

Abstract
Helicobacter pylori causes one of the most common, chronic bacterial infections and is a primary cause of severe gastric disorders. To unravel the bacterial factors necessary for the process of gastric colonization and pathogenesis, signature tagged mutagenesis (STM) was adapted to H. pylori. The Mongolian gerbil (Meriones unguiculatus) was used as model system to screen a set of 960 STM mutants. This resulted in 47 H. pylori genes, assigned to 9 different functional categories, representing a set of biological functions absolutely essential for gastric colonization, as verified and quantified for many mutants by competition experiments. Identification of previously known colonization factors, such as the urease and motility functions validated this method, but also novel and several hypothetical genes were found. Interestingly, a secreted collagenase, encoded by hp0169, could be identified and functionally verified as a new essential virulence factor for H. pylori stomach colonization. Furthermore, comB4, encoding a putative ATPase being part of a DNA transformation-associated type IV transport system of H. pylori was found to be absolutely essential for colonization, but natural transformation competence was apparently not the essential function. Thus, this first systematic STM application identified a set of previously unknown H. pylori colonization factors and may help to potentiate the development of novel therapies against gastric Helicobacter infections.
Keywords: Helicobacter pylori, in vivo essential genes, signature tagged mutagenesis, gastric adaptation, collagenase
Introduction
Colonization of the gastric mucosa by Helicobacter pylori results in an acute inflammatory response and damage to the gastric epithelium. Inflammation can then progress to several disease states, ranging in severity from superficial gastritis, chronic atrophic gastritis, peptic ulceration, to mucosa-associated lymphoma and gastric cancer (1), and the World Health Organization classified H. pylori as a Group 1 carcinogen (2).
Although the current treatments against H. pylori have generally good efficacies, relapses and reinfections do occur, and most significantly, the increasing rate of antibiotic resistance will probably result in rising treatment failures. Therefore, there is a need to search for novel therapies and drug targets. Two complete H. pylori genome sequences are currently available (3, 4). Of the 1590 predicted genes in H. pylori two thirds were assigned biological roles, but about one third did not show any database match. Despite the description of essential and nonessential genes in vitro (5, 6), our understanding of the genes necessary for the specific adaptation of the pathogen to the gastric mucosa is still very limited. There is a need to exploit the information from the H. pylori genome sequences by high throughput functional genomics approaches, as performed for other pathogens (7, 8). Besides the urease enzyme and genes involved in motility of H. pylori, only few further genes have been shown to be essential for colonization in animal models (9). An interesting strategy for this purpose is the identification of specific gene products essential for colonization in the host by signature tagged mutagenesis (STM).* So far only small scale animal-based screening studies for H. pylori colonization factors have been reported (10), which certainly reflects the difficulties in an efficient genome-wide genetic manipulation of these fastidious bacteria.
In the present study, we report about a large scale screen to identify genes in H. pylori essential for gastric colonization using a modified STM procedure. STM was originally established in bacterial pathogens for the detection of genes that are essential for Salmonella typhimurium infection of mice (11). It is a negative selection method allowing large numbers of mutants to be analyzed simultaneously. We improved and adapted the STM method to H. pylori. We generated and analyzed a set of 960 independent H. pylori transposon (Tn) insertion mutants and finally identified 47 genes, which proved to be absolutely essential for gastric colonization of H. pylori in the well-suited gerbil model. In addition, a secreted collagenase activity could be verified as a novel virulence factor of H. pylori for stomach colonization.
Materials and Methods
Bacterial Strains and Growth Conditions.
H. pylori strain G1.1, a human isolate (original named CPY3401), was supplemented with a streptomycin resistance cassette (plasmid pEG21; reference 12) and termed Q1.1 (cagA +, cag-PAI partially deleted). After four animal passages the recovery was optimal and the strain was designated P149. H. pylori cells E. coli Top10 (Invitrogen) and DH5α were grown as described (12), and supplemented with selective antibiotics, as required.
General DNA Manipulations and Construction of the pTnHK9 In Vitro Tn.
