

Abstract
Syncytia arising from the fusion of cells expressing a lymphotropic human immunodeficiency virus (HIV)-1–encoded envelope glycoprotein complex (Env) gene with cells expressing the CD4/CXCR4 complex undergo apoptosis through a mitochondrion-controlled pathway initiated by the upregulation of Bax. In syncytial apoptosis, phosphorylation of p53 on serine 15 (p53S15) precedes Bax upregulation, the apoptosis-linked conformational change of Bax, the insertion of Bax in mitochondrial membranes, subsequent release of cytochrome c, caspase activation, and apoptosis. p53S15 phosphorylation also occurs in vivo, in HIV-1+ donors, where it can be detected in preapoptotic and apoptotic syncytia in lymph nodes, as well as in peripheral blood mononuclear cells, correlating with viral load. Syncytium-induced p53S15 phosphorylation is mediated by the upregulation/activation of mammalian target of rapamycin (mTOR), also called FKBP12-rapamycin-associated protein (FRAP), which coimmunoprecipitates with p53. Inhibition of mTOR/FRAP by rapamycin reduces apoptosis in several paradigms of syncytium-dependent death, including in primary CD4+ lymphoblasts infected by HIV-1. Concomitantly, rapamycin inhibits p53S15 phosphorylation, mitochondrial translocation of Bax, loss of the mitochondrial transmembrane potential, mitochondrial release of cytochrome c, and nuclear chromatin condensation. Transfection with dominant negative p53 has a similar antiapoptotic action as rapamycin, upstream of the Bax upregulation/translocation. In summary, we demonstrate that phosphorylation of p53S15 by mTOR/FRAP plays a critical role in syncytial apoptosis driven by HIV-1 Env.
Keywords: cell death, envelope glycoprotein complex, human immunodeficiency virus, mitochondria, rapamycin
Introduction
HIV-1 infection can cause apoptosis via a variety of mechanisms, some of which rely on the intricate virus host cell interaction (in vitro) and some of which involve activation of the host's inflammatory and immune systems (in vivo; references 1 and 2). The envelope glycoprotein complex (Env)(gp120/gp41) appears to be one of the dominant apoptosis-inducing molecules encoded by the HIV-1 genome. Env expressed on the plasma membrane of infected cells can interact with CD4 and a suitable coreceptor (e.g., CXCR4) to trigger cell-to-cell fusion; the resulting syncytia subsequently undergo apoptosis. This applies to primary CD4+ T lymphocytes inoculated by HIV-1 (references 3 and 4), as well as to cell lines stably transfected with human CD4 cocultured with cells expressing a lymphotropic HIV-1 Env gene 5 6 7. A positive correlation between CD4+ T cell decline and infection by syncytium-inducing HIV-1 or SIV-1 variants has been established in vitro 3 4 8 9 and, more importantly, in vivo, in humans with AIDS 10 11, humanized severe combined immunodeficient mice 12, and monkeys 13. This suggests that fusion-induced apoptosis is relevant to AIDS pathogenesis, although syncytia are relatively infrequent in lymphoid tissues of HIV-1+ donors 14. In lymph nodes from HIV-1–infected individuals, syncytia express markers of early apoptosis such as tissue transglutaminase (tTGase; reference 15). The scarcity of syncytia in vivo may thus be attributed to their rapid apoptotic degeneration and phagocytic removal, yet does not argue against the pathophysiological importance of fusion-induced apoptosis.
The apoptosis-associated activation of caspases can be triggered by two pathways 16 17. In the “extrinsic” pathway, the ligation of plasma membrane death receptors (e.g., CD95, TNF-R) leads to the recruitment of caspase-8 to the receptor complex, culminating in its proteolytic autoactivation 16. In the “intrinsic” pathway, mitochondrial membrane permeabilization (MMP) results in the release of cytochrome c (Cyt. c) from the mitochondrial intermembrane space. Once in the cytosol, Cyt. c triggers the oligomerization of Apaf-1, which in turn recruits procaspase-9 and procaspase-3 into the apoptosome, the caspase activation multiprotein complex 18. MMP may be elicited by a variety of different proapoptotic second messengers (e.g., Ca2+, ganglioside GD3, reactive oxygen species), as well as proapoptotic members of the Bcl-2 family such as Bax, Bak, Bid, Bim, etc. 17. Nuclear DNA damage is one of the best studied initiators of the intrinsic pathway of apoptosis 19 20. DNA double-strand breaks are sensed by two enzymes from a protein family termed the phosphatidylinositol kinase-related kinases (or PIKKs), in particular ataxia teleangiectasia mutated (ATM) and DNA-dependent protein kinase (DNA-PK). UV-induced damage is mainly sensed by ataxia teleangiectasia Rad3 related (ATR), yet another member of the PIKK family. DNA-PK, ATM, ATR, as well as the downstream DNA-damage-induced kinases Chk1 (activated by UV) and Chk2 (activated by ionizing radiation), phosphorylate serine residues within the NH2 terminus of p53, a modification that by itself may increase its transactivating function and/or inhibits the interaction of p53 with MDM2 and so prevents degradation of the p53 protein, which accumulates and can act as a transcription factor, stimulating the expression of several MMP-inducing proteins including Bax 21.
Syncytial apoptosis induced by the interaction between Env and CD4/CXCR4, in the absence of viral infection, involves a mitochondrial pathway characterized by the following sequence of events: (i) translocation of Bax from the cytosol to mitochondria; (ii) Bax-mediated MMP with loss of the mitochondrial transmembrane potential (ΔΨm), release of apoptogenic intermembrane proteins, in particular apoptosis-inducing factor and Cyt. c, (iii) caspase activation, and (iv) nuclear chromatin condensation 6. This sequence of events has been confirmed, in principle, for HIV-1–infected HeLa cells transfected with CD4, as well as for primary CD4+ lymphoblasts infected with lymphotropic HIV-1 6 22. No information was available on the premitochondrial proapoptotic signal transduction pathways involved in Env-induced syncytial apoptosis. Therefore, we have employed a systematic approach, based on proteomics and inhibitor screening assays, to define such upstream signals accounting for the induction and mitochondrial translocation of Bax. Here we show that phosphorylation of p53, via a pathway not related to DNA damage, is involved in Env-induced syncytial apoptosis. Indeed, it appears that p53 is phosphorylated on serine 15 by mammalian target of rapamycin (mTOR), also called FKBP12-rapamycin-associated protein (FRAP), a protein from the PIKK family that previously has been suggested to serve as a master regulator of the balance between protein synthesis and degradation 23, thereby participating in the control of cell size 24 25 and perhaps in oncogenic transformation 26.
Materials and Methods
Cells Lines, Transfection, Microinjection, and Comet Assays.
HeLa cells transfected with the Env gene of HIV-1 LAI (HeLa Env, reference 27) and HeLa cells transfected with CD4 (HeLa CD4, reference 5) were cultured alone or together (1:1 ratio) in medium supplemented with 10% FCS, as described previously 5 6 27. Mouse embryonic fibroblasts (MEFs) with different genotypes (p53+/+, p53−/+, p53−/−) were a gift from T. Jacks, MIT, Boston, MA. MEFs were cultured on coverslips and were fused with polyethylene glycol (PEG) (45 s of incubation with prewarmed [37°C] 50% wt/vol PEG from Sigma-Aldrich, 1,450, in Ca2+-free PBS, pH 7.2; preceded by two washings with FCS-free medium), followed by four washings with complete medium. N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z-VAD. fmk, used at 100 μM) was added each 24 h, whereas other inhibitors were added only once, from the beginning of the culture. Transfection with pcDNA3.1 vector only, pBR322 plasmid constructs containing wild-type p53 or mutant (H175, H273) p53 28 (gift of T. Soussi, Institut Curie, Paris, France), expressed under the control of the cytomegalovirus promoter, luciferase, terminal oligopyrimidine (TOP)-luciferase constructs 29 (gift of J. Chen, Illinois University, Urbana, IL), or a p53-responsive enhanced green fluorescent protein (GFP) plasmid (gift from K. Wiman, Karolinska Cancer Center, Stockholm, Sweden) was performed by electroporation 30 24 h before coculture of HeLa CD4 and HeLa Env cells. The p53-responsive GFP plasmid was generated by replacing the luciferase gene in PG13PY Luc (a luciferase construct containing 13 repeats of the p53-binding oligonucleotide 5′-CCTGCCCTGGGACTTGCCTGG-3′, gift from Bert Vogelstein, Howard Hughes Medical Institute, Baltimore, MD) with an EcoA7III-MluI fragment from pEGFP-C1. In some experiments, all syncytia growing on a premarked V-shaped area of a coverslip (>200 per experiment) were microinjected into the cytoplasm 31, with PBS only, pH 7.2, recombinant human Bcl-2 (amino acids 1–218; 500 ng/μl; reference 32), a mAb specific for mTOR/FRAP (BD Transduction Laboratories) or an isotype-(IgG2a) matched LAMP-1 mAb (Oncogene Research Products) (both at 50 ng/μl). Comet assays were performed using a kit from Trevigen, on HeLa Env cells (untreated or treated with 100 μM H2O2 for 30 min) or 24-h-old HeLa Env/CD4 syncytia (nonapoptotic adherent or apoptotic cells).
Patients' Samples.
