

Abstract
We have previously shown that interferon and tumor necrosis factor noncytopathically abolish hepatitis B virus (HBV) replication from the hepatocyte and kidney tubular epithelial cells in vivo. Here we show that a persistent lymphocytic choriomeningitis virus (LCMV) infection is cleared from the hepatocyte noncytopathically when the same cytokines are induced in the liver by antigen-nonspecific stimuli. These results indicate that, like HBV, LCMV is also susceptible to intracellular inactivation by cytokine-induced antiviral mechanisms that are operative in the hepatocyte. In contrast, LCMV is not cleared from intrahepatic nonparenchymal cells or splenocytes, indicating that, unlike the hepatocyte, these cells do not produce the factors required to inactivate LCMV. Antiviral mechanisms like these may have evolved to maintain the functional integrity of vital organs in the face of massive infection.
Keywords: lymphocytic choriomeningitis virus, noncytopathic clearance, hepatocyte, cytokines
Cytokines appear to play an effector role in the antiviral immune response during hepatitis B virus (HBV)1 infection. Recently we have shown that HBV-specific CTLs can abolish viral gene expression and replication in the liver of HBV transgenic mice by noncytopathic mechanisms that are mediated by IFN-γ and TNF-α (1). Similar cytokine-mediated antiviral events occur in these animals after the administration of IL-12 (2) and also during unrelated hepatotropic infections with lymphocytic choriomeningitis virus (LCMV [3]), adenovirus (4), and CMV (4), which suppress HBV replication by inducing type I IFN as well as IFN-γ and TNF-α. These results suggest that the intrahepatic cytokine profile of the antiviral immune response may determine the outcome of HBV infection in humans, e.g., viral clearance versus viral persistence. In support of this notion, we have recently shown that viral clearance coincides with the kinetics of intrahepatic induction of IFN-γ and TNF-α and precedes the onset of hepatitis in chimpanzees acutely infected with HBV (4a).
The relative contribution of cytodestructive versus curative functions of the immune response is less clear in the case of other viral infections, including LCMV, the prototypic noncytopathic virus that requires CD8+ T cells for clearance (5). For example, perforin-deficient mice are unable to clear LCMV (6, 7), although virus replication is less efficiently controlled in mice that lack IFN-γ receptors (8) or that have been treated with IFN-γ–specific antibodies (9–11). Additionally, persistent LCMV infection can be cleared by very small numbers of adoptively transferred CTLs (12) and in the absence of severe immunopathology (13). Conversely, viral clearance does not occur when relatively high numbers of IFN-γ–deficient CTLs are passively transferred (14).
This study was undertaken to test whether the clearance of persistent LCMV infection from liver and spleen observed after adoptive transfer of LCMV-immune spleen cells could be reproduced by the local induction of cytokines known to control HBV replication (i.e., IFN-γ, TNF-α, and IFN-α/β). Intrahepatic and intrasplenic cytokine induction was achieved in persistently LCMV– infected mice by superinfection with a replication-deficient adenovirus or by repetitive injections of IL-12. The results indicate that LCMV can be eliminated from the hepatocytes in a cytokine-dependent, noncytopathic bystander manner, but that other events, presumably killing, are needed to eliminate LCMV from other cell types.
Materials and Methods
Mice
C57BL/6 and BALB/c mice were purchased from The Jackson Laboratory or from our breeding colony at The Scripps Research Institute.
Viruses and Infection of Mice
LCMV strain Armstrong (ARM) 53B, a clone triple plaque purified from ARM CA 1371 (15), was used in this study. Viral stocks were prepared by growth in BHK-21 cells. Viral titers were measured by plaque assay as described (15). In brief, monolayers of Vero cells were infected with different dilutions of mouse sera or tissue homogenates, and plaques were counted 6 d later. To establish a persistent infection, C57BL/6 and BALB/c mice were infected within 24 h of birth by intracardiac inoculation of 103 PFU of LCMV ARM. LCMV-immune mice were obtained by injecting 8–10-wk-old mice intraperitoneally with 2 × 105 PFU of LCMV ARM. Immune mice were used at >60 d after infection. BALB/c-derived LCMV-immune splenocytes (5 × 107 cells) were injected intraperitoneally into persistently infected BALB/c mice that were irradiated (350 rads) a few hours before transfer and killed at multiple time points thereafter. A recombinant, replication-deficient adenovirus, designated Ad.CBlacZ, provided by Dr. James Wilson (University of Pennsylvania Medical Center, Philadelphia, PA [16]), was also used to infect LCMV-carrier BALB/c mice. Stocks of Ad.CBlacZ were grown in 293 cells (17), and were purified by two rounds of CsCl density centrifugation, as described previously (18). Viral titers were determined by plaque assay on 293 cells, and a single stock was used throughout this study. Mice were injected intravenously with 1.5 × 109 PFU/mouse, a dose of Ad.CBlacZ known to infect 100% of the hepatocytes and to cause a CD8-dependent liver disease (4). Control mice were injected with the same volume of saline. Animals were killed at multiple time points after infection.
IL-12
Recombinant murine IL-12 was provided by Dr. Maurice Gately (Hoffmann-La Roche, Nutley, NJ). C57BL/6 mice were injected intraperitoneally with IL-12 (1 μg/d/mouse). Control animals were injected with saline diluent (saline containing 1% serum) only. Animals were killed 24 h after the last injection of IL-12, and their sera, livers, and spleens were harvested for subsequent analyses.
RNA Analysis
Northern Blot Analysis.
Frozen tissues were mechanically pulverized, and RNA was extracted by the acid-guanidium phenol-chloroform method (19). Total RNA (20 μg) was analyzed for 2′,5′-oligoadenylate synthetase (2′5′ OAS) and glyceraldehyde-3-phosphate (GAPDH) expression by Northern blot as described previously (3).
RNase Protection Assay.
The RNase protection assay for quantitation of mRNA was performed exactly as described (20). The mouse IL-1α(B), mIL-1β(A), mIL-2(A), mIL-3(B), mIL-4(B), mIL-5(C), mIL-6(B), mIFNγ(B), mTNFα(A), mTNFβ(A), and mL32(A) subclones in the pGEM-4 transcription vector were described in a previous report (20). The mCD4(IC), mCD3γ(IC), mCD8α(DM), and F480 subclones in the pGEM-4 vector were described previously (1).