General genetic manipulation was performed following standard procedures. Variable TAG-sequences were generated by oligonucleotide RH171 (5′-cta gggatcc AGATCTNNNNNNNNNNNNNNNNNNNNAGATCTATCCACGTT-GAAAATCTCC-3′), which fuses a 20-mer random nucleotide sequence (20 × N; see Fig. 1 B) to a primer binding downstream of the chloramphenicolacetyltransferase gene (cat) in the mini-Tn TnMax5 (13). The variable sequence is flanked by two BglII restriction sites (italics) and carries a 5′-BamHI site (underlined) for cloning. A second primer, RH172, was designed, which binds upstream of the cat gene in pTnMax5. Thus, a PCR amplification using RH171 and RH172 as primers and pTnMax5 as template resulted in a PCR fragment consisting of the cat gene and a set of variable TAG sequences fused to the sequence downstream of cat (13). The variable fragments were digested with HindIII and XbaI and cloned into the corresponding sites of the plasmid pMOD, the EZ::TN Tn vector (Epicentre), which carries the Tn5 inverted repeat sequences. The obtained clone was named pTnHK9 (see Fig. 1 A).
Figure 1.

Construction of the TnHK9 Tn and outline of the procedure for identification of in vivo essential genes. (A) Construction of the in vitro Tn pTnHK9. Primer RH171, carrying the variable sequence tags and RH172 were used to PCR-amplify the cat resistance gene from pTnMax5 Tn used as template. The obtained fragment was cloned into the EZ::Tn vector pMOD to obtain pTnHK9. (B) The STM Tn TnHK9 carries the variable TAG sequence between the gene encoding a chloramphenicolacetyltransferase (cat) and the inverted repeat (IR). The binding sites for PCR primers to identify specific TAGs in chromosomal DNA of H. pylori mutants are indicated. (C) Overview on the total procedure of H. pylori mutant construction, including the in vitro mutagenesis step of a H. pylori plasmid library, the amplification step via E. coli and the generation of mutants carrying defined STM tags in H. pylori. bla; β-lactamase gene.
For cloning of the hp0169 gene, the DNA was PCR-amplified using primers hp0169-start (5′-ggaattcAGTTGAATTACTCTCTCC) and hp0169-stop (ccgctcgagACCTAATCTCTAAACGCC) with H. pylori 26695-DNA as template. The PCR fragment was digested with EcoRI/XhoI and ligated into compatible sites of the vector pGEX4T3 (Amersham Biosciences). Escherichia coli BL21 was transformed and expression of the gst-hp0169-fusion was induced with 1 mM IPTG at 30°C for 2 h.
H. pylori Gene Library Construction and In Vitro Mutagenesis.
The H. pylori genomic library was constructed by partial digest of chromosomal DNA of H. pylori strain P1 with Sau3A and HpaII, followed by subsequent ligation into cloning vector pSO50 (BglII/ClaI), a derivative of pMin2 (reference 13; see Fig. 2 C). Random in vitro Tn mutagenesis was performed essentially as described by the supplier of the EZ::Tn Tn (Epicentre). After in vitro transposition and transformation into E. coli DH5α, plasmid pools from each TAG were extracted (QIAGEN). Transfer of suicide plasmids from E. coli to H. pylori P149 by natural transformation and allelic exchange was performed as described previously (12). After 4–5 d of growth, 40 individual colonies from each TAG were selected, amplified, and stored at –80°C.
Figure 2.

(A and B) Southern blot analysis of 24 H. pylori STM mutants. The enzyme (PvuII) cuts once within the cat gene resulting in two bands hybridizing with the Tn. Some bands represent double bands. Shown are mutants from TAG groups no 2 (C) and 12 (D). (C) Summary of the procedure to generate the H. pylori STM mutants and the screening procedure resulting in 47 essential genes.
Infection and Screening of Tagged Mutant Bank.
