− | +/+ | 0.5 | 0.75 ± 0.07 | |
| LTα−/− | LTα−/− | 0.5 | 0.08 ± 0.02 | |
| Nonirradiated control | +/+ | 0.5 | 1.14 ± 0.17 | |
| LTα−/− | 0.5 | 0.18 ± 0.06 | ||
| +/+ | 0 | 0.17 ± 0.05 | ||
| 2 | +/+ | +/+ | 1.0 | 0.92 ± 0.27 |
| +/+ | LTα−/− | 1.0 | 0.07 ± 0.02 | |
| LTα−/− | +/+ | 1.0 | 0.52 ± 0.12 | |
| LTα−/− | LTα−/− | 1.0 | 0.09 ± 0.04 | |
| Nonirradiated control | +/+ | 0 | 0.06 ± 0.01 | |
| Nonirradiated control | LTα−/− | 0 | 0.09 ± 0.02 | |
Mice were lethally irradiated (1,100 rad) and reconstituted within 4 h with 2 × 106 BM cells from the indicated mice. Mature T cells in the BM cell suspension were depleted using anti-Thy1.2 mAb plus complement. The CH experiments were performed 6 wk later, using DNFB as the hapten, and three mice per group. Footpad swelling is thickness at site of challenge − thickness of contralateral control.
In experiments using another hapten (TMA), we observed that the delayed type hypersensitivity (DTH) response of LTα1/+ and LTα−/− mice to subcutaneous injections were indistinguishable (Table ). This suggested that dermal APCs were triggering T cell activation despite the lack of LNs or of LT ligands. We tested if excessive hapten doses applied to the epidermis would breach epidermal integrity and lead to a mixed epidermal/dermal CH/DTH response. LTα−/− mice sensitized with as much as 150 μl 2% oxazalone failed to respond to subsequent challenge (Table ). Similarly, LTα−/− mice sensitized with as much as 20 μl of 1% DNFB did not respond to subsequent challenge (data not shown). Thus, LTα−/− mice did not mount a CH response even when excessive doses of hapten were used for sensitization. Finally, we examined the ability of LTα−/− mice to respond to chemical irritation using a croton oil assay. The response to irritant in this model is T cell independent 24. In contrast to the defect seen in the CH models, the irritant response of LTα−/− mice was similar to wild type (Table ). Therefore, LTα−/− were capable of an epidermal inflammatory response which does not require T cell activation.
BM-derived Cells from LTα− /− Mice Are Capable of Mediating CH Responses.
The initial experiments in LTα−/− mice suggested that the CH response to epidermal antigen was defective despite normal T cell priming and immune response to antigens presented within the dermis. To assess whether the inability of lymphocytes to make surface LTαβ or secrete LTα was responsible for the defective CH response in LTα−/− mice, BM cells from LTα−/− mice were transferred into irradiated wild-type mice. Mice were used in CH experiments 8–12 wk after BM transfer. Such chimeric mice, which retain a full complement of LNs, had a slightly attenuated response to the hapten DNFB (Table ), indicating that LT expression by lymphocytes does not play a dominant role in the CH response. Immunohistochemical analyses showed that no FDCs, germinal centers, or MZ markers could be detected in the spleen of these chimeric mice, indicating that LTα expressing cells were absent as expected 3.
When wild-type BM was transferred into irradiated LTα−/− mice and tested in the DNFB CH model, the chimeric mice were unable to respond to DNFB challenge (Table ). The wild type into LTα−/− BM transfer does not restore LN development, as LN organogenesis is developmentally regulated 12 22; however, splenic function is restored 3. These results show that expression of LTα by circulating cells is not essential for CH, but rather suggests a critical role for LNs in the generation of the CH response.
FITC-mediated CH Is Restored in LTα− /− Mice that Have LNs.
The BM transfer experiments suggested that LNs were required for CH responses. To formally address this hypothesis, we induced LN genesis in LTα−/− mice by using the agonist anti–LTβ-R mAb AF.H6 to signal LTβ-R during development 12. Progeny of litters which received this maternal transfer treatment were examined for the presence of LNs, and LTα−/− mice from litters demonstrated to have pLNs (brachial, axillary, mandibular, iliac) and mucosal LNs (mesenteric, cervical, sacral) (Table ) were assayed in the CH model. All mice were examined at necropsy to confirm which LNs were present. The transient exposure to the anti–LTβ-R mAb during gestation did not alter the lymphoid microenvironment of the spleen of LTα−/− mice, as immunohistochemical examination revealed that these mice still had disrupted MZs, lacked T/B cell segregation, and had no FDCs (data not shown). The results showed that LTα−/− mice having LNs generated a CH response that was comparable to the response of LTα1/+ mice, whereas the response of LTα−/− mice without LNs was grossly impaired (Table ).
Table 4.
LTα−/− Mice Treated In Utero with Anti–LTβ-R to Induce LN Genesis Have a Normal CH Response to Hapten
| Percent swelling | |||||||
|---|---|---|---|---|---|---|---|
| Strain | Treatment in utero | LN formed | Sensitization | Elicitation | Expt. 1 | Expt. 2 | Expt. 3 |
| LTα2/− | None or Ha4/8 | None | FITC | FITC | 14 ± 9 | nd | 9 ± 7 |
| LTα2/− | AF.H6 | Brachial, axillary, mandibular, iliac, mesenteric, cervical, sacral | FITC | FITC | 40 ± 19 | 37 ± 7 | 23 ± 7 |
| LTα1/+ | None | All | FITC | FITC | 40 ± 13 | 34 ± 10 | 25 ± 11 |
| LTα1/+ | None | All | FITC | Vehicle | 5 ± 1 | 1 ± 4 | 1 ± .4 |
For sensitization, mice were painted with 100 μl FITC. 10 d later, CH was elicited by painting the ears with10 μl FITC. Ear thickness was measured and the average percent increase in thickness (n = 6) was determined for each set of mice. Expt., experiment; nd, not done.