In Situ Hybridization.
This procedure was carried out exactly as described (21). The 33P-labeled RNA probe used in this study was prepared by transcription from the T7 promoter of plasmid nucleoprotein (NP) Bluescript, a plasmid created by cloning the 1,164-bp BglII fragment from a cDNA of the LCMV ARM S RNA segment (22) into the plasmid Bluescript KS (Stratagene, Inc.). Transcription from the T7 promoter of pNP Bluescript generates a single-stranded RNA probe complementary to the viral NP mRNA and antigenomic sequence.
RNA PCR Assay for the Detection of LCMV ARM and the Variant Clone 13.
Total liver RNA (1 μg) was reverse transcribed into cDNA and amplified by PCR using LCMV glycoprotein– specific primers exactly as described (23). Quantitation of LCMV ARM and clone 13 RNA was carried out by densitometric analysis (NIH Image software) of the amplified PCR products after MnlI digestion, gel electrophoresis, and ethidium bromide staining, exactly as described (23).
Biochemical and Histological Analysis of Liver Disease
Hepatocellular injury was monitored by measuring serum alanine aminotransferase (sALT) activity (1). Results were expressed as mean sALT activity ± SEM. Tissue samples were fixed in 10% zinc-buffered formalin (Anatek, Ltd.), embedded in paraffin, sectioned (3 μm), and stained with hematoxylin and eosin as described (1).
Immunohistochemical Analysis
The intracellular distribution of LCMV NP was analyzed by immunohistochemical analysis based on a method described by Surh et al. (24). 3-amino-9-ethyl carbazole (red) was used as coloring substrate for LCMV NP, exactly as described (3).
β-Galactosidase Histochemistry
The in vivo expression of β-galactosidase in the livers of Ad.CBlacZ-infected animals was quantitated by 5-bromo-4-chloro-3-indolyl-β-d-galactosidase (X-gal) histochemistry exactly as described (4).
Results
Persistent LCMV Infection in C57BL/6 and BALB/c Mice.
C57BL/6 and BALB/c mice were infected at birth with LCMV ARM as described in Materials and Methods. At 7–8 wk of age, all animals were bled and serum viral titers were analyzed by plaque assay. Mice with a serum titer between 104 and 105 PFU/ml were selected, and 2–4 wk later were used as recipients of saline, LCMV-immune “memory” cells, adenovirus, or IL-12. Table I shows the viral titers in serum, liver, and spleen of control BALB/c and C57BL/6 mice that were killed at the indicated time points after saline injection. The hepatic content of LCMV RNA in the saline-injected controls was monitored by Northern blot analysis (see Figs. 1, 5, and 7) and by in situ hybridization analysis (see Figs. 2 A, 6 A, and 8 A) in which hepatic LCMV RNA was detected in ∼30–40% of the hepatocytes (Figs. 2 A, 6 A, and 8 A) and in nonparenchymal cells in the hepatic sinusoids (Fig. 2 A, arrows). Next, we monitored the hepatic content of mRNA for CD3, CD4, CD8, various cytokines, and 2′5′ OAS (a marker of IFN-α/β induction) in these control animals. In keeping with the massive LCMV infection, high levels of 2′5′ OAS mRNA were detected in the liver, illustrating that type I IFN is not sufficient to clear LCMV from the organ. Furthermore, all T cell markers and T cell–dependent cytokines were absent, and only low levels of mRNA for the monokines TNF-α, IL-1α, and IL-1β were detected (see Figs. 1, 5, and 7), reflecting the absence of histological (Fig. 3 A) or biochemical (Table I) evidence of liver disease in these persistently infected animals.
Table I.
sALT Activity and LCMV Titers in Serum, Liver, and Spleen
| Treatment | Mouse strain | No. of Mice | Time of autopsy | sALT (day 0) | sALT (autopsy) | Serum LCMV | Liver LCMV | Spleen LCMV | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (PFU/ml) | (PFU/g) | (PFU/g) | ||||||||||||||
| Saline | BALB/c | 3 | day 3 | 66 ± 11 | 100 ± 10 | 7.3 ± 1.0 × 104 | 6.9 ± 3.0 × 106 | 7.0 ± 5.2 × 106 | ||||||||
| Saline | BALB/c | 3 | day 24 | 63 ± 15 | 56 ± 35 | 2.6 ± 0.1 × 104 | 1.1 ± 0.4 × 107 | 1.2 ± 0.7 × 107 | ||||||||
| Memory | BALB/c | 3 | day 3 | 50 ± 10 | 590 ± 527 | 5.8 ± 2.3 × 104 | 7.5 ± 12 × 106 | 3.7 ± 0.2 × 105 | ||||||||
| Memory | BALB/c | 3 | day 7 | 52 ± 11 | 953 ± 826 | 1.1 ± 0.2 × 104 | <400 | 6.1 ± 5.0 × 104 | ||||||||
| Memory | BALB/c | 3 | day 14 | 64 ± 15 | 283 ± 137 | 3.2 ± 2.4 × 103 | <400 | 6.5 ± 5.5 × 103 | ||||||||
| Memory | BALB/c | 3 | day 24 | 65 ± 14 | 63 ± 20 | <400 | <400 | <400 | ||||||||
| Adenovirus | BALB/c | 4 | day 3 | 63 ± 14 | 80 ± 14 | 8.6 ± 3.5 × 104 | 6.3 ± 3.3 × 106 | 1.8 ± 0.7 × 107 | ||||||||
| Adenovirus | BALB/c | 4 | day 7 | 60 ± 10 | 2,485 ± 739 | 2.9 ± 1.7 × 105 | 3.9 ± 1.5 × 106 | 1.8 ± 0.7 × 107 | ||||||||
| Adenovirus | BALB/c | 4 | day 14 | 54 ± 12 | 2,085 ± 521 | 9.1 ± 2.7 × 104 | 1.0 ± 13 × 107 | 1.0 ± 5.0 × 107 | ||||||||
| Adenovirus | BALB/c | 4 | day 21 | 63 ± 11 | 612 ± 795 | 6.3 ± 2.7 × 104 | 1.9 ± 26 × 106 | 1.4 ± 4.2 × 107 | ||||||||
| Adenovirus | BALB/c | 4 | day 24 | 58 ± 14 | 97 ± 63 | 3.3 ± 1.9 × 104 | 4.4 ± 47 × 106 | 1.2 ± 5.4 × 107 | ||||||||
| Saline | C57BL/6 | 3 | day 4 | 67 ± 16 | 70 ± 27 | 1.3 ± 8.7 × 104 | 3.1 ± 23 × 106 | 1.7 ± 13 × 106 | ||||||||
| IL-12 (1 μg × 3) | C57BL/6 | 5 | day 4 | 60 ± 3 | 78 ± 16 | 2.1 ± 1.3 × 104 | 4.8 ± 35 × 106 | 1.1 ± 8.5 × 106 | ||||||||
| Saline | C57BL/6 | 6 | day 11 | 49 ± 13 | 76 ± 22 | 2.7 ± 1.0 × 104 | 9.2 ± 6.8 × 106 | 2.6 ± 20 × 106 | ||||||||
| IL-12 (1 μg × 10) | C57BL/6 | 8 | day 11 | 54 ± 19 | 140 ± 68 | 8.4 ± 11 × 104 | 2.7 ± 17 × 106 | 3.7 ± 28 × 106 |
sALT and PFU are expressed as mean ± SEM.
