

Abstract
The rae28 gene (rae28), also designated as mph1, is a mammalian ortholog of the Drosophila polyhomeotic gene, a member of Polycomb group genes (PcG). rae28 constitutes PcG complex 1 for maintaining transcriptional states which have been once initiated, presumably through modulation of the chromatin structure. Hematopoietic activity was impaired in the fetal liver of rae28-deficient animals (rae28 −/−), as demonstrated by progressive reduction of hematopoietic progenitors of multilineages and poor expansion of colony forming units in spleen (CFU-S12) during embryonic development. An in vitro long-term culture-initiating cell assay suggested a reduction in hematopoietic stem cells (HSCs), which was confirmed in vivo by reconstitution experiments in lethally irradiated congenic recipient mice. The competitive repopulating units (CRUs) reflect HSCs supporting multilineage blood-cell production. CRUs were generated, whereas the number of CRUs was reduced by a factor of 20 in the rae28 − / − fetal liver. We also performed serial transplantation experiments to semiquantitatively measure self-renewal activity of CRUs in vivo. Self-renewal activity of CRUs was 15-fold decreased in rae28 − / −. Thus the compromised HSCs were presumed to reduce hematopoietic activity in the rae28 − / − fetal liver. This is the first report to suggest that rae28 has a crucial role in sustaining the activity of HSCs to maintain hematopoiesis.
Keywords: Polycomb group genes, rae28, hematopoiesis, hematopoietic stem cells, self-renewal
Introduction
The Polycomb group genes (PcG)* were originally identified from Drosophila mutants with impaired anteroposterior patterning which is elicited by ectopic homeotic complex (HOMC) expression. Spatial regulation of HOMC genes in these mutants is correctly initiated, but it is not maintained properly, indicating that PcG are required, not for the initiation of transcription, but for the maintenance of repression (1–3). PcG products form multimeric protein complexes and epigenetically maintain the repressed state through mitosis. PcG are conserved from Drosophila to mammals. The rae28 gene (rae28) is a mammalian homologue of the Drosophila polyhomeotic gene, which is a member of PcG (4, 5). Mammalian PcG protein complexes can be classified into two distinct sets of complexes, Polycomb complex 1 and 2 (6). rae28 is a constituent of Polycomb complex 1 with M33, bmi1 or mel18, and Scmh1 (7–10). Drosophila Polycomb repressive complex 1, which possibly corresponds to mammalian PcG complex 1, have an ability to competitively inhibit chromatin remodeling complex, SWI/SNF (11), and also interacts with >30 proteins including general transcription factors, sequence specific-DNA binding factor, zeste, and histone deacetylase (HDAC) to repress transcription from the target loci (12, 13). On the other hand, Polycomb complex 2 may include eed, Enx1 or Enx2, Vav, HDAC, and YY1, a mammalian homologue of Pleiohomeotic (14–17). HDAC may constitute the repressive domain, while zeste and YY1 the DNA binding domain (12, 13, 16, 17). There may be transition from PcG complex 2 to 1 during development, which is suggested in early Drosophila development (18).
As we previously reported rae28-deficient mice (rae28 − / −) displayed abnormal anteroposterior patterning in the skeletons and rhombomeres, which is reflected in altered Hox gene expression in the maintenance phase (19, 20). The similar abnormal anteroposterior patterning of the skeletons was observed in mutant mice deficient in the other mammalian PcG genes, M33, bmi1, and mel18 genes (21–24), indicating that PcG are conserved in the function as well as in the structure. Interestingly, studies of all the mutant mice deficient in one of PcG showed that PcG have an important function in lymphopoiesis (19, 21, 23, 25–28). B cell maturation was arrested between the pro- and preB cell stages in bmi1-, mel18-, M33-, and rae28-deficient mice and T cell development was also impaired in some of these mice, while preB lymphoid progenitors increased in eed mutant mice and lymphoproliferative disorders appeared in the later stages of the mutants (29). Although evidence has been presented that bmi1 and eed respectively act as positive and negative regulators of the proliferative activity of lymphohematopoietic cells (29), the role of PcG in hematopoiesis has not been well characterized.
Accumulating evidence suggests that self-renewal and commitment of hematopoietic stem cells (HSCs) to a distinct lineage is governed by complex external signals that modulate gene expression patterns through activation of specific transcription factors (30). However, much less is known about the intrinsic genetic factors regulating HSCs (31). In this context, PcG have recently attracted attention (6).
In this study, since rae28 − / − were lethal at the perinatal period, we performed detailed examinations of hematopoiesis in rae28 − / − embryos and provided in vivo as well as in vitro evidence indicating that HSC activity is not sufficient to maintain hematopoiesis in rae28 − / − during embryonic development. These findings strongly suggest that PcG have a crucial role in the regulation of HSCs.
Materials and Methods
rae28-deficient Mice.
The generation of mice deficient in rae28 by homologous recombination has been described previously. Neonates and embryos obtained by the Cesarean sections were analyzed by PCR to identify the genotype and were subjected to further hematological examinations (19).
Hematological Analysis of Peripheral Blood and Spleen Cells.
The peripheral blood of embryos was obtained by heart puncture and the counts were determined using hemocytometer. Cell suspensions from the spleen were prepared using metal mesh. Manual differential leukocyte counts of the blood cells were performed on smears or cytocentrifuge preparations stained with May-Grünwald-Giemsa.
Methylcellulose Assay.
To detect myeloid colony-forming cells (colony-forming units culture [CFU-Cs]) including erythroid CFU (CFU-E), erythroid burst-forming units (BFU-E), granulocyte/macrophage CFU (CFU-GM) and multilineage CFU (CFU-Mix), fetal liver cells were suspended in MethoCult M3230 (Stem Cell Technologies) supplemented with 3 U/ml human erythropoietin (Chugai), 10 ng/ml mouse stem cell factor (Genzyme), 10 ng/ml mouse GM-CSF, and 10 ng/ml mouse IL-3 (BD PharMingen), and then plated in 35-mm Petri dishes. Colonies were scored after 14 d of incubation at 37°C in a humidified incubator with 5% CO2 in air (32, 33).
Long-Term Culture-initiating Cell (LTC-IC) Assay.
Fetal liver cells were suspended in the myeloid long-term culture media (Methocult M5300; Stem Cell Technologies) containing 10−6 M freshly dissolved hydrocortisone sodium hemisuccinate (Sigma-Aldrich) and placed on S17 feeder layer in 60-mm Petri dishes. Cultures were incubated at 33°C in humidified incubator with 5% CO2 in air and fed weekly by half-media exchange. The cultures were recovered by exposure to 0.25% trypsin solution in EDTA for 5 min. The cells were washed once and were assayed for the presence of myeloid CFU-Cs (33, 34).
Day 12 CFU-Spleen (CFU-S12) Assay.