Peripheral blood samples were obtained from 49 HIV-infected individuals from the National Institute for Infectious Diseases (IRCCS Lazzaro Spallanzani, Rome, Italy). Patients (mean age 34 ± 11 y, n = 49) were included in this study according to the following criteria: asymptomatic HIV-1 infection (A, CDC 1993), under at least 2 mo of antiretroviral therapy interruption, CD4+ T lymphocytes in peripheral blood in the range of 300–600/μl, plasma HIV-RNA levels >80 copies per milliliter, no treatment with IFNs or corticosteroids, absence of infection with hepatitis C virus, hepatitis B virus, and autoimmune disease. All patients provided informed written consent according to the Ethical Committee. PBMCs were isolated by Ficoll/Hypaque (Amersham Pharmacia Biotech) centrifugation of heparinized blood from either healthy donors and HIV-seropositive individuals and fixed with 4% formaldehyde in PBS, pH 7.2. Plasma HIV-1 RNA levels were determined by the Nucleic Acid Sequence Based Amplification (NASBA) procedure (HIV-1 RNA QT assay; Organon Teknika). Plasma levels of HIV-1 RNA were expressed as HIV-RNA copies per milliliter with a threshold level of 80 copies per milliliter. Biopsies of axillary lymph nodes, obtained from healthy donors and HIV-1+ asymptomatic patients, naive for antiretroviral therapy, were immediately fixed with 10% formalin neutral buffered, dehydrated, and paraffin embedded.
HIV-1 Infection.
5 × 106 HeLa CD4 cells were cultured in the presence of 2.5 × 106 chronically HIV-1–infected H9/IIIB cells (obtained from R.C. Gallo, National Institutes of Health, Bethesda, MD) in 5 ml of RPMI 1640 supplemented with 10% heat inactivated FCS and 2 mM glutamine. Alternatively, CEM cells containing a plasmid encoding GFP driven by the HIV-1 long terminal repeat 33; clone 8D6; obtained from J. Corbel, University of California at San Diego, San Diego, CA) were cocultured (1:1 ratio) with HIV-1–infected H9/IIIB cells. Cell-to-cell fusion was assessed by determining the GFP-dependent fluorescence (excitation 458 nm, emission 515 nm) in a Fluoroscan plate fluorometer (Labsystems). Alternatively, cells were resuspended in a solution containing 3.4 mM sodium citrate, 0.05 mg/ml propidium iodide, 0.1 mM EDTA, 1 mM Tris, pH 8, and 0.1% Triton X-100 34, incubated for 60 min at reverse transcription, and analyzed for DNA content in a FACScalibur™ (Becton Dickinson). This method allows for the quantitation of all apoptotic nuclei, including those of syncytia, which are disrupted by the hypotonic buffer. CD4+ T cells were purified from freshly isolated PBMCs by immunomagnetic negative selection (Stem Cell Technologies). Cells were cultured for 2 d in RPMI 1640 medium supplemented with 10% FCS (GIBCO BRL) containing 3 μg/ml PHA (Sigma-Aldrich) and 6 U/ml IL-2 (IL-2; Boehringer Mannheim). For HIV-1 infection, 10 × 106 cells were incubated with the T cell line–adapted HIV-1 strain IIIB (500 ng of p24) for 4 h at 37°C. After washing out unabsorbed virus, cells were cultured at a density of 106 cells per milliliter in RPMI 1640 medium containing 20% FCS and 10 UI/ml IL-2 in 24-well plates. 2 d after infection, when the first syncytia appeared in the infected cultures, infected and uninfected cultures were treated with 5 μg/ml AMD3100, 100 μM ZVAD.fmk, or the indicated dose of rapamycin. Infection was allowed to proceed for 2 more d. At this time mitochondrial function and nuclear morphology were evaluated after JC1/Hoechst staining as described below. Viral replication was evaluated by measuring the level of HIV-1 core protein p24 in the supernatant (Innogenetics ELISA kit). Promonocytic U937 cell line were infected at a multiplicity of infection of 0.5 TCID50 per cell, for 2 h at 37°C washed three times in PBS.
Quantitation of Protein Expression Levels.
Protein samples were simultaneously prepared from HeLa Env and HeLa CD4 single cells (SCs) mixed at a 1:1 ratio in lysis buffer (0 control) or from HeLa Env/CD4 syncytia obtained after 18 or 36 h of coculture. These samples were then processed by the PowerBlot facility (Becton Dickinson), which determined the expression level of ∼800 different signal-transducing proteins using a combination of SDS-PAGE (5–15% gradient), immunoblotting with specific monoclonal antibodies revealed by a secondary goat anti–mouse horseradish peroxidase, capture of chemiluminesence data by a CDD camera, and computerized processing of densitometric data (for details consult www.translab. com/TOC.shtml). Data were normalized by dividing the signal obtained for each protein by the sum of signals obtained for all 800 proteins for one given sample. For proteins revealing a variation in the expression of at least 20%, determined in two initial runs, expression levels were verified in a third independent experiment.
Immunoblots and Coimmunoprecipitation.
Aliquots of total protein extracts (40 μg), were run on SDS-PAGE and electroblotted overnight at 4°C onto nitrocellulose membrane. Immunodetection involved antibodies specific for p53, Bax, Bcl-2, mTOR/FRAP (BD Transduction Laboratories), phosphorylation of p53 on serine 15 (p53S15P) (Cell Signaling Technology), appropriate secondary antibodies (goat anti–rabbit or goat anti–mouse; Bio-Rad Laboratories) conjugated to horseradish peroxidase, and the enhanced ECL chemiluminescence detection system (Amersham Pharmacia Biotech). Equal loading and transfer was monitored by Ponceau red staining of nitrocellulose membranes. For immunoprecipitation, lysates from sonicated, Triton X-100-solublized cells (60 μg protein in 100 μl PBS with protease inhibitors) were incubated for 90 min at 37°C with 500 ng affinity-purified rabbit polyclonal antibodies specific for p53, p53S15P, or Bcl-2, followed by addition of 10 μl-packed protein A/G-agarose beads (30 min, 37°C; Santa Cruz Biotechnology, Inc.), vigorous washing of the pellet (10 min at 10,000 g, 3 ×) in PBS, 5% SDS PAGE, and immunodetection with an mTOR/FRAP-specific mAb.
Fluorescence Staining of Live Cells, Immunofluorescence, and Immunocytochemistry.
For the simultaneous assessment of mitochondrial and nuclear features of apoptosis, live cells were stained with the potentiometric dye 5,5′,6,6′-tetrachloro-1,1′, 3,3′-tetraethylbenzimidazolylcarbocyanine iodide (2 μM JC-1; Molecular Probes), as well as Hoechst 33342 (2 μM; Sigma-Aldrich) for 30 min at 37°C in complete culture medium 6. A rabbit antiserum specific for p53S15P was used on paraformaldehyde (4% wt:vol) and picric acid-fixed (0.19% vol:vol) cells and revealed with a goat anti–rabbit IgG conjugated to PE (Southern Biotechnology Associates, Inc.). Cells were also stained for the detection of Cyt-c (mAb 6H2.B4 from BD PharMingen), Hsp60 (mAb H4149 from Sigma-Aldrich), Bax (mAb 6A7; BD PharMingen), p21 (mAb EA10; Oncogene Research Products), MDM2 (mAb IF2; Oncogene Research Products), mTOR/FRAP (mAb 3O; BD Transduction Laboratories), all revealed by a goat anti–mouse IgG ALEXA488 conjugate; Molecular Probes), and/or chromatin (Hoechst 33342; reference 35). Immunocytochemical staining was performed using polyclonal anti-p53S15P or monoclonal tTG (Neomarkers) antibodies. A biotinylated goat anti–mouse or anti–rabbit IgG, as a secondary antibody, was used, followed by a preformed horseradish peroxidase-conjugated streptavidin (Biogenex). The reaction was developed using aminoethylcarbazole and 3-3′ diaminobenzidine (Biogenex) as chromogenic substrates and 0.01% H2O2. Cells were counterstained in Mayer's acid hemalum. Endogenous peroxidase activity was blocked by preincubation with 3% H2O2. Results were quantified by two independent investigators on an average of 500 cells.
Results and Discussion
Early Phosphorylation of p53 on Serine 15 in HIV-1 Env-induced Syncytial Apoptosis.