Figure 1.
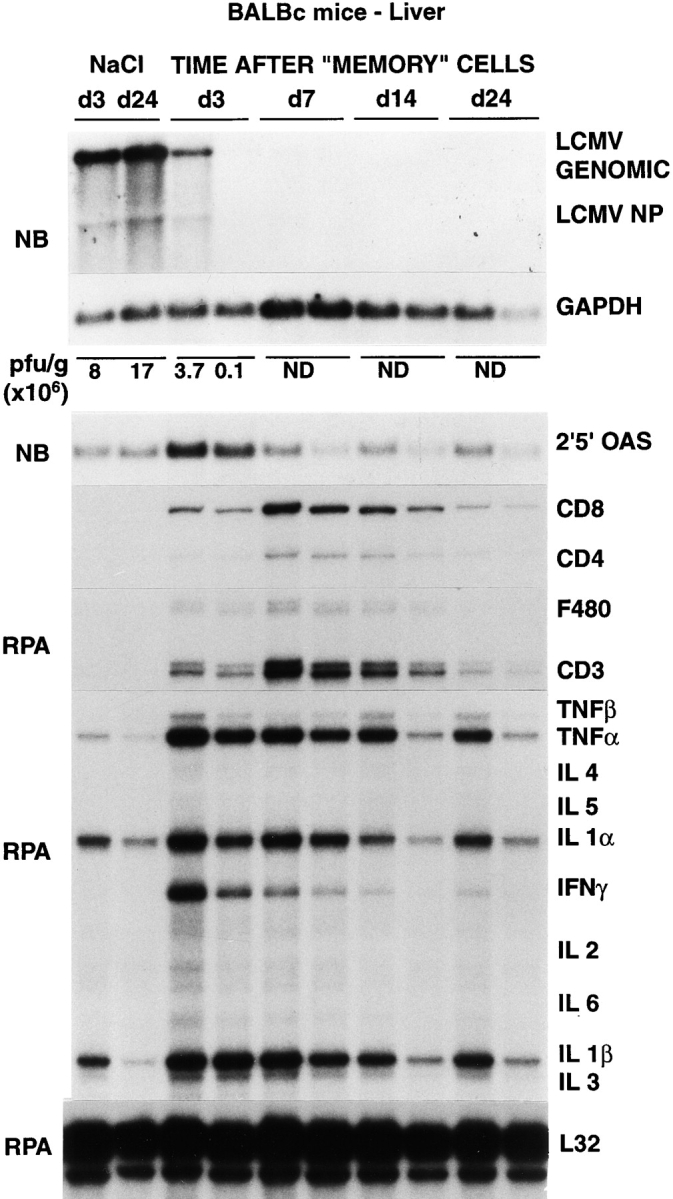
Intrahepatic LCMV, T cell marker, and cytokine gene expression after adoptive transfer of memory cells. Age- and serum LCMV titer–matched BALB/c mice were injected intraperitoneally either with saline or with 2 × 107 memory cells, and livers were harvested from mice killed on days 3, 7, 14, and 24 after injection, as indicated. Northern blot (NB) analysis (top panel) was performed with 20 μg of total liver RNA from two representative mice per group. The membrane was hybridized with 32P-labeled LCMV-, 2′5′ OAS– (a marker of IFN-α/β induction), and GAPDH-specific DNA probes. Total hepatic RNA (10 μg) from the same mice was also analyzed by RNase protection assay (RPA) for the expression of various cytokine transcripts, and for the expression of CD3, CD4, CD8, and F480, as indicated. The mRNA encoding the ribosomal protein L32 was used to normalize the amount of RNA loaded in each lane. The results of the plaque assay (PFU per gram) of individual liver tissues are also indicated. ND, not detectable.
Figure 5.

Intrahepatic LCMV, T cell marker, and cytokine gene expression after adenovirus infection. Age- and serum LCMV titer–matched BALB/c mice were injected intravenously either with saline or with 1.5 × 109 PFU of adenovirus (Ad.CBlacZ), and livers were harvested from mice killed on days 3, 7, 14, 21, and 24 after injection, as indicated. Northern blot (NB) analysis (top panel) was performed with 20 μg of total liver RNA from two representative mice per group. The membrane was hybridized with 32P-labeled LCMV-, 2′5′ OAS– (a marker of IFN-α/β induction), and GAPDH-specific DNA probes. Total hepatic RNA (10 μg) from the same mice was also analyzed by RNase protection assay (RPA) for the expression of various cytokine transcripts, and for the expression of CD3, CD4, CD8, and F480, as indicated. The mRNA encoding the ribosomal protein L32 was used to normalize the amount of RNA loaded in each lane. The results of the plaque assay (PFU per gram) of individual liver tissues are also indicated.
Figure 7.
