

Abstract
Cytotoxic T lymphocyte antigen (CTLA)-4 plays an essential role in immunologic homeostasis. How this negative regulator of T cell activation executes its functions has remained controversial. We now provide evidence that CTLA-4 mediates a cell-intrinsic counterbalance to restrict the clonal expansion of proliferating CD4+ T cells. The regulation of CTLA-4 expression and function ensures that, after ∼3 cell divisions of expansion, most progeny will succumb to either proliferative arrest or death over the ensuing three cell divisions. The quantitative precision of the counterbalance hinges on the graded, time-independent induction of CTLA-4 expression during the first three cell divisions. In contrast to the limits imposed on unpolarized cells, T helper type 1 (Th1) and Th2 effector progeny may be rescued from proliferative arrest by interleukin (IL)-12 and IL-4 signaling, respectively, allowing appropriately stimulated progeny to proceed to the stage of tissue homing. These results suggest that the cell-autonomous regulation of CTLA-4 induction may be a central checkpoint of clonal expansion of CD4+ T cells, allowing temporally and spatially restricted growth of progeny to be dictated by the nature of the threat posed to the host.
Keywords: lymphocyte, CTLA-4, cell cycle, CD4+, CCR7
Introduction
Proliferation and differentiation of CD4+ T cells are regulated by numerous extrinsic signaling pathways. Variables such as the amount of antigen, inflammation, costimulatory ligands, and cytokine milieu all serve to multiply the potential outcomes of an immune response 1. Despite the seemingly limitless possibilities, there are certain behaviors of activated CD4+ T cells that suggest a level of invariant, autonomous control 2 3. One strikingly uniform feature in the activation of helper T cells is the finding that clonal expansion seems invariably accompanied by clonal contraction 4. Activation-induced cell death is generally regarded as the major mechanism for lymphocyte homeostasis during an immune response 5 6 7. CD4+ T cells can be induced to undergo apoptosis through a mechanism that is linked to their activation, but how the processes are coupled is not well understood. Whether nonapoptotic mechanisms exist to limit lymphocyte expansion is also uncertain 8.
The phenotype of cytotoxic T lymphocyte antigen 4 (CTLA-4) deficiency in mice 9 10 11 suggests that this receptor plays a central role in homeostasis. Together with other gain- and loss-of-function experiments, CTLA-4 has been implicated as a regulator of cell cycle 12 13 14, anergy, tolerance 15 16 17, autoimmunity 18, transplantation 18, effector choice 19 20 21, and immunity to tumors 22 and foreign pathogens 23 24 25. One explanation for how negative regulation by CTLA-4 might be involved in many disparate immunologic reactions is that its regulation and function are coupled to the proliferative program. It has become evident that the cell cycle organizes some changes in gene expression in CD4+ T cells that are critical for effector subset choice 26 27 28 and effector-memory homing 29 30. This suggests that many immunologic reactions, despite being cued by extrinsic variables, have levels of cell-autonomous control that are determined by the lineage relationship of proliferating cells 2 3 4.
We wished to determine the precise lineage relationship between cells that were engaged in a proliferative response when well-characterized variables of activation were experimentally manipulated. Using the dilution of carboxy-fluorescein diacetate succinimidyl ester (CFSE) to assess cell division 31, in conjunction with quantitation of cellularity and apoptosis, CD4+ T cells were found to have a cell-intrinsic mechanism to control their proliferation. The control mechanism can be ascribed to the graded, cell cycle–coupled regulation of the expression and function of the CTLA-4 receptor. These findings suggest that vastly different immunologic outcomes mediated by CD4+ T cells result from the integration of extrinsic stimuli with a cell-autonomous program of gene regulation that is linked to cell division.
Materials and Methods
Mice.
Wild-type C57BL/6 and BALB/c mice were obtained from The Jackson Laboratory. CTLA-4−/− 9, CD28−/− 32, BCl-xL (under the control of the CMV promoter and Eμ enhancer) transgenic 33, and DO11.10 TCR transgenic 34 mice were generated as described. CTLA-4−/− DO11.10 TCR transgenic mice were used to delay onset of disease in CTLA-4−/− animals 17 35, and wild-type DO11.10 TCR transgenic, CD28−/− DO11.10 TCR transgenic, and Bcl-xL transgenic DO11.10 TCR transgenic mice were used as their controls. All mutant mice were backcrossed to the BALB/c or B10.D2 background six times before intercross with mice from the same background. Animals were genotyped using PCR and flow cytometry. All animal work was done in accordance with guidelines of the University of Pennsylvania.
Cell Culture.
In experiments with nontransgenic mice, splenocytes were depleted of CD8+ cells using magnetic beads (PerSeptive Biosystems), and stimulated (2 × 106 cells/ml) using soluble anti-CD3 mAb (2.0 μg/ml; BD PharMingen) as described 26, unless indicated. Anti-CD28 mAb (BD PharMingen), human rIL-2 (Life Sciences), murine rIL-4 (Roche), rIL-12 (BD PharMingen), anti–IL-4 mAb (BD PharMingen), and anti–IL-12 mAb (BD PharMingen) were used at concentrations indicated in the figure legends. Stimulations of DO11.10 transgenic splenocytes were performed as described for wild-type cells, except without prior CD8 depletion. All cells were labeled with CFSE (Molecular Probes) as described previously 26. Briefly, cells (2 × 107 cells/ml) were incubated in PBS with CFSE (5 μM) for 9 min at room temperature. Labeling was quenched with an equal volume of FCS, and then cells were washed twice with HBSS supplemented with 10% FCS.
Flow Cytometry.
Flow cytometric analysis was performed on fixed cells as described previously 26. Briefly, cells were washed with PBS and fixed in 4% paraformaldehyde for 11 min at room temperature. Fixed cells were stained in permeabilization buffer (PBS with 0.2% saponin, 1% FCS, and 0.1% sodium azide). Phycoerythrin-conjugated mAbs against CTLA-4, CD25, CD44, and allophycocyanin-conjugated mAbs against IFN-γ and IL-4 were used where indicated. For intracellular cytokine staining only, cells were restimulated with PMA (50 ng/ml; Sigma-Aldrich) and ionomycin (500 ng/ml; Sigma-Aldrich) for 4 h, with brefeldin A (2.0 μg/ml; Sigma-Aldrich) added for the final 2 h, before fixation.
Live, unfixed cells were washed in HBSS 1% FCS before staining. All antibodies were obtained from BD PharMingen, unless indicated. Flow cytometry was performed using a Becton Dickinson FACSCalibur™ instrument and CELLQuest™ software. Throughout the article, data represent only CD4+ events, based on specific staining with fluorochrome-conjugated anti-CD4 mAbs (Caltag). Unless specified, analysis includes only live (as determined by forward and side light scatter), CD4+ events. Gates for specific staining of surface or intracellular proteins were determined using fluorochrome-conjugated species-matched and, where possible, isotype-matched, control mAbs.
Cell Death and DNA Content.