Syncytia formed by coculture of two different HeLa cell lines expressing HIV-1 Env or CD4/CXCR4 spontaneously undergo apoptosis 6 7. Among a panel of different apoptosis-regulatory proteins, we found only one major alteration, namely an upregulation of Bax that is well detectable 36 h after syncytium formation (Fig. 1 A). Significant alterations were also found in the expression level of the proapoptotic proteins Bid (which is downregulated) and caspase-7 (which is upregulated by ∼50%). Whereas total p53 levels did not change after formation of syncytia, an increased phosphorylation of serine 15 (but not serines 6, 9, 20, 37, and 397) of p53 (p53S15) was detected, using a panel of phosphoserine epitope-specific antibodies (Fig. 1 B). Phosphorylated p53S15 (p53S15P) was confined to the nuclei of syncytia (Fig. 1 C), and was not detected in SCs (Fig. 1 D). p53S15P could be detected early (6 h) after syncytium formation, well before Bax was overexpressed (Fig. 1 A and B) and apoptotic chromatin condensation became detectable (Fig. 1 D). Among p53S15P+ syncytia, only a fraction exhibited a conformational change in Bax linked to mitochondrial translocation (Fig. 1 E) as well as the release of Cyt. c from mitochondria (Fig. 1 F). Conversely, the translocation of Bax to mitochondria and of Cyt. c from mitochondria was only found among p53S15P+ (not p53S15P−) syncytia (Fig. 1 G), indicating that these changes occur after p53S15 phosphorylation. Accordingly, inhibition of Cyt. c-mediated caspase activation by Z-VAD.fmk (Fig. 1 H) or suppression of Bax translocation by microinjection of recombinant Bcl-2 protein 36 into syncytia (Fig. 1 I) failed to affect p53S15 phosphorylation. Both Z-VAD.fmk and Bcl-2 did, however, prevent nuclear apoptosis as an internal control of their efficacy (Fig. 1 H and I). Altogether, these data place p53S15 phosphorylation upstream of Bax upregulation/translocation, MMP, and subsequent caspase activation.
Figure 1.

Expression levels of apoptosis-regulatory proteins and p53S15 phosphorylation in syncytia. (A) Alteration in protein expression levels determined by quantitative immunoblot analysis. The percentage changes of the expression levels in Bax, Bid, and caspase-7 are shown after 18 and 36 h of coculture of HeLa Env and HeLa CD4 cells. Results are means of three independent determinations ± SEM. *P < 0.01. The right panel lists apoptosis-regulatory proteins which were detectable in HeLa cells and whose expression level did not change significantly (by <20%) within 36 h after syncytium formation. (B) Immunoblots of p53, p53Ser15P, Bax, and Bcl-2 at different intervals of syncytium formation. (C) Immunofluorescence detection of p53S15P (red fluorescence, left) in syncytia counterstained with the nuclear dye Hoechst 33342 (blue fluorescence, right). (D) Frequency of p53S15P+ and apoptotic cells among syncytia (Syn) and SCs contained in cocultures of HeLa Env and HeLa CD4 cells. (E) Absence or presence of the conformational change of Bax detectable with a monoclonal antibody specific for the NH2-terminal Bax domain (green fluorescence) among 24-h-old p53S15P+ syncytia (red nuclear fluorescence). Note that some cells lack Bax immunoreactivity (left) whereas others exhibit a cytoplasmic, in part punctate staining pattern (right). (F) Subcellular localization of Cyt. c (green) in 24-h-old p53S15P+ syncytia (red). Note the mitochondrial (punctate) staining pattern (left) or the diffuse pattern (right) indicative of mitochondrial Cyt. c release. (G) Quantitation of Bax and Cyt. c translocation among p53S15P− and p53S15P+ syncytia, 24 or 48 h after initiation of coculture. (H) Effect of the pancaspase inhibitor Z-VAD.fmk on p53S15 phosphorylation. Hela Env and HeLa CD4 cells were cocultured for 24 h in the presence or absence of Z-VAD.fmk, followed by determination of the frequency of p53S15P+ cells and of cells exhibiting chromatin condensation. (I) Effect of Bcl-2 on p53S15 phosphorylation. Syncytia (6 h) were microinjected with recombinant Bcl-2 protein or sham injected, and the frequency of p53S15P+ cells and nuclear apoptosis was determined at 24 h. Values in H and I are means of triplicates ± SEM. *P < 0.01 inhibition of chromatin condensation.
p53S15 Phosphorylation Stimulates the Expression of p53-transactivated Genes in HIV-1 Env-induced Syncytia.
p53S15 phosphorylation is known to increase the transactivating function of p53 37 and thus may contribute to transcriptional activation of the bax gene 38 and apoptosis induction 21. p53 is not mutated in HeLa cells 39, yet has been reported to be functionally inactive due to the presence of E6 from papilloma virus 40. However, evidence that the low amount of p53 contained in HeLa cells is functional has been reported 41 42. It appears that p53S15 phosphorylation of HeLa syncytia elicited by the Env–CD4 interaction does result in transcriptional activation of p53 target genes, based on three lines of evidence. First, expression of the Bax protein increases (Fig. 1 B), as does that of Bax mRNA (data not shown). Second, the expression levels of two other prominent p53-activated gene products, namely MDM2 (Fig. 2 A and C) and p21 (Fig. 2 B and C), increase upon syncytium formation, with a similar delay, and this enhanced expression is restricted to the p53S15P+ subpopulation of syncytia (Fig. 2 A and B). Third, we employed a p53-inducible GFP construct that, when transiently transfected into HeLa Env cells, cultured in the absence of HeLa CD4 cells, fails to be expressed, unless it is cotransfected with p53 (but not with a dominant negative p53 mutant, p53H175) (Fig. 2 D). Upon coculture with HeLa CD4 cells, this p53-inducible GFP construct was strongly expressed in a subpopulation of p53S15P+ syncytia (but not in p53S15P− syncytia nor in individual cells) (Fig. 2 E).
Figure 2.

Activation of p53 transcriptional activity in syncytia. (A) Induction of MDM2 protein expression. Syncytia (18 h) were stained for p53S15P and MDM2. Representative cells are shown. Note that strongly MDM2-positive (green) nuclear structures were only detected in p53S15P+ (red) syncytia, whereas only a fraction of p53S15P+ heterokarya were MDM2+ (B). Induction of p21 protein expression as detected by immunofluorescence. Again p21+ cells were a subpopulation of p53S15P+ syncytia. (C) Kinetics of positive immunostaining in syncytia for p53S15P, MDM2, p21, and Bax. Syncytia were subjected to immunofluorescence staining as in B and C. Results are mean values ± SD of three independent cultures. (D) Expression of a p53-inducible reporter gene in HeLa cells. HeLa Env cells were transfected with the p53-inducible GFP construct alone (control, Co.) or together with p53 or a p53 dominant negative mutant (p53H175) and cultured alone for 48 h. Representative cells stained with Hoechst 33342 are shown. The percentage of control or p53H175-transfected cells exhibiting positive GFP fluorescence was <1%, that of p53-transfected cells 38 ± 5% (n = 3). (E) Induction of p53-inducible GFP in syncytia. HeLa Env cells were transfected with a p53-inducible GFP construct (transfection efficiency ∼40%) and fused 24 h later with HeLa CD4 cells. A subpopulation of 24-h-old p53S15P+ syncytia (70%) exhibited a diffuse GFP fluorescence.
p53S15 Phosphorylation in HIV-1 Infection In Vitro and In Vivo.
To validate our initial observations, obtained in HeLa syncytia, we investigated whether p53S15P is induced by HIV infection in vitro and in vivo. p53S15P was induced in heterokaryons generated by coculturing HeLa CD4 cells with a lymphoid cell line chronically infected with a syncytium-inducing HIV-1 isolate (Fig. 3 A), as well as in HIV-1–infected U937 myelomonocytary cells (Fig. 3 B), thus confirming that HIV-1 infection in vitro suffices to induce p53S15P 22. Importantly, p53S15P was also found among syncytia localized in the T cell area of lymph nodes from HIV-1+ patients (Fig. 3 C). Such cells also expressed the preapoptotic marker tTgase (Fig. 3 C). p53S15P was detected in PBMCs of HIV-1+ donors but not in PBMCs from HIV-1− controls (<0.1% positive cells) (Fig. 3 D). The frequency of p53Ser15P+ PBMCs correlated with the percentage of tTgase+ cells (Fig. 3 E), as well as with viral load (Fig. 3 F). In conclusion, p53S15P is associated with HIV-1 infection, both upon in vitro infection of human cells as and vivo, in patient-derived samples. p53S15P expression correlates with HIV-1 viremia and preapoptosis.
Figure 3.

p53S15P in HIV-1 infection. (A) p53S15P in HIV-1 infected syncytia. CD4-expressing HeLa cells were cocultured with chronically HIV-1-infected H9/IIIB cells at a 3:1 ratio and stained for p53Ser15 (red), as well as the mitochondrial matrix protein hsp60 (green). The dominant phenotypes obtained 12 and 48 h after coculture are shown. (B) p53S15P in uninfected (Co.) or HIV-1–infected U937 cells, as determined by immunoperoxidase staining (brown) and hematoxylin counterstaining (blue) and confirmed by immunoblot (inserts). (C) p53S15P+ syncytium in the apical light zone of lymph node derived from an HIV-1+ donor (arrow). On a consecutive section tTgase expression was detected. The insert show a p53S15P+ syncytium with condensed nuclei. Similar pictures were obtained for five different HIV-1+donors, whereas no p53S15P+ cells are detected in HIV-1− controls (bottom). (D) 53S15P in PBMC derived from representative HIV-1− (Co.) or HIV-1+ donors. Immunoperoxidase staining and immunoblots (inserts) are shown. (E) Correlation between p53S15P and tTgase expression in PBMC from 49 HIV-1+ patients. The frequency of p53S15P+ and tTgase+ cells was determined by immunocytochemical analysis of PBMC from 49 HIV-infected individuals, at least 2 mo after interruption of antiretroviral therapy (as in D). Each dot represents one HIV-infected patient. Statistics were calculated by means of the Pearson method for parametric data using the GraphPad Prism statistical program. The correlation coefficient (Pearson r) is 0.7107. (F) Correlation between p53S15P and viral load of the same patients as in E. The correlation coefficient is 0.3922.
