

Abstract
The pre-T cell receptor (TCR), which consists of a TCR-β chain paired with pre–TCR-α (pTα) and associated with CD3/ζ components, is a critical regulator of T cell development. For unknown reasons, extremely low pre-TCR levels reach the plasma membrane of pre-T cells. By transfecting chimeric TCR-α–pTα proteins into pre-T and mature T cell lines, we show here that the low surface expression of the human pre-TCR is pTα chain dependent. Particularly, the cytoplasmic domain of pTα is sufficient to reduce surface expression of a conventional TCR-α/β to pre-TCR expression levels. Such reduced expression cannot be attributed to qualitative differences in the biochemical composition of the CD3/ζ modules associated with pre-TCR and TCR surface complexes. Rather, evidence is provided that the pTα cytoplasmic tail also causes a reduced surface expression of individual membrane molecules such as CD25 and CD4, which are shown to be retained in the endoplasmic reticulum (ER). Native pTα is also observed to be predominantly ER localized. Finally, sequential truncations along the pTα cytoplasmic domain revealed that removal of the COOH-terminal 48 residues is sufficient to release a CD4-pTα chimera from ER retention, and to restore native CD4 surface expression levels. As such a truncation in pTα also correlates with enhanced pre-TCR expression, the observed pTα ER retention function may contribute to the regulation of surface pre-TCR expression on pre-T cells.
Keywords: human pre–T cell receptor, pTα cytoplasmic tail, surface expression, endoplasmic reticulum retention, CD3 complex
Introduction
Development of mature α/β T cells inside the thymus is controlled at two distinct check-points by the sequential expression of the pre-TCR and the mature TCR-α/β 1 2 3. Early in T cell development, pre-T cells that succeed in productive TCR-β gene rearrangements express a functional TCR-β chain which pairs with the invariant pre–TCR-α (pTα) chain and associates with CD3 components to form the pre-TCR 4 5 6 7 8. Signaling through this pre-TCR complex triggers a process, known as TCR-β selection, which induces the cellular expansion and maturation of CD4− CD8− double negative (DN) pre-T cells into CD4+CD8+ double-positive (DP) thymocytes 2 3 9 10 11, and results in the induction of a high rate of TCR-α gene rearrangements 12. On productive TCR-α gene rearrangements and substitution of pTα by TCR-α, the TCR-α/β is expressed associated with CD3, and DP thymocytes can undergo a second step of selection, known as TCR-α/β selection, during which thymocytes are rescued from programmed cell death and induced to differentiate into conventional single positive (SP) thymocytes, upon binding to self-peptide–MHC complexes expressed on thymic stromal cells 3.
In contrast to TCR-α/β selection, current data support the view that TCR-β selection is independent of binding to an extracellular (EC) ligand 13 14 and no evidence for the existence of such a ligand has so far been provided. However, exit from the endoplasmic reticulum (ER) and expression at the cell surface is mandatory for the pre-TCR complex to exert its function 15, although rules controlling the assembly and intracellular transport of pre-TCR and TCR complexes may differ markedly, as the pre-TCR is expressed only transiently during thymocyte development, and at extremely low levels, ∼50–100-fold lower than those of the TCR-α/β on mature T cells 1 13 16. The inefficient expression of pre-TCR complexes on pre-T cells is reminiscent of the inefficient expression of the pre–B cell receptor (BCR) on pre-B cells 1 17, and might be related to the particular signaling function of pre-TCR/BCR complexes during lymphoid development. The question is, therefore, whether such differences in surface expression levels are determined by specific structural properties of the receptors or are intrinsic to the particular developmental stage at which the receptors are expressed.
Although the precise biochemical composition of the pre-TCR has remained elusive until recently, evidence has now been provided that, in the mouse, it has the same subunit composition as does the TCR-α/β complex, differing only in that the TCR-α subunit has been replaced by pTα and that ζ association has been significantly weakened relative to its association with the mature TCR-α/β 4 7 8 18. Because ζ is required for efficient surface expression of the other subunits of the mature TCR-α/β including the CD3 molecules 19 20 21 22 23 24, its weak biochemical association with the pre-TCR could well result in a decreased stability of the receptor complex at the cell surface. An alternative possibility is that, as reported for the pre-BCR, surface pre-TCR expression is controlled by a retention mechanism that is not selective for the pre-TCR, but is inherent to the pre-T cell stage 1 17. In this study, we have examined this issue by performing transfections of human chimeric TCR-α–pTα molecules comprising distinct domains of TCR-α and pTα into either pre-T or mature T cell lines, and provide evidence that low surface expression of the human pre-TCR is intrinsic to the pTα chain. Particularly, our results show that the cytoplasmic (Cyto) domain of pTα is sufficient to promote retention in the ER and to reduce surface expression levels of individual transmembrane (TM) proteins. The possibility that the pTα Cyto tail functions as an ER retention signal that contributes to the regulation of pre-TCR expression levels on pre-T cells is discussed.
Materials and Methods
Isolation of Thymocyte Subsets.
Postnatal thymocytes isolated from thymus samples removed during corrective cardiac surgery of patients aged 1 mo to 3 yr were fractionated by centrifugation on Percoll (Amersham Pharmacia Biotech) density gradients as described 25. pre-TCR/CD3low pre-T cells were isolated from the large-sized cell fraction by immunomagnetic sorting (Dynal) as described 16.
Flow Cytometry.
PE-labeled anti-CD3 (Leu-4-PE), biotin-conjugated anti-CD25 (7D4), and Cy5-PE-labeled anti-CD4 mAbs were obtained from Becton Dickinson, BD PharMingen, and Caltag Laboratories, respectively. The BMA031 mAb, raised against a monomorphic determinant of TCR-α/β 26 was provided by Dr. R. Kurrle (Behringwerke AG, Marburg, Germany), and the 6D6 mAb, against the human Vα12.1 TCR-α domain 27, was the gift of Dr. M. Brenner (Brigham and Women's Hospital, Boston, MA). Surface expression of the human pTα chain was determined by staining with a rabbit antiserum (ED-1) raised against a synthetic peptide present in the pTα EC domain 16. PE-labeled streptavidin and goat anti–mouse fluorescein-, PE-, or PE-Cy5–coupled F(ab′)2 Igs were purchased from Caltag Laboratories. Stained cells were analyzed in a flow cytometer (EPICS XL; Coulter Corp.) as described 25. For flow cytometry, COS cells were removed from culture 48 h after transfection with PBS containing 0.02% EDTA.
Generation of cDNA Constructs.
Full-length cDNAs encoding the human pTα and the AV12S1 TCR α chain, respectively, were cloned into the BamHI site of the pcDNA3 plasmid vector (Invitrogen), as described previously 16. TCR-α–pTα chimeric constructs (see Fig. 2 A) comprising the Vα12.1 and Jα domains of the AV12S1 TCR α chain fused to pTα cDNA lacking the leader sequence (αI), or encoding a AV12S1 TCR α chain in which either both the TM and Cyto domains (αII) or exclusively the Cyto domain (αIII) of TCR-α have been replaced with homologous domains from pTα, were generated by PCR using TCR-α and pTα cDNAs as templates, and specific oligonucleotides. The sense 5′-ATG GAT CCT CTA GAT GAT TTT TGC CAG CCT GTT G-3′ primer (with a BamHI site), corresponding to the NH2-terminal region of the AV12S1 TCR α chain 16, was used in combination with three distinct antisense primers: 5′-CCC GGA TCC TGG TGC CTG TTC CTG TTC-3′ (with a BamHI site), 5′-CGG AAT TCG GTT TTG AAA GTT TAG GTT CG-3′ (with an EcoRI site), or 5′-CGG AAT TCC CAG CGT CAT GAG CAG ATT AAA-3′ (with an EcoRI site), which correspond to the COOH-terminal region of either the Jα, the EC, or the TM domains of the AV12S1 TCR α chain, respectively. PCR products were either BamHI or BamHI/EcoRI digested, and ligated into pcDNA3 plasmid containing, respectively, a BamHI/XhoI PCR fragment coding for pTα lacking the leader sequence, an EcoRI/XhoI PCR fragment encoding both the TM and Cyto pTα domains, or an EcoRI/XhoI PCR fragment encoding the Cyto tail of pTα. Such pTα PCR products were amplified from pTα cDNA with the antisense 5′-CCG CTA ACG AGT CAG GCA GCA GCT CCA GCC TGC AG-3′ primer, corresponding to the COOH-terminal region of the pTα Cyto domain, and containing an XhoI site, in combination with the sense primers 5′-CCC-GGA TCC ATA TGC TAC CCA CAG GTG TGG GC-3′, including a BamHI site, or 5′-CGG AAT TCT GTG GCT GGG GGT CCT GCG-3′, with an EcoRI site, or 5′-CGG AAT TCT TAC CTG CAG CTG CCT GTG CG-3′, including an EcoRI site, respectively.
Figure 2.
