

Abstract
An allograft is often considered an immunologically inert playing field on which host leukocytes assemble and wreak havoc. However, we demonstrate that graft-specific physiologic responses to early injury initiate and promulgate destruction of vascularized grafts. Serial analysis of allografts showed that intragraft expression of the three chemokine ligands for the CXC chemo-kine receptor CXCR3 was induced in the order of interferon (IFN)-γ–inducible protein of 10 kD (IP-10, or CXCL10), IFN-inducible T cell α-chemoattractant (I-TAC; CXCL11), and then monokine induced by IFN-γ (Mig, CXCL9). Initial IP-10 production was localized to endothelial cells, and only IP-10 was induced by isografting. Anti–IP-10 monoclonal antibodies prolonged allograft survival, but surprisingly, IP-10–deficient (IP-10−/−) mice acutely rejected allografts. However, though allografts from IP-10+/+ mice were rejected by day 7, hearts from IP-10−/− mice survived long term. Compared with IP-10+/+ donors, use of IP-10−/− donors reduced intragraft expression of cytokines, chemokines and their receptors, and associated leukocyte infiltration and graft injury. Hence, tissue-specific generation of a single chemokine in response to initial ischemia/reperfusion can initiate progressive graft infiltration and amplification of multiple effector pathways, and targeting of this proximal chemokine can prevent acute rejection. These data emphasize the pivotal role of donor-derived IP-10 in initiating alloresponses, with implications for tissue engineering to decrease immunogenicity, and demonstrate that chemokine redundancy may not be operative in vivo.
Keywords: chemokine, transplantation, rejection, IP-10, endothelium
Introduction
Chemokine expression during allograft rejection, first documented since 1993 1, has since been demonstrated in many experimental and clinical transplant studies 2. Though the methods for chemokine detection have ranged from immunohistology to reverse transcription PCR, the recent advent of commercial RNase protection assay kits has provided a standardized means for the detection of chemokine and chemokine receptor mRNA, and has proven a boon to the field. However, there are still very few mechanistic studies concerning the role of chemokine pathways in alloresponses 2.
In the first mechanistic study of the role of a chemokine receptor in the pathogenesis of allograft rejection, analysis of cardiac allograft rejection in CC chemokine receptor 1–deficient (CCR1−/−) versus CCR1+/+ mice showed that CCR1 contributed to the development of acute and chronic allograft rejection 3. Though by itself, CCR1 deficiency had only modest effects of rejection of fully MHC-mismatched allografts, an absolute requirement for CCR1 in the development of chronic rejection was shown in recipients treated with subtherapeutic courses of cyclosporin A or CD4 mAb. Beneficial effects on allograft survival were also noted using CCR2- and CCR5-deficient allograft recipients 2. However, the most profound effects reported so far involved targeting of the CXC chemokine receptor, CXCR3, which is expressed by activated Th1 cells and NK cells 4. Use of a blocking anti-CXCR3 mAb or CXCR3−/− mice was shown to prevent acute rejection, with a mean allograft survival of almost 60 d in CXCR3−/− recipients versus 7 d in CXCR3+/+ controls 5.
In vitro studies have documented a plethora of chemokines and receptors, with many chemokines binding to the same receptor and individual chemokines binding to multiple receptors, such that notions of chemokine redundancy and doubts as to the value of ligand targeting, in particular, are widespread 6. Given the importance of CXCR3 to alloresponses and detection of intragraft mRNA expression of each of its three ligands, IFN-γ–inducible protein (IP)-10 7, IFN-inducible T cell α-chemoattractant (I-TAC; reference 8), and Mig 9, during development of acute rejection 5, we asked whether these ligands might show temporal or spatial differences in their expression that would help explain their biologic roles. The results of our studies lead to a rationale for targeting selected chemokines in vivo.
Materials and Methods
Transplantation.
IP-10−/− mice were generated in the laboratory of A.D. Luster. Heterotopic vascularized cardiac allografting was performed using fully MHC-mismatched BALB/c (H-2d) and C57BL/6 × 129Sv/J (H-2b) mice 10. B6/129 mice were IP-10−/− versus age- and sex-matched littermate IP-10+/+ mice. BALB/c and additional B6/129, plus control C57BL/6 (H-2b) and 129 (H-2b) mice, were obtained commercially (The Jackson Laboratory); all mice used were between 6 and 10 wk of age. Additional recipients were treated with neutralizing hamster 11 or rat (BD PharMingen) anti–mouse IP-10 mAbs, or corresponding hamster or rat IgG (Jackson ImmunoResearch Laboratories) by intraperitoneal injection of 200 μg every second day; therapy was begun at engraftment and stopped at 14 d after transplant. In preliminary studies, this dose of mAb resulted in serum trough levels at 48 h which exceeded the IC50 for in vitro neutralization of IP-10–mediated chemotaxis of mouse CXCR3+ spleen cells (data not shown).