LTβ− /− Mice that Lack LNs Cannot Respond to Hapten Challenge.
LTα−/− mice and LTβ−/− mice share defects in LN and PP genesis, spleen and LN cellular organization, and humoral responses, but differ in regard to the presence of LTα homotrimers and of mucosa-associated LNs 3. In contrast to LTα−/− mice, LTβ−/− mice responded at the same level as LT+/+ or C57Bl/6 control mice to both FITC or oxazalone challenge in the CH model (Table , and data not shown). Importantly, the LTβ−/− mice used for these studies lacked all LNs except mesenteric, sacral, and cervical, none of which directly drain the sensitized area of the skin. We next investigated the capacity for CH responses in LTβ−/− mice that lacked all LNs.
Table 5.
The Loss of LNs from LTβ−/− Mice Results in the Loss of the CH Response to Hapten
| Percent swelling | ||||||
|---|---|---|---|---|---|---|
| Strain | LN found | Sensitization | Elicitization | Expt. 1 | Expt. 2 | Expt. 3 |
| LT+/+ | All | Vehicle | FITC | nd | 3.1 ± 3.2 | 2.5 ± 2.9 |
| LT+/+ | All | FITC | Vehicle | 3.2 ± 3.8 | nd | 2.3 ± 2.7 |
| LT+/+ | All | FITC | FITC | 52.2 ± 20 | 24 ± 9.5 | 23 ± 11 |
| LTβ−/− | Mucosal | FITC | FITC | 62.2 ± 3.3 | 29 ± 8.4 | 43 ± 18 |
| LTβ−/− | None | FITC | FITC | 8.8 ± 2.7 | 8.5 ± 4.4(n = 2) | 9.3 ± 6.8 |
Mice were painted on the upper trunk with 100 μl FITC then rechallenged 10 d later on the ears with 10 μl FITC.
Ear thickness was determined and values shown are percent increase. “Mucosal” refers to the presence of one or more mesenteric, cervical, or sacral LNs. n = 6 in each experiment shown, except LTβ−/− mice, no LNs, in experiment 2 (n = 2 as noted). Expt., experiment; nd, not done.
The original colonies of LTβ−/− mice retained mLNs, cervical LNs, and sacral LNs, which was true regardless of whether the gene deletion was on a pure C57BL/6 or mixed 129/C57BL/6 backgrounds (15 16; and unpublished data). The presence of various mucosa-associated LNs in LTβ−/− mice versus their absence in LTα−/− mice has been interpreted as evidence for additional signaling components in the LT system, e.g., via LTα−/− or LIGHT 15. However, a complete lack of mLNs was observed in up to 25% of the LTβ−/− mice in three separate colonies (15 16; and unpublished data). Furthermore, considerable variation in the LN content of LTα−/− mice was noted in different colonies with mLNs being present in 5–25% of LTα−/− mice in three separate colonies (13 14; and unpublished data). Mice lacking mLNs were only rarely observed in the original colony of LTβ−/− mice 15; however, after sterile rederivation and housing in our specific pathogen-free facility, we now find LTβ−/− mice which have no LNs (40%), retain only a single mLN or several mLNs and cervical LNs, (40%), or have all mLNs, sacral LNs, and cervical LNs (20%). A similar change in phenotype has been reported in a separately derived colony of LTβ−/− mice 19. It now appears that LN formation is largely defective in these mice.
To determine if the ability of LTβ−/− mice to respond to hapten challenge depended on the presence of LNs, LTβ−/− mice were tested in the FITC or oxazalone CH models then examined at necropsy to determine which LNs were present. In contrast to LTβ−/− mice that retained LNs, those without LNs failed to mount a CH response to either FITC or oxazalone (Table , and data not shown). Strikingly, a single remaining mLN was sufficient to generate a CH response. In agreement with the BM transfer and LTα−/− studies, these results suggest that the ability of LTβ−/− mice to generate a CH response depends on the presence of LNs.
Hapten-mediated CH Responses Are Blocked in Normal Mice that Lack All LNs.
As the presence of LNs in LTα−/− mice restored the ability to mount a CH response, we next asked if the loss of LNs from wild-type mice would ablate the CH response. Using either LTβ-R-Ig or a combination of LTβ-R-Ig and TNF-R55-Ig, we can reproducibly block the development of only pLNs, or of peripheral and mucosal LNs, respectively 12. By varying the time at which dosing with antagonists is initiated, the extent of LN development can be manipulated 12. Furthermore, one or two mLNs are occasionally “spared” in some pups treated with LTβ-R-Ig and TNF-R55-Ig starting at day 11 of gestation 12. Using these methods we have engineered normal mice having variable numbers of mLNs, but lacking all other LNs (Table ). We tested the immunocompetence of these mice in CH models, and evaluated at necropsy which LNs were present.
Table 6.
Balb/c Mice which Selectively Lack Peripheral and mLNs Have a Normal CH Response to Hapten whereas Balb/c Mice which Lack All LNs Do Not Respond to Hapten Challenge
| Percent swelling | |||||
|---|---|---|---|---|---|
| LN | Sensitization | Elicitation | Expt. 1 | Expt. 2 | Expt. 3 |
| All | FITC | FITC | 75 ± 13 | 62 ± 13 | 30 ± 6 |
| All | FITC | Vehicle | 0 ± 3 | 5 ± 2 | nd |
| All | Vehicle | FITC | nd | nd | 3 ± 3 |
| Mucosal LNs | FITC | FITC | 51 ± 12 | 43 ± 19 | 25 ± 10 |
| mLNs only | FITC | FITC | nd | nd | 33 ± 4 |
| None | FITC | FITC | nd | nd | 9 ± 5 |
For the CH experiments, the adult progeny were painted with 100 μl FITC then rechallenged 10 d later on the ears with 10 μl FITC. Change in ear thickness was determined. All mice were examined for the presence of LNs at necropsy. Values shown are percent increase in ear thickness (n = 6).