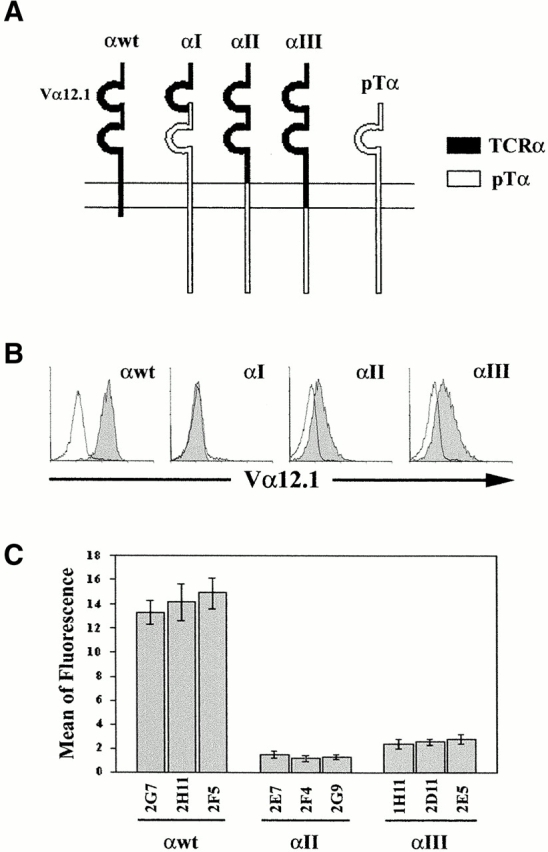
The Cyto tail of pTα is sufficient to reduce levels of surface expression of a conventional TCR α chain to pTα expression levels. (A) Schematic representation of the three chimeric TCR-α–pTα chains (αI, αII, and αIII), and of the wild-type TCR-α (αwt) and pTα (pTα) chains. (B) Stable transfectants derived from SUP-T1 human pre-T cells were selected in the presence of G-418, and analyzed by flow cytometry for surface expression levels of the αI, αII, or αIII TCR-α–pTα chimeric chains or the wild-type TCR α chain with an anti-Vα12.1 mAb plus a PE-labeled goat anti–mouse IgG1 (shaded histograms). Background staining (unshaded histograms) was determined with a nonreactive mouse IgG1 mAb plus PE-coupled goat anti–mouse IgG1. Results are representative of at least 10 different stable transfectants. (C) Mean fluorescence values ± SD of surface expression of TCR-α (αwt) and αII and αIII chimeric chains on three distinct stable transfectants of each type. Results are representative of 60–70 distinct transfectants of each type and summarize the data from six experiments.
Full-length cDNAs encoding murine CD25 (provided by Dr. L.R. Borlado, Centro Nacional de Biotecnología, Madrid, Spain) or human CD4 28 were subcloned into the BamHI site of pcDNA3. CD25-pTα and CD4-pTα chimeric constructs, comprising the complete EC and TM domains of CD25 or CD4 fused to the pTα Cyto tail, were generated by PCR amplification. CD25 PCR products obtained with the sense 5′-ATG GAT CCA AGA TGG AGC CAC GTC TGC TG-3′ primer and the antisense 5′-GCG AAT TCC CCA GGT GAG CCC GCT CAG G-3′ primer were digested with BamHI/EcoRI, and CD4 PCR products obtained with the sense 5′-AGA GAG AGA GAG AAG CTT TCG GCA AGG CCA CAA TGA AC-3′ primer and the antisense 5′-GCG AAT TCC GAA GAA GAT GCC TAG CCC AAT G-3′ primer were digested with HindIII/EcoRI. Digested PCR products were independently ligated into pcDNA3 containing the EcoRI/XhoI PCR fragment encoding the pTα Cyto tail described above. Truncated CD4-pTα chimeras lacking either the COOH-terminal 22 (CD4-pTαt22) or 48 (CD4-pTαt48) residues of the pTα Cyto tail were generated essentially as described for CD4-pTα chimeric constructs, except that pTα PCR was performed with the sense primer corresponding to the NH2-terminal region of the pTα Cyto domain described above, in combination with the antisense 5′-AGA GAG AGA GAG GAT ATC TCA AGC CCT GAG GCG AGA TCT TG-3′ or 5′-AGA GAG AGA GAG GAT ATC TCA TAC TGG GCT CCC GGG CTT C-3′ primers, for CD4-pTαt22 and CD4-pTαt48, respectively. EcoRI/EcoRV-digested PCR products were then ligated into pcDNA3 containing the HindIII/EcoRI-digested CD4 product described above. These antisense primers were independently used with a previously described sense primer 16 corresponding to the NH2-terminal pTα region to generate truncated pTα proteins with identical deletions in the Cyto tail (pTαt22 and pTαt48). EcoRI/EcoRV-digested products were independently ligated into pcDNA3. Both cDNAs as well as full-length pTα and TCR-α cDNAs 16 were subsequently subcloned into the bicistronic expression vector pCIGFP. To construct the pCIGFP plasmid, a NotI cassette containing an internal ribosomal entry site (IRES) sequence followed by the enhanced green fluorescent protein (EGFP) cDNA was transferred from the pLZRS retroviral vector 29, provided by Dr. H. Spits (Netherlands Cancer Institute, Amsterdam, The Netherlands), into the NotI site in the pcDNA3 vector. Tailless pTα cDNA was generated by amplification with the sense primer described previously 16 in combination with the 5′-GGG GGA TCC TAC AGG AGC AGG TCA AAC AG-3′ antisense primer, BamHI digested, and ligated into the pCIGFP bicistronic plasmid. Each construct was sequenced directly in pcDNA3. The pTα-EGFP construct has been described previously 16.
Cell Lines and Transfection.
The TCR-α–deficient JR3.11 mutant, derived from the mature human T cell line Jurkat (30; provided by Dr. B. Rubin, Centre National de la Recherche Scientifique, Toulouse, France), the human pre-T cell line SUP-T1 31, and the murine lymphoid cell line BW, were grown in RPMI 1640 (Biowhittaker) supplemented with 10% FCS (GIBCO BRL). COS cells were grown in DMEM (Biowhittaker) supplemented with 5% FCS (GIBCO BRL). Transfections were carried out by electroporation as described 16.
Generation of Polyclonal Anti–Human pTα Abs.
A rabbit antiserum (CT-1) was generated against a synthetic peptide corresponding to the human pTα sequence 200–212 present in the Cyto domain 25, essentially as described previously for the ED-1 antiserum 16. The specificity of the CT-1 antiserum was assayed by immunofluorescence microscopy of COS cells transfected with a pTα cDNA tagged with a c-myc epitope recognized by the specific 9E10 mAb (unpublished results).
Cell Surface Radioiodination, Immunoprecipitation, and N-Glycosidase F Digestion.
Cells (107) were washed with PBS, resuspended in 0.5 ml of PBS containing 0.5 mg/ml of sulfo-succinimidyl-3-(4-hydroxyphenyl) propionate (SHPP) (Boltont-Hunter reagent; Pierce Chemical Co.), and incubated on ice for 30 min. The reaction was stopped by diluting the cells with 10 ml of 10 mM l-lysine (Sigma-Aldrich) in PBS. Cells were centrifuged, resuspended in 150 μl of PBS, and 125I-labeled by the lactoperoxidase method. In brief, 1 mCi Na125I (Amersham Pharmacia Biotech) and 30 μl of 140 IU/ml lactoperoxidase (Sigma-Aldrich) solution were added to the cells, and 10 μl aliquots of 0.06% H2O2 solution were then added three times, at 7-min intervals. The reaction was stopped by adding 20 mM KI (Sigma-Aldrich) and 1 mM l-tyrosine (Sigma-Aldrich) in PBS. Subsequently, the cells were lysed in 1% Brij 96–containing (Sigma-Aldrich) lysis buffer (10 mM Tris-HCl, pH 7.6, 150 mM NaCl, 10 mM iodoacetamide, 1 mM PMSF, and 1 μg/ml each of leupeptin, pepstatin, and aprotinin) and centrifuged for 14,000 rpm for 30 min at 4°C. The supernatants were precleared three times with normal mouse serum (NMS) Igs coupled to protein A/G-Sepharose beads (Amersham Pharmacia Biotech) and subjected to immunoprecipitation with the indicated Abs coupled to protein A/G-Sepharose beads. The following Abs were used: the 6D6 anti–human TCR Vα12.1 mAb 27; the UCHT1 anti–human CD3ε mAb 32; the 448 anti–human TCR-ζ rabbit antiserum, generously provided by Dr. B. Alarcón (Centro de Biología Molecular Severo Ochoa, Madrid, Spain); and the CT-1, anti–human pTα rabbit antiserum, generated in this study. The immunoprecipitates were washed five times with 1% Brij 96–containing lysis buffer and, when indicated, digested overnight with N-glycosidase F (N-Gly; 0.2 U/sample; Roche Diagnostics). For analysis, the immunoprecipitates were separated by SDS-12% PAGE under nonreducing conditions or, alternatively, by two-dimensional gels using SDS-10% polyacrilamide tube gels in the first dimension (nonreduced), which were subsequently resolved by SDS-12% PAGE in the second dimension (reduced).
Immunofluorescence and Confocal Microscopy.
48 h after transfection, COS cells were fixed with 2% paraformaldehyde, permeabilized with 0.1% saponin in PBS containing 1% BSA, and stained either with the PC61 fluorescein-labeled anti–mouse CD25 mAb (BD PharMingen), or with the HP2.6 anti–human CD4 mAb (provided by Dr. F. Sánchez-Madrid, Hospital de la Princesa, Madrid, Spain), followed by goat anti–mouse fluorescein-coupled F(ab′)2 Igs (Caltag Laboratories). The coverslips were viewed using a Radiance 2000 confocal microscope (Bio-Rad Laboratories).