Immunopathology.
Histologic evaluation or allografts was undertaken using hematoxylin and eosin (H&E)-stained paraffin sections. Infiltrating host leukocytes and chemokine expression were detected in serially harvested allografts by immunoperoxidase staining of cryostat sections using rat anti–mouse cell lineage–specific mAbs (BD PharMingen), rat anti–mouse CXCR3 mAb 5, and polyclonal anti–I-TAC and anti-Mig Abs (R&D Systems; reference 12), with quantitative analysis of labeled cells 3. Control sections were stained with isotype-matched mAbs or normal IgG.
Cellular and Molecular Studies.
Splenocytes were activated with Con A and cultured in IL-2 to generate CXCR3+ T cell blasts for use in chemotaxis studies 13. Methods for the production of cDNA probes for murine IP-10, I-TAC, and Mig, and an RNase protection assay probe for murine CXCR3; methods for RNA isolation, use of Northern blot analysis, and RNase protection assays for intragraft cytokines, chemokines, and their receptors (BD PharMingen), with normalization to glyceraldehyde 3-phosphate dehydrogenase or L32 genes, have been reported previously 3 5.
Results and Discussion
Given the importance of CXCR3 expression to the development of acute allograft rejection in the current model 5, we undertook a serial study of mRNA expression of the three known CXCR3 chemokine ligands. Serial Northern blot analyses showed that no CXCR3 chemokine ligand mRNA expression was seen in normal hearts and only IP-10 was induced in allografts or isografts within 24 h of transplantation (Fig. 1 a). Subsequent intracardiac expression of I-TAC and Mig mRNA was detected at day 3 after transplant, in conjunction with IFN-γ mRNA expression (Fig. 1 a). Immunohistologic studies of corresponding samples showed that IP-10 expression was initially localized to graft endothelial cells, whereas by day 3 (and later) IP-10 was also expressed by infiltrating leukocytes (Fig. 1 b). I-TAC was detected focally within microvascular endothelial cells and some leukocytes at day 3, whereas Mig staining was largely confined to infiltrating large inflammatory macrophages (Fig. 1 b).
Figure 1.

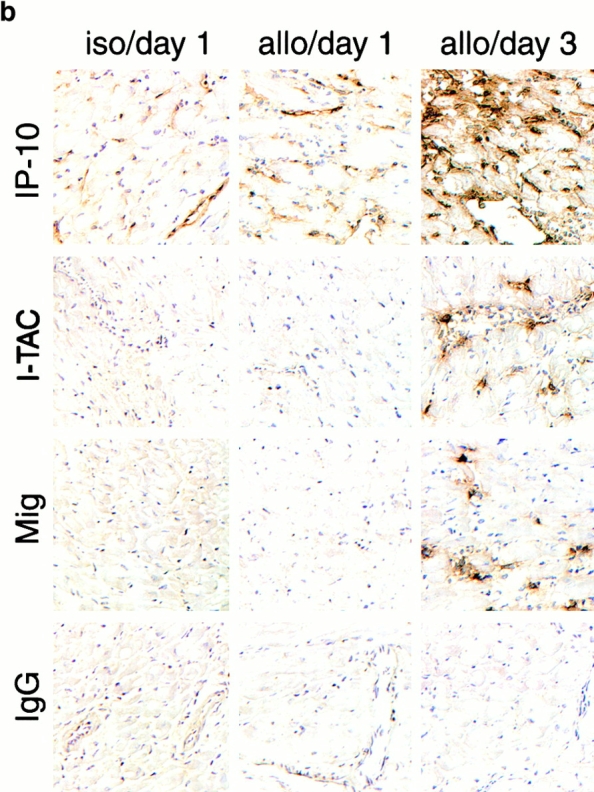
Rationale for targeting of selected CXCR3 ligands in allograft recipients. (a) Northern blot analysis of cardiac expression of the CXCR3 ligands, IP-10, I-TAC, and Mig, after isografting (H2b→H2b; 1 d) or allografting (H2d→H2b; 1, 3, and 7 d). IP-10 mRNA was induced in both isografts and allografts, whereas I-TAC and Mig mRNA were restricted to allografts and were first seen at 3 d after transplant. (b) Immunohistologic localization of CXCR3 ligands in portions of the same isografts or allografts as shown in panel a. IP-10 production in isografts and allografts was initially confined to cardiac endothelial cells, whereas by day 3, IP-10 was widely expressed by graft endothelial cells and infiltrating leukocytes; I-TAC was focally expressed by endothelial cells and leukocytes, and Mig was confined to leukocytes, especially large inflammatory macrophages. Cryostat sections, hematoxylin counterstain; original magnifications: ×300. For both Northern blot analysis and immunohistologic studies, data are representative of the results from three samples per group per time point.