Mice retaining mucosal LNs but no pLNs were capable of efficient CH responses, whereas mice which lacked all LNs failed to respond to challenge with either FITC or oxazalone (Table and data not shown). These results strongly suggested that the ability of individual mice to respond to hapten challenge was due to the presence of LNs.
The minimal LNs required to support a CH response was examined using mice from litters given dual treatment in utero beginning on day 11 of gestation and having progressively fewer peripheral or mucosal LNs. All mice which retained a single LN were capable of responding to hapten challenge whereas those which had no LNs failed to respond to the challenge (Table ). This was true within litters having some pups with a single mLN and littermates having no LNs. Therefore, even a single nondraining LN was sufficient to enable a CH response.
Trafficking of LCs in LT− /− Mice and in Normal Mice Lacking pLNs.
As mentioned earlier, immunohistochemical examination of the skin of wild-type and LTα−/− mice had not revealed any difference in the extent or pattern of LC staining (unpublished data). However, it was critical to demonstrate that LCs could traffic from the skin to LNs in the LT−/− mice, as the increase in LC number isolated after hapten painting correlates with T cell proliferative activity in the LNs 26. Therefore, various LT−/− mice were painted with FITC, and the draining brachial and axillary LNs (if present), the mLNs (if present), and the spleen were harvested 24 h later. Mice painted with vehicle alone had no FITC reactivity in the draining LNs (data not shown). Little or no FITC staining was found in the spleens of painted control, LTα−/−, or LTβ−/− mice (data not shown). FITC reactivity in LNs taken from LT+/+, LTα−/−, or LTβ−/− mice 24 h after painting was primarily limited to the T cell areas and adjacent sinus (Fig. 1, and data not shown). The source of FITC reactivity in the LNs of FITC-painted mice was confirmed to be LCs using manual disruption of LNs followed by metrizamide isolation and FACS® analyses. LC purity after isolation was ∼70%, with the remainder being primarily B cells and T cells. LCs, defined based on large size and complexity, were FITC+, CD11c+, NLDC145+, MHC class II bright, and B7-2 bright (data not shown). From these data we concluded that the fluorescent staining in LNs detected by immunohistochemistry was due to the influx of LCs carrying FITC, as reported previously 26 27 28 29 30 31.
Figure 1.
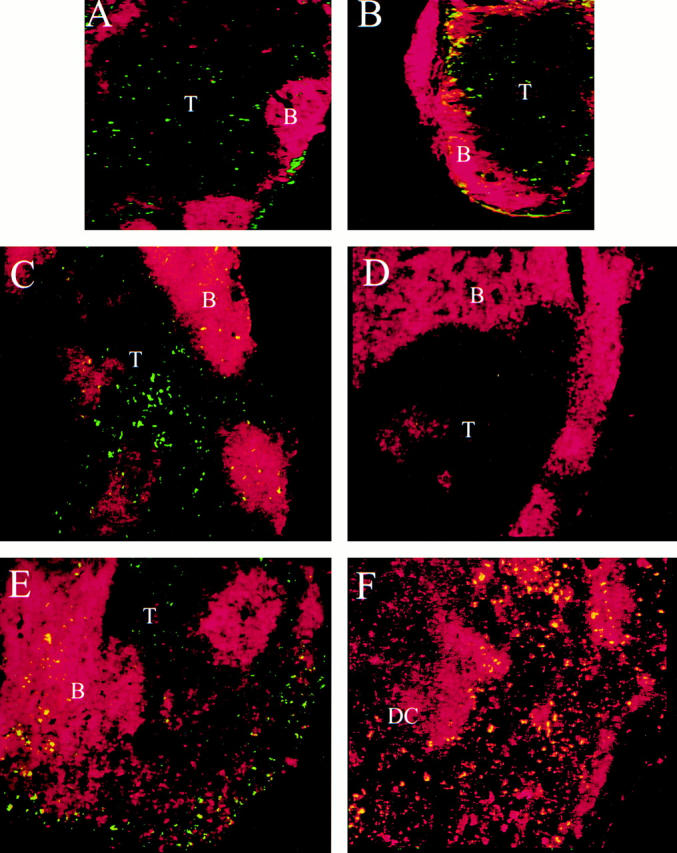
Immunohistochemical characterization of FITC-laden LCs within LNs. (A–E) Immunohistochemical images of LNs taken from mice 24 h after FITC painting: brachial LNs from LTα1/+ (A) and LTα−/− mice (B). Original magnification: ×50. FITC reactivity is present in the subcapsular sinus and in the T cell area; counterstaining is PE–anti-B220. mLNs are from Balb/c mice which lack peripheral LNs (C and F), from a Balb/c mouse which has pLNs (D), and from an LTβ−/− mouse (E). Original magnification: ×100. FITC reactivity is in the T cell area of the Balb/c mLN (C, red staining is PE–anti-B220; and F, red staining is PE–anti-CD11c). FITC-laden cells are also CD11c+, as are resident DCs (F). Trafficking LCs are both FITC+ and CD11c+ and so appear yellow. In the LTβ−/− mouse, FITC reactivity is present in the T cell area, as well as the disrupted B cell areas and adjacent sinus (E, red staining is PE–anti-B220). T, T cell area; B, B cell area.
These results suggested that LCs could traffic to remaining LNs to trigger T cell activation, and thereby support a CH reaction upon rechallenge. When pLNs were absent, FITC reactivity was readily observed in the T cell areas of the nondraining mLNs (Fig. 1B–E), whereas little or no FITC reactivity could be found in the white pulp zones of the spleen (data not shown). When draining pLNs were present, FITC reactivity was not observed in nondraining LNs or in mLNs (Fig. 1 D). Therefore, LCs preferentially accumulate in the first LNs they encounter, which is normally the draining LNs, as noted previously 10. When the draining LNs are absent, LCs can home to any LNs. In contrast, either LCs do not home to splenic white pulp, or the density of LCs that reach the spleen is insufficient to trigger a CH response. To further demonstrate that hapten specific responses were generated in mLNs but not spleen, we performed adoptive transfer experiments. Balb/c mice lacking all pLNs, but retaining mLNs and spleen, were sensitized with oxazalone. 4 d later mLN cells and splenocytes were isolated and transferred to normal, naive Balb/c mice, which were then challenged by ear painting with oxazalone. The CH response was measured after 24 h, and the results (Fig. 2) demonstrate that cells isolated from mLNs, but not spleen, were capable of adoptively transferring the capacity for CH to naive recipients. Therefore LN-specific homing by LCs is essential for the T cell priming underlying the CH response.