Binding of PE-conjugated Annexin V (BD PharMingen) was performed on unfixed cells according to the manufacturer's instructions. Propidium iodide (P.I.) exclusion was performed by resuspending unfixed cells in PBS with P.I. (1.0 μg/ml; Sigma-Aldrich) 5 min before flow cytometry. ToPro-3 (Molecular Probes) was used to assess DNA content in fixed, permeabilized cells. Cells were first stained with mAbs, and then washed, fixed, and incubated in PBS with saponin (0.3%), RNase A (50 μg/ml), EDTA (5 mM), and ToPro-3 (1.0 μM) for 30 min at 4°C before analysis by flow cytometry.
Reverse Transcription PCR of CC Chemokine Receptor 7 Expression.
CD8-depleted, CFSE-labeled cells were stimulated for 4 d in the cytokine conditions indicated in the figure legend, and then stained with fluorochrome-conjugated anti-CD4 mAb, before sorting of live-gated, CD4+ events into separate generations by flow cytometry, using a MoFlo instrument (Cytomation). Cells were washed and total RNA was extracted using Trizol Reagent (Life Technologies) and reverse transcribed using random hexamer primers (Amersham Pharmacia Biotech), as described 36. cDNA levels were equalized by reverse transcription (RT)-PCR, using a competitive template of hypoxanthine guanine phosphoribosyl transferase (HPRT) cDNA as an internal standard, as described 36. The competitive template, because of its higher molecular weight, migrates slower than authentic HPRT cDNA during electrophoresis. PCR for CC chemokine receptor (CCR)7 was performed on equalized cDNA samples. Each reaction was performed at least three times. The following primer sets were used: CCR7 sense CTACAGCCCCCAGAGCACCAT, CCR7 antisense GAAGGGAAGAATTAGGAGGAAAAG, HPRT sense GTTGGAGACAGGCCAGACTTTGTTG, and HPRT antisense GAGGGTAGGCTGGCC-TATAGGCT.
Results
Limits on Clonal Expansion Are Regulated by the Cell Division Cycle.
CFSE labeling was used to resolve individual cell divisions by the quantitative dilution of green fluorescence that accompanies cytokinesis 31. CD8-depleted splenocytes were stimulated with soluble anti-CD3 mAb, using endogenous accessory cells to cross-link the anti-CD3 mAb and provide a natural source of ligands for CD28 and CTLA-4. We first performed a quantitative analysis of relative cellularity as a function of cell division in an asynchronously dividing population that encompassed at least five cell divisions. By comparing the relative number of live CD4+ T cells in each successive division, we found that the dynamics of clonal expansion follow a highly reproducible pattern over a range of conditions (Fig. 1). The greatest number of cells was typically found in the population having completed the third or fourth cell division. Although many cells continued through 1–3 more cell divisions, cellular expansion was limited not only by proliferative arrest, but apoptosis (see below). As a result, fewer than 5% of the viable cells in culture completed the sixth cell division.
Figure 1.
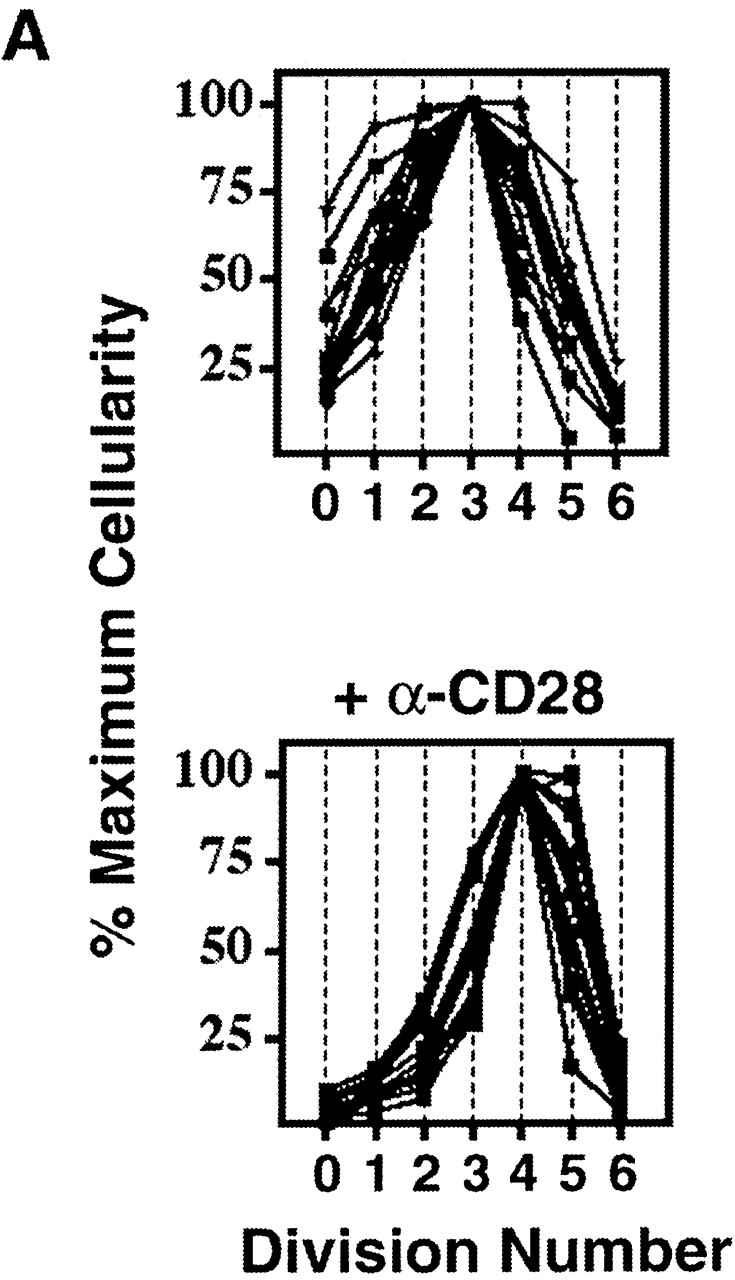

Uniformity in the clonal expansion of CD4+ T cells. (A) CD8-depleted, CFSE-labeled splenocytes were stimulated (see Materials and Methods) with varying concentrations of anti-CD3 mAb (0.05–5.0 μg/ml), and analyzed on day 3, 4, 5 or 6 (top panel). Cellularity of live, CD4+ cells, as determined by specific staining with anti-CD4 mAb, in relation to cell division number is depicted graphically. For comparative purposes the number of cells in each cell division as determined by CFSE dilution (x-axis) was expressed as a percentage of the number of cells in the cell division having the largest number of cells (y-axis) within each experimental condition. Top panel includes 10 experiments (with each line representing one experimental condition and time point). Bottom panel depicts 10 experiments (with each line representing one experimental condition and time point) using varying concentrations of both anti-CD3 (0.01–2.0 μg/ml) and anti-CD28 (0.3–10.0 μg/ml) mAbs, all analyzed on day 4. Cells from both C57BL/6 and BALB/c mice were tested. Dose responses of anti-CD3 and anti-CD28 were tested at least twice. (B) CD8-depleted, CFSE-labeled splenocytes were stimulated with anti-CD3 mAb (1.0 μg/ml), anti-CD28 mAb (2.0 μg/ml), and rIL-2 (2 U/ml) for 3 d, and stained with fluorochrome-conjugated Annexin V and anti-CD4 mAb. Binding of Annexin V (“% Apoptotic,” y-axis) among CD4+ cells of indicated division number (x-axis) is depicted in bar graph (left panel), and flow cytometric data of CD4+ events is displayed (right panel) as cell division (x-axis) versus Annexin V binding (y-axis). Polygonal gate indicates upper limit of background fluorescence. Results are representative of four separate experiments.