The day 12 CFU-spleen (CFU-S12) content of cell suspensions was determined as described originally. Appropriate dilutions of cells were injected into congenic recipients that has been irradiated with 9.0 Gy given in a single dose from a 137Cs γ-ray source. The spleen was excised 12 d later, fixed in Telleyesniczky's solution, and macroscopic surface colonies were counted (32, 35).
Long-Term Reconstitution Assay.
C57BL/6 mice were used as recipients. The fetal liver cells (4 × 105) were injected into lethally irradiated recipient mice and their survival was monitored. To quantitatively measure competitive repopulating units (CRUs) test cells were prepared from 14.5-d post coitus (dpc) fetal liver genetically labeled by EGFP. Lethally irradiated recipients were cotransplanted with the test cells and 105 competitor cells from the bone marrow of normal congenic mice. Reconstitution by the test HSCs was assessed for each recipient 3 mo after transplantation by analysis of peripheral blood. Peripheral blood was obtained by retroorbital sinus puncture. Animals showing >1% of EGFP+-multilineage cells 3 mo after transplantation, as determined by two-color FACS® analysis with mAbs against Gr-1, Mac-1, B220, and CD3 (BD PharMingen), were considered to be positive for repopulation by the test HSCs. The frequency of CRUs in the test cells was determined by limiting dilution analysis. A line of best fit was generated by the maximal-likelihood method, and the frequency of CRUs determined using standard statistical methods by interpolation of the number of test cells required to obtain a 37% negative response (36, 37).
For serial bone marrow transplantation, EGFP-labeled fetal liver cells containing predetermined number of CRUs were injected into lethally irradiated first recipient mice. Secondary transplantation was performed 1 mo after the primary transplantation to estimate expansion of CRUs in the primary recipients. Increasing number of bone marrow cells from the primary recipients were coinjected with competitor bone marrow cells. Animals showing both EGFP+-lymphoid and myeloid cells at a frequency of >1% were considered to be repopulated by CRUs present in the primary recipients as described above. Expansion of CRUs in the primary recipients was calculated by CRU frequency in the recipients, which was estimated by limiting dilution analysis described above.
FACS® Analysis.
Single cell suspensions of the fetal liver and bone marrow were incubated with specific mAbs to murine cell surface antigens: Mac-1, Gr-1, Ter-119, CD3, CD4, CD8, B220, Sca-1, and c-kit (BD PharMingen). The antibodies were directly coupled to FITC or biotin, the latter being visualized with R-phycoerythrin-streptoavidin. Analysis was performed on a FACSCalibur™ (Becton Dickinson) and dead cells were excluded by PI (1 μg/ml) staining and gating forward angle and side scatter of light (25).
Preparation of Subpopulations of Hematopoietic Cells.
10–14-wk-old C57BL/6 mice and 14.5 dpc embryos were dissected to obtain bone marrow and fetal liver cells. These cells were pooled, washed twice with PBS, made into single-cell suspension by repeating pipetting, and filtered through 40-μm nylon mesh. Cells reacting to antibodies against Gr-1, B220, CD4, CD8, and Ter-119 were removed by using immunomagnet beads. The resulting Lin− cells were stained with FITC-conjugated anti–Sca-1 antibodies. Cell sorting was performed on a FACSVantage™ (Becton Dickinson), to purify Lin− Sca-1+ and Lin−Sca-1− fractions.
Cell-Cycle Analysis.
Cells were pulse labeled with bromodeoxyuridine (BrdU) (Sigma-Aldrich) at 10 μg/ml for 15 min at 37°C, harvested, washed with PBS, and fixed in cold 70% ethanol. Cells were treated with 1 mg/ml RNase at 37°C for 20 min. Cells were subjected to ice-cold 0.1 M HCl for 10 min. The washed cells were heated to 95°C. The tubes were suspended in ice-cold PBS containing 0.5% Tween 20 (PBST). The cells were stained with anti-BrdU antibody (BD PharMingen) diluted 1:100 in PBST containing 0.5% BSA at room temperature for 30 min. After washing twice with PBST, the cells were stained in 0.4 ml FITC-conjugated goat anti–mouse IgG antibody (BD PharMingen) diluted 1:100 in PBST containing 0.5% BSA. After 20 min the cells were washed and resuspended in PBS containing 10 μg/ml PI to stain DNA. Cells were analyzed on a FACSCalibur™ (reference 38; Becton Dickinson).
Total cDNA Amplification and Southern Blot Analysis of the cDNA.
Total cellular RNAs were extracted from fetal liver cells and purified hematopoietic cells by the acid guanidinium thiocyanate-phenol-chloroform method. cDNA was synthesized with a 60-mer primer containing a polythymidine stretch. A short polyadenosine tail was added to the first strand of the cDNA using terminal deoxynucleotidyltransferase (GIBCO BRL). Second-strand synthesis and PCR amplification were done with the same primers. Amplified total cDNA was size fractionated on a 1% agarose gel, transferred to nylon membranes, and subsequently hybridized with 32[P]-labeled probes (39).
Results
Expression of rae28 in Hematopoietic Cells.
As we reported previously, expression of rae28 is predominant in the embryonic stage and almost disappears after birth except for the genital organs, brain, and thymus (4). We performed detailed examinations of rae28 expression in the hematopoietic organs and subpopulations of the fetal liver and bone marrow cells. Total cellular RNAs were extracted from the fetal liver, spleen, thymus, and from the FACS®-purified subpopulations of the fetal liver and bone marrow cells. PCR-amplified total cDNAs were generated from mRNAs extracted from these organs and subpopulations, and it was confirmed that the amplified cDNAs preserved quantitative differences in mRNA abundance (data not shown).
The amplified cDNAs were subjected to Southern blot hybridization for analysis of rae28 expression. This expression was detected in abundance in the fetal liver at 12.5 and 14.5 dpc, while it decreased at 18.5 dpc and was undetectable in the adult liver probably due to emigration of the hematopoietic cells (Fig. 1). rae28 expression levels were much higher in the Lin− Sca-1+ subpopulation enriched for primitive hematopoietic cells including HSCs than in the whole fetal liver and bone marrow cells (Fig. 1). It should be noted that a relatively high expression of rae28 was preserved in the HSC-enriched subpopulation of the hematopoietic cells.
Figure 1.

Expression of rae28 in hematopoietic tissues and purified subpopulations of fetal liver and bone marrow cells. PCR-amplified total cDNAs were prepared from total RNAs extracted from the hematopoietic tissues and fractionated cells and were analyzed by Southern blot analysis with a rae28 probe. Note that high rae28 expression was detected in Lin−, Sca-1+ subpopulations. BM, bone marrow; FL, fetal liver; Whole, whole fetal liver or bone marrow cells; Lin−, Sca-1+, Lin−, Sca-1+ subpopulations of fetal liver and bone marrow cells. Expression of β-actin is shown to confirm equal amount of cDNA on the filter.