Figure 2.

mLN cells, but not splenocytes, can adoptively transfer CH to naive recipients. Balb/c mice having mLNs, but no pLNs, were sensitized with oxazalone. 4 d later cell suspensions of mLN (“LN cells”) and spleen (“spleen cells”) were harvested and injected intravenously into normal, naive, recipient mice, which were challenged to induce CH. “+ control” refers to mLN+/pLN− mice which were sensitized and challenged with oxazalone. “− control” refers to mLN cells isolated from nonsensitized mice. *P < 0.001 for values significantly less than the positive control (n = 6).
Discussion
We have demonstrated that LNs are required to initiate a CH response by using the LT system to manipulate the development of LNs on wild-type and LT-deficient backgrounds. Our critical finding is that CH can occur in LTα−/− mice in which LNs have been induced to develop, regardless of the lack of LTα expression by circulating lymphocytes. LNs were essential for the CH response to haptens which induce cytokines typical of both Th1- (DNFB) and Th2- (FITC and oxazalone) dominated responses (30, 31, and references therein). This observation was consistent in all experimental settings when LTα−/− mice and LTβ−/− mice with or without LNs were contrasted, and further supported by the BM transplantation studies using LTα−/− mice. These results are fully consistent with the paradigm that LNs are a critical site of T cell activation in response to epidermal antigens.
The earliest modern investigations of CH grew out of efforts to understand the pathology associated with human responses to poison ivy, Japanese lacquer, and other substances which caused contact reactions. Early work focused on identifying the inducing agents, which led to the discovery of a variety of sensitizing compounds. Some investigators of the period hypothesized that contact sensitization was due to the diffusion of antigen or conjugates via epidermal channels, whereas others stressed the importance of passage via lymphatic vessels or the bloodstream (6, and references therein). A functional role for the draining LNs was specifically postulated somewhat later 32, but the anatomy of the sensitization reaction remained controversial for many years, with the mechanistic focus remaining on the site of sensitization, that is, the skin itself (33, and references therein). The relationship between epidermal sensitization and the lymphatic system became clearer as a understanding of lymphocyte and dendritic cell populations developed, particularly with reference to presence of LCs in the epidermis 26 34. Finally, it was determined that the induction of CH can be blocked by antagonism of various LC properties: activation, migration to LNs, or ability to activate T cells 4 5 8 9 28 31. Thus, data accumulated over the course of many decades supported the hypothesis that LCs migrated to LNs, which hosted the T cell–dependent immune response to epidermal antigen, and therefore the development of CH.
We formally tested the hypothesis that LN structure provides a critical environment which supports CH responses by using in utero blockade of the LTβ-R and TNF-R pathways to eliminate LNs from wild-type mice. In utero antagonism of the signaling pathways is transient, and our previous studies have indicated that although the organogenic defects are irreversible, secondary lymphoid architecture, including B cell localization, integrity of splenic MZ populations, and LN addressin expression, is restored in the adult progeny 22 23. Furthermore, we have recently shown that wild-type mice treated in utero with LTβ-R-Ig are capable of antigen-specific T cell responses 35. Therefore, the simplest model to explain the loss of CH responsiveness in wild-type mice lacking LNs is that LNs provide the proper environment to ensure productive interaction of LCs with circulating T cells. Furthermore, because LCs do not appear to access the splenic white pulp, the spleen is incapable of compensating for the loss of LNs. Finally, any other encounter of LCs with potentially responsive T cells is either very rare or functionally irrelevant 2.
Surprisingly, the CH response can be triggered in LNs that do not directly drain the site of antigen exposure; instead, any LN within the intact lymphatic network is sufficient to support the required LC/T cell interaction. We have demonstrated that antigen-bearing LCs traffic to distant LNs; the route of traffic, however, cannot be determined from these studies. Lymph typically drains into the bloodstream via the thoracic and right subclavian ducts. It is possible that antigen-bearing LCs are carried by normal circulation to tissue beds and then drain to remaining LNs via afferent lymphatics; however, it is also possible that they are entering LNs directly from the bloodstream by crossing HEVs. Regardless of the route of traffic, the adoptive transfer experiments clearly show that T cell priming to hapten has occurred in the mLNs, but not the spleen, of pLN–deficient mice.
Several recent studies have addressed the role of different secondary lymphoid compartments in mediating immune responsiveness. An essential role for LNs in antiviral immunity was suggested by studies using the LN-deficient alymphoplastic (aly/aly) mice. Aly mice were unable to control LCMV infections, and BM transfer studies supported the hypothesis that the impaired antiviral response was due to the lack of LNs 36. This hypothesis was supported by the observation that the Hox11−/− mice, which lack the spleen but have LNs, retained a normal antiviral response 36. LTβ−/− mice also had a defective antiviral responses, with significantly delayed kinetics of lymphocytic choriomeningitis virus (LCMV) clearance 37. The LTβ−/− mice used in this LCMV study retained mucosal LNs; moreover, it appeared that the LTβ-dependent defect was manifested because it caused disrupted splenic organization, particularly of the MZ. However, this conclusion is at odds with the Hox11−/− mouse study. In addition, some antiviral responses can be manipulated in a manner which may be independent of the status of secondary lymphoid tissue 38. Wild-type mice lacking LNs and/or spleen should provide a useful model to study antiviral responses and address these issues.