Changes in mitogenic dose of anti-CD3 mAb (0.05–5.0 μg/ml), duration of culture (3–6 d), and genetic background of T cells did not alter the pattern of clonal expansion (Fig. 1 A), suggesting that this regulatory mechanism is independent of both responder frequency and time. Addition of varying concentrations of anti-CD28 mAb (0.3–10.0 μg/ml) under the same mitogenic conditions shifted the point of maximal cellularity to a later cell division (Fig. 1 A). The increase in cell expansion that resulted from costimulation, however, did not result from expansion of cells past the fifth division, and, again, fewer than 5% of cells successfully completed the sixth division.
The distribution of cell death within discrete cell divisions was examined by analyzing the binding of Annexin V to apoptotic membranes (Fig. 1 B). As expected, there was significant cell death among activated, proliferating cells. We consistently observed, however, that the majority of apoptosis occurred after the third cell division, and increased substantially in later cell divisions (Fig. 1 B). Identical results were obtained using exclusion of propidium iodide as a parameter of survival (see below). Thus, over a wide range of conditions, clonal expansion of CD4+ T cells is limited by a combination of cell cycle arrest and apoptosis.
CTLA-4 Limits Clonal Expansion.
The fact that each experiment followed the same fundamental pattern was quite unexpected since the incorporation of radioactive thymidine yielded orders-of-magnitude differences between the experimental variables we employed (data not shown). Although CD28 costimulation could increase the percentage of cells that complete the fifth division before arresting and dying, there appeared to be an absolute barrier past which cells did not proceed, or if they did, underwent apoptosis. This reproducible pattern under different stimulatory conditions and at different time points suggests the possibility that clonal expansion is limited by a division-dependent counterbalance that is metered in a cell-autonomous manner.
We observed both a failure to proceed past five divisions and increasing cell death in later divisions even in cells that were deficient in FAS (data not shown), suggesting that FAS, although potentially important to subsequent cell elimination, was not required for the termination of proliferative expansion. CTLA-4−/− mice were bred to DO11.10 TCR transgenic mice, to delay the onset of disease seen in CTLA-4–deficient animals 17 35. Although CTLA-4 −/− cells from mice of this age may contain up to fivefold more activated cells than wild-type cells (9), we used only younger CTLA-4−/− DO11.10 animals and excluded from analysis any mice with grossly enlarged spleens or lymph nodes. CTLA-4−/− DO11.10 TCR transgenic cells were compared with those from DO11.10 TCR transgenic mice, and CD28−/− DO11.10 TCR transgenic mice. CD4+ T cells from CD28−/− mice had a profound proliferative defect compared with normal cells (Fig. 2 A). CD28−/− cells did not achieve significant cellular expansion, despite many precursors undergoing at least one division, because many of the cells in culture underwent apoptosis (Fig. 2 A).
Figure 2.


CTLA-4 limits clonal expansion of CD4+ T cells. (A) CFSE-labeled splenocytes from 3–4-wk-old B10.D2 wild-type (top), CD28−/− (middle), and CTLA-4−/− (bottom) DO11.10 TCR transgenic mice were stimulated with anti-CD3 mAb (2.0 μg/ml) for 4 d. For each genotype, computation of relative cellularity of live-gated, CD4+ cells within each cell division was determined as in Fig. 1 A, and is depicted graphically (left column). At the time of harvest, and anti-CD4 staining, cells were also resuspended in propidium iodide for determination of cell death (right column). Flow cytometric data of CFSE dilution (“Cell Division”, x-axis) versus exclusion (lower gate)/inclusion (upper gate) of propidium iodide (y-axis) among all CD4+ events is displayed to the right of cellularity graphs. Numbers above horizontal gates indicate the percentage of dead cells. Results are representative of three separate experiments. (B) CFSE-labeled splenocytes from 5-wk-old littermate BALB/c CTLA-4+/+ (top) and CTLA-4+/− (bottom) DO11.10 TCR transgenic mice were stimulated with anti-CD3 mAb (2.0 μg/ml) for 4 d and analyzed identically to the manner described for part A. Identical results were obtained with cells from B10.D2 littermate-matched pairs of CTLA-4+/+ and CTLA-4+/− mice. Results are representative of four separate experiments.
In contrast, T cells from CTLA-4−/− mice divided at least 2–3 divisions more than wild-type cells, and did not show any evidence of reaching a clear limit of proliferative cell division (Fig. 2 A). The proliferative arrest of wild-type cells was accompanied by substantial apoptosis of cells that completed multiple rounds of division, manifest as an increase in propidium iodide uptake in the later cell divisions (Fig. 2 A). In contrast, the unchecked clonal expansion of CTLA-4−/− cells correlated with decreased cell death in the later divisions of proliferating cells (Fig. 2 A). The apex of cellularity in CTLA-4−/− cells occurred at even later divisions than what had been achieved in wild-type cells cultured with anti-CD28 (Fig. 1 A), suggesting that intact CTLA-4 functions even in the presence of maximal CD28-mediated costimulation.
Genetic confirmation that CTLA-4 regulates cell division was recently observed in vivo using recombination activating gene (RAG)−/−CTLA-4−/− TCR transgenic cells 17. RAG-proficient CTLA-4−/− TCR transgenic cells, however, might proliferate differently from normal cells because they often contain higher percentages of activated cells 17 35. To control for such secondary effects within CTLA-4−/− cells, and to determine whether the mechanism limiting clonal expansion might be quantitative, we also studied CTLA-4-haplo-insufficient cells (Fig. 2 B), which do not have increased numbers of activated cells (data not shown). We found that CTLA-4+/− cells had both increased number of cells and decreased frequency of apoptosis in later divisions compared with cells from CTLA-4+/+ littermates. Among CTLA-4-haplo-insufficient cells, a greater percentage completed six divisions than did wild-type cells maximally costimulated through CD28 (Fig. 2 B and 1 A, respectively). Thus, CTLA-4, in a dose-dependent manner, is a critical regulator of clonal expansion of CD4+ T cells.