For confocal analysis, pTα-GFP SUP-T1 stable transfectants were adhered to Poly L-Lys (Sigma-Aldrich) precoated coverslips (5 × 105 cells/coverslip). Coverslips were then washed in PBS, fixed with 2% paraformaldehyde in PBS for 10 min, permeabilized for 5 min with 0.05% Triton X-100 (Sigma-Aldrich), and blocked with 2% BSA/PBS. Cells were then consecutively stained with the anti–TCR-β βF1 mAb (provided by Dr. M. Brenner) in combination with Cy5-conjugated goat anti–mouse Igs (Jackson ImmunoResearch Laboratories), and with a rabbit antiserum against the ER resident protein PDI (protein disulfide isomerase; StressGen Biotechnologies) plus Cy3-conjugated goat anti–rabbit Igs (Jackson ImmunoResearch Laboratories). Confocal microscopy was performed on a Radiance 2000 (Bio-Rad Laboratories) system coupled to an Axiovert S100TV inverted microscope (ZEISS). Fluorescein and EGFP, Cy3, and Cy5 fluorescence were detected using bandpass filter HQ515/30, longpass filter HQ600/50, and longpass filter HQ660LP, respectively.
Metabolic Labeling, Immunoprecipitation, and Endoglycosidase H Treatment.
48 h after transfection, COS cells were rinsed in PBS twice and incubated for 30 min in DMEM without l-cysteine and l-methionine. Then, 500 μCi [35S]methionine/cysteine (Amersham Pharmacia Biotech) were added, and cells were pulsed for 30 min followed by chase periods, in the presence of DMEM supplemented with 10% FCS. Finally, the cells were washed in PBS, lysed in 1% Triton X-100 (Sigma-Aldrich) lysis buffer, and immunoprecipitated either with the PC61 anti–mouse CD25 mAb (BD PharMingen), with the CT-1 rabbit antiserum against the human pTα tail, or with the HP2.6 anti–human CD4 mAb. The immunoprecipitates were digested overnight with endoglycosidase H (endo-H; Roche) or left undigested, and resolved by SDS-12% PAGE under reducing conditions. Signal intensity was quantitated by densitometry (Bio-imaging BAS 1500; Fujifilm).
Results
Low Surface Pre-TCR Expression Is pTα Chain Dependent.
As shown by flow cytometry using an anti-pTα antiserum and an anti-CD3 mAb 16, the human pre-TCR is expressed on the surface of primary pre-T cells at very low levels, compared with expression levels of the mature TCR-α/β on SP thymocytes (Fig. 1 A) or peripheral T cells (not shown). Low pre-TCR expression such as that found on primary pre-T cells was detected also on a pre-T cell line, SUP-T1 (Fig. 1 B), which expresses TCR-β and pTα chains but lacks TCR-α 31. It is thus possible that, as proposed for the pre-BCR 1 17, the pre-TCR is expressed at low surface levels due to regulatory mechanisms that operate selectively at early developmental stages. In that case, one would expect that only limited amounts of a conventional mature TCR-α/β could reach the plasma membrane of pre-T cells. In contrast, we found that the introduction of a conventional TCR α chain (Vα12.1) into SUP-T1 pre-T cells (αwt) resulted in a reproducible increase (∼15-fold) in surface expression levels of CD3, presumably through the formation of heterodimers composed of the transfected TCR-α paired with the endogenous TCR-β and associated with CD3. Accordingly, CD3 was found coexpressed in stoichiometric amounts with the TCR-α/β on stable SUP-T1 αwt transfectants (Fig. 1 B).
Figure 1.

Flow cytometry analysis of human thymocytes and SUP-T1 transfectants. (A) Primary human pre-T cells, isolated ex vivo as described in Materials and Methods, were analyzed by flow cytometry for surface expression of pTα and CD3 using a rabbit anti–human pTα Ab plus a fluorescein-conjugated second reagent and a PE-labeled anti-CD3 mAb. Phenotypic analysis of SP thymocytes was performed on electronically gated CD3bright large-sized thymocytes stained for surface expression of a mature CD3-associated TCR-α/β with an anti–TCR-α/β mAb plus a fluorescein-labeled second reagent and a PE-labeled anti-CD3 mAb. (B) SUP-T1 pre-T lineage cells were mock transfected (SUP-T1) or transfected either with a human TCR-α (Vα12.1) cDNA (αwt) or with a pTα-GFP cDNA (pTα-GFP). Stable transfectants were analyzed by flow cytometry for the coexpression of pTα and CD3 or TCR-α/β and CD3 as described above. (C) Relative levels of surface CD3 expressed on mock-transfected SUP-T1 (shaded histogram) are compared with those expressed on TCR-α (αwt) transfectants (thin line) or pTα–GFP (bold line) transfectants. Background values (dotted line) were obtained with an isotype-matched irrelevant PE-coupled mAb.
It remains possible that one component of the low level pre-TCR expression could be limiting amounts of the endogenous pTα chain, whereas increased TCR-α/β surface density on αwt transfectants could be the result of TCR-α overexpression. To rule out that possibility, a pTα–GFP chimeric construct 16 was introduced into SUP-T1 cells, and surface expression of CD3 was analyzed by flow cytometry on stable pTα–GFP transfectants traced by their GFP expression. As shown in Fig. 1 C, GFP+ cells displayed surface CD3 levels that were indistinguishable from those observed on nontransfected SUP-T1 cells, but still 15-fold lower than CD3 levels on αwt transfectants. Moreover, surface CD3 was coexpressed with the pTα–GFP chimeric protein (Fig. 1 B) and with the endogenous TCR-β chain 16 in stoichiometric amounts, indicating that pTα overexpression does not alleviate endogenous pre-TCR components from the regulatory mechanisms that control their limited expression on the surface of pre-T cells. We thus concluded that pTα amounts are not rate limiting for the assembly and expression of the pre-TCR on the surface of SUP-T1 pre-T cells.
Reciprocal experiments aimed at analyzing surface pre-TCR expression levels on mature T cells were then performed by introduction of pTα or TCR-α into a TCR-α–deficient mutant (JR3.11) derived from the mature T cell line Jurkat 30. As shown in Table , low pre-TCR and high TCR-α/β surface expression levels, identical to those observed on SUP-T1 pre-T cells, were consistently detected on JR3.11 T cells. These data indicate that differences in surface expression levels of the pre-TCR and the TCR are not intrinsic to the particular cell type in which these receptors are expressed, but may be due to the presence of a pTα chain in the former and of a TCR α chain in the latter.
Table 1.
Relative Levels of Surface pre-TCR, Mature TCR-α/β, and Chimeric TCR Expression on Stable Transfectants Derived from Human Pre-T Cell and Mature T Cell Lines
| SUP-T1 (mean of FL ± SD) | JR3.11 (mean of FL ± SD) | |
|---|---|---|
| pTα | 1.2 ± 0.5 | 1.5 ± 0.7 |
| αwt | 14.1 ± 2.0 | 13.3 ± 2.0 |
| αII | 1.3 ± 0.4 | 1.4 ± 0.3 |
| αIII | 2.2 ± 0.5 | 2.4 ± 0.8 |
TCR-α–deficient pre-T cell (SUP-T1) and mature T cell (JR3.11) lines were stably transfected either with a wild-type TCR-α construct (αwt) or with the TCR-α–pTα αII or αIII chimeric constructs described in Materials and Methods. SUP-T1 and JR3.11 transfectants were analyzed by flow cytometry for the surface expression levels of the conventional TCR-α/β and those of the chimeric TCR by staining with either a mAb against the Vα12.1 domain shared by the TCR-α and TCR-α–pTα chains, or with an anti-CD3 mAb, respectively. Levels of surface expression of endogenous pre-TCR on SUP-T1 pre-T cells were compared to pre-TCR levels on pTα transfectants derived from JR3.11 mature T cells by staining with a rabbit anti–human pTα Ab (reference 16) and flow cytometry. Data are presented as the mean of fluorescence intensity (FL) ± SD of four to six individual transfectants.
The Cyto Tail of pTα Is Sufficient to Reduce Surface Expression of a Conventional TCR α Chain to pTα Expression Levels.
To uncover the structural properties of the pTα molecule that might account for the low surface expression of the pre-TCR, TCR-α–pTα chimeric chains were generated as illustrated in Fig. 2 A, by replacing the Cyto, TM, and EC Ig-like constant (C) domains of a TCR α chain bearing a Vα12.1 domain with homologous domains from the pTα chain. The chimeric constructs were introduced into SUP-T1 pre-T cells, and the resulting stable transfectants were assayed by flow cytometry for surface expression of the chimeric TCRs using a mAb against their shared Vα12.1 domain. Cells stably transfected with the wild-type TCR α chain (αwt) were analyzed in parallel to control surface expression levels of conventional TCR-α/βs. After screening of up to 70 stable transfectants of each type, no surface expression of the αI chimera consisting of a pTα chain lacking the peptide leader fused to the Vα12.1 and Jα domains of TCR-α was found (Fig. 2 B). However, the αI chimeric protein could be detected intracellularly in COS transfectants (data not shown), thus confirming that the TCR-α modification had not adversely affected protein expression. Therefore, it remains possible that the αI chimera does not associate properly with TCR-β, preventing its surface expression and/or detection. Surprisingly, TCR-α–pTα chimeras in which both the TM and Cyto domains (αII) or exclusively the Cyto domain (αIII) of TCR-α were replaced with the equivalent pTα domains were found expressed at the cell surface, although at levels that were reproducibly 10–15-fold lower than those of αwt (Fig. 2 C), but comparable to those of endogenous pTα or transfected pTα–GFP chimeric chains (Fig. 1 C). Importantly, when the αwt and the αII and αIII chimeric chains were introduced into the TCR-α–deficient JR3.11 mature T cell line, their relative surface expression levels were identical to those expressed on SUP-T1 pre-T cells (Table ). Taken together, these data provide evidence that replacing the Cyto domain of TCR-α with the pTα Cyto tail is sufficient to reduce TCR α chain surface expression (and, hence, expression of the chimeric TCR, see below) to conventional pTα chain expression levels, whereas the TM domains of the TCR-α and pTα chains were equivalent for surface expression of the TCR-α–pTα chimera.