p53S15 Phosphorylation Is Mediated by mTOR/FRAP.
p53S15 is known to be phosphorylated by stress kinases, in particular ERK and p38 kinase 43, as well as by several members of the PIKK family, in particular DNA-PK 44, ATM 45, ATR 46, and mTOR/FRAP 47. Among a large panel of serine kinases, phosphatases, and serine kinase activators, the only two proteins whose expression level changed in Env-induced syncytia were mTOR/FRAP, which is upregulated, and the protein phosphatase PP2A, which is downregulated (Fig. 4 A). PP2A is known to be inhibited by mTOR/FRAP 48, suggesting that its late (36 h) downregulation may be secondary to the upregulation/activation of mTOR/FRAP detectable at 18 h. Rapamycin (a specific inhibitor of mTOR/FRAP) and LY294002 (a general inhibitor of PI-3 kinases including mTOR/FRAP) significantly inhibited p53S15 phosphorylation (Fig. 4 B). In contrast, the ERK inhibitor PD98059, the p38 inhibitor SB203580, the ATM/DNA-PK inhibitor wortmannin 49 50 and the ATM/ATR inhibitor caffeine 46 51 failed to prevent the syncytium-specific p53S15 phosphorylation (Fig. 4 B). Accordingly, no phosphorylation of ERK (motif: pTEpY) or p38 kinase (motif: pTGpY) was detectable in syncytia (data not shown), indicating that these kinases are not activated. Moreover, comet assays did not detect double- strand breaks among nonapoptotic p53S15P+ syncytia (Fig. 4 C and D), further arguing against the implication of DNA-PK, ATM, and ATR (all of which are, in principle, activated by double-strand breaks; references 44–46) in p53S15 phosphorylation. As an alternative to rapamycin, microinjection of a mAb specific for mTOR/FRAP into newly formed syncytia caused a significant inhibition of p53S15 phosphorylation (Fig. 4 E). In conclusion, p53S15 phosphorylation is mediated via mTOR/FRAP.
Figure 4.

Mechanism of p53S15 phosphorylation. (A) Alterations in the expression level of serine kinases and phosphatases as determined by quantitative immunoblot analysis. The percentage changes of the expression levels in mTOR/FRAP and PP2a are shown (X ± SEM, n = 3). *P < 0.01. The right panel lists serine kinases and phosphatases, as well as related proteins, which were detectable in HeLa cells and whose expression level varied <20% within 18 or 36 h after syncytium formation. (B) Inhibitory profile of p53S15 phosphorylation. HeLa Env and HeLa CD4 cells were cocultured for 24 h in the presence of the indicated agents, followed by determination of the frequency of p53S15P+ syncytia. Results are means of three to five independent determinations ± SEM. *P < 0.01, Student's t test inhibitory effects. (C) Representative comet assay results obtained for untreated HeLa CD4+ SC (negative control), H2O2-treated HeLa CD4+ cells (positive control), adherent Env/CD4-induced syncytia (18 h), or nonadherent, apoptotic Env/CD4-induced syncytia. (D) Quantitation of comets (which reveal double-strand breaks), as determined by image analysis (X ± SD, n = 100). (E) Effect of microinjected mAb specific for mTOR/FRAP on p53S15P. 6 h after fusion, syncytia were microinjected with mAbs specific for mTOR/FRAP or LAMP-1 (negative control), cultured for further 24 h, and subjected to p53S15P staining. The asterisk indicates a significant (P < 0.01) inhibitory effect. Results are mean values ± SEM of three experiments.
Cellular Alterations Induced by Activated mTOR/FRAP.
mTOR/FRAP mediates translational control via phosphorylation of the p70s6k protein kinase and the 4E-binding protein (4EBP) 1 23 24 52. In a control experiment performed on SCs, serum withdrawal (which inactivates mTOR/FRAP) was found to cause the dephosphorylation of p70s6k and 4EBP1 (lane 4 in Fig. 5 A), which were phosphorylated again upon readdition of serum (lane 5). This serum-stimulated phosphorylation was fully inhibited by rapamycin (lane 6) as an internal control of its efficacy. In contrast, both SCs (lane 1) and syncytia (lane 2) cultured in complete, serum-containing medium exhibited constitutive phosphorylation of p70s6k and 4EBP (Fig. 5 A), as well as constitutively high expression of a mTOR/FRAP-controlled 5′-TOP-luciferase reporter construct (Fig. 5 B). Accordingly, the expression of several mTOR/FRAP-controlled gene products including c-Myc, HIF-1, Rb, Rb2, and signal transducer and activator of transcription 3 was not increased upon syncytium formation (data not shown). Neither the constitutive phosphorylation of p70s6k and 4EBP nor the translation of TOP-luciferase were inhibited by a 24-h exposure to rapamycin (lane 3 in Fig. 5 A and B), and rapamycin did not block the cell cycle of HeLa cells (data not shown). In contrast, within the same time frame, the fusion-dependent p53Ser15 phosphorylation was inhibited by rapamycin (Fig. 5 A). mTOR/FRAP coimmunoprecipitated with p53 and p53S15P in syncytia, not in SCs (Fig. 5 C). Moreover, we found that in syncytia mTOR/FRAP was enriched in the nucleus, as compared with SCs. This alteration was observed both in p53S15P− and p53S15P+ syncytia (Fig. 5 D), and kinetic analyses revealed that the nuclear accumulation of mTOR/FRAP precedes the phosphorylation of p53S15P (Fig. 5 E). Altogether, these data suggest that mTOR/FRAP is activated in syncytia, where it directly phosphorylates p53 within the nucleus. In contrast, other cytoplasmic targets of mTOR/FRAP such as p70s6k and 4EBP1 are unlikely to explain the rapamycin effects on syncytia.
Figure 5.
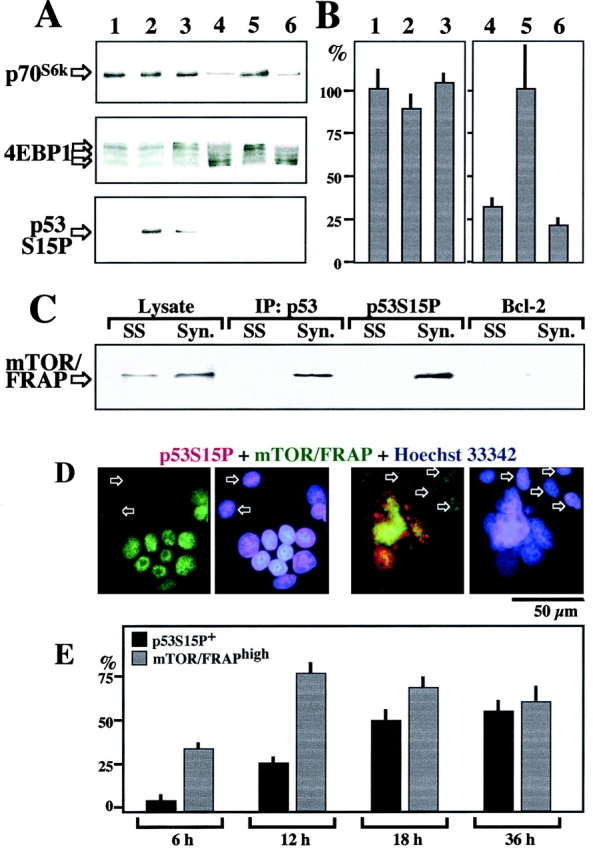
Biological effects of FRAP on syncytia. (A) Phosphorylation of the p70s6k protein kinase, 4EBP1, and p53S15 in syncytia. A 1:1 mixture of HeLa Env and HeLa CD4 SCs (lane 1), 24-h-old syncytia cultured in the absence (lane 2) or presence (lane 3) of 1 μM rapamycin, 30 h-serum-deprived Env cells before (lane 4), or after addition of serum for 3 h (lanes 5 and 6), in the presence (lane 5) or absence of 1 μM rapamycin (lane 6) were subjected to SDS-PAGE followed by immunodetection of phosphorylated p70s6k (using a phosphospecific mAb), 4EBP1 (note the reduced electrophoretic mobility of the phosphorylated protein), or p53S15P. (B) TOP-dependent translation in syncytia. HeLa Env cells were transiently transfected with a luciferase construct containing TOP in the promoter region or a control construct lacking TOP. These cells were mixed with an equivalent amount of HeLa CD4 cells without coculture 1, or cocultured with HeLa CD4 cells for 24 h, in the absence 2 or presence 3 of 1 μM rapamycin. Alternatively, the cells were serum-deprived for 30 h and then cultured for an additional 3 h in the absence 4 or in the presence (5 and 6) of serum, with 6 or without 5 1 μM rapamycin. Cellular extracts (50 μg per point) were then prepared and luciferase activities was determined. TOP-dependent luciferase activity was divided by the TOP-independent luciferase activity and normalized assuming that SCs cultured in the presence of serum and in the absence of rapamycin (1 and 5) represent 100% TOP-dependent translation. (C) Coimmunoprecipitation of mTOR/FRAP and p53S15P+ in syncytia. Ad hoc mixtures of HeLa Env and HeLa CD4 SCs, 24-h-old syncytia (Syn) were lysed and then subjected to immunoprecipitation with antisera specific for p53, p53S15P, or Bcl-2 (negative control), SDS-PAGE, and immunodetection of mTOR/FRAP. (D) Enhanced expression of mTOR/FRAP in syncytia. As compared with SCs (arrows), syncytia manifest an enhanced, mainly nuclear staining for mTOR/FRAP. Two 12-h-old syncytia representing the two major phenotypes are shown: mTOR/FRAPhighp53Ser15P− and mTOR/FRAPhighp53Ser15P+. Note that all p53Ser15P+ syncytia were mTOR/FRAPhigh, whereas mTOR/FRAPhigh syncytia were either p53Ser15P+ or p53Ser15P−. (E) Quantitation of the frequency of mTOR/FRAPhigh and p53Ser15P+ cells at different time points.