Recently, it was demonstrated that an immune response to intradermally administered tetanus toxoid in an immunocompetent SCID-hu model required an engrafted human fetal LNs 39, suggesting the mouse spleen could not trigger an immune response in this setting. Another recent study demonstrated functional overlap of spleen and LN responses, in which transplant rejection could be mediated in the absence of LNs (and PPs), or spleen, but not in the absence of all secondary lymphoid tissue 40. Functional overlap of mLNs and PPs for gut immune responses was also recently demonstrated 35. In contrast, skin transplant rejection required LNs, but not spleen 40, a requirement similar to that described in this study. Finally, the spleen is known to be essential for immunity to encapsulated bacteria 41, and the splenic MZ compartment has recently been shown to be critical for the immune response to T cell–independent antigens 42. These observations suggest that each secondary lymphoid compartment has evolved, at least in part, to respond to specific types of antigen presentation.
Our results invite a reinterpretation of several other studies of immune responsiveness in LT−/− mice. For example, LTα−/− mice and LTβ−/− mice responded differently to myelin oligodendrocyte glycoprotein (MOG) peptide immunization in a model of experimental autoimmune encephalomyelitis (EAE) development, suggesting that LTα was required for this response 43. However, a second study suggested that genetic background, immune incompetence, and/or altered lymphoid structure in LTα−/− mice accounted for their failure to respond in an EAE model 44. This hypothesis was supported by results demonstrating that robust disease developed upon MOG peptide immunization after transfer of LTα−/− BM cells to recombination activating gene (RAG)−/− recipients. These observations suggest that it is difficult to compare LTα−/− mice, LTβ−/− mice, or TNF−/− mice in such disease models without taking into account additional variables such as LN status, cellular organization, and other developmental defects, e.g., on hematopoiesis 45. The data presented here support the interpretation that the LTα−/− and LTβ−/− mice are relatively similar in immune competence, and differences in their disease susceptibility stem from the presence and absence of LNs. Recent work suggesting a critical role for draining LNs in diabetes 46 indicates that there will be similar complexity in interpreting the role of the LT axis, separate from the role of LNs, in several disease models.
The fact that LT ligands did not play a more dominant role in the CH response is perhaps surprising in light of recent data. LTα−/− mice have reduced numbers of resident DCs within secondary lymphoid organs, probably due to reduced expression of chemokines, including secondary lymphoid tissue chemokine (SLC; CCL21) and EBV-induced molecule 1 ligand chemokine (ELC; CCL19) 47 48 49 50. This reduction in resident DC number reflects defective traffic into the T cell zones of LNs, rather than the failure of DCs to develop 48. The reduction in resident DC number may be partially offset by the loss of LNs and PPs in LT-deficient or wild-type mice, such that DCs may accumulate in tissues such as spleen or remaining LNs; however, this has not been investigated. Certainly the localization of DCs, and of LCs which reach LNs in LT-deficient mice appears disrupted, being less restricted than normal to the T cell zone.
The concept of “T zone tropism” 1 is well illustrated by the paucity of LN T cells (plt) mouse, in which both T cells and DCs fail to migrate into the T cell zone of spleen and mLNs, due to the lack of expression of SLC and ELC in LNs 51 52. When plt mice were challenged on the skin with hapten, 75% fewer LCs accumulated in draining LNs than in wild-type mice 51. Therefore, LCs traffic into LNs is also influenced by expression of these chemokines. However, CH responses in these mice appeared normal, and, although the site of priming was not identified, labeled epidermal DCs could not be found in the spleen 53. It does appear that supraoptimal doses of hapten were used in this study 54, which may complicate interpretation of the CH result. Indeed, knockout of the SLC/ELC receptor CC chemokine receptor (CCR)7 produced somewhat different results, as the CCR7−/− mice had essentially no T cells or DCs in LNs and did not respond to hapten challenge in the FITC CH model 29. SLC was absent and ELC expression was reduced by 75% in the spleen of LTα−/− mice 49, and it may be the continued expression of ELC in LT−/− mice that supports sufficient accumulation of LCs into LNs to support the CH response. Alternatively, unknown mechanisms may be operating. Given that multiple genes for SLC have been identified, and that there is some evidence for differential expression of SLC and ELC in secondary lymphoid organs, the epidermis, and lymphatics, the roles of SLC and ELC in the traffic of LCs and DCs into secondary lymphoid organs will require additional investigation 52 55 56.
It has been demonstrated that LCs form a continuous reticular barrier which has evolved to trap epidermal antigens 57. Our results highlight not only the critical role that LCs play in mediating epidermal immune responses, but also the essential nature of LNs in supporting LC-driven T cell activation. The mechanisms which allow preferential LC traffic to distant LNs, but not splenic white pulp, are not understood, but may involve specific chemokine responsiveness. The reliance on LNs appears to be unique to LCs residing in the epidermal compartment, as our results indicate that subcutaneous hapten injection results in an intact hypersensitivity response in LTα−/− mice (Table ). This suggests that presentation of subepidermal antigen does not rely solely on LNs, possibly because antigen which reaches the dermal layer will encounter additional populations of APCs 58. Our model predicts that dermal APCs have trafficking properties that allow access to other lymphoid compartments such as spleen in addition to LNs. Indeed, such a mechanism was recently proposed to explain the enhanced response to subcutaneously presented antigen in the plt mouse, whereby the T cell response shifted from draining LNs to the spleen 53.
Our studies have conclusively identified an essential role for LNs in a T cell–dependent immune response. Furthermore, we have demonstrated that LCs specifically home to LNs, and will traffic until a LN is encountered, bypassing the spleen. This specific trafficking of LCs to LNs comprises a unique and critical component of the secondary lymphoid system, and illustrates the importance of functional anatomy for immune responses. The signals that support LC access to LNs will be a important area for future study, and may yield therapeutic targets to allow highly specific intervention in CH and other disorders.
Acknowledgments
We wish to thank the following individuals who created, characterized, and produced mAbs and receptor-Ig fusion proteins: Chris Benjamin, Irene Sizing, Mohammad Zafari, Werner Meier, Konrad Miatkowski, Joe Amatucci, Gerard Majeau, Cathy Hession, and Apinya Ngam-Ek. Danelle James, Maxine Guay, Apinya Ngam-Ek, Ponsri Lawton, and Sandy Cho provided valuable technical help. We thank J. Sedjwick and H. Korner for information regarding LT-deficient mice on the C57BL/6 background.