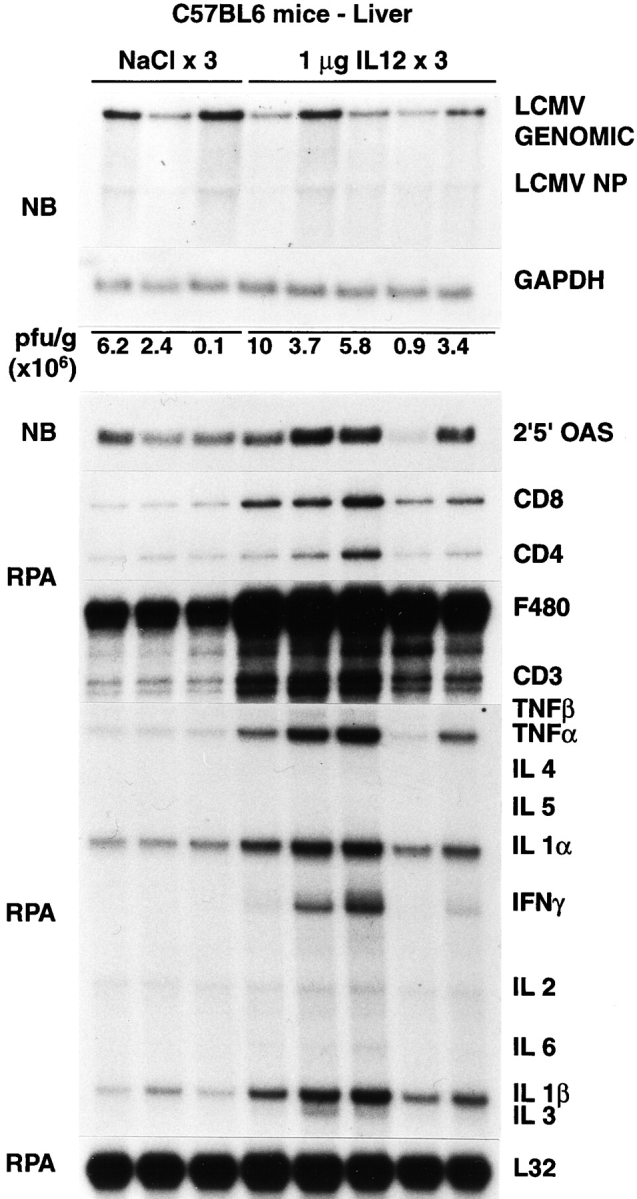
Intrahepatic LCMV, T cell marker, and cytokine gene expression after IL-12 administration. Age- and serum LCMV titer–matched C57BL/6 mice were injected intraperitoneally either with saline or with IL-12 (1 μg/d/mouse) and killed on day 3 after injection, as indicated. Northern blot (NB) analysis (top panel) was performed with 20 μg of total liver RNA from two representative mice per group. The membrane was hybridized with 32P-labeled LCMV-, 2′5′ OAS– (a marker of IFN-α/β induction), and GAPDH-specific DNA probes. Total hepatic RNA (10 μg) from the same mice was also analyzed by RNase protection assay (RPA) for the expression of various cytokine transcripts, and for the expression of CD3, CD4, CD8, and F480, as indicated. The mRNA encoding the ribosomal protein L32 was used to normalize the amount of RNA loaded in each lane. The results of the plaque assay (PFU per gram) of individual liver tissues are also indicated.
Figure 2.

Intrahepatic distribution of LCMV RNA after adoptive transfer of memory cells. The same livers described in the legend to Fig. 1 were analyzed for the expression of LCMV RNA by in situ hybridization using a specific 33P-labeled LCMV riboprobe. (A) Detection of hepatic LCMV mRNAs in saline-injected control mice. Note that most LCMV RNA is contained in the hepatocytes compared with nonparenchymal cells (arrows). (B). Detection of hepatic LCMV mRNAs in mice injected with memory cells and killed at day 7. Note that LCMV RNA is no longer detectable in the hepatocytes but remains in a few nonparenchymal cells that resemble endothelial cells and biliary duct epithelial cells in the portal tracts (hematoxylin and eosin; original magnification: ×600).
Figure 3.
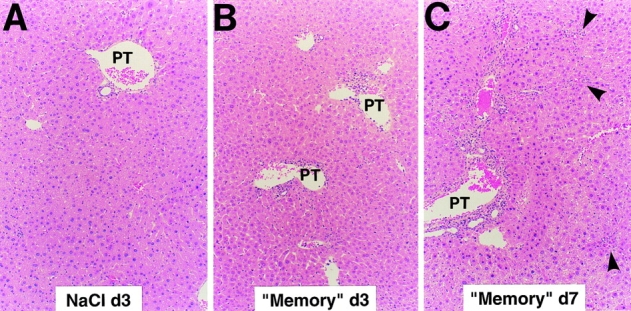
Histopathological features of viral hepatitis in the liver of mice after adoptive transfer of memory cells. Liver sections obtained from persistently LCMV–infected mice killed at the indicated time points after injection of saline (A) or after adoptive transfer of memory cells (B and C) were stained with hematoxylin and eosin as described in Materials and Methods. Note that the vast majority of the hepatic parenchyma is cytologically normal in all panels. A few mononuclear cells are seen in rare portal tracts (PT) 3 d after transfer of memory cells (B) and, in addition to this, small, scattered necroinflammatory foci (arrowheads) are detected in the hepatic parenchyma 7 d after transfer of memory cells (C). Original magnification: ×100.