CD3/ζ Components Are Physically Associated in the Wild-type Pre-TCR as well as in the TCR-α–pTα Chimeric TCRs.
Three-color flow cytometry showed that the wild-type TCR α chain and the αII and αIII (αI was not studied further) chimeric proteins were expressed at the cell surface in stoichiometric amounts with TCR-β (Vβ1.1) and CD3 (data not shown), indicating that surface expression of αwt, αII, and αIII chains must be achieved by pairing them with the endogenous TCR-β chain and CD3/ζ components. Therefore, the cyto tail of pTα appears sufficient to reduce surface expression of the chimeric TCR complex to pre-TCR expression levels. To assess whether this effect could be attributed to defective TCR–CD3/ζ interactions, we directly compared the biochemical composition of chimeric TCRs with that of conventional TCR-α/βs, and pre-TCR complexes containing a wild-type pTα chain. To this end, the αwt, αII, and αIII stable transfectants were surface iodinated, treated with 1% Brij 96–containing lysis buffer, and surface TCRs were immunoprecipitated from cell lysates with a mAb specific for their shared Vα12.1 domain. The untransfected SUP-T1 pre-T cell line was simultaneously labeled and the endogenous pre-TCR complex was immunoprecipitated from cell lysates with a rabbit antisera (CT-1) raised against a synthetic peptide contained in the Cyto region of the human pTα molecule. Specificity of the affinity-purified Ab was confirmed previously by immunofluorescence microscopy of COS cells transfected with a c-myc–tagged pTα chain (data not shown). Subsequently, the immunoprecipitates were mock treated or treated with N-Gly, in order to distinguish between CD3ε and CD3δ chains, and resolved by SDS-PAGE under nonreducing conditions. As shown in Fig. 3, a conventional TCR-α/β heterodimer of 90 kD was coimmunoprecipitated with CD3γδε from the surface of αwt transfectants, whereas fewer CD3γδε-associated chimeric TCRs of an apparent MW of 115 kD were immunoprecipitated from both αII and αIII transfectants. N-Gly treatment of the immunoprecipitated proteins resulted in an increase in their electrophoretic mobility by elimination of their glycosidic component, and revealed no qualitative biochemical differences in the composition of the CD3 modules associated with conventional and chimeric TCRs, as all γ, δ, and ε CD3 components were reproducibly precipitated with both heterodimers. Similarly, CD3γ, CD3δ, and CD3ε chains were all associated with a pTα-containing complex of 100 kD immunoprecipitated from the surface of untransfected SUP-T1 pre-T cells, corresponding to the endogenous pre-TCR. However, coprecipitation of ζ chain components either with the pre-TCR or with the chimeric or the conventional TCR-α/βs was not detected under these experimental conditions, likely reflecting poor ζ chain iodination due to its short EC domain 33.
Figure 3.
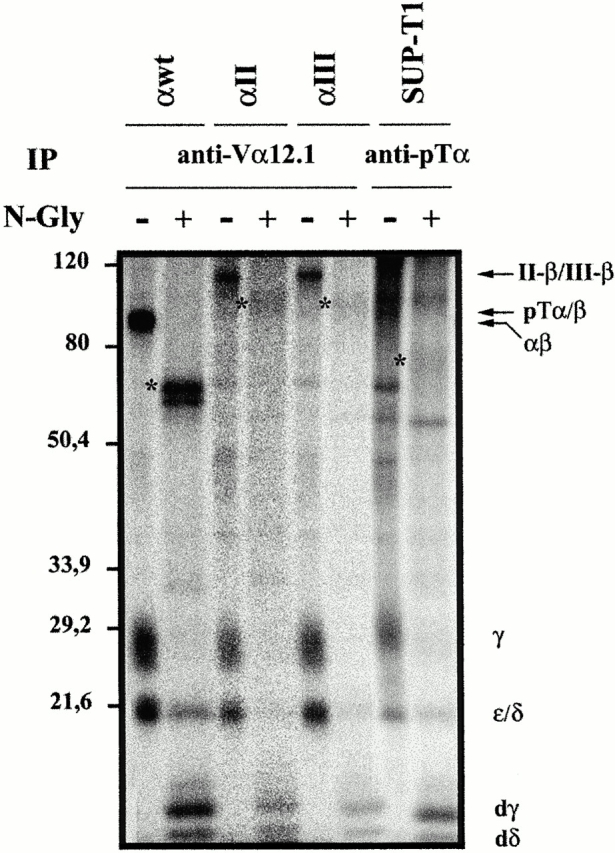
Biochemical composition of pre-TCR and chimeric TCR complexes expressed on SUP-T1 transfectants. Surface 125I-labeled SUP-T1 pre-T cells either mock transfected (SUP-T1) or transfected with wild-type TCR-α (αwt) or with αII and αIII TCR-α–pTα chimeric chains were lysed in 1% Brij 96–containing buffer, and the surface receptors were immunoprecipitated with an anti-Vα12.1 mAb or with a rabbit antiserum against the pTα CY domain. Immunoprecipitates (107 cell equivalents per lane) were either digested with N-Gly (+) or left untreated (−), and resolved by 12% SDS-PAGE under nonreducing conditions. The positions of the pTα–TCR-β, TCR-α/β, αII–TCR-β and αIII–TCR-β heterodimers, CD3δε, and CD3γ chains, and deglycosyl-ated CD3γ (dγ) and CD3δ (dδ) are indicated on the right. The migration positions of the molecular weight standards (in kD) are indicated on the left. *, N-deglycosylated heterodimers.
To directly investigate whether the ζ chain was physically associated with the pre-TCR and the chimeric TCRs as with the conventional TCR-α/βs, cell lysates from surface-iodinated SUP-T1 pre-T cells and αwt and αIII transfectants were immunoprecipitated in parallel either with a rabbit anti–human ζ chain antiserum or with an anti-CD3ε mAb, and the immunoprecipitates resolved by two-dimensional nonreducing/reducing SDS-PAGE. As shown in Fig. 4, the anti-CD3ε mAb coprecipitated the TCR-α/β, as well as the αIII chimeric TCR, and the pre-TCR with CD3γδε, whereas no ζ chain was detected. However, the ζ chain was detected as an individual protein migrating out of the diagonal with an apparent MW of 16 kD in anti-ζ immunoprecipitates. Interestingly, in all samples the anti-ζ Ab coprecipitated an heterodimeric complex together with CD3γδε, indicating that the ζ chain is physically associated at the cell surface with the pre-TCR and with the αIII chimeric TCR, as observed with the mature TCR-α/β. Furthermore, it is worth noting that the wild-type pre-TCR was precipitated with both the anti-CD3ε and anti-ζ Abs not only from the untransfected SUP-T1 cells, but also from the surface of αwt and αIII SUP-T1 transfectants (Fig. 4), indicating that the conventional TCR α chain (or the αIII chimeric chain) does not prevent surface expression of the endogenous pre-TCR but, rather, both the pre-TCR and the TCR-α/β can be coexpressed at the cell surface.
Figure 4.

The ζ chain is physically associated to the pre-TCR as well as to the αIII chimeric TCR. Surface iodinated SUP-T1 cells either mock transfected (SUP-T1) or transfected with the αIII TCR-α–pTα chimera (αIII) or with the wild-type TCR α chain were lysed in 1% Brij 96–containing buffer and surface receptors were then immunoprecipitated with a rabbit anti–human ζ chain antiserum or with an anti-CD3ε mAb. The immunoprecipitates (IP) were resolved by two-dimensional nonreducing (NR)/reducing (R) SDS-PAGE. The positions of the individual pre-TCR or TCR components migrating out of the diagonal are shown, and the relative positions of the molecular weight standards are indicated on the right.
The Cyto Tail of pTα Functions As an ER Retention Signal.
As biochemical studies provided evidence that low surface expression of the pre-TCR and the chimeric TCRs does not result from their impaired association with CD3/ζ components (although quantitative differences can not be ruled out), we next investigated whether the pTα Cyto tail by itself could be directly responsible for controlling surface expression levels of the whole receptor complex. To test this possibility, we first analyzed the effect of the pTα Cyto domain on the surface expression levels of individual plasma membrane reporter proteins. Murine CD25 (IL-2 receptor α chain) is a particularly useful marker in this type of study, because transfection of the wild-type protein results in high surface expression level, as assessed by flow cytometry with an anti-CD25 mAb (Fig. 5). Interestingly, we found that replacement of the CD25 Cyto domain with that of pTα results in a >50-fold reduction of surface CD25 expression on COS cells transfected with the CD25–pTα chimera, compared with surface expression levels of wild-type CD25. A comparable reduction of CD25 expression was observed in SUP-T1 pre-T cells transfected with the CD25–pTα chimera (Fig. 5). In both cell types, the relative levels of CD25 expression were not altered by deletion of the CD25 cyto tail (CD25 tailless), confirming that impaired CD25 surface expression was indeed due to the presence of the pTα Cyto tail.