Since IP-10 was the first CXCR3 ligand detected after allografting and was the sole CXCR3 ligand induced by isografting, we tested the effects of ongoing intraperitoneal administration of neutralizing mAbs to IP-10, beginning at the time of transplantation. Both anti–IP-10 mAbs tested resulted in a doubling of cardiac allograft survival time over recipients treated with control IgG (P < 0.01; Fig. 2 a). This occurred whether H2d→H2b transplantation or the reverse donor to recipient combination was used, suggesting the importance of this ligand to allograft rejection independent of the genetic background of the mouse strain and associated predisposition to develop Th1 or Th2 predominant responses. Also, we tested the effects of a complete lack of IP-10 on alloresponses using IP-10−/− mice generated by homologous recombination. To our surprise, IP-10−/− recipients rejected cardiac allografts at the same rate as IP-10+/+ recipients (Fig. 2 b). However, reasoning that anti–IP-10 mAb therapy may have prolonged allograft survival by neutralizing chemokine being produced by graft endothelial cells, we tested the survival of cardiac allografts from IP-10−/− versus IP-10−/− donors. Again surprisingly, whereas wild-type grafts were rejected normally, IP-10−/− donor hearts survived for >40 d (P < 0.001).
Figure 2.


Effects of IP-10 targeting on allograft survival. (a) Cardiac allografts across a full MHC disparity were rejected in ∼7 d, regardless of the choice of donor or recipient, but allograft survival was prolonged by use of neutralizing rat anti–mouse IP-10 mAb (*P < 0.01). Comparable prolongation was seen using a hamster anti–mouse IP-10 mAb. (b) IP-10−/− mice reject cardiac allografts at the same rate as IP-10+/+ recipients; however, the survival of allografted hearts from IP-10−/− mice is markedly prolonged compared with hearts from IP-10+/+ mice. Data from n = 6 transplants/group for each of the 10 groups shown; *P < 0.001.
To determine how the absence of a single chemokine in donor tissue might have such profound effects on allograft survival, we undertook immunohistologic studies of relevant intragraft cell populations during the initial period of engraftment. In hearts from IP-10+/+ donors, small numbers of CXCR3+ cells began to accumulate along the graft vasculature within 1 d of transplantation, were doubled in number by 3 d, and peaked at the time of rejection ∼7 d after transplant (Fig. 3). Since CXCR3 is primarily expressed by NK cells and activated T cells 14, we assessed the numbers of each of these cell types. NK cells infiltrated IP-10+/+ allografts rapidly after transplant, with a peak by day 3 and decline towards baseline thereafter, whereas CD3+ T cells were first detected in substantial numbers by day 3 and continued to increase thereafter. In contrast to the events in IP-10+/+ donor grafts, allografts from IP-10−/− donors showed significantly less infiltration by CXCR3+ leukocytes (Fig. 3), and largely lacked the early wave of infiltrating leukocytes, primarily NK cells, that was seen in control grafts. An influx of NK cells within 2–3 d of allografting has long been noted in clinical transplant recipients 15, though the mechanisms responsible have not been explored. Data from rodent skin 16 and cardiac 17 allograft models indicate that an early influx of NK or CD8+ T cells can promote allograft chemokine production as a result of IFN-γ expression, but the key role of IP-10 in the initiation of this cascade of events was not demonstrated previously.
Figure 3.