Clearance of persistent LCMV infection after adoptive transfer of memory cells occurs very rapidly in the liver without massive liver cell injury. Splenocytes derived from mice that have cleared an acute LCMV infection (i.e., memory cells) are known to clear LCMV when injected into persistently infected animals (5). To better understand the mechanisms responsible for this effect, 2 × 107 BALB/c LCMV-immune memory splenocytes were adoptively transferred into 12 BALB/c mice, and groups of 3 mice were killed on days 3, 7, 14, and 24 after transfer (Table I). Predictably, LCMV was undetectable on day 24 in serum, liver, and spleen by plaque assay (Table I). Interestingly, the viral titer dropped very rapidly in the liver (>6 logs in 4 d), becoming undetectable by day 7 after memory cell transfer (Table I). At this time point, viral RNA was also undetectable by Northern blot analysis of total liver RNA (Fig. 1). However, traces of viral RNA remained detectable at day 7 in a few nonparenchymal cells that resemble endothelial cells and biliary duct epithelial cells in the portal tracts (Fig. 2 B). Interestingly, these cells remained LCMV RNA–positive even at the latest time point examined (day 24; not shown), suggesting that they are relatively resistant to viral clearance. Along with LCMV RNA, LCMV NP was undetectable immunohistochemically in the hepatocytes by day 7 (not shown). Importantly, LCMV titer dropped much more slowly (<2 logs in 14 d) in serum and spleen, although LCMV was eventually cleared from these compartments between days 14 and 24 (Table I). sALT activity (a marker of hepatocellular necrosis [25]) reached values of ∼600, ∼700, and ∼900 U/l at days 3, 5, and 7 (Table I), respectively. After its peak at day 7, sALT activity returned to normal levels (∼60 U/l) between days 14 and 24 (Table I). sALT activity was only modestly elevated in these mice, compared with previously reported mouse models of liver disease (26). In particular, by comparing these ALT profiles with known standards (1), we estimate that no more than 5–10% of the hepatocytes were killed during the first week after adoptive transfer, despite the fact that at least 30–40% of hepatocytes were infected (Figs. 2 A, 6 A, and 8 A). This notion is also supported by histological analysis of these livers, which shows that 3 and 7 d after transfer of memory cells, the vast majority of the hepatic parenchyma is cytologically normal (Fig. 3, B and C). At day 3, few mononuclear cells are seen in portal tracts (PT), and at day 7 the hepatic parenchyma contains small, scattered necroinflammatory foci (Fig. 3 C, arrowheads); and, in keeping with the relatively mild liver disease, no liver cell regeneration was observed at the peak of sALT activity (day 7; Fig. 3 C), at which time LCMV RNA had disappeared from the hepatocytes (Fig. 2 B). The kinetics of liver disease and the kinetics of viral clearance coincided with the appearance of T cell (especially CD8), macrophage, and cytokine (2′5′ OAS, IFN-γ, TNF-α, IL-1α, and IL-1β) markers in the liver (Fig. 1). It is noteworthy that the virus was cleared much more slowly in the spleen despite the fact that the same cytokines were induced with the same kinetics observed in the liver (Fig. 4). Collectively, these results indicate that hepatocytes have the capacity to clear LCMV very rapidly, and this occurs in the presence of antiviral cytokines and in the absence of a commensurate degree of cell death. In contrast, the slower kinetics of clearance of LCMV from nonparenchymal liver cells and from the spleen indicate that these cells are relatively resistant to memory cell– induced clearance.
Figure 4.

Cytokine gene expression in the spleen of mice after adoptive transfer of memory cells. Total RNA (10 μg) extracted from the spleen of the same mice described in the legend to Fig. 1 was analyzed by RNase protection assay (RPA) for the expression of various cytokine transcripts as indicated. The mRNA encoding the ribosomal protein L32 was used to normalize the amount of RNA loaded in each lane. The results of the plaque assay (PFU per gram) of individual spleen tissues are also indicated. ND, not detectable. Uninf., uninfected.
Clearance of Persistent LCMV Infection from the Hepatocyte after Adenovirus Infection or IL-12 Administration.
To monitor the antiviral effect of the induction of these cytokines in the liver of mice persistently infected with LCMV, 20 BALB/c mice were infected intravenously with a dose (1.5 × 109 PFU) of a recombinant, replication-deficient adenovirus (Ad.CBlacZ) that we have previously shown to infect virtually all of the hepatocytes (4), and groups of four mice were killed on days 3, 7, 14, 21, and 24 after infection (Table I). Additionally, 13 C57BL/6 mice were injected intraperitoneally with recombinant murine IL-12 (1 μg/d) for either 3 or 10 d, and all animals were killed 24 h after the last injection (Table I). Results were compared with saline-injected controls as indicated in Table I. These treatments were chosen because they are known to induce IFN type I, IFN-γ, and TNF-α in the liver either with (adenovirus) or without (IL-12) destroying hepatocytes (2, 4). As expected, the adenovirus-infected animals developed a necroinflammatory liver disease that was detectable histologically (not shown) and biochemically as elevated sALT activity (Table I) starting between 3 and 7 d after inoculation and lasting ∼3 wk until lacZ-positive hepatocytes were no longer detectable (not shown). Despite the relatively severe liver disease (compared with that induced by transfer of memory cells), the viral titers in the serum, liver, and spleen did not change at any of the time points examined (Table I). Similarly, no change in viral titer was observed in the IL-12–treated animals in which we observed no biochemical (Table I) and/or histological evidence of liver cell injury (not shown). The absence of viral clearance in the livers of adenovirus-infected and IL-12–injected mice was also confirmed by Northern blot analysis of LCMV RNA, as shown in Figs. 5 and 7.
Because these results appeared to contradict the results of the memory cell transfer experiments, we performed in situ hybridization analyses on the adenovirus-infected and IL-12– injected livers to examine the hepatic distribution of LCMV RNA. Surprisingly, LCMV RNA was dramatically reduced in the hepatocytes (but not in nonparenchymal cells, which include resident nonparenchymal cells and infiltrating inflammatory cells) by days 7 (Fig. 6 B) and 14 (not shown) after adenovirus infection, concomitant with the induction of cytokines in the liver (Fig. 5). In contrast, the intensity and distribution of grains in the hepatocytes on day 24 after adenovirus infection (Fig. 6 C) were similar to those of saline-injected controls (Fig. 6 A), and this occurred at a time point when the liver disease and the adenoviral infection had resolved (not shown) and the cytokines were no longer induced in the liver (Fig. 5). Similarly, LCMV RNA disappeared from the hepatocyte (but not from nonparenchymal cells) after 3 (Fig. 8, B–F) or 10 (not shown) IL-12 injections, and this occurred in livers in which there was inflammatory cell infiltration but little or no histological evidence of hepatocyte injury (not shown) or sALT elevation (Table I). Collectively, these results indicate that LCMV is rapidly cleared from the hepatocyte in a bystander manner concomitant with intrahepatic cytokine induction, and this effect is not dependent on the destruction of hepatocytes.
Figure 6.