Figure 5.

Impaired surface expression of a CD25–pTα chimeric protein bearing the human pTα Cyto tail. Relative levels of surface expression of murine CD25 were analyzed by flow cytometry (shaded histograms) with a biotin-coupled anti-CD25 mAb plus PE-coupled streptavidin on SUP-T1 pre-T cells or COS cells transfected with either the wild-type murine CD25 (CD25), a truncated CD25 molecule lacking the cytoplasmic domain (CD25 tailless), or a chimeric protein consisting of the CD25 EC and TM domains and the pTα Cyto tail (CD25/pTα). Mock-transfected cells were analyzed as controls. Background values (unshaded histograms) were determined by staining with a biotin-coupled isotype-matched irrelevant mAb plus PE-coupled streptavidin.
To assess whether the CD25–pTα chimeric protein was retained intracellularly, COS transfectants were permeabilized and examined for the intracellular distribution of CD25 by immunofluorescence microscopy. As shown in Fig. 6 A, transfection of wild-type CD25 resulted in a predominant localization of the protein in the Golgi apparatus, although CD25 expression was also detected at the plasma membrane. However, a markedly different staining pattern showing strong perinuclear fluorescence and a tubular-vesicular peripheral staining was observed in COS cells transfected with the CD25–pTα chimera, which strongly suggested an ER distribution. To confirm that the pTα tail was in fact conferring ER residence to membrane proteins, we next examined susceptibility to endo-H digestion of wild-type and chimeric CD25–pTα molecules, as protein transport out of the ER to the Golgi apparatus is marked by acquisition of endo-H resistance 34. To this end, COS transfectants were labeled for 30 min with [35S]methionine and chased for various periods of time up to 8 h, and Triton X-100–solubilized molecules were then subjected to immunoprecipitation with an anti-CD25 mAb or with an antiserum against the pTα Cyto tail (CT-1). Mock-treated or endo-H–treated proteins were then analyzed for their glycosylation state by SDS-PAGE, as endo-H sensitivity causes a marked change in the electrophoretic mobility of CD25. As shown in Fig. 6 B (top), a significant fraction (∼40%, as quantified by densitometric analysis) of wild-type CD25 acquired endo-H resistance after 1 h of biosynthesis, and was completely converted after 4 h. The situation was markedly different for the CD25–pTα chimeras analyzed from either anti-pTα (Fig. 6 B, bottom) or anti-CD25 precipitates (data not shown), as none of them acquire endo-H resistance, even after a 4-h chase. In contrast to wild-type CD25, which was partly degraded after 4 h and was hardly detectable after 8 h, up to 20% of pulsed CD25–pTα was still recovered essentially in an immature form after 8 h (Fig. 6 B). Therefore, the lack of maturation of the chimeric protein does not result from their increased degradation rate in the ER. Rather, we can conclude that a major proportion of the CD25–pTα chimeric proteins primarily reside in the ER and, therefore, cannot be processed in the Golgi apparatus. Taken together, the biosynthetic data in conjunction with the flow cytometry and immunofluorescence microscopy analyses provide evidence that the Cyto tail of the human pTα molecule confers ER residency to plasma membrane proteins.
Figure 6.


ER retention of a CD25–pTα chimeric protein bearing the Cyto tail of human pTα. (A) COS cells were transfected with either cDNA encoding the wild-type murine CD25 or with a CD25/pTα chimeric cDNA engineered to encode the CD25 EC and TM domains and the pTα Cyto tail (CD25/pTα). 48 h after transfection, cells were fixed, permeabilized, stained with a fluorescein-conjugated anti-CD25 mAb, and analyzed by immunofluorescence microscopy. Original magnification: ×63. (B) CD25 and CD25-pTα COS transfectants were pulse-chase labeled with [35S]methionine/cysteine and lysed in Triton X-100–containing lysis buffer. CD25 (top) and CD25-pTα (bottom) immunoprecipitates obtained with an anti–mouse CD25 (PC.61) mAb or with a rabbit antiserum against the human pTα tail (CT-1), respectively, were treated with endo-H (+) or left untreated (−) and resolved by 12% SDS-PAGE under reducing conditions. The positions of the mature (m) and immature (im) proteins is indicated on the right.
The physiological relevance of these findings was further confirmed by confocal microscopy studies aimed at analyzing the intracellular distribution of the native pTα protein in pre-T cells displaying poor surface expression of the pre-TCR (Fig. 7). When permeabilized SUP-T1 pre-T cells were stained with the rabbit anti-pTα Ab CT-1 in combination with fluorescein-conjugated goat anti–rabbit Igs, native pTα showed a typical intracellular staining compatible with ER location (data not shown). An identical intracellular pattern was revealed by green fluorescence examination of SUP-T1 pre-T cells transfected with a pTα–GFP chimeric protein (Fig. 7). To provide formal proof that the intracellular compartment containing pTα was in fact the ER, double-color analyses were performed in pTα–GFP transfectants stained with a rabbit Ab recognizing the ER resident PDI protein followed by Cy3-conjugated goat anti–rabbit Igs. As shown in Fig. 7, GFP clearly localized to the same intracellular region as the PDI protein in pTα–GFP transfectants. Moreover, costaining with an anti–TCR-β (βF1) mAb followed by Cy5-conjugated goat anti–mouse Igs revealed that endogenous TCR-β colocalized with GFP and PDI. Thus, from these experiments we concluded that intracellular pTα and TCR-β are predominantly retained in the ER in SUP-T1 pre-T cells.
Figure 7.

Colocalization of pTα-GFP with the ER-resident protein PDI. SUP-T1 cells stably transfected with pTα–GFP were fixed, permeabilized, and stained with an anti–TCR-β mAb followed by Cy5-coupled goat anti–mouse Igs, and an anti-PDI rabbit antiserum followed by Cy3-coupled goat anti–rabbit Igs. Analysis of pTα–GFP and TCR-β intracellular localization was performed by confocal microscopy. (a) Nomarsky analysis of selected individual cells. (b) Expression pattern of the pTα–GFP chain. (c) Intracellular staining of the ER-resident protein PDI. (d) Intracellular TCR-β chain staining. (e–g) Overlays of pTα-GFP and PDI (e), pTα–GFP and TCR-β (f), and TCR-β and PDI (g) stainings. The panels of individual cells shown are representative of all SUP-T1 cells examined. Original magnification: ×63.
Partial Truncation of the pTα Cyto Tail Releases a CD4–pTα Chimeric Protein from ER Retention and Restores Cell Surface Protein Expression.
Collectively, the above experiments provided evidence that the Cyto domain of pTα is directly responsible for retaining proteins in the ER. Thus, it was reasonable to predict that deletion of the pTα tail (and, therefore, the proposed ER retention determinants) would result in enhanced expression of the pre-TCR complex at the plasma membrane. However, the effect of the pTα tail deletion was the opposite, as it prevented surface pre-TCR expression. Indeed, JR3.11 cells transfected with a tailless pTα chain failed to react with anti-CD3 mAbs (Fig. 8 A), as well as with an anti–TCR-β (Vβ8) mAb and an anti-pTα Ab (data not shown). This unexpected discrepancy could be explained if the cytoplasmic domain of pTα contains essential structural information for the proper assembly and/or surface expression of a CD3-associated pre-TCR.
Figure 8.


pTα tailless molecules fail to reach the plasma membrane, but a partial truncation of the pTα Cyto tail releases a CD4–pTα chimeric protein from ER retention and restores cell surface expression. (A) JR3.11 cells transfected with a bicistronic vector encoding GFP and a pTα tailless molecule were stained with a PE-coupled anti-CD3 mAb. Flow cytometry analysis of surface CD3 expression (shaded histogram) was performed 48 h after transfection on electronically gated GFP+ cells. Background values (unshaded histogram) were obtained with a PE-coupled isotype-matched irrelevant mAb. (B) COS and BW cells were transfected with cDNAs encoding human CD4 molecules in which the Cyto domain has been deleted (CD4 tailless) or replaced either with a full pTα Cyto domain (CD4-pTα) or with a truncated pTα tail lacking the COOH-terminal 22 (CD4-pTαt22) or 48 (CD4-pTαt48) residues. 48 h after transfection, COS cells were fixed, permeabilized, and stained with an anti-CD4 mAb plus FITC-coupled goat anti–mouse Igs, and then analyzed by immunofluorescence microscopy (original magnification: ×63). BW cells were stained 24 h after transfection with a Cy5-PE–coupled anti-CD4 mAb (shaded histograms) and analyzed by flow cytometry. Control values (unshaded histograms) were obtained with a Cy5-PE–coupled isotype-matched irrelevant mAb.