For the gastric infection experiments 12-wk-old specific pathogen free Mongolian gerbils (RCC Ltd.) were used. Animals were maintained under standard laboratory conditions. To ensure that all mutants in the pool would be equally represented, individual mutant clones were grown separately on serum plates and pooled before intragastric infection. Two animals were used for each pool, and they were orally inoculated with 300 μl corresponding to 1.0 × 109 (OD550 = 3.3) H. pylori P149 mutants in broth culture (Brucella medium) at three consecutive days. From the bacterial pool of the last infection, aliquots were taken for the preparation of chromosomal DNA for the input pool (QIAamp® mini kit). Animals were killed 21 d after inoculation. Stomachs were opened along the great curvature, homogenized (glass homogenizer; Wheaton) and aliquots were plated onto selective media for quantitative culture. Colonizing mutants were recovered 4–5 d after culture, and the chromosomal DNA extracted using the QIAamp® mini kit for generation of the output pool. Detection of specific mutants in the output pool was achieved using a TAG-specific PCR method (Fig. 1 B). One TAG-specific primer was combined with primer SO38 (5′-GAAGATCTCTAAGGAAGCTAAAATGGAG-3′) binding at the 5′ sequence of the cat gene and the procedure was optimized to use all primer pairs in parallel. In general, thermal cycling was performed at 94°C for 5 min, followed by 30 cycles of 94°C for 30 s, 52°C for 1 min, and 72°C for 1 min, with a terminal extension at 72°C for 5 min
Analysis of Tn Insertion Sites.
Chromosomal DNA was prepared from the attenuated mutants, digested with HindIII, and the fragments ligated into vector pBA. After transformation of ligations into E. coli Top10, plasmid DNA of individual CmR colonies was isolated and sequencing was performed using primers M13-FP (5′-GTAAAACGACGGCCAGT-3′) or TnHK9 out. (5′-GGATCTCTAGAGGATCCCCG-3′) (GATC). Homology searches were performed with the Blast X algorithm (http://www.ncbi.nlm.nih.gov), and against the H. pylori genomes of TIGR and J99 (http://www.tigr.org, and http://scriabin.astazeneca-boston.com), respectively.
Determination of Protease Activity Using the Azocoll Test.
1 mg of insoluble Azocoll was incubated with bacterial crude extract (10–100 μl) together with test buffer (50 mM Tris-HCl, pH 7.5, 5 mM CaCl2) (14 h, 37°C shaking at 1,000 rpm). Insoluble Azocoll was removed by centrifugation (18,000 g, 5 min). The protease-digested, solubilized Azocoll in the supernatant was analyzed by measuring with a photometer (442 nm) and comparing it to the control incubation lacking the bacterial supernatant.
Measuring Collagenase Activity.
Bacterial supernatant was concentrated by precipitation with 50% ammonium acetate and the precipitate was dialyzed against test buffer (100 mM Tris-HCl, pH 7.5, 5 mM CaCl2). Type I collagen from calves skin (Sigma-Aldrich) was solubilized in 100 mM acetic acid (1 mg/ml). 10 μl of the solution was mixed with 70 μl test buffer and 20 μl of concentrated bacterial supernatant or purified recombinant HP0169 at 37°C. After different time points aliquots were taken, and prepared for SDS-PAGE. Analysis of collagenase activity was determined by the densitometric evaluation of the intensity of collagen bands in the immunoblot using a collagen type I–specific antibody (rabbit) (Rockland).
Purification of the GST-HP0169 Fusion Protein.
For heterologous overproduction of HP0169, the glutathion S-transferase system was used (Amersham Biosciences). E. coli BL21 carrying the fusion gene was grown in 100 ml LB to an OD550 of 0.8 and the tac-promoter of the plasmid was induced by the addition of 1 mM isopropylthiogalactosid (IPTG). The bacterial culture was chilled on ice (15 min) and pelleted (6,000 g, 4°C, 10 min). The pellet was suspended in 4 ml ice-cold buffer (50 mM Tris-HCl, pH 7.5, 5 mM CaCl2) and lysed by sonification. The lysate was cleared by centrifugation (15,000 g, 4°C, 15 min) and the supernatant was mixed with 0.4 ml of glutathion S-sepharose 4B solution. After 30 min incubation (4°C) the sepharose was removed by centrifugation (500 g) and washed three times in 2 ml buffer (50 mM Tris-HCl, pH 7.5, 5 mM CaCl2). Finally, the bound fusion protein was eluted by adding 10 mM glutathion. The eluate was dialyzed against 3 liter ice-cold buffer.
Results
Optimization of H. pylori Strain and Selection of Infection Model for Screening of In Vivo Essential Genes.