Hematological Examination of the Peripheral Blood Cells and Spleen.
The hematological findings for the peripheral blood showed no significant differences in myeloid and erythroid lineages in rae28 −/− neonates, although lymphoid cells tended to be reduced (Fig. 2 A). No significant differences were also observed in the 10.5-dpc primitive erythrocytes, peripheral blood and fetal liver cells at the cytological level (data not shown). The spleen in rae28 −/− neonates was hypoplastic, although the severity varied among individuals (Fig. 2 B). The number of nucleated cells was reduced to 12.5% of the wild-type in the spleen of the mutant neonates (data not shown) but cell numbers were not reduced in the fetal liver. FACS® analysis of the spleen cells showed a reduction in B cells, but cells with the other lineage markers were conserved (data not shown).
Figure 2.


Peripheral blood cells and the spleen. (A) Peripheral blood cell counts. The data represent mean values with a SD from at least three independent individuals. (B) Picture of spleen from +/+, wild-type and −/−, rae28 − / − mice.
Expansion of CFU-C and CFU-S12 during Embryonic Development.
Splenic hypoplasia and the reduced number of spleen cells described above suggested the presence of hematopoietic defects in rae28 −/− embryos, because the spleen is one of the major hematopoietic organs in the neonatal period. We first examined the increase in CFU-Cs during fetal development. Although clonogenic progenitors including CFU-Mix, CFU-GM, CFU-E, and BFU-E were generated and increased until 14.5 dpc in rae28 −/− embryos, expansion of all these hematopoietic progenitors was impaired later in the fetal development (Fig. 3 A). We next examined more premature hematopoietic progenitors in rae28 −/− embryos, and found that CFU-S12 was markedly reduced in number to one-tenth of the normal level as early as 12.5 dpc and expansion of CFU-S12 was affected during embryonic development (Fig. 3 B). The size of CFU-S12 colonies was also significantly reduced in rae28 −/− (Fig. 3 C).
Figure 3.


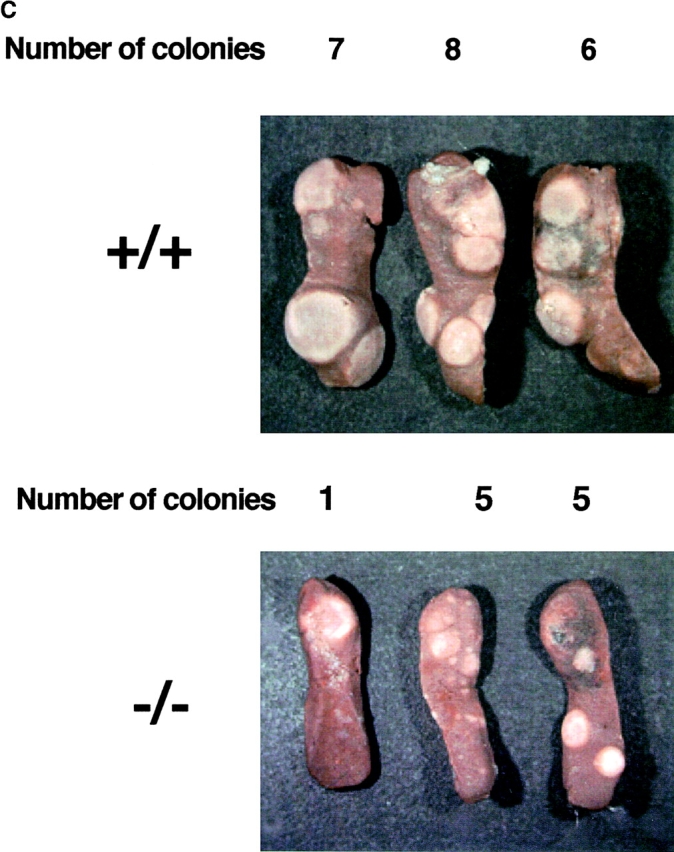

(A) Expansion of CFU-Cs during embryonic development. The number of CFU-Cs in the embryonic fetal liver from 12.5 to 18.5 dpc was examined by methylcellulose assay. The data represent the mean number of CFU-Cs with a SD from at least five individuals. Both BFU-E and CFU-E are shown as erythroid colonies. Note that expansion of CFU-Cs including CFU-mix, CFU-GM, and BFU-E/CFU-E was progressively impaired in rae28 − / − embryos after 14.5 dpc. Black squares, wild-type; black circles, rae28 − / −. (B) Expansion of CFU-S12 during embryonic development. The data represent the mean of CFU-S12 colony numbers with a SD determined from a minimum of five recipients. The magnified panel shows the number of CFU-S12 in 12.5 dpc fetal liver cells. The number of CFU-S12 was already reduced at 12.5 dpc and its expansion during fetal development was also less effective in rae28 − / −. Black squares, wild-type; black circles, rae28 − / −. (C) Picture of CFU-S12 colonies. Fetal liver cells (4 × 105) were injected to sublethally irradiated mice and the number of CFU-S12 was counted after fixation with Telleyesniczky's solution. The size as well as the number of CFU-S12 colonies was reduced in rae28 − / −. +/+, wild-type; −/−, rae28 − / −. (D) The number of LTC-IC in the 14.5 dpc fetal liver. Fetal liver cells were cultured 2 and 3 wk on S17 feeder layer cells and cultured cells were subjected to methylcellulose assay. The data represent the mean of CFU-C colony numbers with a SD determined from at least five individuals. The number is presumed to reflect the frequency and expansion of LTC-IC during the culture. Note that the reduction of LTC-IC was more severe in 3-wk culture than in 2-wk culture. White squares, wild-type; black squares, rae28 − / −.
LTC-IC Assay.
Activity of HSCs in the mutants at 14.5 dpc was examined in vivo as well as in vitro. First, we performed the LTC-IC assay. In this assay we determined the number of clonogenic progenitors in fetal liver cells cultured on an S17 feeder layer for 2–3 wk. Although the limiting dilution assay to determine the frequency of LTC-IC was not performed, the number of CFU-C colonies can be assumed to reflect the frequency and proliferative capacity of LTC-IC on the feeder layer. The reduction in the number of colonies was evident in rae28 −/− (Fig. 3 D). The number was reduced more remarkably in 3-wk than in 2-wk cultured fetal liver cells, suggesting that reduction of LTC-IC is more prominent in the upper level of the hematopoietic hierarchy in rae28 −/−.
Long-Term Reconstitution Assay.