Clonal expansion is regulated at the level of proliferation and survival 8. We, therefore, tested whether proliferative arrest could be experimentally separated from apoptosis as a cause for the decline in cellularity with increasing division. CD4+ T cells expressing a Bcl-xL transgene had substantially reduced apoptosis compared with normal cells, but no significant enhancement of proliferation (Fig. 3 A). In contrast, we could correct the proliferative defect in CD28−/− cells by adding rIL-2 (Fig. 3 A), however, few viable cells remained that could complete the sixth cell division. Thus, CTLA-4 antagonizes both proliferative and survival signals mediated by costimulation from CD28, which can be separately attributed to the actions of IL-2 and Bcl-xL, respectively 37. In addition, we tested the effect of a relative decrease in B7 ligands by stimulating highly purified CD4+ cells with immobilized anti-CD3 plus anti-CD28 and rIL-2. In the absence of antigen-presenting cells, we found that division-dependent changes in apoptosis were substantially attenuated (Fig. 3 B).
Figure 3.

Survival and proliferation are antagonized in clonally expanding CD4+ T cells. 4 d after stimulation, cells were stained with anti-CD4 mAb and resuspended in propidium iodide for determination of cell death. Flow cytometric data of CFSE dilution (“Cell Division”, x-axis) versus exclusion/inclusion of propidium iodide (y-axis) among all CD4+ events are displayed. Numbers above horizontal gates indicate the percentage of dead cells as determined by uptake of propidium iodide. (A) CFSE-labeled splenocytes from 6-wk-old BALB/c (from left to right) wild-type, Bcl-xL-transgenic, and CD28−/− DO11.10 TCR transgenic mice were stimulated with anti-CD3 mAb (2.0 μg/ml) for 4 d. Where indicated (far right panel), human rIL-2 (50 U/ml) was added. (B) In a separate experiment, splenocytes from 6-wk-old C57BL/6 mice were either CD8-depleted (left) or subjected to positive selection (>98% purity) of CD4+ cells (right) before labeling with CFSE and stimulation with either soluble anti-CD3 mAb and anti-CD28 mAb (2.0 μg/ml) plus human rIL-2 (5 U/ml) (left) or plate-bound anti-CD3 mAb and soluble anti-CD28 mAb (2.0 μg/ml) plus human rIL-2 (5 U/ml) (right, “no APCs”). All results are representative of at least two separate experiments.
Induction of CTLA-4 Is Coupled to the Cell Cycle.
The precise, invariant regulation of the limits on clonal cell division (Fig. 1 and Fig. 2) suggested the existence of a cell-autonomous mechanism controlling the action of CTLA-4. Kinetics of CTLA-4 induction 38 39, together with the phenotype of CTLA-4-haplo-insufficiency (Fig. 2 B), further suggested that this counterbalance might be regulated at the level of receptor dosage. We, therefore, examined the total cellular levels of CTLA-4 in stimulated, proliferating CD4+ T cells. We found that the levels of CTLA-4 were low in undivided cells, and that levels increased progressively within the initial cell divisions, achieving maximal expression after ∼3 cell divisions (Fig. 4 A). Addition of anti-CD28 and rIL-2, positive regulators of CTLA-4 expression 39, did not influence the pattern of induction per cell division but did accelerate its induction in relation to time, by causing more cells to be represented in later divisions over a fixed time (data not shown).
Figure 4.

Cell cycle–coupled induction of CTLA-4. (A–E) C57BL/6, CD8-depleted, CFSE-labeled splenocytes were stimulated with anti-CD3 (2.0 μg/ml) for 3 d. All flow cytometric plots depict only live-gated, CD4+ events. Polygonal gates are drawn around the upper limit of background staining, as illustrated in part A. (A) After culture in the absence (left) or presence (right) of mimosine (300 μM), cells were fixed, permeabilized, and stained (see Materials and Methods) with anti-CD4 mAb and either fluorochrome-conjugated hamster control mAb (top row) or hamster anti–mouse CTLA-4 mAb (lower row), before analysis of cell division (x-axis) versus control staining (top) or total cellular CTLA-4 expression (bottom) (y-axis). (B) Cells were washed and stained (without prior fixation or permeabilization) with either anti-CD25, anti-CD44, or control mAb, before analysis of cell division (x-axis) versus surface expression (y-axis) of CD25 (left panels) and CD44 (right panels). (C) Cells were washed, fixed and either stained directly (“surface,” left panel) or permeabilized before staining (“total,” right panel) as above. (D) In the same experiment as C, a group of cells was stimulated in the presence of sodium butyrate (600 μM), to inhibit histone deacetylases, and analyzed for total cellular CTLA-4 expression (y-axis). (E) Cytochalasin B (3.5 μg/ml) was used to prevent cytokinesis in stimulated cells (left panel) before analysis of both DNA content (x-axis, right panel) and CTLA-4 expression (y-axis). (F) Splenocytes from 4 week-old BALB/c CTLA-4+/+ (filled symbols) and CTLA-4+/− (open symbols) littermate mice were stimulated and analyzed as in part A. Geometric mean fluorescence intensity of CTLA-4 expression (y-axis) for each cell generation (x-axis) is displayed. All experiments were performed at least twice.
To further discriminate between time- and division-dependent controls of expression, we used inhibitors of cell cycle and monitored CTLA-4 expression in proliferating or arrested cells cultured for the same length of time. Cells arrested in G1 using mimosine 40 had defective induction of CTLA-4 expression (Fig. 4 A). Cells arrested in S or G2/M had higher levels than cells that had undergone an earlier arrest, but less than the peak level achieved by proliferating cells (data not shown). To control for the specificity of this regulatory pattern, we compared the pattern of expression of CTLA-4 with other molecules associated with T cell activation. In contrast to CTLA-4, induction of both CD25 and CD44 was maximal before the first division and did not achieve any increase with further division (Fig. 4 B). Moreover, induction of both CD25 and CD44, in contrast to CTLA-4, was unimpeded by cell cycle arrest (Fig. 4 B).
CTLA-4 can be recycled between intracellular pools and the cell surface 39 41. We, therefore, compared staining of CTLA-4 in permeabilized cells and unpermeabilized cells. The intensity of surface CTLA-4 expression in unpermeabilized cells was in a lower range than intensity of total cellular expression in permeablized cells. Nonetheless, surface expression still increased in a step-wise fashion from lowest levels in undivided cells to highest levels after the third division (Fig. 4 C).
The division-dependent induction of CTLA-4 in naive cells, and its more rapid reiteration in immunologically experienced cells 42, suggested that gene expression might be regulated by epigenetic mechanisms of repression. To test this, we cultured cells in the presence of sodium butyrate, an inhibitor of histone deacetylase 43 that is capable of derepressing some silent genes. The graded pattern of total cellular CTLA-4 expression in the initial cell divisions was substantially increased (Fig. 4C and Fig. D) by the addition of butyrate. Trichostatin A, a more specific inhibitor of histone deacetylation, and 5-aza-2-deoxycytidine, an inhibitor of cytosine methylation, also caused increases in the level of CTLA-4 induction within the first three cell divisions (data not shown). Thus, chromatin structure might be a rate-limiting factor in the induction of CTLA-4.