Intrahepatic distribution of LCMV RNA after adenovirus infection. The same livers described in the legend to Fig. 5 were analyzed for the expression of LCMV RNA by in situ hybridization using a specific 33P-labeled LCMV riboprobe. (A) Detection of hepatic LCMV mRNAs in saline- injected control mice. (B) Detection of hepatic LCMV mRNAs in mice injected with adenovirus and killed at day 7. Note that LCMV RNA is virtually no longer detectable in the hepatocytes but remains in nonparenchymal and inflammatory cells (arrows). (C) Detection of hepatic LCMV mRNAs in mice injected with adenovirus and killed at day 24. Note that most LCMV RNA is detected in the hepatocytes, similar to saline-injected controls shown in A (hematoxylin and eosin; original magnification: ×600).
Figure 8.
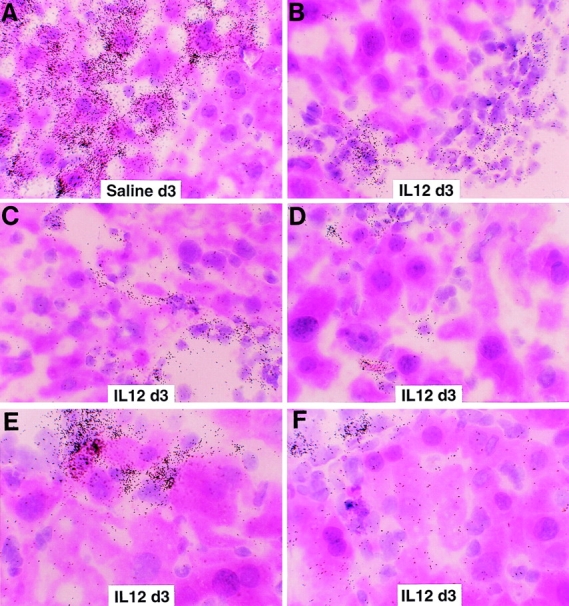
Intrahepatic distribution of LCMV RNA after IL-12 administration. The same livers described in the legend to Fig. 7 were analyzed for the expression of LCMV RNA by in situ hybridization using a specific 33P-labeled LCMV riboprobe. (A) Detection of hepatic LCMV mRNAs in saline- injected control mice. (B–F) Detection of hepatic LCMV mRNAs in mice injected with IL-12. Note that LCMV RNA is virtually no longer detectable in the hepatocytes but remains in nonparenchymal and inflammatory cells (hematoxylin and eosin; original magnification: ×600).
Lack of Clearance of Persistent LCMV Infection from Intrahepatic Nonparenchymal Cells and from Splenocytes after Adenovirus Infection or IL-12 Administration.
As shown in Figs. 6 and 8, the local induction of cytokines observed after adenovirus infection and IL-12 administration was not associated with clearance of LCMV RNA from nonparenchymal liver cells or the spleen. This probably explains why the virus was not eliminated from these organs or the serum as measured by plaque assay (Table I). Indeed, the number of splenocytes positive for LCMV RNA by in situ hybridization was similar in the saline-injected controls and in mice injected with IL-12 (not shown), despite the fact that the cytokines were strongly induced in these organs (not shown). Collectively, these results indicate that LCMV clearance from nonparenchymal cells does not occur in the absence of a LCMV-specific immune response, suggesting that other mechanisms are probably required to clear the virus from these cells.
LCMV Variants Are Selected in Persistently Infected Livers and They Are Not Eliminated from Intrahepatic Nonparenchymal Cells after Adenovirus Infection or IL-12 Administration.
After neonatal infection with LCMV ARM, variants such as clone 13, which are known to be more resistant to control by IFNs (11), have been isolated from persistently infected livers (23). To test whether these variants are present in the livers of control mice (in which most viral RNA is in the hepatocytes) and IL-12–treated or adenovirus-infected mice (in which most viral RNA is in the nonparenchymal cells), we performed a PCR assay for the detection of a point mutation in the viral RNA that results in a phenylalanine versus a leucine change at position 260 in the viral glycoprotein (23). The phenylalanine/leucine change is diagnostic for the ARM strain of LCMV versus variants such as clone 13. Roughly equimolar amounts of RNA encoding a phenylalanine versus a leucine residue were detected in the liver of saline-injected controls and IL-12–treated or adenovirus-infected mice (not shown). This suggests that the ARM as well as the variant strains of LCMV are resistant to the antiviral effects induced by IL-12 or adenovirus when they replicate in the nonparenchymal cells. Whether the hepatocytes were infected with ARM or the variant strains or both, and whether these viruses would be differently sensitive to clearance by IL-12 injection or adenovirus infection, remain to be determined.
Discussion
In this study, we demonstrate that LCMV can be cleared from hepatocytes by noncytopathic, cytokine-associated antiviral mechanisms that are not operative in splenocytes or in nonparenchymal cells (which include resident nonparenchymal cells and infiltrating inflammatory cells) in the liver. Several lines of evidence support these conclusions. After transfer of memory cells, viral clearance from the hepatocytes occurs very rapidly, between 3 and 7 d after transfer and 4 d (or less) after the peak of LCMV replication (Table I). This is associated with a relatively mild liver disease, and it occurs in the context of intrahepatic cytokine induction. Indeed, the liver disease in these animals would probably be much more severe if viral clearance was due primarily to the destruction of the infected cells, based on comparison with previously reported mouse models of liver disease (1, 26). Conversely, viral clearance from the intrahepatic nonparenchymal cells and from the spleen of the same animals is a much slower process that follows the peak of local cytokine induction by >10 d, suggesting either that these cells are less responsive to cytokines or that other events, including killing, are required to clear the virus from these cell types. Furthermore, LCMV clearance from the hepatocytes (but not from the intrahepatic nonparenchymal cells and from the spleen) can also occur when cytokines are locally induced after adenovirus infection or IL-12 administration. Again, this effect occurs rapidly (by day 3 for IL-12 and by day 7 for adenovirus), concomitant with the peak of intrahepatic cytokine induction and, in the case of IL-12, independently of hepatocyte destruction. This strongly suggests that cytokines can activate intracellular antiviral pathways in the hepatocytes (but not in the intrahepatic nonparenchymal cells or in the spleen). In support of this hypothesis, LCMV RNA reappears in the hepatocytes of adenovirus-infected and IL-12–injected animals when the intrahepatic cytokine induction subsided, in contrast to the memory cell recipients. This is presumably due to the absence of anti-LCMV antibodies and LCMV-specific T cells in the adenovirus-infected and IL-12–injected animals, since both B and T cells are required to clear a persistent LCMV infection (27).