The most rigorous test of the function of HSCs is the ability to reconstitute the hematopoietic system of recipients in which endogenous hematopoiesis has been ablated. Since the methylcellulose clonogenic, CFU-S12 and LTC-IC assays showed reduction of premature hematopoietic cells in rae28 − / − as mentioned earlier, we assessed the ability of the fetal liver cells to reconstitute the hematopoietic system in lethally irradiated mice. The fetal liver cells (4 × 105) from 14.5 dpc wild-type and rae28 − / − embryos were injected into lethally irradiated congenic recipient mice and their survival was monitored. All the recipients injected with rae28 −/− fetal liver cells died within 3 wk, while 70% of those with wild-type cells survived >12 wk, indicating that the ability to reconstitute the hematopoietic system was severely reduced in rae28 −/− fetal liver cells (Fig. 4).
Figure 4.

Kaplan-Meier survival curve of lethally irradiated congenic mice transplanted with fetal liver cells. Fetal liver cells (4 × 105) prepared from 14.5-dpc embryos were injected into congenic recipient mice and survival of the mice was investigated in the conventional facility. Black squares, wild-type; black circles, rae28 − / −.
Quantitative measurement of CRUs by limiting dilution analysis was then performed to allow direct comparison of the frequency and activity of HSCs. First, we intercrossed rae28 + / − heterozygous mice with a transgenic line ubiquitously expressing EGFP (40) and prepared rae28 − / − homozygous and wild-type embryos expressing EGFP to distinguish the test cells from the competitors. Increasing doses of fetal liver cells from the EGFP-labeled 14.5 dpc rae28 − / − and wild-type embryos were mixed with 105 wild-type bone marrow cells as competitors and transplanted into lethally irradiated congenic recipients. Limiting dilution analysis revealed >20-fold reduction in CRU frequency in rae28 − / − compared with the wild-type (1 per 4 × 105 vs. 1 per 2 × 104, respectively, P < 0.01; Fig. 5 A). Thus total numbers of CRUs in the rae28 − / − and wild-type fetal liver were estimated to be 50 and 1,000, respectively, because the number of fetal liver cells was 2 × 107 in either of rae28 − / − and wild-type.
Figure 5.



Competitive repopulation assay. (A) Frequency of CRUs in the fetal liver. The peripheral blood cells in the congenic recipient mice injected with 14.5-dpc fetal liver cells were examined to assess the long-term reconstitution capacity of HSCs. 3 mo after transplantation, the proportion of mice exhibiting >1% EGFP+ cells was used to calculate the frequency of CRU. Black squares, wild-type; black circles, rae28 − / − (B and C) Serial transplantation experiments. Lethally irradiated recipient mice were injected with fetal liver cells including 20 CRUs, and 1 mo after injection frequency of CRUs in the primary recipient mice was determined by CRU assay. Frequency of CRUs derived from the wild-type and rae28 − / − cells was calculated from Figs. B and C, respectively. Note the number of injected cells shown in the top column of the figures is 10 times different between Figs. B and C.
Serial Transplantation Experiment.
To further assess the self-renewal ability of rae28 −/− HSCs, we performed serial transplantation experiments. Fetal liver cells containing 20 CRUs, as calculated in terms of the frequency of CRUs determined as described above, were transplanted into lethally irradiated primary recipient mice. 1 mo after transplantation, bone marrow cells from the femurs and tibias of the primary recipient mice were assayed for CRU number by limiting dilution analysis in secondary recipients. The frequency of CRUs was 1 per 2 × 105 cells in primary recipients reconstituted with wild-type CRUs (Fig. 5 B) compared with 1 per 3 × 106 in those with rae28 −/− (Fig. 5 C), indicating a markedly impaired regenerative capacity for CRUs in the absence of rae28 (Table I). The number of total bone marrow cells was 2.4 × 107 in the femurs and tibias of primary recipients reconstituted with either the wild-type or rae28 −/− CRUs. Since bone marrow cells in the femurs and tibias are estimated to correspond to 18.7% of total marrow cells (41), 1 mo after reconstitution the numbers of CRUs in the total marrow of primary recipients reconstituted with the wild-type and rae28 −/− CRUs were ∼640 and 43, respectively. The increase in CRUs was estimated to be 32 times within 1 mo in the wild-type, but that in rae28 −/− was, surprisingly, limited to no more than 2.2 times (Table I). These in vivo results clearly demonstrated that the self-renewal ability of CRUs was severely impaired in rae28 −/−.
Table I.
Semiquantitative Analysis of Self-Renewal Ability of CRUs
| Genotype | +/+ | −/− |
|---|---|---|
| Injected CRUs | 20 | 20 |
| BM cells in the femurs and tibias | 2.4 × 107 | 2.4 × 107 |
| Frequency of CRUs | 1/2 × 105 | 1/3 × 106 |
| CRUs in the femurs and tibias | 120 | 8 |
| CRUs in the entire body | 640 | 43 |
| Increase of CRUs | 32× | 2.2× |
The numbers of bone marrow cells and CRUs in the primary recipients were examined one month after transplantation. The numbers of bone marrow cells represent mean values from four independent individuals. Frequency of CRUs was estimated by competitive repopulating unit assay shown in Figure 5. The numbers of CRUs in the entire body were calculated based on the previous report that bone marrow cells in the femurs and tibias correspond to 18.7 % of those in the entire body. BM, bone marrow; +/+, wild-type; −/−, rae28−/−.
Cell-Cycle Analysis of Fetal Liver Cells.
To examine whether hematopoietic defects in the rae28 −/− fetal liver reflected an impaired proliferation of fetal liver cells, we examined the cell cycle status of 14.5 dpc fetal liver cells. The relative DNA content of the fetal liver cells was assessed with PI staining, whereas BrdU incorporation by the cells was measured after in vitro BrdU pulse labeling for 1 h of the isolated cells. As shown in Fig. 6, no significant difference in cycling parameters was evident.
Figure 6.

Cell cycle status of fetal liver cells. Cells were pulse-labeled with 10 μg/ml BrdU after harvest, stained with PI, and subjected to FACS® analysis. Representative BrdU-FITC/PI fluorescence profiles of wild-type and rae28 − / − fetal liver cells are shown in the Figure. +/+, wild-type; −/−, rae28 − / −.
Expression of Genes Involved in Hematopoiesis.
To identify possible downstream target genes causing the HSC defects in rae28 − / −, we analyzed the expression of Hox and genes which could potentially be involved in hematopoiesis. Total cDNAs were synthesized from mRNAs extracted from fetal liver cells at 14.5 dpc, which proved to predominantly consist of hematopoietic cells (>95%; data not shown) and Hox expression was detected by Southern blot analysis. We focused on a- and b-clustered Hox genes which are suspected to be involved in myelopoiesis. Although the expressions of Hoxa1, a2, a3, a4, a7, a9, a10, b1, b2, b3, b4, b5, and b8 were all examined, no significant differences in the expression levels of these genes could be detected (Fig. 7 A).