Inhibition of mTOR/FRAP or p53S15 Phosphorylation Prevents Env-induced Syncytial Apoptosis.
When added to HeLa Env/HeLa CD4 cocultures, rapamycin and LY294002 inhibited all signs of syncytial apoptosis, including the loss of the ΔΨm (indicated by a red→green shift of the fluorescence emitted by the ΔΨm-sensitive fluorochrome JC-1), chromatin condensation (detected with the blue fluorochrome Hoechst 33342) (Fig. 6 A and B), the translocation of Bax to mitochondria, and the Bax-mediated release of Cyt. c from mitochondria (Fig. 6 B). Concomitantly, rapamycin and LY294002 allowed for the formation of larger syncytia (Fig. 6 C), an effect quantitatively similar to what could be seen with the pancaspase inhibitor Z-VAD.fmk (Fig. 6 C and reference 6). The apoptosis-inhibitory effect of rapamycin was durable, with a strong inhibitory effect on apoptosis of 6-d-old syncytia (81 ± 4% of viable, ΔΨm high syncytia with 1 μM rapamycin versus only 25 ± 3% ΔΨm high syncytia in untreated controls, n = 3). The antiapoptotic effects of rapamycin and LY294002 were observed at doses similar to those required for inhibition of p53S15 phosphorylation (Fig. 4 B). In contrast, wortmannin, caffeine, PD98059, SB203580, CsA, or FK506, which do not affect p53S15 phosphorylation (Fig. 4 B), also failed to prevent apoptosis (Fig. 6 D). Thus, a large panel of agents which inhibit apoptosis in other experimental systems failed to prevent syncytial apoptosis, with the notable exception of pyrrolidine dithiocarbamate (PDTC, Fig. 6 G), a pleiotropic agent known to interfere with p53 function 53 54. Of note, PDTC also reduced the phosphorylation of p53S15 (Fig. 4 B), underlining the intimate relationship between p53S15P and syncytial apoptosis.
Figure 6.

Apoptosis inhibition by rapamycin and LY294002 in Env/CD4-induced syncytia. (A) Representative JC-1/Hoechst 33342 staining of syncytia generated by 48 h of coculture in the absence (control) or presence of 1 μM rapamycin. Cells were stained with Hoechst 33342 (blue fluorescence) and the ΔΨm-sensitive dye JC-1 (red fluorescence of mitochondria with a high ΔΨm, green fluorescence of mitochondria with a low ΔΨm). Note that rapamycin antagonizes the ΔΨm loss, as well as chromatin condensation. (B) Inhibition of mitochondrial and nuclear signs of apoptosis. Cocultures (48 h) of HeLa Env and HeLa CD4 cells were performed in the presence of rapamycin or LY294002, followed by quantitation of the ΔΨm loss and chromatin condensation on live cells (determined as in A) or, alternatively, permeabilization, fixation and immunostaining with an anti-Bax or anti-Cyt. c antibody (as in Fig. 1 E and F). Asterisks indicate significant (P < 0.01) inhibitory effects. (C) Quantitation of the number of nuclei per syncytium (n = 100) after 72 h of coculture. The pancaspase inhibitor Z-VAD.fmk (100 μM, added at the beginning of coculture) was included as a positive control. *P < 0.01). (D) Effects of apoptosis-regulatory compounds on syncytia. The effect of different agents on nuclear apoptosis were assessed as in A. “No effect” denotes <20% inhibition or enhancement of chromatin condensation. “Toxicity” and “protection” denote >20% increase or reduction of apoptosis, respectively. PDTC inhibited apoptosis by 52 ± 6% in three independent experiments.
p53 Participates in Syncytial Apoptosis.
To directly assess the role of p53 in syncytial apoptosis, Env and CD4/CXCR4-expressing cells were transiently transfected with two dominant negative (DN) p53 constructs (p53H175 and p53H273) 24 h before fusion. As compared with vector-only control transfections, DN-p53 conferred protection against all the mitochondrial and nuclear hallmarks of syncytial apoptosis including the translocation of Bax to mitochondria (Fig. 7). This protection was partial (∼50%), in agreement with the transfection efficiency (∼50%). These data indicate that the mTOR/FRAP→p53S15P pathway delineated here dictates the fate of Env-induced syncytia at the premitochondrial level, upstream of the upregulation of Bax. To address the involvement of p53 in syncytial apoptosis in a completely different experimental setting, MEFs with different p53 genotypes were subjected to short-term exposure to PEG, resulting in their fusion. In wild-type MEFs (p53+/+), syncytium formation led to phosphorylation of p53S15, positive staining for Bax (Fig. 8 A), and apoptotic cell death (Fig. 8 B), whereas syncytia formed from p53−/− MEFs (which obviously do not stain for p53S15P) failed to activate Bax (Fig. 8 A) and to undergo apoptosis (Fig. 8 B). In conclusion, p53 is rate-limiting for apoptosis triggered by syncytium formation.
Figure 7.

Apoptosis inhibition by dominant negative p53. Both HeLa Env and HeLa CD4 cells were transfected by p53H175 or p53 H273, as well as a vector control only (Co.). 24 h after the transfection the cells were cocultured for 48 h and the mitochondrial and nuclear parameters indicative of apoptosis were assessed by JC1/Hoechst 33342 or immunofluorescence staining, as detailed in Materials and Methods. Asterisks indicate significant (P < 0.01) inhibitory effects, as compared with vector-only-transfected cells. The insert demonstrates the transfection-enforced overexpression of mutant p53, as determined by immunoblotting. Results are representative of four independent determinations.
Figure 8.

p53 is required for syncytial apoptosis. Wild-type (p53+/+), p53+/−, and p53−/− cells were fused by transient exposure to PEG. (A) Syncytia (9 h) arising from p53+/+ MEF showing positive immunoreactivity after staining for p53S15P or Bax. Note that Bax+ cells were also positive for p53S15P. As a control, a representative p53−/− syncytium (which stays Bax−) is shown. (B) Quantitation of apoptosis. Syncytia arising from the fusion of p53+/+, p53+/−, or p53−/− cells were stained at the indicated time points with JC1/Hoechst 33342 (as in Fig. 6 A) and the frequency of cells exhibiting a low ΔΨm and chromatin condensation was determined. Results are representative of three independent experiments.
mTOR/FRAP Inhibition Prevents Syncytial Apoptosis Induced by HIV-1 Infection.
HeLa CD4 cells cocultured with H9 lymphoid cells chronically infected with the HIV-1 strain IIIb can form syncytia, which undergo apoptosis. Syncytial apoptosis was inhibited by rapamycin, both at the mitochondrial and the nuclear levels (Fig. 9 A and B). To further substantiate the rapamycin-mediated inhibition of syncytial cell death, CEM T lymphoma cells engineered to contain a GFP gene under the control of the HIV-1 LTR promoter (normally inactive) were cocultured with HIV-1 IIIb-infected H9 cells, in the presence of absence of rapamycin. Upon formation of syncytia, HIV-1–encoded Tat transactivates LTR and induces GFP expression 33. In the presence of rapamycin (or Z-VAD.fmk, as a positive control) the frequency of viable syncytia was increased, leading to an enhanced GFP-dependent fluorescence (Fig. 9 C). When individual nuclei from such syncytia were subjected to cytofluorometric DNA content analysis, the frequency of hypoploid (apoptotic) nuclei was found to be strongly reduced by rapamycin (Fig. 9 D). In a further series of experiments, primary CD4+ lymphoblasts from healthy donors were infected by HIV-1 IIIb in vitro, a manipulation that induced syncytium formation and subsequent apoptosis. Addition of rapamycin 48 h after infection did not affect viral replication, as determined by measuring the production of p24 (Fig. 9 E). However, rapamycin did inhibit chromatin condensation and led to an increase in the number of nuclei per syncytium, similar to Z-VAD.fmk (Fig. 9 E). Thus, the cytoprotective effects of rapamycin on lymphoblasts infected with HIV-1 cannot be attributed to an inhibition of viral replication or syncytium formation. Rapamycin inhibited the phosphorylation of p53S15 in HIV-infected cells (Fig. 9 B). Taken together, these data confirm that the rapamycin-inhibited mTOR/FRAP→p53S15P pathway selectively controls apoptosis of syncytia in human primary CD4+ lymphocytes infected by HIV-1.
Figure 9.