These results indicate that cytokine-dependent activation of hepatocytes triggers intrahepatocellular antiviral events that are absent or not operative in certain other cell types. This is reminiscent of the unique role that hepatocytes play in host defense mechanisms after tissue damage. Indeed, during the acute phase response, the interaction of a wide variety of cytokines (including IFN-γ, IL-1α, IL-1β, IL-6, TNF-α, and TNF-β), glucocorticoids, and growth factors with hepatocytes results in a complex response characterized by activation of the acute phase plasma proteins (APPs) and profound changes in most metabolic pathways (28). The regulation of APP gene expression depends on transcriptional and posttranscriptional events (28) that are most likely operative in hepatocytes but not in nonhepatic cells. This study indicates that cytokines may activate hepatic cells not only to regulate APP genes but also to induce cell-specific antiviral pathways.
It has been suggested that specific destruction of infected cells (including hepatocytes) is required to control LCMV replication in the liver and other organs, since perforin- deficient mice fail to clear acute LCMV infection (6, 7). However, it is important to note that LCMV clearance was monitored by plaque assay in those studies which might not have detected the selective clearance of LCMV from the hepatocytes. Experiments designed to monitor the cytokine profile and the cellular distribution of viral RNA in the liver of these animals are needed to examine the possibility that LCMV clearance may have occurred from the hepatocytes but not from nonparenchymal cells in the perforin-deficient mice, as we observed after adenoviral infection and IL-12 administration. Indeed, we have previously shown that 2 d after acute LCMV infection, most of the virus replicates within nonparenchymal cells rather than hepatocytes (3). In addition, passive transfer of IFN-γ–deficient memory cells into persistently LCMV ARM–infected animals fails to clear the virus in liver and spleen (14), and mice genetically deficient for IFN-γ or its receptor have been shown to control LCMV replication less efficiently in various organs, including the liver, despite normal immune responses (8). Finally, in vivo treatment of neutralizing antibodies to this cytokine results in higher levels of LCMV replication during acute infection (11).
In conclusion, the results of this report show that LCMV, like HBV, is susceptible to noncytopathic antiviral control mechanisms that most likely depend on local cytokine induction, provided that the virus replicates within the hepatocyte. We have recently shown that HBV replication is completely abolished in the hepatocytes of HBV transgenic mice by cytokine-dependent pathways that do not require cell death (29), and that intrahepatic induction of the IFNs and TNF-α mediates the antiviral effect (1), irrespective of the stimuli that trigger their induction. Whether similar intracellular events may interfere with the life cycle of additional viruses remains to be determined. Nevertheless, viral clearance of these and other viruses might rely very heavily on noncytopathic curative mechanisms, especially for viruses that infect a large number of parenchymal cells in vital organs, like the liver. Indeed, it has been recently suggested that the control of murine CMV infection in the liver may depend on IFN-γ–dependent noncytopathic mechanisms (30). Conversely, viral clearance from nonparenchymal cells might rely mostly on immune-mediated cytodestructive mechanisms.
Acknowledgments
We thank Dr. James Wilson for providing the recombinant adenovirus Ad.CBlacZ; Dr. Maurice Gately for providing the recombinant murine IL-12; Dr. Claire Evans for providing the primers for the PCR RNA analysis of LCMV ARM and the variant clone 13; Dr. Monte Hobbs for providing the cytokine gene and T cell marker probe sets used in the RNase protection assays; Ms. Margie Chadwell and Ms. Josan Chung for excellent technical assistance; and Ms. Jennifer Newmann for help with manuscript preparation.
This work was supported by grants AI40696 and CA40489 from the National Institutes of Health. This is manuscript number 12087-MEM from The Scripps Research Institute.
Abbreviations used in this paper
- APP
acute phase protein
- ARM
Armstrong strain
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HBV
hepatitis B virus
- LCMV
lymphocytic choriomeningitis virus
- NP
nucleoprotein
- 2′5′ OAS
2′,5′-oligoadenylate synthetase
- sALT
serum alanine aminotransferase
Footnotes
P. Borrow's present address is The Edward Jenner Institute for Vaccine Research, Compton, Newbury, Berkshire RG20 7NN, UK.
References
- 1.Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 1996;4:25–36. doi: 10.1016/s1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- 2.Cavanaugh VJ, Guidotti LG, Chisari FV. Interleukin-12 inhibits hepatitis B virus replication in HBV transgenic mice. J Virol. 1997;71:3236–3243. doi: 10.1128/jvi.71.4.3236-3243.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guidotti LG, Borrow P, Hobbs MV, Matzke B, Gresser I, Oldstone MBA, Chisari FV. Viral cross talk: intracellular inactivation of the hepatitis B virus during an unrelated viral infection of the liver. Proc Natl Acad Sci USA. 1996;93:4589–4594. doi: 10.1073/pnas.93.10.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavanaugh VJ, Guidotti LG, Chisari FV. Inhibition of hepatitis B virus replication during adenovirus and cytomegalovirus infections in HBV transgenic mice. J Virol. 1998;72:2630–2637. doi: 10.1128/jvi.72.4.2630-2637.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4a.Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Francis, Chisari V. Viral clearance without destruction of infected cells during acute HBV infection. Science. 1999;284:825–829. doi: 10.1126/science.284.5415.825. [DOI] [PubMed] [Google Scholar]
- 5.Borrow, P., and M.B.A. Oldstone. 1997. Lymphocytic choriomeningitis virus. In Viral Pathogenesis. N. Nathanson, editor. Lippincott-Raven Publishers, Philadelphia. 593–627.