Figure 7.


Expression of a- and b-clustered Hox genes (A) and genes for receptors, signaling molecules and transcription factors (B) involved in hematopoiesis. Expression of the p16, p19, and p21 genes were also shown in the Figure. PCR-amplified total cDNAs were prepared from 14.5 dpc fetal liver cells and expression of the genes was analyzed by Southern blot analysis. Expression of β-actin is shown as an internal marker to confirm the equal amount of cDNAs on the filter. +/+, wild-type; −/−, rae28 − / −.
We similarly analyzed expression levels of representative receptors and transcription factors which are involved in hematopoiesis in the mutant fetal liver, including gp130, Epo receptor, G-CSF receptor, GM-CSF receptor, c-erbAα, c-Mpl, c-jun, c-fos, c-myc, c-myb, tal1/SCL, AML1, GATA1, GATA2, and PU.1. Expression of none of these genes was found to have significantly changed in the rae28 − / − fetal liver cells (Fig. 7 B). We also examined expression of the ink4a-ARF genes (p16 ink4A and p19 ARF), p21 CDK inhibitor, B-raf, bax, and Ubiquitin genes. No significant differences could be detected in the expression of these genes in the rae28 − / − fetal liver (Fig. 7 B). ink4a-ARF gene products were further detected by Western blot analysis to confirm the findings (data not shown).
Discussion
We have previously shown that rae28 is involved in the early stage of B cell differentiation in a gene dosage–dependent manner (25). rae28 is, however, predominantly expressed during early ontogeny (4) and rae28 expression is preserved at high levels even in the adult stage in cell populations that are enriched in HSCs (Fig. 1). The detailed analysis of premature hematopoietic cells including HSCs in rae28 − / − reported here reveals a crucial role for rae28 in sustaining HSC activity. Fetal liver cellularity and mature cell components were, however, relatively conserved in rae28 − / −, probably because premature hematopoietic cells, although limited in number, can compensate for the reduction. The progressive reduction of clonogenic progenitors may thus be compatible with functional failure of HSCs to supply the progenitors. The number of CRUs in rae28 − / − was only one-twentieth of the wild-type (1 per 4 × 105 cells) (Fig. 5 A), suggesting that no more than 50 CRUs were generated in the rae28 − / − fetal liver during the developmental period. Furthermore, rae28 − / − CRUs underwent a net expansion of no more than 2.2-fold in transplant recipients in 1 mo, while wild-type CRUs expanded 32-fold (Table I), indicating that the transplanted rae28 − / − CRUs were amplified much less efficiently under the conditions of this assay.
Although we could not determine whether wild-type HSCs can reconstitute the hematopoietic system in rae28 − / − mice because rae28 − / − embryos were lethal at the perinatal period, the result of a series of reconstituting experiments described in this study suggests that HSC defects in rae28 − / − are manifest within the HSC population rather than in the nonhematopoietic accessory cells of fetal liver and spleen. This is also supported by the evidence that a secondary CFU-S12 assay with wild-type congenic recipients demonstrated a reduction in the self-renewal ability of rae28 − / − CFU-S12 (data not shown). The reduced activity of rae28 − / − HSCs is presumed to be at least in part responsible for the poor expansion of CFU-S12 and the progressive reduction of clonogenic progenitors during fetal development. However, the size of CFU-S12 was significantly reduced and their self-renewal ability was diminished in the secondary CFU-S12 assay as described earlier, suggesting that the progenies derived from rae28 − / − HSCs were also affected in their ability.
Thus, abrogated HSCs thus appear to cause a systemic defect in the hematopoietic system in rae28 − / −, which can be expected to result in hyposplenia in rae28 − / − neonates. Our study supports the notion that rae28 is indispensable for sustaining the activity of HSCs. The function of rae28 in anteroposterior patterning of the rhombomeres and paraxial mesoderm is mediated by its sustaining effect on Hox expression once initiated (19). Expression of multiple Hox genes from the three different clusters (Hoxa3, a4, a5, b3, b4, and d4) was found to be affected in the paraxial mesoderm of rae28 − / − embryos and that of Hoxb3 and b4 was also affected in the rhombomeres during the maintenance phase (19, 20). Since Hox overexpression results in myelo- or lymphoproliferation (42–46), we examined the effect of rae28 deficiency on the expression of Hox genes in the a and b clusters in the fetal liver cells, because these Hox genes are expressed in the myeloid cells (47). However, none of the Hox genes examined showed significant differences in their expression in rae28 − / − fetal liver cells (Fig. 7 A).
We further analyzed expression of representative cytokine receptors (gp130, Epo receptor, G-CSF receptor, GM-CSF receptor, c-erbAα, and c-Mpl), transcription factors (c-jun, c-fos, c-myc, c-myb, tal1/SCL, AML1, GATA1, GATA2, PU.1, and Ikaros; unpublished data), a tumor suppressor (p19) and a cyclin-dependent kinase inhibitor (p16 and p21) in the rae28 − / − fetal liver. gp130, a common signal transducer of LIF, IL-6, and oncostatin M, and c-MPL, a receptor for thrombopoietin, are assumed to have an important function in inducing self-renewal of HSCs (48–50). c-kit, a receptor for SCF, synergizes with other cytokines to promote the formation of all lineages from HSCs (51). Exaggerated expression of p16 and p19 has been reported to cause abnormal lymphopoiesis in mutant mice overexpressing and lacking bmi1 (52, 53), and p21 has been shown to play a part in governing the entry of HSCs into the cell cycle (54). However, our study did not detect any significantly altered expressions of mRNAs for these molecules in rae28 − / − fetal liver cells (Fig. 7 B). FACS® analysis confirmed that there were no differences in c-kit, sca-1, and IL-7Rα expression in the fetal liver cells (data not shown).
These findings strongly suggest that development of hematopoietic cells with the lineage markers is well conserved in the rae28 − / − fetal liver and that the affected cells are restricted to a small subpopulation of premature hematopoietic cells including HSCs. While it is not likely that abnormal expression of these genes mediates the functional defects of HSCs in rae28 − / −, we cannot exclude the possibility that rae28 deficiency affects expression of the genes exclusively in the specific subpopulations of hematopoietic cells such as HSCs. Furthermore, TUNEL staining could not detect increased apoptosis in the 14.5 dpc fetal liver from rae28 − / − (data not shown) and cell cycle status of rae28 − / − fetal liver cells remained unaffected at 14.5 dpc (Fig. 6). Although compensatory mechanisms might preserve cell cycling, it is also unlikely that defective hematopoiesis in rae28 − / − resulted from abnormal cell cycling and apoptosis in fetal liver cells.