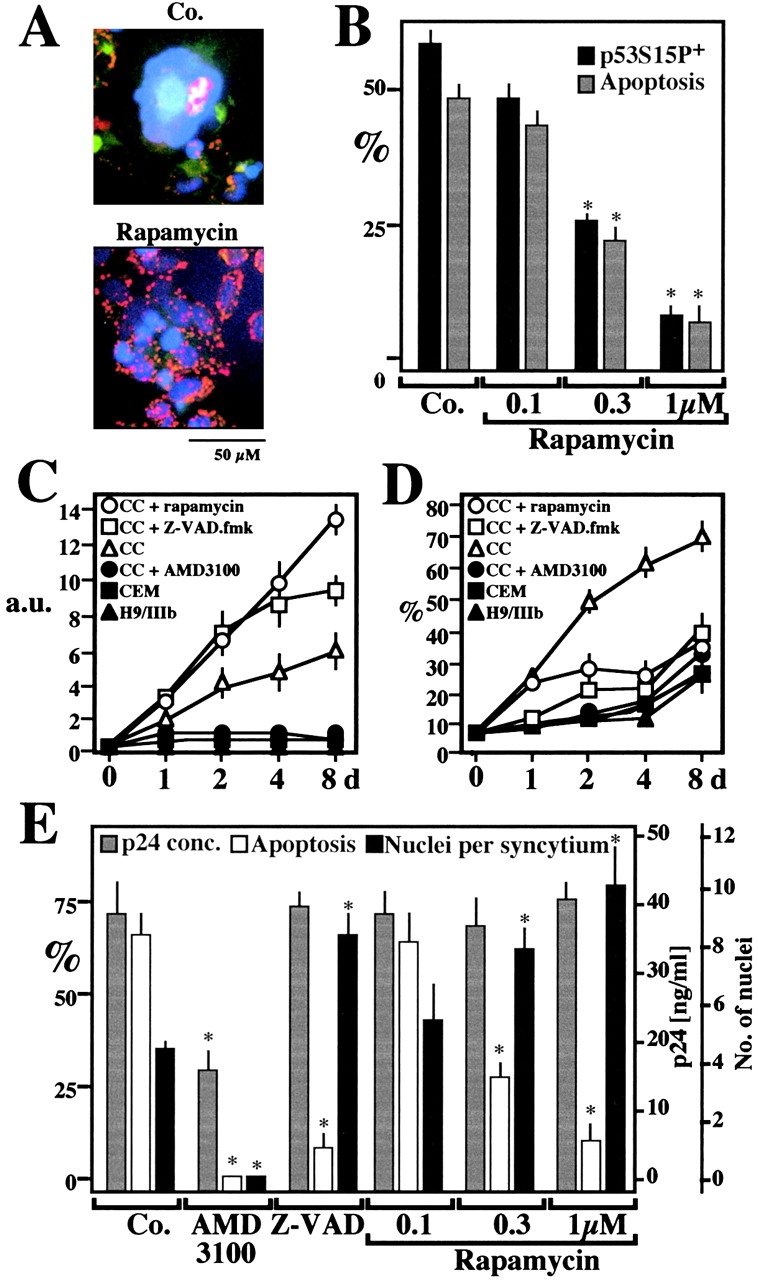
Rapamycin effects on syncytial apoptosis induced by HIV-1 infection. (A) Fluorescence micrographs of HeLa CD4 cells cocultured with HIV-1–infected H9/IIIB cells for 48 h in the absence (Co.) or the presence of 1 μM rapamycin. Cells were stained with Hoechst 33342 and the ΔΨm-sensitive dye JC-1 (as in Fig. 6 A). Primary CD4+ lymphoblasts infected with HIV-1 IIIb are also shown. (B) Rapamycin effects on p53S15P and apoptosis in HeLa CD4 cells cocultured with H9/IIIB cells. p53S15P was determined by immunofluorescence as in Fig. 1 C and apoptosis was measured by staining with JC-1/Hoechst. Values refer to the percentage of syncytia that are p53S15P+ or apoptotic. Asterisks denote significant (P < 0.01) effects of rapamycin, as compared with untreated cocultures. (C and D) Rapamycin-enhanced viability of syncytia associated with apoptosis inhibition. CEM cells stably transfected with a Tat-inducible GFP or HIV-1–infected H9/IIIB cells were cultured alone or cocultured (CC), in the absence or presence of AMD3100 (1 μg/ml), Z-VAD.fmk (50 μM), or rapamycin (1 μM). GFP-dependent fluorescence (arbitrary units, a.u.) was measured to assess the total syncytial mass (C). Parallel cultures were subjected to lysis, staining with propidium iodine, and cytofluorometric determination of the percentage (X ± SEM, n = 3) of hypoploid (sub-G1) nuclei (D). (E) Rapamycin effects on apoptosis of HIV-1 infected primary CD4+ lymphoblasts. 2 d after infection with HIV-IIIB, the inhibitors AMD3100 (which prevents the Env/CD4 interaction, 5 μg/ml), 100 μM Z-VAD.fmk, or rapamycin were added to the cultures. At day 4, the production of p24 (<0.5 ng/ml on day 2) was determined. In addition the frequency of syncytia with condensed nuclei and the number of nuclei per syncytium were determined. Results are mean values ± SEM of three independent experiments. Asterisks denote significant (P < 0.01) effects as compared with control cultures.
Concluding Remarks.
As shown here, HIV-1-Env–induced syncytium formation leads to apoptosis via a pathway that involves p53S15 phosphorylation by mTOR/FRAP. This corroborates the role of p53 as a major point of integration of proapoptotic signaling and damage sensing 21. The data presented here unravel that p53 is rate limiting for the apoptotic demise of syncytia. It is tempting to speculate that p53 could be generally required for the clearance of nonphysiological polyploid cells (syncytia or plasmodia). Indeed, HeLa cells transfected with antisense p53 spontaneously form giant multinucleate cells 41; antisense oligonucleotides suppressing p53 expression induce polyploidy in regenerating rat liver 55; and p53−/− mice accumulate multinucleate cells in the testis 56 as well as in the prostate epithelium 57. However, further studies are required to distinguish the relative impact of p53 on the prevention of nuclear endoreplication and the illicit survival of polyploid cells in these model systems.
The role of mTOR/FRAP in apoptosis control is far less established than p53. The mTOR/FRAP analogues of S. cerevisae and Drosophila are known to play a major role in the control of cell size 24 25. External constraints on cell size and shape have a profound effect on HIV-1-Env–induced syncytial apoptosis 58. It thus may be possible that the abnormal size or the distorted volume/surface ratio of syncytia, sensed via yet unknown mechanisms, activate the mTOR/FRAP pathway and consequent cellular demise. Alternatively or in addition, signals triggered via the Env-CD4/CXCR4 interaction might stimulate the mTOR/FRAP pathway. In some cell types, the mTOR/FRAP antagonist rapamycin induces apoptosis 59 via a p53-independent pathway 60, indicating that mTOR/FRAP can also act as an endogenous cell death antagonist. Indeed, mTOR/FRAP mediates survival signals received through the receptors for IL-3, insulin, and insulin-like growth factor 61 62 and may participate in oncogenic transformation 26. mTOR/FRAP is constitutively active in mammalian cells, at least in the presence of serum and nutrients 24 52, suggesting that the cellular context rather than the expression level of mTOR/FRAP dictates whether its proapoptotic or antiapoptotic function prevails.
In the context of HIV-1-Env–induced syncytia, mTOR/FRAP-mediated p53S15 phosphorylation appears to contribute to apoptosis by activating Bax (and perhaps other proapoptotic proteins) and consecutive MMP. Intriguingly, a fraction of lymph node cells from HIV-1+ donors are p53S15P+ (this paper), express (p53-inducible, reference 63) tTgase 15, and exhibit a ΔΨm loss 64 65, suggesting that they die via a pathway similar to that delineated for Env-induced syncytial apoptosis. Pharmacological targeting of the mitochondrial or premitochondrial events involved in HIV-1–associated apoptosis may thus offer a novel opportunity to intervene on AIDS pathogenesis.
Acknowledgments
We thank Drs. Jie Chen (Illinois University), Thierry Soussi (Institut Curie), Bert Vogelstein (Howard Hughes Medical Institute), Klas Wiman (Karolinska Cancer Center) for plasmids, John C. Reed (The Burnham Institute, La Jolla, CA) for recombinant Bcl-2, Jacques Corbeil (University of California at San Diego), Tyler Jacks (MIT), and the National Institutes of Health AIDS reagents program (Bethesda, MD) for cell lines.
This work has been supported by a special grant from the Ligue Nationale contre le Cancer, Comité Val de Marne de la Ligue contre le Cancer, as well as grants from Agence Nationale de Recherches sur le Sida, Fondation pour la Recherche Médicale (to G. Kroemer), the European Commission (QLG1-CT-1999-00739) (to G. Kroemer and M. Piacentini), Ricerca Corrente e Finalizzata from the Italian Ministry of Health (to M. Piacentini), Fundació irsiCaixa, MCYT BFM2000-1382 (to J.A. Este), FIS 00/0893 (to J. Barretina), and the Picasso Program (to J.A. Este and G. Kroemer). K.F. Ferri receives a fellowship from the French Ministry of Science. J. Blanco is a researcher at the Fundació per a la Recerca Biomèdica Germans Trias i Pujol, FIS 98/3047.