Early graft infiltration by host leukocytes is modulated in IP-10−/− donor hearts, as shown by quantitative immunohistologic analysis of intragraft CXCR3+ cells, mAb DX5+ NK cells, and CD3+ T cells present at days 0, 1, 3, and 7 after transplant. Data (mean ± SD) from counting of 20 consecutive fields/graft and 3 allografts/group. *Significantly different cell numbers in IP-10+/+ donor hearts versus IP-10−/− samples (*P < 0.05; **P < 0.01; ***P < 0.005).
Histologic and immunohistologic comparisons at day 7 after transplant were performed to assess the effects of lack of donor IP-10 on key histopathologic and immunopathologic manifestations of allograft rejection. Whereas H&E-stained sections of allografts from IP-10+/+ donors showed widespread myocardial necrosis and leukocyte infiltration, allografts from IP-10−/− were essentially normal (Fig. 4 a). IP-1+/+ donor grafts contained extensive labeling for all three CXCR3 ligands, but no staining was seen in grafts from IP-10−/− donors (Fig. 4 b). Consistent with the H&E findings, immunohistologic analysis showed that lack of IP-10 expression in donor hearts led to significantly reduced CD45+ leukocyte infiltration, involving significant reductions in CD4+ and CD8+ T cells subsets, macrophages, and intragraft immune activation, as reflected by expression of IL-2R (CD25; Fig. 4 c). At day 7, both groups showed only negligible NK cell, B cell, or neutrophil infiltration.
Figure 4.

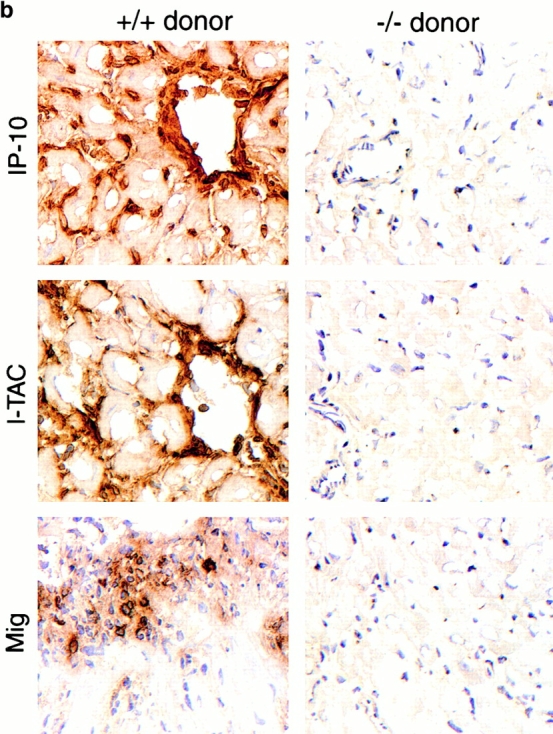

Mechanisms underlying the beneficial effects of targeting donor tissue IP-10 expression. (a) Histology showing acute rejection with extensive mononuclear cell infiltration, vascular injury, and myocyte necrosis in grafts from IP-10+/+ donors versus almost normal histology in grafts from IP-10−/− donors (day 7 after transplant). Paraffin sections, hematoxylin; original magnifications: ×300. (b) Immunohistologic detection of IP-10, I-TAC, and Mig in cardiac allografts from IP-10+/+ but not IP-10−/− donors (day 7 after transplant). Cryostat sections, hematoxylin; original magnifications: ×450. (c) Immunohistologic analysis showed significant reduction in recruitment of intragraft CD45+ cells (all leukocytes), CD4 and CD8 T cell subsets, macrophages, and IL-2R+ (CD25+) cells in allografts from IP-10−/− versus IP-10+/+ donors at day 7 after transplantation. Data (mean ± SD) from counting of 20 consecutive fields/graft and 3 grafts/group. *Significantly reduced cell numbers versus controls (*P < 0.05; **P < 0.01; ***P < 0.005).