To determine whether the hematopoietic defects in rae28 − / − resulted from a defect in the development of HSCs, we examined hematopoiesis in the early stage of development. Histoanatomical examinations showed no significant abnormalities in primitive hematopoiesis in the rae28 − / − yolk sac (data not shown). Since we could not detect CFU-S12 even in the wild-type aorta-gonad-mesonephros region, development of premature hematopoietic cells in rae28 − / − embryos at 10.5 dpc remains uncharacterized. CFU-S12 was, however, already reduced in the 12.5 dpc fetal liver of the mutants and CRUs were remarkably underdeveloped at 14.5 dpc (Fig. 3 B). Although it appears difficult to make a clear distinction as to whether or not rae28 deficiency affects the development of HSCs, we detected CRUs, even at a low frequency, in the rae28 − / − fetal liver. These CRUs were able to reconstitute the hematopoietic system by producing multilineage mature blood cells for >3 mo in the congenic recipients. Thus, it can be assumed that rae28 is not essential for the development of HSCs but is required for sustaining the HSC activity for the amplification of HSCs after they are developed.
eed and bmi1 were shown to regulate the proliferative activity of the mature and premature hematopoietic progenitors in, respectively, a positive and negative manner (29). However, HSCs were reported to be unaffected critically in bmi1-deficient mice, because erythropoiesis was not altered (23). The hematopoietic defect in bmi1-deficient mice was explained by the hypothesis that parallel pathways exist to compensate for the lack of bmi1 in the fetal period and that the hematopoietic disorder becomes overt later on after the disappearance of the compensatory pathway (23). And another explanation of the hematopoietic defects was that it results from progressive overexpression of p16 CDK inhibitor from the ink4A-ARF locus (52). Abnormal expression of p16 and p19 from the ink4A-ARF locus, however, was not detected in rae28 − / − fetal liver cells and impaired activity of HSCs is assumed to result in inefficient hematopoiesis during fetal development of rae28 − / −. Since expression levels of transcription factors regulating lineage determination and those of lineage specific genes are conserved as far as we examined in this study, differentiation potential of HSCs seem to be relatively conserved except for B cell differentiation (25). The defect appears to be mainly focused on self-renewal ability of HSCs, which involves regulation of the cell cycle machinery and maintenance of the pluripotency.
Because PcG complex 1 appears to consist of multiple complexes varying in the constituents (6), that may reflect the functional specificity, each of the PcG members may display a different specific function in hematopoiesis. It is now possible to speculate about some plausible mechanisms underlying HSC defects in rae28 − / − embryos. First, a PcG complex with rae28 may play a part in maintaining the repressive state of genes which preserves the activity of HSCs, i.e., transcription of genes crucial for regulation of proliferation, commitment, and apoptosis of HSCs may be dysregulated, causing dysfunction of HSCs. This assumption is highly compatible with the expression profile of rae28, which shows that high rae28 expression in primitive hematopoietic cells is progressively reduced in the course of hematopoietic cell differentiation. Second, since B cell defects in mice deficient in PcG family members are known to result at least partly from unresponsiveness to IL-7 (23, 25, 27), it may be possible that rae28 − / − HSCs show no response or a poor response to cytokines crucial for the regulation of HSCs. In any case, since PcG-mediated repression system is presumed to play a crucial part in the regulation of HSCs, rae28 −/− mutant mice can be expected to facilitate the clarification of molecular mechanisms underlying the regulation of HSCs.
Acknowledgments
We thank Drs. H.W. Brock, A. Hakura, T. Nakano, Y. Niho, and K. Yamauchi-Takihara for their encouragement, Drs. M. Nishikawa and T.W. Mak for discussion, and R. Hasegawa for secretarial help. We also thank Drs. D. Duboule, P. Gruss, R. Krumlauf, O. Chisaka, H. Kondoh, H. Koseki, T. Nakano, R. Fukunaga, I. Matsumura, and M. Yamamoto for providing probes for detecting expression of Hox genes, various receptors, and transcription factors regulating hematopoiesis described in the text. We are grateful for the offer of the extensive use of the Genome Information Research Center, Osaka University.
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan.
Footnotes
Abbreviations used in this paper: BFU-E, erythroid burst-forming unit; CFU-C, CFU culture; CFU-E, erythroid CFU; CFU-GM, granulocyte/macrophage CFU; CFU-Mix, multilineage CFU; CFU-S12, day-12 CFU spleen; CRU, competitive repopulating unit; dpc, d post coitus; HDAC, histone deacetylase; HSC, hematopoietic stem cell; LTC-IC, long-term culture-initiating cell; PcG, Polycomb group.
References
- 1.Simon, J. 1995. Locking in stable states of gene expression: transcriptional control during Drosophila development. Curr. Opin. Cell Biol. 7:376–385. [DOI] [PubMed] [Google Scholar]
- 2.Gould, A. 1997. Functions of mammalian Polycomb group and trithorax group related genes. Curr. Opin. Genet. Dev. 7:488–494. [DOI] [PubMed] [Google Scholar]
- 3.Brock, H.W., and M. van Lohuizen. 2001. The polycomb group-no longer an exclusive club? Curr. Opin. Genet. Dev. 11:175–181. [DOI] [PubMed] [Google Scholar]
- 4.Nomura, M., Y. Takihara, and K. Shimada. 1994. Isolation and characterization of retinoic acid-inducible cDNA clones in F9 cells: one of the early inducible clones encodes a novel protein sharing several highly homologous regions with a Drosophila polyhomeotic protein. Differentiation. 57:39–50. [DOI] [PubMed] [Google Scholar]
- 5.Ohta, H., S. Tokimasa, Z. Zou, S. Funaki, H. Kurahashi, Y. Takahashi, M. Kimura, R. Matsuoka, M. Horie, J. Hara, et al. 2000. Structure and chromosomal localization of the RAE28/HPH1 gene, a human homologue of the polyhomeotic gene. DNA Seq. 11:61–73. [DOI] [PubMed] [Google Scholar]
- 6.Takihara, Y., and J. Hara. 2000. The Polycomb-group genes and hematopoiesis. Int. J. Hematol. 72:165–172. [PubMed] [Google Scholar]
- 7.Alkema, M.J., M. Brock, E. Verhoeven, A. Otte, L.J.V.T. Veer, A. Berns, and M. van Lohuizen. 1997. Identification of Bmi1-interacting proteins as constituents of a multimeric mammalian Polycomb complex. Genes Dev. 11:226–240. [DOI] [PubMed] [Google Scholar]