Footnotes
M. Castedo and K.F. Ferri contributed equally to this paper.
Abbreviations used in this paper: 4EBP, 4E-binding protein; ATM, ataxia teleangiectasia mutated; ATR, ataxia teleangiectasia Rad3; Cyt. c, cytochrome c; DNA-PK, DNA-dependent protein kinase; Env, envelope glycoprotein complex; FRAP, FKBP12-rapamycin-associated protein; GFP, green fluorescent protein; MEF, mouse embryonic fibroblast; MMP, mitochondrial membrane permeabilization; mTOR, mammalian target of rapamycin; PEG, polyethylene glycol; PIKK, phosphatidylinositol kinase-related kinases; SC, single cell; TOP, terminal oligopyrimidine; tTGase, tissue transglutaminase.
References
- Gougeon M.L., Montagnier L. Programmed cell death as a mechanism of CD4 and CD8 T cell depletion in AIDSmolecular control and effect of highly active anti-retroviral therapy. Ann. NY Acad. Sci. 1999;887:199–212. doi: 10.1111/j.1749-6632.1999.tb07934.x. [DOI] [PubMed] [Google Scholar]
- Badley A.D., Pilon A.A., Landay A., Lynch D.H. Mechanisms of HIV-associated lymphocyte apoptosis. Blood. 2000;96:2951–2964. [PubMed] [Google Scholar]
- Sodroski J.G., Goh W.C., Rosen A., Campbell K., Haseltine W.A. Role of the HTLV/LAV envelope in syncytia formation and cytopathicity. Nature. 1986;322:470–474. doi: 10.1038/322470a0. [DOI] [PubMed] [Google Scholar]
- Lifson J.D., Reyes G.R., McGrath M.S., Stein B.S., Engleman E.G. AIDS retrovirus-induced cytopathologygiant cell formation and involvement of CD4 antigen. Science. 1986;232:1123–1127. doi: 10.1126/science.3010463. [DOI] [PubMed] [Google Scholar]
- Dragic T., Charneau P., Clavel F., Alizon M. Complementation of murine cells for human immunodeficiency virus envelope/CD4-mediated fusion in human/murine heterokaryons. J. Virol. 1992;66:4794–4802. doi: 10.1128/jvi.66.8.4794-4802.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri K.F., Jacotot E., Blanco J., Esté J.A., Zamzami A., Susin S.A., Brothers G., Reed J.C., Penninger J.M., Kroemer G. Apoptosis control in syncytia induced by the HIV-1-envelope glycoprotein complexrole of mitochondria and caspases. J. Exp. Med. 2000;192:1081–1092. doi: 10.1084/jem.192.8.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri K.F., Jacotot E., Geuskens M., Kroemer G. Apoptosis and karyogamy in syncytia induced by HIV-1-ENV/CD4 interaction. Cell Death Differ. 2000;7:1137–1139. doi: 10.1038/sj.cdd.4400748. [DOI] [PubMed] [Google Scholar]
- Sylwester A., Murphy S., Shutt D., Soll D.R. HIV-induced T cell syncytia are self-perpetuating ant the primary cause of T cell death in culture. J. Immunol. 1997;158:3996–4007. [PubMed] [Google Scholar]
- Scheller C., Jassoy C. Syncytium formation amplifies apoptotic signalsa new view on apoptosis in HIV infection in vitro. Virology. 2001;30:48–55. doi: 10.1006/viro.2000.0811. [DOI] [PubMed] [Google Scholar]
- Blaak H., van't Wout A.B., Brouwer M., Hoolbrink B., Hovenkamp E., Schuitemaker H. In vivo HIV-1 infection of CD45RA+ CD4+ T cells is established primarily by syncytium-inducing variants and correlates with the rate of CD4+ T cell decline. Proc. Natl. Acad. Sci. USA. 2000;97:1269–1274. doi: 10.1073/pnas.97.3.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas J.J., Gange S.J., Schuitemaker G., Coutinho R.A., van Leeuwen R., Margolick J.B. Strong association between failure of T cell homeostasis and the syncytium-inducing phenotype among HIV-1-infected men in the Amsterdam Cohort Study. AIDS. 2000;16:1155–1161. doi: 10.1097/00002030-200006160-00012. [DOI] [PubMed] [Google Scholar]
- Camerini D., Su H.P., Gamez-Torre G., Johnson M.L., Zack J.A., Chen I.S. Human immunodeficiency virus type 1 pathogenesis in SCID-hu mice correlates with syncytium-inducing phenotype and viral replication. J. Virol. 2000;74:3196–3204. doi: 10.1128/jvi.74.7.3196-3204.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etemad-Moghadam B., Sun Y., Nicholson E.K., Fernandes M., Liou K., Gomila R., Lee J., Sodroski J. Envelope glycoprotein determinants of increased fusogenicity in a pathogenic simian-human immunodeficiency virus (SHIV-KB9) passaged in vivo. J. Virol. 2000;74:4433–4440. doi: 10.1128/jvi.74.9.4433-4440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dargent J.L., Lespagnard L., Kornreich A., Hermans P., Clumeck N., Verhest A. HIV-associated multinucleated giant cells in lymphoid tissue of the Waldeyer's ringa detailed study. Mod. Pathol. 2000;13:1293–1299. doi: 10.1038/modpathol.3880237. [DOI] [PubMed] [Google Scholar]
- Amendola A., Gougeon M.L., Poccia F., Bondurand A., Fesus L., Piacentini M. Induction of “tissue” transglutaminase in HIV pathogenesisevidence for high rate of apoptosis of CD4+ T lymphocytes and accessory cells in lymphoid tissues. Proc. Natl. Acad. Sci. USA. 1996;93:11057–11062. doi: 10.1073/pnas.93.20.11057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer P.H. CD95's deadly mission in the immune system. Nature. 2000;407:789–795. doi: 10.1038/35037728. [DOI] [PubMed] [Google Scholar]
- Kroemer G., Reed J.C. Mitochondrial control of cell death. Nat. Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- Budijardjo I., Oliver H., Lutter M., Luo X., Wang X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- Zhou B.-B.S., Elledge S.J. The DNA damage responseputting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- Rich T., Allen R.L., Wyllie A.H. Defying death after DNA damage. Nat. Cell Biol. 2000;407:777–783. doi: 10.1038/35037717. [DOI] [PubMed] [Google Scholar]
- Vogelstein B., Lane D., Levine A.J. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Genini D., Sheeter D., Rought S., Zaunders J.J., Susin S.A., Kroemer G., Richman D.D., Carson D.A., Corbeil J., Leoni L.M. HIV induced lymphocyte apoptosis by a p53-iniated, mitochondrion-mediated mechanism. FASEB J. 2001;15:5–6. doi: 10.1096/fj.00-0336fje. [DOI] [PubMed] [Google Scholar]
- Raught B., Gingras A.-C., Sonenberg N. The target of rapamycin (TOR) proteins. Proc. Natl. Acad. Sci. USA. 2001;98:7037–7044. doi: 10.1073/pnas.121145898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis P.B., Fumagalli S., Thomas G. Target of rapamycin (TOR)balancing the opposing forces of protein synthesis and degradation. Curr. Op. Genet. Dev. 1999;9:49–54. doi: 10.1016/s0959-437x(99)80007-0. [DOI] [PubMed] [Google Scholar]
- Zhang H., Stallock J.P., Ng J.C., Reinhard C., Neufeld T.P. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev. 2000;14:2712–2724. doi: 10.1101/gad.835000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume-Jensen P., Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- Schwartz O., Alizon M., Heard J.M., Danos O. Impairment of T cell receptor-dependent stimulation in CD4+ lymphocytes after contact with membrane-bound HIV-1 envelope glycoprotein. Virology. 1994;198:360–365. doi: 10.1006/viro.1994.1042. [DOI] [PubMed] [Google Scholar]
- Lassus P., Bertrand C., Zugasti O., Chambon J.P., Soussi T., Marthieur-Mahul D., Hibner U. Anti-apoptotic activity of p53 maps to the COOH-terminal domain and is retained in the highly oncogenic natural mutant. Oncogene. 1999;18:4699–4709. doi: 10.1038/sj.onc.1202841. [DOI] [PubMed] [Google Scholar]
- Kim J.E., Chen J. Cytoplasmic-nuclear shuttling of FKBP12-rapamycin-associated protein is involved in rapamycin-sensitive signaling and translation initiation. Proc. Natl. Acad. Sci. USA. 2000;97:14340–14345. doi: 10.1073/pnas.011511898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler M., Daugas E., Susin S.A., Zamzami N., Métivier D., Nieminen A.-L., Brothers G., Penninger J.M., Kroemer G. Dominant cell death induction by extramitochondrially targeted apoptosis inducing factor. FASEB J. 2001;15:758–767. doi: 10.1096/fj.00-0388com. [DOI] [PubMed] [Google Scholar]
- Jacotot E., Ferri K.F., El Hamel C., Brenner C., Druillenec S., Hoebeke J., Rustin P., Métivier D., Lenoir C., Geuskens M. Control of mitochondrial membrane permeabilization by adenine nucleotide translocator interacting with HIV-1 Vpr and Bcl-2. J. Exp. Med. 2001;193:509–520. doi: 10.1084/jem.193.4.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z.H., Schendel S., Matsuyama S., Reed J.C. Acidic pH promotes dimerization of Bcl-2 family proteins. Biochemistry. 1998;37:6410–6418. doi: 10.1021/bi973052i. [DOI] [PubMed] [Google Scholar]