Since allografts from IP-10+/+ but not IP-10−/− donors were rejected by 7–8 d after transplant, we used RNase protection assays to compare intragraft cytokine, chemokine, and chemokine receptor mRNA expression at day 7 (Fig. 5). Compared with IP-10+/+ donors, use of IP-10−/− hearts significantly decreased intragraft expression of IFN-γ mRNA, as well as that of multiple chemokine mRNAs (and that of their receptors), including macrophage inflammatory protein (MIP)-1α (CCR1, CCR5), MIP-1β (CCR5), and regulated upon activation normal T cell expressed and secreted (RANTES; CCR1, CCR5). IP-10−/− hearts also had significantly decreased CXCR3 mRNA expression, consistent with the immunohistologic data (Fig. 3), and decreased CCR2 mRNA. Given that mRNA levels of the main CCR2 ligand, monocyte chemotactic protein 1, showed a trend towards increased rather than decreased levels in IP-10−/− hearts, the decreased intragraft CCR2 mRNA levels likely reflect secondary effects, e.g., decreased recruitment of cells that happen to bear multiple chemokine receptors, including CCR2, and CXCR4 whose mRNA levels were also decreased.
Figure 5.

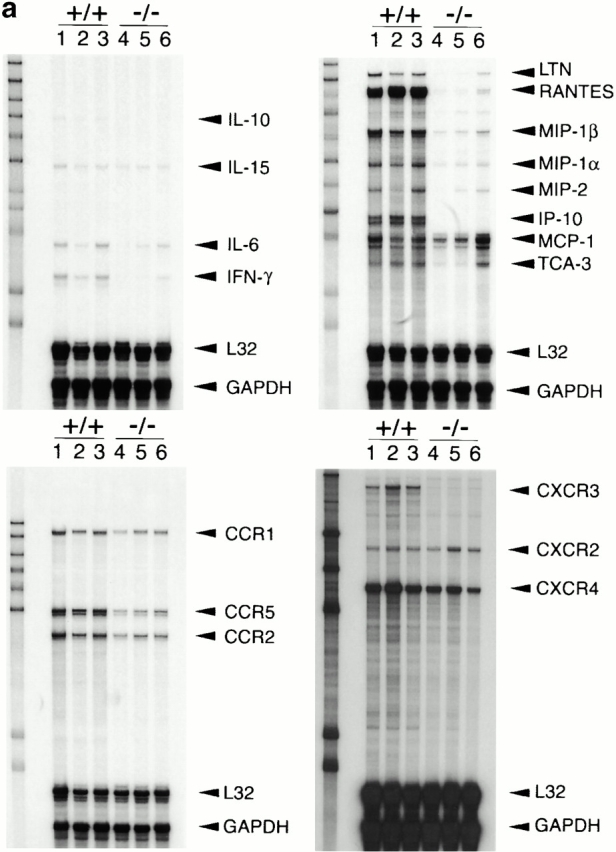
Effects of lack of donor-derived IP-10 on intragraft events. (a) RNase protection assays of intragraft cytokine, chemokine, and chemokine receptor mRNA expression, comparing grafts from IP-10+/+ (lanes 1–3) to IP-10−/− (lanes 4–6) donors at day 7 after transplant. (b) Quantitative analysis of RNase protection assay data, showing the significant depression of intragraft IFN-γ, chemokine, and CC and CXC chemokine receptor mRNA expression in IP-10−/− versus IP-10+/+ donor hearts. Data are from three transplants/group, as shown, and experiments were performed twice with comparable findings. *P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.001.
There may well be multiple mechanisms responsible for the remarkable prolongation of survival of allografts from IP-10−/− donors, in view of the decreased early posttransplant graft infiltration, absence of acute rejection, and prolonged allograft survival. Our data suggest that IP-10–dependent effects on initial NK production of IFN-γ 18, T cell activation and cytokine production 19, T cell homing and recirculation 20, and development of multiple effector cell pathways 21 may be involved, though whether any one mechanism is paramount is unknown. However, there are reports concerning the proximal role of IP-10 in other inflammatory responses, and data relevant to understanding why IP-10 appears to be selectively induced after transplantation. Both mechanical injury 22 and ischemia/reperfusion injury 23 to blood vessels induce IP-10 production, and the IP-10 promoter has a functional nuclear factor κB binding site 24, whereas I-TAC and Mig do not. Moreover, in studies to be reported separately, we have found that inhibition of nuclear factor κB activation prevents intragraft expression of IP-10 and prolongs allograft survival.