The augmentation of CTLA-4 expression with agents that modify chromatin structure and the requirement of cell cycling to induce CTLA-4 expression suggested that activation of this gene might be coupled to DNA replication. To dissect the contributions of cytoplasmic from nuclear division, cells were cultured with cytochalasin B, an inhibitor of actin polymerization 44. Cytochalasin B–treated cells were unable to undergo cytokinesis (Fig. 4 E), but, in stark contrast to cells arrested in G1 (Fig. 4 A), were able to express near-maximal levels of CTLA-4 (Fig. 4 E). When we analyzed which cells had undergone gene induction, we found that the level of CTLA-4 expression was directly linked to DNA content. Lowest levels of CTLA-4 expression (4 geometric mean-fluorescence-intensity units above background) were found in cells with 2N DNA content, and highest levels (61 geometric mean-fluorescence-intensity units above background) were found in cells with 8N DNA content (Fig. 4 E). It is unlikely this 15-fold increase is due simply to the quadrupling of alleles as, in a separate analysis, we found that levels of CD25 and CD44 increased only 4-fold while levels of CTLA-4 increased 13-fold. Thus, CTLA-4 gene expression may be progressively induced in the first three cell divisions because of a mechanism coupled to DNA replication. That such small changes in protein expression between the initial cell divisions might indeed mediate significant biological effects is further suggested by the observation that the differences in levels of CTLA-4 expression in CTLA-4+/+ and CTLA-4+/− cells (Fig. 4 F) were found to closely parallel their differences in survival and cell division (Fig. 2 B).
Growth and Maturation Signals Can Counteract the Limits on Clonal Expansion.
During analysis of proliferating CTLA-4+/+ and CTLA-4−/− cells, we noted that CTLA-4 functions in activated cells to limit their size as well as their proliferation (data not shown). Comparison of cells at day 4 demonstrated that the over-representation of CTLA-4−/− cells in later cell divisions was accompanied by increased cell size, as measured by forward light scatter, in comparison to control cells (Fig. 5 A). This suggested that CTLA-4 function was restricting not only proliferation but also cell growth.
Figure 5.

Growth signals can counteract intrinsic limits on clonal expansion. In all experiments, T cells were stimulated for 4 d. All flow cytometric plots depict only live-gated, CD4+ events. Vertical gates are drawn before the fifth cell division, and percentage of cells that achieved five or more divisions is indicated in left top or bottom corner. (A) CFSE-labeled splenocytes from 3–4-wk-old B10.D2 CTLA4+/+ (top panel) and CTLA-4−/− (bottom panel) DO11.10 TCR transgenic mice were stimulated with anti-CD3 (2.0 μg/ml), before staining with anti-CD4 mAb, and analysis of CFSE dilution (“Cell Division,” x-axis) versus forward light scatter (“Cell Size,” y-axis). (B) CD8-depleted, CFSE-labeled splenocytes were stimulated with anti-CD3 in the indicated doses of human rIL-2 before analysis as in part A. (C) CD8-depleted, CFSE-labeled splenocytes were stimulated with anti-CD3 (2.0 μg/ml), anti-CD28 (0.5 μg/ml), rIL-2 (10 U/ml), and either rIL-12 (5 ng/ml) plus anti–IL-4 (10 μg/ml) (“Th1”conditions, left) or rIL-4 (5 U/ml) plus anti–IL-12 (10 μg/ml) (“Th2” conditions, right) before analysis of intracellular cytokine staining as described in Materials and Methods. Cell division (x-axis) and intracellular accumulation (y-axis) of IFN-γ (left) or IL-4 (right) are depicted in the top row inset. Horizontal gates indicate level of background staining. Cells that were unambiguously cytokine-negative (middle panels) or cytokine-positive (bottom panels) were further analyzed for cell division (x-axis) and cell size (y-axis) as in part A. (D) Expression of CCR7 (top panels) and HPRT (bottom panels) was determined by RT-PCR among indicated cell division numbers (“Div. No.”), as described in Materials and Methods. All results are representative of at least three separate experiments.
Extrinsic growth factor signaling can permit nutrient utilization in T lymphocytes 45. We, therefore, used the parameters of cell division and cell size to determine whether the clonal growth arrest mediated by CTLA-4 was a general checkpoint in the maturation of CD4+ T cell immunity. Because IL-2 expression begins to be downregulated after approximately three cell divisions 26, we tested whether saturating levels of rIL-2 could alter the pattern of growth arrest. Indeed, cells cultured in 200 U/ml of rIL-2 could remain larger and divide more than those cultured in 2 U/ml of rIL-2 (Fig. 5 B). Thus, in the course of an immune response, the presence of survival signals might regulate the outcome of clonal expansion.
IL-12 46 47 and IL-4 48 49 promote growth and maturation of committed Th1 and Th2 subsets, respectively. We, therefore, examined the effects of IL-12 and IL-4 on cell division and cell size during polarized immune responses. In the presence of rIL-12, newly differentiating, IFN-γ–positive, Th1 cells were able to undergo more division and grow to substantially larger size than undifferentiated, IFN-γ–negative cells within the same culture (Fig. 5 C). In the presence of rIL-4, newly differentiating, IL-4–positive, Th2 cells were able to undergo more division and grow to substantially larger size than undifferentiated, IL-4–negative cells in the same culture. A large proportion of differentiated cells progressed past the fifth cell division, while only a small fraction of undifferentiated cells could do so (Fig. 5 C). Thus, in subsets of cells that are specifically responsive, growth signals can successfully counteract CTLA-4–mediated proliferative- and growth-arrest.
In human CD4+ T cells, expression of CCR7 protein begins to be downregulated after five cell divisions 30, giving progeny that achieved greater cell division the potential to migrate from lymphoid organs to peripheral tissues 29. To determine whether the pattern of expression in relation to cell division was similar in the mouse and, further, whether this was the result of transcriptional repression, we analyzed mRNA levels of CCR7 using RT-PCR among individual sorted cell generations of CD4+ T cells that had been stimulated in Th1- and Th2-polarizing conditions (Fig. 5 D). In either cytokine milieu, the expression of CCR7 was highest in cells that had divided less than five times. After ∼5 cell divisions levels of CCR7 mRNA became low-to-undetectable in both cytokine conditions (Fig. 5 D). Therefore, as is the case in human cells 30, expression of murine CCR7 in Th cells is regulated by cell division. Thus, cytokines that mediate polarization of immunity might function to ensure that subsets of effector T cells can overcome clonal arrest and selectively achieve division numbers (Fig. 5 C) that allow their emigration from lymph nodes 2 29.
Discussion
The present results suggest that the pattern of clonal expansion of CD4+ T cells has a remarkable, and previously uncharacterized, uniformity in response to a broad range of stimuli (Fig. 6). After nonpolarizing stimulation, CD4+ T cells clonally expand, reaching an apex of cellularity at approximately the third cell division. Less than 5% of viable cells are likely to complete the sixth cell division. This control over proliferation appears dependent on CTLA-4, and in its absence, CD4+ T cells display unbridled expansion, due to both excessive cell division and enhanced survival. Although death receptors may be the final executioner of those cells undergoing attrition 5 6 7, our genetic experiments indicate that CTLA-4 alone is sufficient to discriminate the divisional history of a cell and mark generations for death or arrest.
Figure 6.