The failure to observe surface expression of a pre-TCR with a complete deletion of the pTα tail prompted us to define more precisely the location of the potential ER retention signal in the pTα Cyto domain. To this end, we examined the effect of successive truncations along the pTα tail on the intracellular location and surface expression levels of individual CD4–pTα chimeric proteins which could be readily detected by staining with a mAb against CD4. As shown in Fig. 8 B, immunofluorescence microscopy analyses of COS cells transfected with CD4–pTα chimeric constructs where the Cyto domain of human CD4 was replaced with the full pTα tail (CD4–pTα) revealed a clear tubular-vesicular staining of the ER that was virtually identical to that recorded for the CD25–pTα chimera (Fig. 6 A), and correlated with low surface CD4 expression on both COS (data not shown) and BW cells (Fig. 8 B). No loss of the CD4 ER staining pattern was noted with the removal of the 22 COOH-terminal pTα residues by a truncation at position 243 (CD4-pTαt22), although this truncation resulted in a partial increase of surface CD4 expression on the corresponding BW transfectants (Fig. 8 B). In contrast, when an additional 26 amino acids (aa) were removed, the resulting CD4–pTαt48 truncated chimera was expressed on the surface of BW at levels that were as high as those expressed on BW cells transfected with a CD4-tailless (Fig. 8 B) or a wild-type CD4 (data not shown) construct. As this enhanced expression of surface CD4 correlated with loss of the ER staining pattern and appearance of fluorescence staining at the plasma membrane, we concluded that a region in the pTα Cyto domain between aa 217 and 243 is responsible for the observed ER retention function of the pTα tail. Accordingly, the CD4–pTαt48 chimera as well as the CD4-tailless protein were efficiently transported from the ER to the Golgi apparatus, and displayed similar kinetics, as assessed by pulse-chase metabolic labeling of COS transfectants, anti-CD4 immunoprecipitation, and endo-H treatment. As shown in Fig. 9 A, both proteins acquired endo-H resistance after a 90-min chase, and were mostly converted after 4 h. In contrast, the CD4–pTαt22 chimera remained essentially endo-H sensitive after 90 min and up to 50% of pulsed chimeric protein failed to acquire endo-H resistance, even after an 8-h chase.
Figure 9.

Truncation of the COOH-terminal 48 aa of the pTα Cyto domain restores transport out of the ER of a CD4–pTα chimeric protein and correlates with enhanced expression of the pre-TCR (A) COS cells transfected with the CD4 tailless, CD4-pTαt48, or CD4-pTαt22 constructs described in the legend to Fig. 8 were pulse-chased labeled and lysed as described in the legend to Fig. 6. Anti-CD4 immunoprecipitates were treated with endo-H (+) or left untreated (−) and resolved by 12% SDS-PAGE under reducing conditions. (B) CD3 surface expression was analyzed by flow cytometry, as described in the legend to Fig. 8 A, on JR3.11 cells transiently transfected with bicistronic vectors encoding GFP and either the wild-type pTα (shaded histogram) or a truncated pTα lacking the COOH-terminal 48 residues (pTαt48; bold line), or a wild-type TCR-α (thin line). Background values (dotted line) were obtained with a PE-coupled isotype-matched irrelevant mAb. The positions of the mature (m) and immature (im) proteins is indicated on the right.
Finally, experiments were performed to analyze the effect of equivalent truncations along the pTα protein on pre-TCR surface expression. Bicistronic vectors containing such pTα constructs allowed tracing of transiently transfected JR3.11 cells by EGFP expression as a separate but not as a chimeric protein. As shown in Fig. 9 B, although the pTα deletions were not sufficient to reach TCR-α expression levels, JR3.11 cells transfected with a pTα chain lacking the terminal 48 residues (pTαt48) consistently showed a modest increase of surface CD3 levels, compared with CD3 expression on wild-type pTα transfectants. Thus, as observed with the CD4–pTαt48 chimera, this particular truncation may release pTα from ER retention, resulting in enhanced surface pre-TCR expression. In contrast, surface expression of a pre-TCR composed of the pTαt22 truncated protein was indistinguishable from expression levels of a wild-type pre-TCR. Collectively, our data suggest that the observed pTα ER retention function contributes to the regulation of surface pre-TCR expression on pre-T cells.
Discussion
Complementary approaches in this study allowed us to conclude that differences between plasma membrane levels of the pre-TCR and the TCR are not intrinsic to the particular cell type in which they are expressed, but depend on the presence of a pTα chain in the former and of a TCR α chain in the latter. We have first shown that introduction of a conventional TCR α chain into pre-T cells increases surface receptor expression to levels similar to those of TCR-α/β on transfected mature T cells. Interestingly, although it has been reported that expression of TCR-α precludes the formation of a TCR-β–pTα dimer in a murine pre-T cell line 35, TCR-α transfection does not impair endogenous pre-TCR surface expression on human pre-T cells, indicating that TCR-β chains are in large excess and that TCR-α and pTα are not competing each other for pairing with TCR-β. Conversely, transfection of a TCR-α–deficient mature T cell line with a pTα chain results in low surface expression of the pre-TCR, relative to TCR-α/β surface levels induced upon TCR-α transfection. Because low levels of pre-TCR expression is a common feature of both cell types, we concluded that this is an intrinsic property of the pre-TCR complex, and therefore, the pTα chain itself might play a direct role in controlling pre-TCR expression levels.
In an effort to identify the structural domain/s of pTα responsible for such a role, we took advantage of the similarities and differences between the EC, TM, and Cyto domains of pTα and TCR-α and produced TCR-α–pTα chimeric proteins that were analyzed for their ability to bring a CD3-associated chimeric TCR complex to the cell surface. We initially focused on the EC pTα region, which lacks a covalently associated Ig-like V domain 25, and asked the question of whether replacement of the pTα EC domain with the EC region of TCR-α could provide a symmetrical shape to the TCR-β–pTα heterodimer that could result in an increased expression at the cell surface. Unexpectedly, however, such chimeric TCR-α–pTα chains (αII) did not affect the levels of expression of the pre-TCR, indicating that low surface pre-TCR expression is not dependent on the pTα EC domain. From these data one could infer that the lack in pTα of a partner domain capable of pairing with the hydrophobic surface of the Vβ domain 36 does not impair expression of the complex at the cell surface. Alternatively, the pre-TCR, by analogy with the pre-BCR, may contain an additional not yet identified VpreT component that fulfils that function, and that might thus be rate limiting for proper assembly and transport of the TCR-β–pTα heterodimer to the cell surface 2 6. Expression of such a putative VpreT is expected to be developmentally regulated, such that its absence would account for the impaired surface pre-TCR expression described in murine mature T cells 6. However, human mature T cells proved to be as efficient as pre-T cells in expressing a functional TCR-β–pTα heterodimer on their surface (this study and unpublished results), thus supporting the alternative hypothesis that the pTα chain is sufficient to promote surface expression of the pre-TCR complex in the absence of an additional component.
The finding that the chimeric chain in which the pTα EC domain was replaced with the EC TCR-α domain behaves as a wild-type pTα chain in terms of surface receptor expression was not obvious, because the TCR-α C domain displays little homology with the equivalent pTα domain 9 25. However, as the TCR-α C domain residues involved in polar interactions with the TCR-β C domain are all conserved in the pTα C domain, it has been proposed that association between pTα and TCR-β C domains may occur in a way similar to that observed in the TCR CαCβ module 3 37. These data are against an essential function of TCR-β–pTα assembly in controlling surface expression levels of the pre-TCR, and suggest that the CD3/ζ subunits associated with the TCR-β–pTα heterodimer probably account for such a regulatory function. Of them, the ζ chain would merely be a dispensable amplification module for the pre-TCR, which increases its assembly rate and stability 19 20 21 22 24, and CD3δ seems dispensable for pre-TCR assembly and function 8 13 38. In contrast, the CD3γε pair, which may associate with the pre-TCR EC domain module in a manner similar to that suggested for its association with the TCR-α/β 39, has proved mandatory for proper assembly of a functional pre-TCR 8 40 41 42. Therefore, it is not surprising that the wild-type pTα chain and the chimeric chains containing both the EC and TM domains, or exclusively the EC domain of TCR-α, are equally capable of mediating association with CD3γ, CD3δ, and CD3ε subunits. What is more surprising is that all the wild-type pre-TCR and the chimeric TCR complexes are equally found physically associated with the ζ chain, given that the weak association of the ζ chain with the pre-TCR has recently been shown to map to the connecting peptide (CP) of pTα lying between its TM and Ig-like EC domains 43. Therefore, one would expect that the presence of the TCR-α CP domain in both chimeras would confer an increased stability to their corresponding receptors that might result in higher levels of surface expression. In contrast, their expression levels were as low as those of the wild-type pre-TCR. Taken together, our results allowed us to conclude that qualitative differences in the contribution of the CD3/ζ components to pre-TCR or TCR assembly do not influence surface receptor expression levels.
A striking finding was that replacing the Cyto domain of TCR-α with the equivalent pTα domain is sufficient for inducing low surface expression of the chimeric TCR. It is thus obvious that the pTα Cyto tail is directly responsible for low surface pre-TCR expression. A strong argument in favor of the attractive possibility that the pTα Cyto tail functions as an ER retention signal was the observation that individual plasma membrane proteins are mostly retained in the ER and fail to reach the Golgi compartment when appended to the pTα tail. Accordingly, pTα chains are primarily found in internal structures which colocalize with the ER. The demonstration that truncation of the terminal 48 aa of pTα (and particularly deletion of the region between residues 217 and 243) releases CD4–pTα chimeric proteins from ER retention lends further support to that notion, and suggests that pTα ER retention determinants localize primarily in the deleted region. However, the pTα tail sequence from position 217 to 243 does not harbor the consensus dilysine cytoplasmic motif for ER retention of TM proteins 34 or the reported CD3ε ER retention sequence 44, suggesting the existence of alternative ER retention signals. An extensive series of point mutations will have to be introduced into that particular pTα sequence to unravel the precise requirements for its retention in the ER.