R. Flavell and D. Chaplin are investigators of the Howard Hughes Medical Institute. Y. Fu acknowledges the support of the National Institutes of Health.
Footnotes
Abbreviations used in this paper: BM, bone marrow; CH, contact hypersensitivity; DC, dendritic cell; DNFB, dinitrofluorobenzene; ELC, EBV-induced molecule 1 ligand chemokine; FDC, follicular dendritic cell; LC, Langerhans cell; LT, lymphotoxin; mLN, mesenteric LN; MZ, marginal zone; pLN, peripheral LN; plt, paucity of LN T cells; PP, Peyer's patch; SLC, secondary lymphoid tissue chemokine; TMA, phenyltrimethylammoaniline.
References
- Goodnow C.C. Chance encounters and organized rendezvous. Immunol. Rev. 1997;156:5–10. doi: 10.1111/j.1600-065x.1997.tb00954.x. [DOI] [PubMed] [Google Scholar]
- Zinkernagel R.M., Ehl S., Aichele P., Oehen S., Kundig T., Hengartner H. Antigen localisation regulates immune responses in a dose- and time-dependent fashiona geographical view of immune reactivity. Immunol. Rev. 1997;156:199–209. doi: 10.1111/j.1600-065x.1997.tb00969.x. [DOI] [PubMed] [Google Scholar]
- Fu Y.-X., Chaplin D.D. Development and maturation of secondary lymphoid tissues. Ann. Rev. Immunol. 1999;17:399–433. doi: 10.1146/annurev.immunol.17.1.399. [DOI] [PubMed] [Google Scholar]
- Cella M., Sallusto F., Lanzavecchia A. Origin, maturation and antigen presenting function of dendritic cells. Curr. Opin. Immunol. 1997;9:10–16. doi: 10.1016/s0952-7915(97)80153-7. [DOI] [PubMed] [Google Scholar]
- Grabbe S., Schwarz T. Immunoregulatory mechanisms involved in elicitation of allergic contact hypersensitivity. Immunol. Today. 1998;19:37–44. doi: 10.1016/s0167-5699(97)01186-9. [DOI] [PubMed] [Google Scholar]
- Landsteiner K., Jacobs J. Studies on the sensitization of animals with simple chemical compounds. II. J. Exp. Med. 1936;64:625–639. doi: 10.1084/jem.64.4.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker C.F., Billingham R.E. The role of afferent lymphatics in the rejection of skin homografts. J. Exp. Med. 1968;128:197–221. doi: 10.1084/jem.128.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalina M.D., Carroll M.C., Arizpe H., Takashima A., Estess P., Siegelman M.H. The route of antigen entry determines the requirement for L-selectin during immune responses. J. Exp. Med. 1996;184:2341–2351. doi: 10.1084/jem.184.6.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engeman T.M., Gorbachev A.V., Gladue R.P., Heeger P.S., Fairchild R.L. Inhibition of functional T cell priming and contact hypersensitivity responses by treatment with anti-secondary lymphoid chemokine antibody during hapten sensitization. J. Immunol. 2000;164:5207–5214. doi: 10.4049/jimmunol.164.10.5207. [DOI] [PubMed] [Google Scholar]
- Pugh C.W., MacPherson G.G., Steer H.W. Characterization of nonlymphoid cells derived from rat peripheral lymph. J. Exp. Med. 1983;157:1758–1779. doi: 10.1084/jem.157.6.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futterer A., Mink K., Luz A., Kosco-Vilbois M.H., Pfeffer K. The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity. 1998;9:59–70. doi: 10.1016/s1074-7613(00)80588-9. [DOI] [PubMed] [Google Scholar]
- Rennert P.D., James D., Mackay F., Browning J.L., Hochman P.S. Lymph node genesis is induced by signaling through the lymphotoxin β receptor. Immunity. 1998;9:71–79. doi: 10.1016/s1074-7613(00)80589-0. [DOI] [PubMed] [Google Scholar]
- De Togni P., Goellner J., Ruddle N.H., Streeter P.R., Fick A., Mariathasan S., Smith S.C., Carlson R., Shornick L.P., Strauss-Schoenberger J. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin alpha. Science. 1994;264:703–707. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- Banks T.A., Rouse B.T., Kerley M.K., Blair P.J., Godfrey V.L., Kuklin N.A., Bouley D.M., Thomas J., Kanangat S., Mucenski M.L. Lymphotoxin-alpha-deficient miceeffects on secondary lymphoid development and humoral immune responsiveness. J. Immunol. 1995;155:1685–1693. [PubMed] [Google Scholar]
- Koni P.A., Sacca R., Lawton P., Browning J.L., Ruddle N.H., Flavell R.A. Distinct roles in lymphoid organogenesis for lymphotoxins α and β revealed in lymphotoxin β-deficient mice. Immunity. 1997;6:491–500. doi: 10.1016/s1074-7613(00)80292-7. [DOI] [PubMed] [Google Scholar]
- Alimzhanov M.B., Kuprash D.V., Kosco-Vilbois M.H., Luz A., Turetskaya R.L., Tarakhovsky A., Rajewsky K., Nedospasov S.A., Pfeffer K. Abnormal development of secondary lymphoid tissues in lymphotoxin beta-deficient mice. Proc. Natl. Acad. Sci. USA. 1997;94:9302–9307. doi: 10.1073/pnas.94.17.9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay F., Browning J.L. Turning off follicular dendritic cells. Nature. 1998;395:26–27. doi: 10.1038/25630. [DOI] [PubMed] [Google Scholar]
- Endres R., Alimzhanov M.B., Plitz T., Futterer A., Kosco-Vilbois M.H., Nedospasov S.A., Rajewsky K., Pfeffer K. Mature follicular dendritic cell networks depend on expression of lymphotoxin beta receptor by radioresistant stromal cells and of lymphotoxin beta and tumor necrosis factor by B cells. J. Exp. Med. 1999;189:159–168. doi: 10.1084/jem.189.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuprash D.V., Alimzhanov M.B., Tumanov A.V., Anderson A.O., Pfeffer K., Nedospasov S.A. TNF and lymphotoxin beta cooperate in the maintenance of secondary lymphoid tissue microarchitecture but not in the development of lymph nodes. J. Immunol. 1999;163:6575–6580. [PubMed] [Google Scholar]