The Mongolian gerbil (Meriones unguiculatus), a well-established H. pylori animal model was used for infection studies. The gerbil-adapted strain G1.1 was transformed to carry a defined chromosomal streptomycin resistance, which did not interfere with its colonization properties and allowed a quantitative recovery of H. pylori from the animal by antibiotic selection. After four consecutive passages a reproducible recovery rate of 105 bacteria/g of stomach was obtained (data not depicted). The adapted strain was designated P149. Comparison of P149 with the recovery of noncolonizing H. pylori strains revealed that a 3-wk gerbil infection period was sufficient to remove all strains not actively colonizing the stomach.
Construction of an H. pylori Signature Tagged Mutant Library.
The original STM approach was modified by selecting 24 defined 20-mer sequence TAGs, based on their efficiency of amplification and lack of crosshybridization with each other. The TAGs were incorporated close to the left inverted repeat (IR) of TnHK9 (see Materials and Methods, and Fig. 1, A and B). A plasmid gene library was constructed, statistically covering the H. pylori genome ∼20-fold (Figs. 1 C and 2 C). In separate in vitro reactions a random mutagenesis of the plasmid library was performed, using the respective pTnHK9-TAG1–24 constructs and the hyperactive mutant transposase (EZ::Tn, Epicentre; Fig. 1 C). After transposition and subsequent transformation of individual TAG groups into E. coli strain DH5α (amplification step), total plasmids were extracted from the resulting transformants (100–5,000 transformants per in vitro mutagenesis reaction) and analyzed by restriction for random Tn insertion (unpublished data). Transfer of plasmid pools containing defined TAGs into H. pylori P149 by natural transformation resulted in the final library of H. pylori mutants (Fig. 1 C). For a single TAG we obtained at least 40 independent H. pylori mutants, resulting in 960 (24 × 40) H. pylori mutants in total, which were stored at –80°C. To confirm that transformants harbored random insertions of a single Tn, 12 mutant clones were selected from TAG groups 2 and 12 each and the chromosomal DNA was subjected to restriction enzyme (PvuII) digestion. Southern analysis using TnHK9 DNA as probe revealed that all mutants tested had single Tn insertions in apparently different loci (Fig. 2, A and B) .
Validation of the Gerbil Model for Reproducible Screening of STM Mutants.
To test the assumption that it is feasible to consistently identify in a mixed H. pylori mutant population genes as either essential or nonessential for colonization, we chose a set of 10 STM mutants carrying different TAGs for oral infection in 10 gerbils (Table I). TnHK9 insertions in the urease gene (ureB, hp0072) and in the flagellin gene (flaA, hp0601) served as candidates known to be colonization-defective in other animal models. Eight further random clones from the library (Table I) acted as candidates with unknown colonization behavior in the screen to validate the model. Quantitative reisolation of the mutants after three weeks and identification of the individual mutants by a TAG-specific PCR (Figs. 1 B and 3) revealed that the ureB and flaA mutants were not reisolated from any gerbil, but all other mutants could be reisolated in 8/10, 9/10, or 10/10 animals after 3 wk (Table I). The loss of mutants in established colonization-relevant genes verified the model and the experiment demonstrated that the screening of mixed populations of mutants is reproducible.
Table I.
Statistical Validation of the STM Approach
| Hp P149 mutant | hp0072 | hp0601 | hp0094 | hp0138 | hp0407 | hp0635 | hp0755 | hp0755 | hp1225 | 23S-rrnB |
|---|---|---|---|---|---|---|---|---|---|---|
| Mutants reisolated/gerbils | 0/10 | 0/10 | 9/10 | 9/10 | 8/10 | 8/10 | 9/10 | 10/10 | 10/10 | 10/10 |
hp0755 was chosen twice with two different insertions within the gene.
Screening Procedure of H. pylori Signature Tagged Mutants.
The 960 STM mutants were applied in successive rounds of gerbil infections. According to the 24 defined sequence TAGs, the number of independent mutants screened as a single pool (input pool) was fixed to 24 (Fig. 3) . This number was found to be low enough to reduce the complexity of the experiment to obtain reproducible results, and high enough to manage the screening of 960 H. pylori mutants. Two animals were infected with the same input pool and H. pylori mutants were reisolated from the stomach after 21 d of infection (see Materials and Methods). Chromosomal DNA was prepared from these and stored as the output pool (Fig. 3). To detect the presence or absence of a respective mutant in the output pool, we used a specific PCR amplification procedure applying the mixed population of H. pylori mutant DNA as template.