In summary, we have begun to explore the significance in rejecting allografts of three ligands for the one receptor that, based on data using gene targeting and neutralizing mAbs, appears of greatest importance in mediating leukocyte recruitment to rejecting allografts. Of the three chemokine ligands for CXCR3, only IP-10 is induced by surgical manipulation, though the relative roles of ischemia/reperfusion injury, hypoxia, and other potential factors in mediating this rapid expression by endothelial cells of both isografts and allografts are not yet known. We found that endothelial IP-10 expression promotes the initial local recruitment of NK cells and shortly thereafter, host T cells. The current findings suggest that the production of IFN-γ by these rapidly recruited CXCR3+ cells serves to amplify and extend chemokine production, leading to increasing IP-10 and induction of I-TAC and Mig production by graft endothelial cells and host leukocytes. Though the effects of targeting of I-TAC and Mig in this system remain to be determined, our data emphasize the pivotal role of donor-derived IP-10 induction in initiation and amplification of the host alloresponses which lead to acute rejection. Further understanding of this sequence of events may be of therapeutic importance in allotransplantation, as well as in the fields of tissue engineering and xenotransplantation. In the latter case, development of donors deficient in production of IP-10 may facilitate the control of host T cell responses in xenograft recipients 25.
Acknowledgments
This work was supported by grants from the National Institutes of Health to W.W. Hancock and A.D. Luster.
References
- Wieder K.J., Hancock W.W., Schmidbauer G., Corpier C.L., Wieder I., Kobzik L., Strom T.B., Kupiec-Weglinski J.W. Rapamycin treatment depresses intragraft expression of KC/MIP-2, granzyme B, and IFN-γ in rat recipients of cardiac allografts. J. Immunol. 1993;151:1158–1166. [PubMed] [Google Scholar]
- Hancock W.W., Gao W., Faia K.L., Csizmadia V. Chemokines and their receptors in allograft rejection. Curr. Opin. Immunol. 2000;12:511–516. doi: 10.1016/s0952-7915(00)00130-8. [DOI] [PubMed] [Google Scholar]
- Gao W., Topham P.S., King J.A., Smiley S.T., Csizmadia V., Lu B., Gerard C.J., Hancock W.W. Targeting of the chemokine receptor CCR1 suppresses development of acute and chronic cardiac allograft rejection. J. Clin. Invest. 2000;105:35–44. doi: 10.1172/JCI8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loetscher M., Gerber B., Loetscher P., Jones S.A., Piali L., Clark-Lewis I., Baggiolini M., Moser B. Chemokine receptor specific for IP-10 and Migstructure, function, and expression in activated T-lymphocytes. J. Exp. Med. 1996;184:963–969. doi: 10.1084/jem.184.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock W., Lu B., Gao W., Csizmadia V., Faia K., King J., Smiley S., Ling M., Gerard N., Gerard C.J. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J. Exp. Med. 2000;192:1515–1519. doi: 10.1084/jem.192.10.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy P.M., Baggiolini M., Charo I.F., Hebert C.A., Horuk R., Matsushima K., Miller L.H., Oppenheim J.J., Power C.A. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol. Rev. 2000;52:145–176. [PubMed] [Google Scholar]
- Luster A.D., Unkeless J.C., Ravetch J.V. Gamma-interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nature. 1985;315:672–676. doi: 10.1038/315672a0. [DOI] [PubMed] [Google Scholar]
- Cole K.E., Strick C.A., Paradis T.J., Ogborne K.T., Loetscher M., Gladue R.P., Lin W., Boyd J.G., Moser B., Wood D.E. Interferon-inducible T cell α chemoattractant (I-TAC)a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J. Exp. Med. 1998;187:2009–2021. doi: 10.1084/jem.187.12.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber J.M. A macrophage mRNA selectively induced by γ-interferon encodes a member of the platelet factor 4 family of cytokines. Proc. Natl. Acad. Sci. USA. 1990;87:5238–5242. doi: 10.1073/pnas.87.14.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock W.W., Sayegh M.H., Zheng X.G., Peach R., Linsley P.S., Turka L.A. Costimulatory function and expression of CD40 ligand, CD80, and CD86 in vascularized murine cardiac allograft rejection. Proc. Natl. Acad. Sci. USA. 1996;93:13967–13972. doi: 10.1073/pnas.93.24.13967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan I.A., MacLean J.A., Lee F.S., Casciotti L., DeHaan E., Schwartzman J.D., Luster A.D. IP-10 is critical for effector T cell trafficking and host survival in Toxoplasma gondii infection. Immunity. 2000;12:483–494. doi: 10.1016/s1074-7613(00)80200-9. [DOI] [PubMed] [Google Scholar]