- 8.Gunster, M.J., D.P.E. Satijn, K.M. Hamer, J.L. van den Blauwen, D. de Bruijn, M.J. Alkema, M. van Lohuizen, R. van Driel, and A.P. Otte. 1997. Identification and characterization of interactions between the vertebrate Polycomb-Group protein BMI1 and human homologs of Polyhomeotic. Mol. Cell. Biol. 17:2326–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hashimoto, N., H.W. Brock, M. Nomura, M. Kyba, J. Hodgson, Y. Fujita, Y. Takihara, K. Shimada, and T. Higashinakagawa. 1997. Rae28, Bmi1, and M33 are members of heterogeneous multimeric mammalian Polycomb group complexes. Biochem. Biophys. Res. Comm. 245:356–365. [DOI] [PubMed] [Google Scholar]
- 10.Tomotsune, D., Y. Takihara, J. Berger, D. Duhl, S. Joo, M. Kyba, M. Shirai, H. Ohta, Y. Matsuda, B.M. Honda, et al. 1999. A novel member of murine Polycomb-group proteins, Sex comb on midleg homolog protein, is highly conserved, and interacts with RAE28/mph1 in vitro. Differentiation. 65:229–239. [DOI] [PubMed] [Google Scholar]
- 11.Shao, Z., F. Raible, R. Mollaaghababa, J.R. Guyon, C.T. Wu, W. Bender, and R.E. Kingston. 1999. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell. 98:37–46. [DOI] [PubMed] [Google Scholar]
- 12.Breiling, A., B.M. Turner, M. Bianchi, and V. Orlando. 2001. General transcription factors bind promoters repressed by Polycomb group proteins. Nature. 412:651–655. [DOI] [PubMed] [Google Scholar]
- 13.Saurin, A.J., Z. Shao, H. Erdjument-Bromage, P. Tempst, and R.E. Kingston. 2001. A Drosophila Polycomb group complex includes Zeste and dTAFII proteins. Nature. 412:655–660. [DOI] [PubMed] [Google Scholar]
- 14.van Lohuizen, M., M. Tijms, J.W. Voncken, A. Schumacher, T. Magnuson, and E. Wientjens. 1998. Interaction of mouse polycomb-group (Pc-G) proteins Enx1 and Enx2 with Eed: indication for separate Pc-G complexes. Mol. Cell. Biol. 18:3572–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sewalt, R.G., J. van der Vlag, M.J. Gunster, K.M. Hamer, J.L. den Blaauwen, D.P. Satijn, T. Hendrix, R. van Driel, and A.P. Otte. 1998. Characterization of interactions between the mammalian polycomb-group proteins Enx1/EZH2 and EED suggests the existence of different mammalian polycomb-group protein complexes. Mol. Cell. Biol. 18:3586–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Vlag, J., and A.P. Otte. 1999. Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat. Genet. 23:474–478. [DOI] [PubMed] [Google Scholar]
- 17.Satijn, D.P., K.M. Hamer, J. den Blaauwen, and A.P. Otte. 2001. The Polycomb group protein EED interacts with YY1, and both proteins induce neural tissue in Xenopus embryos. Mol. Cell. Biol. 19:1360–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poux, S., R. Melfi, and V. Pirrotta. 2001. Establishment of Polycomb silencing requires a transient interaction between PC and ESC. Genes Dev. 15:2509–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takihara, Y., Y. Katoh-Fukui, K. Nishii, M.A. Motaleb, R. Tsuchiya, Y. Fujita, M. Nomura, Y. Shibata, T. Higashinakagawa, and K. Shimada. 1997. Targeted replacement of the mouse homologue of the Drosophila polyhomeotic gene leads to altered anteroposterior patterning and neural crest defects. Development. 121:3673–3682. [DOI] [PubMed] [Google Scholar]
- 20.Tomotsune, D., M. Shirai, Y. Takihara, and K. Shimada. 2000. Regulation of Hoxb3 expression in the hindbrain and pharyngeal arches by rae28, a member of the mammalian Polycomb group genes. Mech. Dev. 98:165–169. [DOI] [PubMed] [Google Scholar]
- 21.Córe, N., S. Bel, S.J. Gaunt, M. Aurrand-Lions, J. Pearce, A. Fisher, and M. Djabali. 1997. Altered cellular proliferation and mesoderm patterning in Polycomb-M33-deficient mice. Development. 124:721–729. [DOI] [PubMed] [Google Scholar]
- 22.Katoh-Fukui, Y., R. Tsuchiya, T. Shiroishi, Y. Nakahara, N. Hashimoto, K. Noguchi, and T. Higashinakagawa. 1998. Male-to-female sex reversal in M33 mutant mice. Nature. 393:688–692. [DOI] [PubMed] [Google Scholar]
- 23.van der Lugt, M.T., J. Domen, K. Linders, M. Roon, E. Robanus-Maandag, H. Riele, M. Valk, J. Deschamps, M. Sofroniew, M. van Lohuizen, and A. Berns. 1994. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 8:757–769. [DOI] [PubMed] [Google Scholar]
- 24.Akasaka, T., M. Kanno, R. Balling, M.A. Mieza, M. Taniguchi, and H. Koseki. 1996. A role for mel-18, a Polycomb group related vertebrate gene, during the anteroposterior specification of the axial skeleton. Development. 122:1513–1522. [DOI] [PubMed] [Google Scholar]
- 25.Tokimasa, S., H. Ohta, A. Sawada, Y. Matsuda, J.-Y. Kim, S. Nishiguchi, J. Hara, and Y. Takihara. 2001. Lack of the Polycomb-group gene rae28 causes maturation arrest at the early B-cell developmental stage. Exp. Hematol. 29:135–146. [DOI] [PubMed] [Google Scholar]
- 26.Alkema, M.J., H. Jacobs, M. van Lohuizen, and A. Berns. 1997. Pertubation of B and T cell development and predisposition to lymphomagenesis in Eμ Bmi1 transgenic mice require the Bmi1 RING finger. Oncogene. 15:899–910. [DOI] [PubMed] [Google Scholar]
- 27.Akasaka, T., K. Tsuji, H. Kawahira, M. Kanno, K. Harigaya, L. Hu, Y. Ebihara, T. Nakahata, O. Tetsu, M. Taniguchi, and H. Koseki. 1997. The role of mel-18, a mammalian Polycomb group gene, during IL-7-dependent proliferation of lymphocyte precursors. Immunity. 7:135–146. [DOI] [PubMed] [Google Scholar]
- 28.Kimura, M., Y. Koseki, M. Yamashita, N. Watanabe, C. Shimizu, T. Katsumoto, T. Kitamura, M. Taniguchi, H. Koseki, and T. Nakayama. 2001. Regulation of Th2 cell differentiation by mel-18, a mammalian Polycomb group gene. Immunity. 15:275–287. [DOI] [PubMed] [Google Scholar]
- 29.Lessard, J., A. Schumacher, U. Thorsteinsdottir, M. van Lohuizen, T. Magnuson, and G. Sauvageau. 1999. Functional antagonism of the Polycomb-group genes eed and Bmi1 in hemopoietic cell proliferation. Genes Dev. 13:2691–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Metcaf, D. 1998. Lineage commitment and maturation in hematopoietic cells. The case for extrinsic regulation. Blood. 92:345–352. [PubMed] [Google Scholar]
- 31.Cross, M.A., and T. Enver. 1997. The lineage commitment of haemopoietic progenitor cells. Curr. Opin. Gen. Dev. 7:609–613. [DOI] [PubMed] [Google Scholar]
- 32.Heyworth, C.M., and E. Spooncer. 1993. In vitro clonal assays for murine multi-potential and lineage restricted myeloid progenitor cells. Haematopoiesis: A Practical Approach. N.G. Testa and G. Molineux, editors. Oxford University Press, NY. 37–52.