As proposed for the mature TCR-α/β 45, ER retention of individual subunits or partial complexes might be an essential mechanism for regulating assembly and levels of expression of the pre-TCR. However, additional structural information may be contained in the pTα Cyto tail that is essential for surface expression of the pre-TCR, as pTα tailless molecules fail to bring a CD3-associated pre-TCR complex to the cell surface. Therefore, efficient surface expression of the pre-TCR complex may be the result of a balance between complex mechanisms controlling retention and assembly of its individual components. This may explain the finding that release of individual chimeras bearing a truncated pTα tail from ER retention correlates with recovery of wild-type protein expression levels, whereas pTα molecules with identical truncations were unable to recover TCR-α/β surface levels, although they were more efficient than wild-type pTα in bringing the pre-TCR to the cell surface. Interestingly, an ER retention mechanism has also been proposed to account for the low plasma membrane expression of the pre-BCR 17, although in that case ER retention seems to be inherent to pre-B cells. Despite the striking functional similarities between the pre-TCR and the pre-BCR, the particular structural features of their individual components (i.e., pTα and λ5) may explain such a discrepancy.
Finally, although one would expect that a retention mechanism similar to that proposed here for the human pre-TCR would be functional also in mice, the poor conservation of the Cyto tail in both species 6 25 would make this possibility unlikely. However, conserved residues in the cytoplasmic domain have been identified 25. In addition, two independent findings strengthen our view: first, DN thymocytes from pTα−/− mice made transgenic for a pTα tailless chain display increased TCR-β surface levels, compared with DN thymocytes from wild-type or pTα−/− nontransgenic mice 46, and second, low CD3 expression is common to DP thymocytes from recombination activating gene (Rag)1−/− mice expressing a transgenic functional pre-TCR consisting of truncated TCR-β and pTα forms that keep their TM and Cyto regions 14. In conclusion, our data provide evidence that the pTα Cyto tail displays a novel ER retention function which may participate in the regulation of surface pre-TCR expression levels on pre-T cells.
Acknowledgments
We thank Drs. M. Brenner, R. Kurrle, F. Sánchez-Madrid, and R. Bragado for the generous gift of Abs, H. Spits for providing us with a bicistronic retroviral vector, and B. Rubin and L. Borlado for the JR3.11 cell line and for the murine CD25 cDNA, respectively; Drs. B. Alarcón and I. Sandoval for helpful discussions, Dr. A. Alcover for helpful advice on confocal microscopy, and Dr. J.R. Regueiro for reading the manuscript.
This work was supported by grants from the Comisión Interministerial de Ciencia y Tecnología (SAF 97-0161), Dirección General de Enseñanza Superior (PB97-1194), Comunidad de Madrid (08.3/0015.1/99), Fondo de Investigación Sanitaria (FIS 00/1044), and Fundación Eugenio Rodríguez Pascual. We thank the Fundación Ramón Areces for an institutional grant to the Centro de Biología Molecular Severo Ochoa.
Footnotes
C. Trigueros' present address is Imperial Cancer Research Fund, London WC2A 3PX, UK.
Abbreviations used in this paper: aa, amino acid(s); BCR, B cell receptor; Cyto, cytoplasmic; DN, double negative; DP, double positive; EC, extracellular; EGFP, enhanced green fluorescent protein; endo-H, endoglycosidase H; ER, endoplasmic reticulum; PDI, protein disulfide isomerase; pTα, pre–TCR-α; SP, single positive; TM, transmembrane.
References
- Borst J., Jacobs H., Brouns G. Composition and function of T-cell receptor and B-cell receptor complexes on precursor lymphocytes. Curr. Opin. Immunol. 1996;8:181–190. doi: 10.1016/s0952-7915(96)80056-2. [DOI] [PubMed] [Google Scholar]
- von Boehmer H., Fehling H.J. Structure and function of the pre-T cell receptor. Annu. Rev. Immunol. 1997;15:433–452. doi: 10.1146/annurev.immunol.15.1.433. [DOI] [PubMed] [Google Scholar]
- Malissen B., Ardouin L., Lin S.-Y., Gillet A., Malissen M. Function of the CD3 subunits of the pre-TCR and TCR complexes during T cell development. Adv. Immunol. 1999;72:103–148. doi: 10.1016/s0065-2776(08)60018-8. [DOI] [PubMed] [Google Scholar]
- Groettrup M., Ungeweiss K., Azogi O., Palacios R., Owen M.J., Hayday A.C., von Boehmer H. A novel disulfide-linked heterodimer on pre-T cells consists of the T cell receptor β chain and a 33 kd glycoprotein. Cell. 1993;75:283–294. doi: 10.1016/0092-8674(93)80070-u. [DOI] [PubMed] [Google Scholar]
- Dudley E.C., Petrie H.T., Shah L.M., Owen M.J., Hayday A.C. T cell receptor β chain gene rearrangement and selection during thymocyte development in adult mice. Immunity. 1994;1:83–93. doi: 10.1016/1074-7613(94)90102-3. [DOI] [PubMed] [Google Scholar]
- Saint-Ruf C., Ungewiss K., Groettrup M., Bruno L., Fehling H.J., von Boehmer H. Analysis and expression of a cloned pre-T cell receptor gene. Science. 1994;266:1208–1212. doi: 10.1126/science.7973703. [DOI] [PubMed] [Google Scholar]
- van Oers N.S.C., von Boehmer H., Weiss A. The pre-T cell receptor (TCR) complex is functionally coupled to the TCR-ζ subunit. J. Exp. Med. 1995;182:1585–1995. doi: 10.1084/jem.182.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger M.A., Davé V., Rhodes M.R., Bosma G.C., Bosma M.J., Kappes D.J., Wiest D.L. Subunit composition of pre-T cell receptor complexes expressed by primary thymocytesCD3δ is physically associated but not functionally required. J. Exp. Med. 1997;186:1461–1467. doi: 10.1084/jem.186.9.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehling H.J., Krotkova A., Saint-Ruf C., von Boehmer H. Crucial role of the pre-T-cell receptor alpha gene in development of alpha beta but not gamma delta T cells. Nature. 1995;375:795–798. doi: 10.1038/375795a0. [DOI] [PubMed] [Google Scholar]
- Hoffman E.S., Passoni L., Cromptom T., Leu T.M.J., Schatz D.G., Koff A., Owen M.J., Hayday A.C. Productive T-cell receptor beta-chain gene rearrangement and selection during thymocyte development in vivo . Genes Dev. 1996;10:948–962. doi: 10.1101/gad.10.8.948. [DOI] [PubMed] [Google Scholar]
- Malissen B., Malissen M. Functions of TCR and pre-TCR subunitslessons from gene ablation. Curr. Opin. Immunol. 1996;8:383–393. doi: 10.1016/s0952-7915(96)80129-4. [DOI] [PubMed] [Google Scholar]
- Levelt C.N., Wang B., Ehrfeld A., Terhost C., Eichmann K. Regulation of T cell receptor (TCR)-β locus allelic exclusion and initiation of TCR-α locus rearrangement in immature thymocytes by signaling through the CD3 complex. Eur. J. Immunol. 1995;25:1257–1261. doi: 10.1002/eji.1830250519. [DOI] [PubMed] [Google Scholar]
- Jacobs H., Vandeputte D., Tolkamp L., de Vries E., Borst J., Berns A. CD3 components at the surface of pro-T cells can mediate pre-T cell development in vivo . Eur. J. Immunol. 1994;24:934–939. doi: 10.1002/eji.1830240423. [DOI] [PubMed] [Google Scholar]
- Irving B.A., Alt F.W., Killeen N. Thymocyte development in the absence of pre-T cell receptor extracellular immunoglobulins domains. Science. 1998;280:905–908. doi: 10.1126/science.280.5365.905. [DOI] [PubMed] [Google Scholar]
- O'Shea C.C., Thornell A.P., Rosewell I.R., Hayes B., Owen M.J. Exit of the pre-TCR from the ER/cis-golgi is necessary for signaling differentiation, proliferation, and allelic exclusion in immature thymocytes. Immunity. 1997;7:591–599. doi: 10.1016/s1074-7613(00)80380-5. [DOI] [PubMed] [Google Scholar]
- Trigueros C., Ramiro A.R., Carrasco Y.R., de Yébenes V.G., Albar J.P., Toribio M.L. Identification of a late stage of small noncycling pTα− pre-T cells as immediate precursors of T cell receptor α/β+ thymocytes. J. Exp. Med. 1998;188:1401–1412. doi: 10.1084/jem.188.8.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouns G.S., de Vries E., Neefjes J.J., Borst J. Assembled pre-B cell receptor complexes are retained in the endoplasmic reticulum by a mechanism that is not selective for the pseudo-light chain. J. Biol. Chem. 1996;271:19272–19278. doi: 10.1074/jbc.271.32.19272. [DOI] [PubMed] [Google Scholar]