Figure 3.

Oral infection of the gerbil with 24 individually tagged H. pylori STM mutants and screening for genes essential for gastric colonization by a PCR based identification of mutants (negative selection). Chromosomal DNA was isolated from the 24 STM mutants before infection (Input pool) and after quantitative reisolation of H. pylori from the stomach of gerbils 3 wk later (Output pool). Two gerbils were infected per mutant cocktail. (Output pool 1; Output pool 2). The PCR gave rise to a 896 bp fragment only, when the tag-specific primer identified the corresponding chromosomal template DNA, as visualized by separation of the specific PCR products on a 0.8% agarose gel (see Fig. 1 B for primers). Those STM mutants which appeared to be colonization-defective were pooled again and went through a second screen. DNA sequencing using TnHK9-based primers (see Fig. 1 A) identified the genes which were inactivated. (See Table II for gene explanations.)
Verification of Colonization-defective Status of Mutants and Their Competition with Wild-Type.
A major challenge was to avoid the appearance of false negative mutants (mutants incorrectly assigned as colonization-defective). Therefore, each noncolonizing mutant of the first screen was rechecked in a second screening round (Fig. 3). DNA sequencing of independent H. pylori mutants identified sometimes Tn insertions at different locations in the same gene (see Table II, column: ind. mutant). Such mutants demonstrated the reproducibility of the screening procedure for a given gene. The sequencing revealed that 19 genes were not represented by more than one independent mutant isolate from the original mutagenesis. These Tn-inactivated genes were isolated and transferred into the wild type P149 strain again, to obtain the mutants independently and to minimize the risk of spontaneous or procedure-induced mutations elsewhere in the genome. The corresponding mutants were screened again in pools. Thus, each of the colonization negative mutant was verified by at least two, sometimes more than five independent isolates in several independent screens (Table II, column ind. mutant). STM mutants of a pool were considered as colonization-defective, when no PCR product was found from their chromosomal DNA isolated from both gerbils, but a clear signal from the input pool PCR was present (Fig. 3). Each output pool was analyzed by PCR separately. From the 960 mutants analyzed applying these rather strict guidelines, 252 noncolonizing H. pylori mutants were isolated (Fig. 2 C). Analysis of the Tn insertions of these noncolonizing mutants by DNA sequencing identified 47 different genes assigned to 9 functional groups, as listed in Table II. From the pool-infection assay, each of these genes was considered as absolutely essential for colonization in the gerbil model.
Table II.
In Vivo Essential Genes of H. pylori Identified by STM
| Mutant-ID | HP-ORF | Gene | Ind. mutant | Predicted function | C.I. |
|---|---|---|---|---|---|
| Motility and chemotaxis | |||||
| STM0217 | 0601a | flaA | >5 | Flagellin A | |
| STM0030 | 0295 | 2 | Flagellin B homologue | ||
| STM0287 | 1558a | flgC | 3 | Flagellar basal-body rod protein | |
| STM0315 | 0907a | flgD | 4 | Hook assembly protein, flagella | |
| STM0247 | 0870a | flgE | 2 | Flagellar hook | |
| STM0106 | 1092 | flgG | 2 | Flagellar basal-body rod protein | |
| STM0015 | 0325a | flgH | 2 | Flagellar basal-body L-ring protein | |
| STM0195 | 1119a | flgK | >5 | Flagellar hook-associated protein 1 | |
| STM0289 | 0752a | fliD | >5 | Flagellar hook-associated protein 2 | |
| STM0040 | 0351a | fliF | 2 | Flagellar basal-body M-ring protein | |