- Hancock W.W., Buelow R., Sayegh M.H., Turka L.A. Antibody-induced transplant arteriosclerosis is prevented by graft expression of anti-oxidant and anti-apoptotic genes. Nat. Med. 1998;4:1392–1396. doi: 10.1038/3982. [DOI] [PubMed] [Google Scholar]
- Topham P.S., Csizmadia V., Soler D., Hines D., Gerard C.J., Salant D.J., Hancock W.W. Lack of chemokine receptor CCR1 enhances Th1 responses and glomerular injury during nephrotoxic nephritis. J. Clin. Invest. 1999;104:1549–1557. doi: 10.1172/JCI7707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S.X., Rottman J.B., Myers P., Kassam N., Weinblatt M., Loetscher M., Koch A.E., Moser B., Mackay C.R. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J. Clin. Invest. 1998;101:746–754. doi: 10.1172/JCI1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock W.W., Gee D., De Moerloose P., Rickles F.R., Ewan V.A., Atkins R.C. Immunohistological analysis of serial biopsies taken during human renal allograft rejection. Changing profile of infiltrating cells and activation of the coagulation system. Transplantation. 1985;39:430–438. doi: 10.1097/00007890-198504000-00018. [DOI] [PubMed] [Google Scholar]
- Kondo T., Morita K., Watarai Y., Auerbach M.B., Taub D.D., Novick A.C., Toma H., Fairchild R.L. Early increased chemokine expression and production in murine allogeneic skin grafts is mediated by natural killer cells. Transplantation. 2000;69:969–977. doi: 10.1097/00007890-200003150-00051. [DOI] [PubMed] [Google Scholar]
- Kapoor A., Morita K., Engeman T.M., Koga S., Vapnek E.M., Hobart M.G., Fairchild R.L. Early expression of interferon-γ inducible protein 10 and monokine induced by interferon-γ in cardiac allografts is mediated by CD8+ T cells. Transplantation. 2000;69:1147–1155. doi: 10.1097/00007890-200003270-00020. [DOI] [PubMed] [Google Scholar]
- Inngjerdingen M., Damaj B., Maghazachi A.A. Expression and regulation of chemokine receptors in human natural killer cells. Blood. 2001;97:367–375. doi: 10.1182/blood.v97.2.367. [DOI] [PubMed] [Google Scholar]
- Bonecchi R., Bianchi G., Bordignon P.P., Dambrosio D., Lang R., Borsatti A., Sozzani S., Allavena P., Gray P.A., Mantovani A., Sinigaglia F. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J. Exp. Med. 1998;187:129–134. doi: 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalor P.F., Adams D.H. Lymphocyte homing to allografts. Transplantation. 2000;70:1131–1139. doi: 10.1097/00007890-200010270-00001. [DOI] [PubMed] [Google Scholar]
- Arakelov A., Lakkis F.G. The alloimmune response and effector mechanisms of allograft rejection. Semin. Nephrol. 2000;20:95–102. [PubMed] [Google Scholar]
- Wang X., Yue T.L., Ohlstein E.H., Sung C.P., Feuerstein G.Z. Interferon-inducible protein-10 involves vascular smooth muscle cell migration, proliferation, and inflammatory response. J. Biol. Chem. 1996;271:24286–24293. doi: 10.1074/jbc.271.39.24286. [DOI] [PubMed] [Google Scholar]
- Wang X., Ellison J.A., Siren A.L., Lysko P.G., Yue T.L., Barone F.C., Shatzman A., Feuerstein G.Z. Prolonged expression of interferon-inducible protein-10 in ischemic cortex after permanent occlusion of the middle cerebral artery in rat. J. Neurochem. 1998;71:1194–1204. doi: 10.1046/j.1471-4159.1998.71031194.x. [DOI] [PubMed] [Google Scholar]
- Ohmori Y., Hamilton T.A. Cooperative interaction between interferon (IFN) stimulus response element and κ-B sequence motifs controls IFN-γ-stimulated and lipopolysaccharide-stimulated transcription from the murine IP-10 promoter. J. Biol. Chem. 1993;268:6677–6688. [PubMed] [Google Scholar]
- Hancock W.W. The past, present, and future of renal xenotransplantation. Kidney Int. 1997;51:932–944. doi: 10.1038/ki.1997.132. [DOI] [PubMed] [Google Scholar]