CTLA-4 as the central checkpoint of clonal expansion. Speculative model in which limits imposed on clonal expansion of a precursor (“P”) CD4+ T cell by CTLA-4 expression and function can mediate various immunologic outcomes. Progressive induction of CTLA-4 during the initial cell divisions (triangle of dose response) would maintain homeostasis (manifest as anergy or tolerance) in the absence of inflammatory signals by limiting the division of clonal progeny. In response to a pathogen, a subset of clonal progeny may receive growth and maturation signals that cause further division, enabling their emigration from lymph nodes. Pathogen-specific clonal progeny that arrest before the fifth division could remain in lymphoid niches that may support long-term survival (memory), perhaps to be rapidly mobilized into further division and emigration during reinfection.
Signal transduction through CD28 is likely to be an important element in this control mechanism for at least two reasons. First, CD28-deficient T cells, despite a limited ability to divide, do not undergo clonal expansion in the presence of an intact CTLA-4 system (Fig. 2 and Fig. 3). Second, genetic experiments reveal that a deficiency of B7 ligands, which bind both receptors, can correct the auto-aggressive phenotype of CTLA-4 deficiency 50. Thus, the gene duplication that gave rise to this pair of receptors represents an evolutionary “yin-yang” 51, in which the locus encoding the receptor pair is wholly epistatic.
The unique regulation of CTLA-4 gene expression allows the CD28 receptor, the nondominant member of the pair for binding B7 52, to become engaged initially without complete interference from CTLA-4. Counterbalance from CTLA-4 is then allotted in a precisely controlled manner by the cell cycle and its ability to permit gene induction in proportion to proliferation. We are still determining whether there is actual repression in cis at the CTLA-4 locus, in trans at the locus of an activator of the CTLA-4 promoter, or some other explanation for the graded, division-dependent gene induction. Nonetheless, cell cycle–coupled regulatory and epigenetic effects might be the mechanistic explanation for what others have recently recognized as a level of cell-autonomous control in the way lymphocytes respond to extrinsic signals 2 4.
There are surely some redundant or cooperative negative controls for CD4+ T cells since CTLA-4−/− cells do not behave as though immortalized. These are likely to be the death receptors and the in vivo limitations or niches that permit survival or growth, such as nonpolarizing and polarizing cytokines. The phenotype of cells with CTLA-4 deficiency that have been stimulated in vitro is strikingly similar to the behavior of wild-type Th1 cells cultured in IL-12 or wild-type Th2 cells cultured in IL-4. These results support a model in which programming of selective growth factor reception 46 47 48 49 is a proximate event in effector lineage commitment 53. Early in differentiation, Th1 cells become uniquely programmed for IL-12Rβ2 expression 53 54 55, and Th2 cells become uniquely competent in IL-4R signal transduction 56 57. Subsets of differentiated cells might, therefore, receive selective signaling from the cytokine milieu to progress to a CCR7-negative stage and thereby mediate immunity in tissues (Fig. 6). This checkpoint, thus, sharpens polarity from cytokines, and also restricts important fate transitions in the effector and memory response 2 29 30.
Our results may provide insight into the elusive basis of long-lived immunologic memory, as well as the unusual auto-aggressive phenotype of CTLA-4–null mice, wherein peripheral tissues are the primary targets of inflammation. The limitation on the number of divisions a cell undergoes, provided by CTLA-4 (Fig. 6), may ensure a long-lived remnant of CCR7-positive clonal progeny in central lymphoid compartments, which can be rapidly mobilized during rechallenge 58 59. If T cells are fated to linear, terminal differentiation as they divide, a proliferative arrest of some progeny may be the only way to maintain self-renewal of clonally selected cells. The substantial increase in division number that is readily achieved by CTLA-4–deficient cells would, therefore, be compatible with predominantly peripheral tissue inflammation (Fig. 6).
In double-positive thymocytes, variations in signal transduction through the antigen receptor are integrated into vastly different outcomes by intrinsic rheostats, such as Grb2 60. In the periphery, where TCR signaling is coupled to proliferation, and perhaps obligatory for survival 61 62 63, CTLA-4 gene induction and function appears to be an intrinsic rheostat that may be a central checkpoint of antigen-driven clonal expansion (Fig. 6). Further maturation and expansion might then become dependent on nonantigenic extrinsic signals, such as those provided by inflammatory cytokines, which can mediate avoidance of clonal arrest.
Acknowledgments
We are grateful to K. Frauwirth and M. Alegre for helpful discussion, and W. DeMuth for cell sorting.
This work was supported by grants from the National Institutes of Health (A1-42370 to S.L. Reiner, AI-35294 to C.B. Thompson, and DK-07006 for A.M. Doyle).
Footnotes
Abbreviations used in this paper: CCR, CC chemokine receptor; CFSE, carboxy-fluorescein diacetate succinimidyl ester; CTLA, cytotoxic T lymphocyte antigen; HPRT, hypoxanthine guanine phosphoribosyl transferase; RT, reverse transcription.
References
- Janeway C.A., Bottomly K. Signals and signs for lymphocyte responses. Cell. 1994;76:275–285. doi: 10.1016/0092-8674(94)90335-2. [DOI] [PubMed] [Google Scholar]
- Lanzavecchia A., Sallusto F. Dynamics of T lymphocyte responsesintermediates, effectors, and memory cells. Science. 2000;290:92–97. doi: 10.1126/science.290.5489.92. [DOI] [PubMed] [Google Scholar]
- Reiner S.L., Seder R.A. Dealing from the evolutionary pawnshophow lymphocytes make decisions. Immunity. 1999;11:1–10. doi: 10.1016/s1074-7613(00)80076-x. [DOI] [PubMed] [Google Scholar]
- Gett A.V., Hodgkin P.D. A cellular calculus for signal integration by T cells. Nat. Immunol. 2000;1:239–244. doi: 10.1038/79782. [DOI] [PubMed] [Google Scholar]
- Ucker D.S., Ashwell J.D., Nickas G. Activation-driven T cell death. I. Requirements for de novo transcription and translation and association with genome fragmentation. J. Immunol. 1989;143:3461–3469. [PubMed] [Google Scholar]
- Lenardo M.J. Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature. 1991;353:858–861. doi: 10.1038/353858a0. [DOI] [PubMed] [Google Scholar]