- Gervaix A., West D., Leoni L.M., Richman D.D., Wong-Staal F., Corbeil J. A new reporter cell line to monitor HIV infection and drug susceptibility in vitro. Proc. Natl. Acad. Sci. USA. 1997;94:4653–4658. doi: 10.1073/pnas.94.9.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti I., Migliorati G., Pagliacci M.C., Riccardi C. A rapid simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Meth. 1991;139:271–280. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- Daugas E., Susin S.A., Zamzami N., Ferri K., Irinopoulos T., Larochette N., Prevost M.C., Leber B., Andrews D., Penninger J., Kroemer G. Mitochondrio-nuclear redistribution of AIF in apoptosis and necrosis. FASEB J. 2000;14:729–739. [PubMed] [Google Scholar]
- Murphy K.M., Streips U.N., Lock R.B. Bcl-2 inhibits Fas-induced conformational change in the Bax N-terminus and Bax mitochondrial translocation. J. Biol. Chem. 2000;275:17225–17228. doi: 10.1074/jbc.C900590199. [DOI] [PubMed] [Google Scholar]
- Dumaz N., Meek D.W. Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J. 1999;18:7002–7010. doi: 10.1093/emboj/18.24.7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita T., Reed J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- Yaginuma Y., Westphal H. Analysis of the p53 in human uterine carcinoma cell lines. Cancer Res. 1991;15:6506–6509. [PubMed] [Google Scholar]
- Kessis T.D., Slebos R.J., Nelson W.G., Kastan M.B., Plunkett B.S., Han S.M., Lorincz A.T., Hedrick L., Cho K.R. Human paillimovirus 16 E6 expression disrupts the p53-mediated cellular response to DNA damage. Proc. Natl. Acad. Sci. USA. 1993;90:3988–3992. doi: 10.1073/pnas.90.9.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iotsova V., Stehelin D. Antisense p53 provokes changes in HeLa cell growth and morphology. Eur. J. Cell Biol. 1995;68:122–132. [PubMed] [Google Scholar]
- Hietanen S., Lain S., Krausz E., Blattner C., Land D.P. Activation of p53 in cervical carcinoma cells by small molecules. Proc. Natl. Acad. Sci. USA. 2000;97:8501–8506. doi: 10.1073/pnas.97.15.8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She Q.B., Chen N., Dong Z. ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J. Biol. Chem. 2000;7:20444–20449. doi: 10.1074/jbc.M001020200. [DOI] [PubMed] [Google Scholar]
- Woo R.A., McLure K.G., Lees-Miller S.P., Rancourt D.E., Lee P.W. DNA-dependent protein kinase acts upstream of p53 in response to DNA damage. Nature. 1998;394:700–704. doi: 10.1038/29343. [DOI] [PubMed] [Google Scholar]
- Canman C.E., Lim D.S., Cimprich K.A., Taya Y., Tamai K., Sakaguchi K., Appella E., Kastan M.B., Siliciano J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- Hall-Jackson C.A., Cross D.A., Morrice N., Smythe C. ATR is a caffeine-sensitive, DNA-activated protein kinases with a substrate specificity distinct from DNA-PK. Oncogene. 1999;18:6707–6713. doi: 10.1038/sj.onc.1203077. [DOI] [PubMed] [Google Scholar]
- Yarosh D.B., Cruz P.D., Dougherty I., Bizios N., Kibitel J., Goodtzova K., Both D., Goldfarb S., Green B., Brown D. FRAP DNA-dependent protein kinase mediates a late signal transduced from ultraviolet-induced DNA damage. J. Invest. Dermatol. 2000;114:1005–1010. doi: 10.1046/j.1523-1747.2000.00964.x. [DOI] [PubMed] [Google Scholar]
- Peterson R.T., Desai B.N., Hardwick J.S., Schreiber S.L. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycin-associated protein. Proc. Natl. Acad. Sci. USA. 1999;96:4438–4442. doi: 10.1073/pnas.96.8.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaria J.N., Tibbetts R.S., Busby E.C., Kennedy A.P., Hill D.E., Abraham R.T. Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res. 1998;58:4375–4382. [PubMed] [Google Scholar]
- Chan D.W., Son S.C., Block W., Ye R., Khanna K.K., Wold M.S., Douglas P., Goodarzi A.A., Pelley J., Taya Y. Purification and characterization of ATM from human placenta. A manganese-dependent, wortmannin-sensitive serine/threonine protein kinase. J. Biol. Chem. 2000;275:7803–7810. doi: 10.1074/jbc.275.11.7803. [DOI] [PubMed] [Google Scholar]
- Sarkaria J.N., Busby E.C., Tibbetts R.S., Roos P., Taya Y., Karnitz L.M., Abraham R.T. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–4382. [PubMed] [Google Scholar]
- Schmelzle T., Hall M.N. TOR, a central controller of cell growth. Cell. 2000;103:253–262. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- Verhaegh G.W., Richard M.J., Hainaut P. Regulation of p53 by metal ions and by antioxidantsdithiocarbamate down-regulates p53 DNA binding activity by increasing the intracellular level of copper. Mol. Cell. Biol. 1997;17:5699–5706. doi: 10.1128/mcb.17.10.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.H., Momand J. Pyrrolidine dithiocarbamate prevents p53 activation and promotes p53 cysteine residue oxidation. J. Biol. Chem. 1998;273:18898–18905. doi: 10.1074/jbc.273.30.18898. [DOI] [PubMed] [Google Scholar]
- Arora V., Iversen P.L. Antisense oligonucleotides targeted to the p53 gene modulate liver regeneration in vivo. Drug Metabolism Disposition. 2000;28:131–138. [PubMed] [Google Scholar]
- Rotter V., Schartz D., Almon E., Goldfinger N., Kapon A., Meshorer A., Donehower L.A., Levine A.J. Mice with reduced levels of p53 protein exhibit the testicular giant-cell degenerative syndrome. Proc. Natl. Acad. Sci. USA. 1993;90:9075–9079. doi: 10.1073/pnas.90.19.9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombel M., Radvanyi F., Blanche M., Abbou C., Buttyan R., Donehower L.A., Chopin D., Thiery J.P. Androgen suppressed apoptosis is modified in p53 deficient mice. Oncogene. 1995;10:1269–1274. [PubMed] [Google Scholar]
- Ferri K.F., Jacotot E., LeDuc P., Geuskens M., Ingber D.E., Kroemer G. Apoptosis of syncytia induced by HIV-1-envelope glycoprotein complex. Influence of cell shape and size. Exp. Cell Sci. 2000;261:119–126. doi: 10.1006/excr.2000.5062. [DOI] [PubMed] [Google Scholar]
- Gottschalk A.R., Boise L.H., Thompson C.B., Quintans J. Identification of immunosuppressant-induced apoptosis in a murine B cell line and its prevention by bcl-x but not bcl-2. Proc. Natl. Acad. Sci. USA. 1994;91:7350–7354. doi: 10.1073/pnas.91.15.7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi H., Dilling M.B., Shikata T., Liu L.N., Shu L., Ashmun R.A., Germain G.S., Abrham R.T., Houghton P.J. Rapamycin causes poorly reversible inhibition of mTOR and induces p53-independent apoptosis in human rhabdomyosarcoma cells. Cancer Res. 1999;59:886–894. [PubMed] [Google Scholar]
- Shinjyo T., Kuribara R., Inukai T., Hosoi H., Kinoshita T., Miyajima A., Houghton P.J., Look A.T., Ozawa K., Inaba T. Downregulation of Bim, a pro-apoptotic relative of Bcl-2 is a pivotal step in cytokine-initiated survival signaling in murine hematopoietic progenitors. Mol. Cell. Biol. 2001;21:854–864. doi: 10.1128/MCB.21.3.854-864.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conejo R., Lorenzo M. Insulin singaling leading to proliferation, survival, and membrane ruffling in C2C12 myoblasts. J. Cell. Physiol. 2001;187:96–108. doi: 10.1002/1097-4652(2001)9999:9999<::AID-JCP1058>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Szegezdi E.E., Szondy Z., Nagy L., Nemes Z., Friis R.R., Davies P.J., Fesus L. Apoptosis-linked in vivo regulation of the tissue transglutaminase gene promoter. Cell Death Differ. 2000;7:1225–1233. doi: 10.1038/sj.cdd.4400751. [DOI] [PubMed] [Google Scholar]
- Macho A., Castedo M., Marchetti P., Aguilar J.J., Decaudin D., Zamzami N., Girard P.M., Uriel J., Kroemer G. Mitochondrial dysfunctions in circulating T lymphocytes from human immunodeficiency virus-1 carriers. Blood. 1995;86:2481–2487. [PubMed] [Google Scholar]
- Carbonari M., Pesce M., Cibati M., Modica A., Dellanna L., Doffizi G., Angelici A., Uccini S., Modesti A., Fiorilli M. Death of bystander cells by a novel pathway involving early mitochondrial damage in human immunodeficiency virus-related lymphadenopathy. Blood. 1997;90:209–216. [PubMed] [Google Scholar]