- 33.Helgason, C.D., J.E. Damen, P. Rosten, G.R.P. Sorensen, S.M. Chappel, A. Borowski, F. Jirik, G. Krystal, and R.K. Humphries. 1998. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 12:1610–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sutherland, H.J., P.M. Lansdorp, D.H. Henkelman, A.C. Eaves, and C. Eaves. 1990. Functional characterization of individual human hematopoietic stem cells cultured at limiting dilution on supportive marrow stromal layers. Proc. Natl. Acad. Sci. USA. 87:3584–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Till, J.E., and E.A. McCulloch. 1961. A direct measure of the radiation sensitivity of normal mouse bone marrow cells. Radiat. Res. 14:213–222. [PubMed] [Google Scholar]
- 36.Szilvassy, S.J., R.K. Humphries, P.M. Lansdorp, A.C. Eaves, and C.J. Eaves. 1990. Quantitative assay for totipotent reconstituting hemopoietic stem cells by a competitive repopulation strategy. Proc. Natl. Acad. Sci. USA. 87:8736–8740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pawliuk, R., C. Eaves, and R.K. Humphries. 1996. Evidence of both ontogeny and transplant dose-regulated expansion of hematopoietic stem cells in vivo. Blood. 88:2852–2858. [PubMed] [Google Scholar]
- 38.Giaretti, W., M. Nüse, S. Bruno, A. Di Vinci, and E. Geido. 1989. A new method to discriminate G1, S, G2, M, and G1 postmitotic cells. Exp. Cell Res. 182:290–295. [DOI] [PubMed] [Google Scholar]
- 39.Brady, G., M. Barbara, and N.N. Iscove. 1990. Representative in vitro cDNA amplification form individual hemopoietic cells and colonies. Methods Mol. Cell. Biol. 2:17–25. [Google Scholar]
- 40.Okabe, M., M. Ikawa, K. Kominami, T. Nakanishi, and Y. Nishimune. 1997. Green mice as a source of ubiquitous green cells. FEBS Lett. 407:313–319. [DOI] [PubMed] [Google Scholar]
- 41.Chervenick, P.A., D.R. Boggs, J.C. Marsh, G.E. Cartwright, and M.M. Wintrobe. 1968. Quantitative studies of blood and bone marrow neutrophils in normal mice. Am. J. Physiol. 215:353–360. [DOI] [PubMed] [Google Scholar]
- 42.Sauvageau, G., U. Thorsteinsdottir, C.J. Eaves, H.J. Lawrence, C. Largman, P.M. Lansdorp, and R.K. Humphries. 1995. Overexpression of HOXB4 in hematopoietic cells causes the selective expansion of more primitive populations in vitro and in vivo. Genes Dev. 9:1753–1765. [DOI] [PubMed] [Google Scholar]
- 43.Sauvageau, G., U. Thorsteinsdottir, M.R. Hough, P. Hugo, H.J. Lawrence, C. Largman, and R.K. Humphries. 1997. Overexpression of HOXB3 in hematopoietic cells causes defective lymphoid development and progressive myeloproliferation. Immunity. 6:13–22. [DOI] [PubMed] [Google Scholar]
- 44.Thorsteinsdottir, U., G. Sauvageau, M. Hough, W. Dragowska, P.M. Lansdorp, H.J. Lawrence, C. Largman, and R.K. Humphries. 1997. Overexpression of HOXA10 in murine hematopoietic cells perturbs both myeloid and lymphoid differentiation and leads to acute myeloid leukemia. Mol. Cell. Biol. 17:495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lawrence, H.J., C.D. Helgason, G. Sauvageau, S. Fong, D.J. Izon, R.K. Humphries, and C. Largman. 1997. Mice bearing a targeted interruption of the homeobox gene HOXA9 have defects in myeloid, erythroid, and lymphoid hematopoiesis. Blood. 89:1922–1930. [PubMed] [Google Scholar]
- 46.Buske, C., and R.K. Humphries. 2000. Homeobox genes in leukemogenesis. Int. J. Hematol. 71:301–308. [PubMed] [Google Scholar]
- 47.Sauvageau, G., P.M. Lansdorp, C.J. Eaves, D.E. Hogge, W.H. Dragowska, D.S. Reid, C. Largman, H.J. Lawrence, and R.K. Humphries. 1994. Differential expression of homeobox genes in functionally distinct CD34+ subpopulations of human bone marrow cells. Proc. Natl. Acad. Sci. USA. 91:12223–12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mukouyama, Y., T. Hara, M. Xu, K. Tamura, P.J. Donovan, H. Kim, H. Kogo, K. Tsuji, T. Nakahata, and A. Miyajima. 1998. In vitro expansion of murine multipotential hematopoietic progenitors from the embryonic aorta-gonad-mesonephros region. Immunity. 8:105–114. [DOI] [PubMed] [Google Scholar]
- 49.Audet, J., C.L. Miller, S. Rose-John, J.M. Piret, and C.J. Eaves. 2001. Distinct role of gp130 activation in promoting self-renewal divisions by mitogenically stimulated murine hematopoietic stem cells. Proc. Natl. Acad. Sci. USA. 98:1757–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kimura, S., A.W. Roberts, D. Metcaf, and W.S. Alexander. 1998. Hematopoietic stem cell deficiencies in mice lacking c-Mpl, the receptor for thrombopoietin. Proc. Natl. Acad. Sci. USA. 95:1195–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morrison, S.J., N. Uchida, and I.L. Weissman. 1995. The biology of hematopoietic stem cells. Annu. Rev. Cell Dev. Biol. 11:35–71. [DOI] [PubMed] [Google Scholar]
- 52.Jacobs, J.J., K. Kieboom, S. Marino, R.A. DePinho, and M. van Lohuizen. 1999. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 397:164–168. [DOI] [PubMed] [Google Scholar]
- 53.Jacobs, J.J.L., B. Scheijen, J.W. Voncken, K. Keiboom, A. Berns, and M. van Lohuizen. 1999. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 13:2678–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng, T., N. Rodrigues, H. Shen, Y. Yang, D. Dombkowski, M. Sykes, and D.T. Scadden. 2000. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 287:1804–1808. [DOI] [PubMed] [Google Scholar]