- Chan K.F., Siegel M.R., Lenardo J.M. Signaling by the TNF receptor superfamily and T cell homeostasis. Immunity. 2000;13:419–422. doi: 10.1016/s1074-7613(00)00041-8. [DOI] [PubMed] [Google Scholar]
- Van Parijs L., Abbas A.K. Homeostasis and self-tolerance in the immune systemturning lymphocytes off. Science. 1998;280:243–248. doi: 10.1126/science.280.5361.243. [DOI] [PubMed] [Google Scholar]
- Waterhouse P., Penninger J.M., Timms E., Wakeham A., Shahinian A., Lee K.P., Thompson C.B., Griesser H., Mak T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- Tivol E.A., Borriello F., Schweitzer A.N., Lynch W.P., Bluestone J.A., Sharpe A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- Chambers C.A., Cado D., Truong T., Allison J.P. Thymocyte development is normal in CTLA-4-deficient mice. Proc. Natl. Acad. Sci. USA. 1997;94:9296–9301. doi: 10.1073/pnas.94.17.9296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummel M.F., Allison J.P. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 1996;183:2533–2540. doi: 10.1084/jem.183.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walunas T.L., Bakker C.Y., Bluestone J.A. CTLA-4 ligation blocks CD28-dependent T cell activation. J. Exp. Med. 1996;183:2541–2550. doi: 10.1084/jem.183.6.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner M.C., Chambers C.A., Chan F.K., Hanke J., Winoto A., Allison J.P. CTLA-4-Mediated inhibition of early events of T cell proliferation. J. Immunol. 1999;162:5813–5820. [PubMed] [Google Scholar]
- Kearney E.R., Walunas T.L., Karr R.W., Morton P.A., Loh D.Y., Bluestone J.A., Jenkins M.K. Antigen-dependent clonal expansion of a trace population of antigen-specific CD4+ T cells in vivo is dependent on CD28 costimulation and inhibited by CTLA-4. J. Immunol. 1995;155:1032–1036. [PubMed] [Google Scholar]
- Perez V.L., Van Parijs L., Biuckians A., Zheng X.X., Strom T.B., Abbas A.K. Induction of peripheral T cell tolerance in vivo requires CTLA-4 engagement. Immunity. 1997;6:411–417. doi: 10.1016/s1074-7613(00)80284-8. [DOI] [PubMed] [Google Scholar]
- Greenwald R.J., Boussiotis V.A., Lorsbach R.B., Abbas A.K., Sharpe A.H. CTLA-4 regulates induction of anergy in vivo. Immunity. 2001;14:145–155. doi: 10.1016/s1074-7613(01)00097-8. [DOI] [PubMed] [Google Scholar]
- Salomon B., Bluestone J.A. Complexities of CD28/B7CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu. Rev. Immunol. 2001;19:225–252. doi: 10.1146/annurev.immunol.19.1.225. [DOI] [PubMed] [Google Scholar]
- Oosterwegel M.A., Mandelbrot D.A., Boyd S.D., Lorsbach R.B., Jarrett D.Y., Abbas A.K., Sharpe A.H. The role of CTLA-4 in regulating Th2 differentiation. J. Immunol. 1999;163:2634–2639. [PubMed] [Google Scholar]
- Khattri R., Auger J.A., Griffin M.D., Sharpe A.H., Bluestone J.A. Lymphoproliferative disorder in CTLA-4 knockout mice is characterized by CD28-regulated activation of Th2 responses. J. Immunol. 1999;162:5784–5791. [PubMed] [Google Scholar]
- Masteller E.L., Chuang E., Mullen A.C., Reiner S.L., Thompson C.B. Structural analysis of CTLA-4 function in vivo. J. Immunol. 2000;164:5319–5327. doi: 10.4049/jimmunol.164.10.5319. [DOI] [PubMed] [Google Scholar]
- Chambers C.A., Kuhns M.S., Egen J.G., Allison J.P. CTLA-4-mediated inhibition in regulation of T cell responsesmechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001;19:565–594. doi: 10.1146/annurev.immunol.19.1.565. [DOI] [PubMed] [Google Scholar]
- McCoy K., Camberis M., Gros G.L. Protective immunity to nematode infection is induced by CTLA-4 blockade. J. Exp. Med. 1997;186:183–187. doi: 10.1084/jem.186.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M.L., Cotterell S.E., Gorak P.M., Engwerda C.R., Kaye P.M. Blockade of CTLA-4 enhances host resistance to the intracellular pathogen, Leishmania donovani . J. Immunol. 1998;161:4153–4160. [PubMed] [Google Scholar]
- Heinzel F.P., Maier R.A. Interleukin-4-independent acceleration of cutaneous leishmaniasis in susceptible BALB/c mice following treatment with anti-CTLA4 antibody. Infect. Immun. 1999;67:6454–6460. doi: 10.1128/iai.67.12.6454-6460.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird J.J., Brown D.R., Mullen A.C., Moskowitz N.H., Mahowald M.A., Sider J.R., Gajewski T.F., Wang C.R., Reiner S.L. Helper T cell differentiation is controlled by the cell cycle. Immunity. 1998;9:229–237. doi: 10.1016/s1074-7613(00)80605-6. [DOI] [PubMed] [Google Scholar]
- Gett A.V., Hodgkin P.D. Cell division regulates the T cell cytokine repertoire, revealing a mechanism underlying immune class regulation. Proc. Natl. Acad. Sci. USA. 1998;95:9488–9493. doi: 10.1073/pnas.95.16.9488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter A., Lohning M., Radbruch A. Instruction for cytokine expression in T helper lymphocytes in relation to proliferation and cell cycle progression. J. Exp. Med. 1999;190:1439–1450. doi: 10.1084/jem.190.10.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F., Lenig D., Forster R., Lipp M., Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- Langenkamp A., Messi M., Lanzavecchia A., Sallusto F. Kinetics of dendritic cell activationimpact on priming of TH1, TH2 and nonpolarized T cells. Nat. Immunol. 2000;1:311–316. doi: 10.1038/79758. [DOI] [PubMed] [Google Scholar]
- Lyons A.B., Parish C.R. Determination of lymphocyte division by flow cytometry. J. Immunol. Methods. 1994;171:131–137. doi: 10.1016/0022-1759(94)90236-4. [DOI] [PubMed] [Google Scholar]
- Shahinian A., Pfeffer K., Lee K.P., Kundig T.M., Kishihara K., Wakeham A., Kawai K., Ohashi P.S., Thompson C.B., Mak T.W. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 1993;261:609–612. doi: 10.1126/science.7688139. [DOI] [PubMed] [Google Scholar]
- Grillot D.A., Merino R., Nunez G. Bcl-XL displays restricted distribution during T cell development and inhibits multiple forms of apoptosis but not clonal deletion in transgenic mice. J. Exp. Med. 1995;182:1973–1983. doi: 10.1084/jem.182.6.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy K.M., Heimberger A.B., Loh D.Y. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science. 1990;250:1720–1723. doi: 10.1126/science.2125367. [DOI] [PubMed] [Google Scholar]
- Chambers C.A., Kuhns M.S., Allison J.P. Cytotoxic T lymphocyte antigen-4 (CTLA-4) regulates primary and secondary peptide-specific CD4(+) T cell responses. Proc. Natl. Acad. Sci. USA. 1999;96:8603–8608. doi: 10.1073/pnas.96.15.8603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner S.L., Zheng S., Corry D.B., Locksley R.M. Constructing polycompetitor cDNAs for quantitative PCR. J. Immunol. Methods. 1993;165:37–46. doi: 10.1016/0022-1759(93)90104-f. [DOI] [PubMed] [Google Scholar]