- 6.Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen J, Podack ER, Zinkernagel R, Hengartner H. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369:31–37. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- 7.Walsh CM, Matloubian M, Liu C-C, Ueda R, Kurahara CG, Christensen JL, Huang MT, Young JD-E, Ahmed R, Clark WR. Immune function in mice lacking the perforin gene. Proc Natl Acad Sci USA. 1994;91:10854–10858. doi: 10.1073/pnas.91.23.10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 9.Leist TP, Eppler M, Zinkernagel RM. Enhanced virus replication and inhibition of lymphocytic choriomeningitis virus disease in anti-gamma interferon-treated mice. J Virol. 1989;63:2813–2819. doi: 10.1128/jvi.63.6.2813-2819.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wille A, Gessner A, Lother H, Lehmann-Grube F. Mechanism of recovery from acute virus infection. VIII. Treatment of lymphocytic choriomeningitis virus- infected mice with anti-interferon-gamma monoclonal antibody blocks generation of virus-specific cytotoxic T lymphocytes and virus elimination. Eur J Immunol. 1989;19:1283–1288. doi: 10.1002/eji.1830190720. [DOI] [PubMed] [Google Scholar]
- 11.Moskophidis D, Battegay M, Bruendler M-A, Laine E, Gresser I, Zinkernagel R. Resistance of lymphocytic choriomeningitis virus to alpha/beta interferon and gamma interferon. J Virol. 1994;68:1951–1955. doi: 10.1128/jvi.68.3.1951-1955.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehmann-Grube F, Assmann U, Loliger C, Moskophidis D, Lohler J. Mechanism of recovery from acute virus infection. I. Role of T lymphocytes in the clearance of lymphocytic choriomeningitis virus from spleens of mice. J Immunol. 1985;134:608–615. [PubMed] [Google Scholar]
- 13.Gegin C, Lehmann-Grube F. Control of acute infection with lymphocytic choriomeningitis virus in mice that cannot present an immunodominant viral cytotoxic T lymphocyte epitope. J Immunol. 1992;149:3331–3338. [PubMed] [Google Scholar]
- 14.Tishon A, Lewiski H, Rall G, Von Herrath M, Oldstone MBA. An essential role for type 1 interferon-γ in terminating persistent viral infection. Virology. 1995;212:244–250. doi: 10.1006/viro.1995.1477. [DOI] [PubMed] [Google Scholar]
- 15.Dutko FJ, Oldstone MB. Genomic and biological variation among commonly used lymphocytic choriomeningitis virus strains. J Gen Virol. 1983;64:1689–1698. doi: 10.1099/0022-1317-64-8-1689. [DOI] [PubMed] [Google Scholar]
- 16.Kozarsky K, Wilson JM. Gene therapy: adenovirus vectors. Curr Opin Genet Dev. 1993;3:499–503. doi: 10.1016/0959-437x(93)90126-a. [DOI] [PubMed] [Google Scholar]
- 17.Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977;36:59–72. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 18.Engelhardt JF, Yang Y, Stratford-Perricaudet LD, Allen ED, Kozarsky K, Perricaudet M, Yankaskas JR, Wilson JM. Direct gene transfer of human CFTR into human bronchial epithelia of xenografts with E1-deleted adenoviruses. Nat Genet. 1993;4:27–34. doi: 10.1038/ng0593-27. [DOI] [PubMed] [Google Scholar]
- 19.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 20.Hobbs MV, Weigle WO, Noonan DJ, Torbett BE, McEvilly RJ, Koch RJ, Cardenas GJ, Ernst DN. Patterns of cytokine gene expression by CD4 T cells from young and old mice. J Immunol. 1993;150:3602–3614. [PubMed] [Google Scholar]
- 21.Guidotti LG, Matzke B, Schaller H, Chisari FV. High level hepatitis B virus replication in transgenic mice. J Virol. 1995;69:6158–6169. doi: 10.1128/jvi.69.10.6158-6169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Southern PJ, Singh MK, Riviere Y, Jacoby DR, Buchmeier MJ, Oldstone MB. Molecular characterization of the genomic S RNA segment from lymphocytic choriomeningitis virus. Virology. 1987;157:145–155. doi: 10.1016/0042-6822(87)90323-0. [DOI] [PubMed] [Google Scholar]
- 23.Evans CF, Borrow P, de la Torre JC, Oldstone MB. Virus-induced immunosuppression: kinetic analysis of the selection of a mutation associated with viral persistence. J Virol. 1994;68:7367–7373. doi: 10.1128/jvi.68.11.7367-7373.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Surh CD, Gao E-K, Kosaka H, Lo D, Ahn C, Murphy DB, Karlsson L, Peterson P, Sprent J. Two subsets of epithelial cells in the thymic medulla. J Exp Med. 1992;176:495–505. doi: 10.1084/jem.176.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feutren G, Lacour B, Bach JF. Immune lysis of hepatocytes in culture: accurate detection by aspartate aminotransferase release measurement. J Immunol Methods. 1984;75:85–94. doi: 10.1016/0022-1759(84)90227-8. [DOI] [PubMed] [Google Scholar]
- 26.Ando K, Moriyama T, Guidotti LG, Wirth S, Schreiber RD, Schlicht HJ, Huang S, Chisari FV. Mechanisms of class I–restricted immunopathology. A transgenic mouse model of fulminant hepatitis. J Exp Med. 1993;178:1541–1554. doi: 10.1084/jem.178.5.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Planz O, Ehl S, Furrer E, Horvath E, Bründler M-A, Hengartner H, Zinkernagel RM. A critical role for neutralizing-antibody-producing B cells, CD4+T cells, and interferons in persistent and acute infections of mice with lymphocytic choriomeningitis virus: implications for adoptive immunotherapy of virus carriers. Proc Natl Acad Sci USA. 1997;94:6874–6879. doi: 10.1073/pnas.94.13.6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baumann H, Gauldie J. The acute phase response. Immunol Today. 1994;15:74–80. doi: 10.1016/0167-5699(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 29.Guidotti LG, Chisari FV. To kill or to cure: options in host defense against viral infection. Curr Opin Immunol. 1996;8:478–483. doi: 10.1016/s0952-7915(96)80034-3. [DOI] [PubMed] [Google Scholar]
- 30.Tay CH, Welsh RM. Distinct organ-dependent mechanisms for the control of murine cytomegalovirus infection by natural killer cells. J Virol. 1997;71:267–275. doi: 10.1128/jvi.71.1.267-275.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]