- Jacobs H., Ossendorp F., de Vries E., Ungewiss K K., von Boehmer H., Borst J., Berns A. Oncogenic potential of a pre-T cell receptor lacking the TCRβ variable domain. Oncogene. 1996;12:2089–2099. [PubMed] [Google Scholar]
- Malissen M., Gillet A., Rocha B., Trucy J., Vivier E., Boyer C., Köntgen C., Brun N.K., Mazza G., Spanopoulou E., Guy-Grand D., Malissen B. T cell development in mice lacking the CD3ζ/η gene. EMBO (Eur. Mol. Biol. Organ.) J. 1993;12:4347–4355. doi: 10.1002/j.1460-2075.1993.tb06119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love P.E., Shores E.W., Johnson M.D., Tremblay M.L., Lee E.J., Grinberg A., Huang S.P., Singer A., Westphal H. T cell development in mice that lack the zeta chain of the T cell antigen receptor complex. Science. 1993;261:918–921. doi: 10.1126/science.7688481. [DOI] [PubMed] [Google Scholar]
- Ohno H., Aoe T., Taki S., Kitamura D., Ishida Y., Rajewski K., Saito T. Developmental and functional impairment of T cells in mice lacking CD3ζ chains. EMBO (Eur. Mol. Biol. Organ.) J. 1993;12:4357–4366. doi: 10.1002/j.1460-2075.1993.tb06120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shores E.W., Huang K., Tran T., Lee E., Grinberg A., Love P.E. Role of TCR ζ chain in T cell development and selection. Science. 1994;266:1047–1050. doi: 10.1126/science.7526464. [DOI] [PubMed] [Google Scholar]
- Bäckström B.T., Milia E., Peter A., Jaureguiberry B., Baldari C.T., Palmer E. A motif within the T cell receptor α chain constant region connecting peptide domain controls antigen responsiveness. Immunity. 1996;5:437–447. doi: 10.1016/s1074-7613(00)80500-2. [DOI] [PubMed] [Google Scholar]
- Ardouin L., Ismaili J., Malissen B., Malissen M. The CD3-γδε and CD3-ζ/η modules are each essential for allelic exclusion at the T cell receptor β locus but are both dispensable for the initiation of V to (D)J recombination at the T cell receptor-β, -γ, and –δ loci. J. Exp. Med. 1998;187:105–116. doi: 10.1084/jem.187.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramiro A.R., Trigueros C., Márquez C., San Millán J.L., Toribio M.L. Regulation of pre-T cell receptor (pTα-TCRβ) gene expression during human thymic development. J. Exp. Med. 1996;184:519–530. doi: 10.1084/jem.184.2.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst J., van Dongen J.J.M., de Vries E., Comans-Bitter W.M., van Tol M.J.D., Vossen J.M., Kurrle R. BMA031, a monoclonal antibody suited to identify the T-cell receptor αβ/CD3 complex on viable human T lymphocytes in normal and disease states. Hum. Immunol. 1990;29:175–188. doi: 10.1016/0198-8859(90)90113-4. [DOI] [PubMed] [Google Scholar]
- DerSimonian H., Band H., Brenner M.B. Increased frequency of T cell receptor Vα12.1 expression on CD8+ T cellsevidence that Vα participates in shaping the peripheral T cell repertoire. J. Exp. Med. 1991;174:639–648. doi: 10.1084/jem.174.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddon P.J., Littman D.R., Godfrey M., Maddon D.E., Chess L., Axel R. The isolation and nucleotide sequence of a cDNA encoding the T cell surface protein T4a new member of the immunoglobulin gene family. Cell. 1985;42:93–104. doi: 10.1016/s0092-8674(85)80105-7. [DOI] [PubMed] [Google Scholar]
- Heemskerk M.H.M., Bloom B., Nolan G., Stegmann A.P.A., Bakker A.Q., Weijer K., Res P.C.M., Spits H. Inhibition of T cell and promotion of natural killer cell development by the dominant negative helix loop helix factor Id3. J. Exp. Med. 1997;186:1597–1602. doi: 10.1084/jem.186.9.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud J., Huchenq A., Vernhes M.-C., Caspar-Bauguil S., Lenfant F., Sancho J., Terhost C., Rubin B. The interchain disulfide bond between TCRαβ heterodimers on human T cells is not required for TCR-CD3 membrane expression and signal transduction. Int. Immunol. 1996;9:615–626. doi: 10.1093/intimm/9.4.615. [DOI] [PubMed] [Google Scholar]
- Reynolds T.C., Smith S.D., Sklar J. Analysis of DNA surrounding the breakpoints of chromosomal translocations involving the beta T cell receptor gene in human lymphoblastic neoplasms. Cell. 1987;50:107–117. doi: 10.1016/0092-8674(87)90667-2. [DOI] [PubMed] [Google Scholar]
- Wallace D.L., MacIntyre E.A., Linch D.C., Beverley P.C.L. Analysis of the activation signals induced by CD3 antibodies and their role in T cell activation 1987. Leucocyte Typing III; Oxford University Press, Oxford, UK: pp. 167–169 [Google Scholar]
- Weissmann A.M., Baniyash M., Hou D., Samelson L.E., Burgess W.H., Klausner R.D. Molecular cloning of the zeta chain of the T cell antigen receptor. Science. 1988;239:1018–1021. doi: 10.1126/science.3278377. [DOI] [PubMed] [Google Scholar]
- Jackson M.R., Nilsson T., Peterson P.A. Identification of a consensus motif for retention of transmembrane proteins in the endoplasmic reticulum. EMBO (Eur. Mol. Biol. Organ.) J. 1990;9:3153–3162. doi: 10.1002/j.1460-2075.1990.tb07513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trop S., Rhodes M., Wiest D.L., Hugo P., Zúñiga-Pflücker J.C. Competitive displacement of pTα by TCRα during TCR assembly prevents surface coexpression of pre-TCR and αβ TCR. J. Immunol. 2000;165:5566–5572. doi: 10.4049/jimmunol.165.10.5566. [DOI] [PubMed] [Google Scholar]
- Bentley G.A., Boulot G., Karjalainen K., Mariuzza R.A. Crystal structure of the β chain of a T cell antigen receptor. Science. 1995;267:1984–1987. doi: 10.1126/science.7701320. [DOI] [PubMed] [Google Scholar]
- Wang J.-H., Lim K., Smolyar A., Teng M.-K., Liu J.-H., Tse A.G.D., Liu J., Hussey R.E., Chishti Y., Thompson C.T. Atomic structure of an αβ T cell receptor (TCR) heterodimer in complex with an anti-TCR Fab fragment derived from a mitogenic antibody. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:10–26. doi: 10.1093/emboj/17.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave P.D., Cao Z., Browne C., Alarcón B., Fernández-Miguel G., Lafaille J., de la Hera A., Tonegawa S., Kappes D.J. CD3 delta deficiency arrests development of the alpha beta but not the gamma delta T cell lineage. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:1360–1370. doi: 10.1093/emboj/16.6.1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolios N., Kemp O., Li Z.G. The T cell antigen receptor α and β chains interact via distinct regions with CD3 chains. Eur. J. Immunol. 1994;24:84–92. doi: 10.1002/eji.1830240114. [DOI] [PubMed] [Google Scholar]
- Malissen M., Gillet A., Ardouin L., Bouvier G., Trucy J., Ferrier P., Vivier E., Malissen B. Altered T cell development in mice with a targeted mutation of the CD3-ε gene. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:4641–4653. doi: 10.1002/j.1460-2075.1995.tb00146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haks M.C., Krimpenfort P., Borst J., Kruisbeek A. The CD3γ chain is essential for development of both the TCRαβ and TCRγδ lineages. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:1871–1882. doi: 10.1093/emboj/17.7.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJarnette J.B., Sommers C.L., Huang K., Woodside K.J., Emmons R., Katz K., Shores E.W., Love P.E. Specific requirement for CD3epsilon in T cell development. Proc. Natl. Acad. Sci. USA. 1998;95:14909–14914. doi: 10.1073/pnas.95.25.14909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trop S., Steff A.M., Denis F., Wiest D.L., Hugo P. The connecting peptide domain of pTα dictates weak association of the pre-T cell receptor with the TCR ζ subunit. Eur. J. Immunol. 1999;29:2187–2196. doi: 10.1002/(SICI)1521-4141(199907)29:07<2187::AID-IMMU2187>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Mallabiabarrena A., Fresno M., Alarcón B. An endoplasmic reticulum retention signal in the CD3ε chain of the T-cell receptor. Nature. 1992;357:593–596. doi: 10.1038/357593a0. [DOI] [PubMed] [Google Scholar]
- Klausner R.D., Lippincott-Schwartz J., Bonifacino J.S. The T cell antigen receptorinsights into organelle biology. Annu. Rev. Cell Biol. 1990;6:403–431. doi: 10.1146/annurev.cb.06.110190.002155. [DOI] [PubMed] [Google Scholar]
- Fehling H.J., Iritani B.M., Krotkova A., Forbush K.N., Laplace C., Perlmutter R.M., von Boehmer H. Restoration of thymopoiesis in pTα−/− mice by anti-CD3ε antibody treatment or with transgenes encoding activated lck or tailless pTα. Immunity. 1997;6:703–714. doi: 10.1016/s1074-7613(00)80446-x. [DOI] [PubMed] [Google Scholar]