- Fu Y.-X., Huang G., Matsumoto M., Molina H., Chaplin D.D. Independent signals regulate development of primary and secondary follicle structure in spleen and mesenteric lymph node. Proc. Natl. Acad. Sci. USA. 1997;94:5739–5743. doi: 10.1073/pnas.94.11.5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koni P.A., Flavell R.A. Lymph node germinal centers form in the absence of follicular dendritic cell networks. J. Exp. Med. 1999;189:855–864. doi: 10.1084/jem.189.5.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennert P.D., Browning J.L., Mebius R., Mackay F., Hochman P.S. Surface lymphotoxin alpha/beta complex is required for the development of peripheral lymphoid organs. J. Exp. Med. 1996;184:1999–2006. doi: 10.1084/jem.184.5.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennert P.D., Browning J.L., Hochman P.S. Selective disruption of lymphotoxin ligands reveals a novel set of mucosal lymph nodes and unique effects on lymph node cellular organization. Int. Immunol. 1997;9:1627–1639. doi: 10.1093/intimm/9.11.1627. [DOI] [PubMed] [Google Scholar]
- Homey B., von Schilling C., Blumel J., Schuppe H.C., Ruzicka T., Ahr H.J., Lehmann P., Vohr H.W. An integrated model for the differentiation of chemical-induced allergic and irritant skin reactions. Toxicol. Appl. Pharmacol. 1998;153:83–94. doi: 10.1006/taap.1998.8535. [DOI] [PubMed] [Google Scholar]
- Cavani A., Albanesi C., Traidl C., Sebastiani S., Girolomoni G. Effector and regulatory T cell in allergic contact dermatitis. Trends Immunol. 2001;22:118–120. doi: 10.1016/s1471-4906(00)01815-9. [DOI] [PubMed] [Google Scholar]
- Macatonia S.E., Knight S.C., Edwards A.J., Griffiths S., Fryer P. Localization of antigen on lymph node dendritic cells after exposure to the contact sensitizer fluorescein isothiocyanate. J. Exp. Med. 1987;166:1654–1667. doi: 10.1084/jem.166.6.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saulnier M., Huang S., Aguet M., Ryffel B. Role of interferon-gamma in contact hypersensitivity assessed in interferon-gamma receptor-deficient mice. Toxicology. 1995;102:301–312. doi: 10.1016/0300-483x(95)03101-k. [DOI] [PubMed] [Google Scholar]
- Xu H., Heeger P.S., Fairchild R.L. Distinct roles for B7-1 and B7-2 determinants during priming of effector CD8+ Tc1 and regulatory CD4+ Th2 cells for contact hypersensitivity. J. Immunol. 1997;159:4217–4226. [PubMed] [Google Scholar]
- Forster R., Schubel A., Breitfeld D., Kremmer E., Renner-Muller I., Wolf E., Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- Dearman R.J., Smith S., Basketter D.A., Kimber I. Classification of chemical allergens according to cytokine secretion profiles of murine lymph node cells. J. Appl. Toxicol. 1997;17:53–62. doi: 10.1002/(sici)1099-1263(199701)17:1<53::aid-jat393>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Chen A.I., McAdam A.J., Buhlmann J.E., Scott S., Lupher M.L., Jr., Greenfield E.A., Baum P.R., Fanslow W.C., Calderhead D.M., Freeman G.J., Sharpe A.H. Ox40-ligand has a critical costimulatory role in dendritic cell:T cell interactions. Immunity. 1999;11:689–698. doi: 10.1016/s1074-7613(00)80143-0. [DOI] [PubMed] [Google Scholar]
- Frey J.R., Wenk P. Experimental studies on the pathogenesis of contact eczema in the guinea pig. Int. Arch. Allergy Appl. Immunol. 1957;11:81–100. doi: 10.1159/000228405. [DOI] [PubMed] [Google Scholar]
- Macher E., Chase M.W. Studies on the sensitization of animals with simple chemical compounds. XII. The influence of excision of allergenic depots on onset of delayed hypersensitivity and tolerance. J. Exp. Med. 1968;129:103–121. doi: 10.1084/jem.129.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberberg I., Baer R.L., Rosenthal S.A. The role of Langerhans cells in allergic contact hypersensitivity. A review of findings in man and guinea pigs. J. Invest. Dermatol. 1976;66:210–217. doi: 10.1111/1523-1747.ep12482139. [DOI] [PubMed] [Google Scholar]
- Yamamoto M., Rennert P., McGhee J.R., Kweon M.N., Yamamoto S., Dohi T., Otake S., Bluethmann H., Fujihashi K., Kiyono H. Alternate mucosal immune systemorganized Peyer's patches are not required for IgA responses in the gastrointestinal tract. J. Immunol. 2000;164:5184–5191. doi: 10.4049/jimmunol.164.10.5184. [DOI] [PubMed] [Google Scholar]
- Karrer U., Althage A., Odermatt B., Roberts C.W., Korsmeyer S.J., Miyawaki S., Hengartner H., Zinkernagel R.M. On the key role of secondary lymphoid organs in antiviral immune responses studied in alymphoplastic (aly/aly) and spleenless (Hox11(−/−)) mutant mice. J. Exp. Med. 1997;185:2157–2170. doi: 10.1084/jem.185.12.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger D.P., Naniche D., Crowley M.T., Koni P.A., Flavell R.A., Oldstone M.B. Lymphotoxin-β-deficient mice show defective antiviral immunity. Virology. 1999;260:136–147. doi: 10.1006/viro.1999.9811. [DOI] [PubMed] [Google Scholar]