- Dahl A.M., Klein C., Andres P.G., London C.A., Lodge M.P., Mulligan R.C., Abbas A.K. Expression of bcl-X(L) restores cell survival, but not proliferation off effector differentiation, in CD28-deficient T lymphocytes. J. Exp. Med. 2000;191:2031–2038. doi: 10.1084/jem.191.12.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walunas T.L., Lenschow D.J., Bakker C.Y., Linsley P.S., Freeman G.J., Green J.M., Thompson C.B., Bluestone J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- Alegre M.L., Noel P.J., Eisfelder B.J., Chuang E., Clark M.R., Reiner S.L., Thompson C.B. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J Immunol. 1996;157:4762–4770. [PubMed] [Google Scholar]
- Lalande M. A reversible arrest point in the late G1 phase of the mammalian cell cycle. Exp. Cell Res. 1990;186:332–339. doi: 10.1016/0014-4827(90)90313-y. [DOI] [PubMed] [Google Scholar]
- Linsley P.S., Bradshaw J., Greene J., Peach R., Bennett K.L., Mittler R.S. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity. 1996;4:535–543. doi: 10.1016/s1074-7613(00)80480-x. [DOI] [PubMed] [Google Scholar]
- Alegre M.L., Shiels H., Thompson C.B., Gajewski T.F. Expression and function of CTLA-4 in Th1 and Th2 cells. J. Immunol. 1998;161:3347–3356. [PubMed] [Google Scholar]
- Kruh J. Effects of sodium butyrate, a new pharmacological agent, on cells in culture. Mol. Cell. Biochem. 1982;42:65–82. doi: 10.1007/BF00222695. [DOI] [PubMed] [Google Scholar]
- Smith G.F., Ridler M.A., Faunch J.A. Action of cytochalasin B on cultured human lymphocytes. Nature. 1967;216:1134–1135. doi: 10.1038/2161134a0. [DOI] [PubMed] [Google Scholar]
- Rathmell J.C., Vander Heiden M.G., Harris M.H., Frauwirth K.A., Thompson C.B. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol. Cell. 2000;6:683–692. doi: 10.1016/s1097-2765(00)00066-6. [DOI] [PubMed] [Google Scholar]
- Kennedy M.K., Picha K.S., Shanebeck K.D., Anderson D.M., Grabstein K.H. Interleukin-12 regulates the proliferation of Th1, but not Th2 or Th0, clones. Eur. J. Immunol. 1994;24:2271–2278. doi: 10.1002/eji.1830241002. [DOI] [PubMed] [Google Scholar]
- Murphy E.E., Terres G., Macatonia S.E., Hsieh C.S., Mattson J., Lanier L., Wysocka M., Trinchieri G., Murphy K., O'Garra A. B7 and interleukin 12 cooperate for proliferation and interferon gamma production by mouse T helper clones that are unresponsive to B7 costimulation. J. Exp. Med. 1994;180:223–231. doi: 10.1084/jem.180.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurt-Jones E.A., Hamberg S., Ohara J., Paul W.E., Abbas A.K. Heterogeneity of helper/inducer T lymphocytes. I. Lymphokine production and lymphokine responsiveness. J. Exp. Med. 1987;166:1774–1787. doi: 10.1084/jem.166.6.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Botran R., Sanders V.M., Mosmann T.R., Vitetta E.S. Lymphokine-mediated regulation of the proliferative response of clones of T helper 1 and T helper 2 cells. J. Exp. Med. 1988;168:543–558. doi: 10.1084/jem.168.2.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelbrot D.A., McAdam A.J., Sharpe A.H. B7-1 or B7-2 is required to produce the lymphoproliferative phenotype in mice lacking cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) J. Exp. Med. 1999;189:435–440. doi: 10.1084/jem.189.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison J.P., Krummel M.F. The Yin and Yang of T cell costimulation. Science. 1995;270:932–933. doi: 10.1126/science.270.5238.932. [DOI] [PubMed] [Google Scholar]
- Linsley P.S., Greene J.L., Brady W., Bajorath J., Ledbetter J.A., Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity. 1994;1:793–801. doi: 10.1016/s1074-7613(94)80021-9. [DOI] [PubMed] [Google Scholar]
- Mullen A.C., High F.A., Hutchins A.S., Lee H.W., Villarino A.V., Livingston D.M., Kung A.L., Cereb N., Yao T.-P., Yang S.Y., Reiner S.L. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science. 2001;292:1907–1910. doi: 10.1126/science.1059835. [DOI] [PubMed] [Google Scholar]
- Szabo S.J., Jacobson N.G., Dighe A.S., Gubler U., Murphy K.M. Developmental commitment to the Th2 lineage by extinction of IL-12 signaling. Immunity. 1995;2:665–675. doi: 10.1016/1074-7613(95)90011-x. [DOI] [PubMed] [Google Scholar]
- Ouyang W., Ranganath S.H., Weindel K., Bhattacharya D., Murphy T.L., Sha W.C., Murphy K.M. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity. 1998;9:745–755. doi: 10.1016/s1074-7613(00)80671-8. [DOI] [PubMed] [Google Scholar]
- Huang H., Paul W.E. Impaired interleukin 4 signaling in T helper type 1 cells. J. Exp. Med. 1998;187:1305–1313. doi: 10.1084/jem.187.8.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurata H., Lee H.J., O'Garra A., Arai N. Ectopic expression of activated Stat6 induces the expression of Th2-specific cytokines and transcription factors in developing Th1 cells. Immunity. 1999;11:677–688. doi: 10.1016/s1074-7613(00)80142-9. [DOI] [PubMed] [Google Scholar]
- Reinhardt R.L., Khoruts A., Merica R., Zell T., Jenkins M.K. Visualizing the generation of memory CD4 T cells in the whole body. Nature. 2001;410:101–105. doi: 10.1038/35065111. [DOI] [PubMed] [Google Scholar]
- Iezzi G., Scheidegger D., Lanzavecchia A. Migration and function of antigen-primed nonpolarized T lymphocytes in vivo. J. Exp. Med. 2001;193:987–993. doi: 10.1084/jem.193.8.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Q., Cheng A.M., Akk A.M., Alberola-Ila J., Gong G., Pawson T., Chan A.C. Disruption of T cell signaling networks and development by Grb2 haploid insufficiency. Nat. Immunol. 2001;2:29–36. doi: 10.1038/83134. [DOI] [PubMed] [Google Scholar]
- Takeda S., Rodewald H.R., Arakawa H., Bluethmann H., Shimizu T. MHC class II molecules are not required for survival of newly generated CD4+ T cells, but affect their long-term life span. Immunity. 1996;5:217–228. doi: 10.1016/s1074-7613(00)80317-9. [DOI] [PubMed] [Google Scholar]
- Kirberg J., Berns A., von Boehmer H. Peripheral T cell survival requires continual ligation of the T cell receptor to major histocompatibility complex–encoded molecules. J. Exp. Med. 1997;186:1269–1275. doi: 10.1084/jem.186.8.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooke R., Waltzinger C., Benoist C., Mathis D. Targeted complementation of MHC class II deficiency by intrathymic delivery of recombinant adenoviruses. Immunity. 1997;7:123–134. doi: 10.1016/s1074-7613(00)80515-4. [DOI] [PubMed] [Google Scholar]