- Puglielli M.T., Browning J.L., Brewer A.W., Schreiber R.D., Shieh W.J., Altman J.D., Oldstone M.B., Zaki S.R., Ahmed R. Reversal of virus-induced systemic shock and respiratory failure by blockade of the lymphotoxin pathway. Nat. Med. 1999;5:1370–1374. doi: 10.1038/70938. [DOI] [PubMed] [Google Scholar]
- Carballido J.M., Namikawa R., Carballido-Perrig N., Antonenko S., Roncarolo M.G., de Vries J.E. Generation of primary antigen-specific human T- and B-cell responses in immunocompetent SCID-hu mice. Nat. Med. 2000;6:103–106. doi: 10.1038/71434. [DOI] [PubMed] [Google Scholar]
- Lakkis F.G., Arakelov A., Konieczny B.T., Inoue Y. Immunologic “ignorance” of vascularized organ transplants in the absence of secondary lymphoid tissue. Nat. Med. 2000;6:686–688. doi: 10.1038/76267. [DOI] [PubMed] [Google Scholar]
- Mond J.J, Lees A., Snapper C.M. T cell-independent antigens type 2. Annu. Rev. Immunol. 1995;13:655–692. doi: 10.1146/annurev.iy.13.040195.003255. [DOI] [PubMed] [Google Scholar]
- Guinamard R., Okigaki M., Schlessinger J., Ravetch J.V. Absence of marginal zone B cells in Pyk-2-deficient mice defines their role in the humoral response. Nat. Immunol. 2000;1:31–36. doi: 10.1038/76882. [DOI] [PubMed] [Google Scholar]
- Suen W.E., Bergman C.M., Hjelmstrom P., Ruddle N.H. A critical role for lymphotoxin in experimental allergic encephalomyelitis. J. Exp. Med. 1997;186:1233–1240. doi: 10.1084/jem.186.8.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riminton D.S., Korner H., Strickland D.H., Lemckert F.A., Pollard J.D., Sedgwick J.D. Challenging cytokine redundancyinflammatory cell movement and clinical course of experimental autoimmune encephalomyelitis are normal in lymphotoxin-deficient, but not tumor necrosis factor–deficient, mice. J. Exp. Med. 1998;187:1517–1528. doi: 10.1084/jem.187.9.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman L. Some misconceptions about understanding autoimmunity through experiments with knockouts. J. Exp. Med. 1997;185:2039–2041. doi: 10.1084/jem.185.12.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoglund P., Mintern J., Waltzinger C., Heath W., Benoist C., Mathis D. Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J. Exp Med. 1999;189:331–339. doi: 10.1084/jem.189.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacca R., Turley S., Soong L., Mellman I., Ruddle N.H. Transgenic expression of lymphotoxin restores lymph nodes to lymphotoxin-alpha-deficient mice. J. Immunol. 1997;159:4252–4260. [PubMed] [Google Scholar]
- Wu Q., Wang Y., Wang J., Hedgeman E.O., Browning J.L., Fu Y.X. The requirement of membrane lymphotoxin for the presence of dendritic cells in lymphoid tissues. J. Exp. Med. 1999;190:629–638. doi: 10.1084/jem.190.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo V.N., Korner H., Gunn M.D., Schmidt K.N., Riminton D.S., Cooper M.D., Browning J.L., Sedgwick J.D., Cyster J.G. Lymphotoxin alpha/beta and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J. Exp. Med. 1999;189:403–412. doi: 10.1084/jem.189.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotnik A., Yoshie O. Chemokinesa new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- Gunn M.D., Kyuwa S., Tam C., Kakiuchi T., Matsuzawa A., Williams L.T., Nakano H. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J. Exp. Med. 1999;189:451–460. doi: 10.1084/jem.189.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther S.A., Tang H.L., Hyman P.L., Farr A.G., Cyster J.G. Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc. Natl. Acad. Sci. USA. 2000;97:12694–12699. doi: 10.1073/pnas.97.23.12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori S., Nakano H., Aritomi K., Wang C.-R., Gunn M.D., Kakiuchi T. Mice lacking expression of the chemokines CCL21-Ser and CCL19 (plt mice) demonstrate delayed but enhanced T cell immune responses. J. Exp. Med. 2001;193:207–218. doi: 10.1084/jem.193.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulliven S., Bergstresser P.R., Streilein J.W. Analysis of dose response of trinitrochlorobenzene contact hypersensitivity induction in micepretreatment with cyclophosphamide reveals an optimal sensitizing dose. J. Invest. Dermatol. 1989;94:711–716. doi: 10.1111/1523-1747.ep12876288. [DOI] [PubMed] [Google Scholar]
- Vassileva G., Soto H., Zlotnik A., Nakano H., Kakiuchi T., Hedrick J.A., Lira S.A. The reduced expression of 6Ckine in the plt mouse results from the deletion of one of two 6Ckine genes. J. Exp. Med. 1999;190:1183–1188. doi: 10.1084/jem.190.8.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbiani D.F, Finch R.A., Jager D., Muller W.A., Sartorelli A.C., Randolph G.J. The leukotriene C4 transporter MRP1 regulates CCL19 (MIP-3β, ELC)-dependent mobilization of dendritic cells to lymph nodes. Cell. 2000;103:757–768. doi: 10.1016/s0092-8674(00)00179-3. [DOI] [PubMed] [Google Scholar]
- Shelley W.B., Juhlin L. Langerhans cells form a reticuloepithelial trap for external contact antigens. Nature. 1976;261:46–47. doi: 10.1038/261046a0. [DOI] [PubMed] [Google Scholar]
- Steinman R., Hoffman L., Pope M. Maturation and migration of cutaneous dendritic cells. J. Invest. Dermatol. 1995;105:2S–7S. doi: 10.1111/1523-1747.ep12315162. [DOI] [PubMed] [Google Scholar]