| STM0099 | 1420a | fliI | 4 | Flagellar export protein ATP synthase | |
| STM0244 | 0685 | fliP | 2 | Flagellar biosynthetic protein | <0.0149 |
| STM0057 | 0753a | fliS | >5 | Flagellar protein | |
| STM0019 | 0797a | hpaA | 2 | Flagellar sheath adhesin hpaA | |
| STM0327 | 0232a | 2 | Secreted protein involved in flagellar motility | ||
| STM0357 | 0392a | cheA | 2 | Histidine kinase | |
| STM0007 | 0393a | cheV | 2 | Chemotaxis protein | |
| Cell envelope and outer membrane proteins | |||||
| STM0025 | 0360 | galE | 2 | UDP-glucose 4-epimerase | |
| STM0208 | 0366 | 2 | Spore coat polysaccharide biosynthesis protein C | ||
| STM0138 | 0788 | omp | 2 | Outer membrane protein | <0.0033 |
| STM0252 | 0254a | omp8 | 3 | Outer membrane protein | |
| Type IV secretion system and other transport systems | |||||
| STM0376 | 0017 |
comB4
|
2 | Natural transformation competence-associated type IV transport system; virB4 homolog |
<0.0010 |
| STM0037 | 1421a | trbB | >5 | virB11 homolog | <0.0075 |
| STM0354 | 0055a | putP | 2 | Proline permease | <0.0006 |
| STM0133 | 0302a | dppF | 2 | Dipeptide ABC transporter, ATP-binding protein | <0.0100 |
| STM0312 | 1091a | kgtP | 2 | Alpha-ketoglutarate permease | |
| STM0368 | 1082a | msbA | 2 | Multidrug resistance protein | |
| STM0341 | 1206a | hetA | 2 | Multidrug resistance protein | <0.0016 |
| STM0308 | 1506a | gltS | 2 | Glutamate permease | |
| Stress response and acid survival | |||||
| STM0190 | 0073a | ureA | 2 | Urease, alpha subunit | |
| STM0334 | 0072a | ureB | 4 | Urease beta subunit (urea amidohydrolase) | |
| STM0101 | 0067a | ureH | 2 | Urease accessory protein | |
| STM0274 | 0071a | ureI | >5 | Urease accessory protein, urea transporter | |
| Regulatory functions | |||||
| STM0020 | 0714 | rpoN | 2 | RNA polymerase sigma-54 factor | |
| STM0045 | 0930a | surE | 2 | Stationary-phase survival protein | <0.0032 |
| Central intermediary metabolism and amino acid biosynthesis | |||||
| STM0374 | 0237a | hemC | 2 | Porphobilinogen deaminase | |
| STM0034 | 0397a | serA | 2 | Phosphoglycerate dehydrogenase | |
| Protein degradation | |||||
| STM0299 | 0169a | prtC | 2 | Collagenase | 0.0123 |
| VacA paralogues | |||||
| STM0324 | 0289a | 4 | VacA-paralogue | <0.0003 | |
| Hypothetical proteins | |||||
| STM0249 | 0245a | 5 | hp | <0.0160 | |
| STM0193 | 0288a | 2 | hp | ||
| STM0297 | 0350a | 2 | hp | ||
| STM0240 | 0486 | 2 | hp | <0.0025 | |
| STM0210 | 0973 | 2 | hp | <0.0009 | |
| STM0259 | 1525 | 2 | hp | ||
| STM0349 | 0758a | 2 | chimp | <0.0026 | |
| STM0256 | 1486a | 2 | chimp | ||
Hp, hypothetical protein; chimp, conserved hypothetical integral membrane protein; C.I., competitive index, defined as the output ratio (mutant/wild type) divided by the input ratio (mutant/wild-type).
Transposon insertion may have polar effects; ind mutants, number of mutants generated and screened independently for a given gene.
To determine the degree of attenuation quantitatively, 14 mutants from different functional groups were chosen and a competition assay was performed. A defined mutant and the wild-type strain were mixed in a 1:1 ratio and used for the oral infection of gerbils as described before. The competitive index (C.I.) is defined as the output ratio (mutant/wild-type) divided by the input ratio (mutant/wild-type). Using these criteria, all tested mutants were identified as heavily attenuated or completely colonization defective (see Table II, C.I.).
The Essential ORF hp0169 Encodes a Secreted Collagenase.
Interestingly, the gene product of hp0169 was annotated as a putative protease of the U32 protein family, which is characterized by the consensus sequence E-x-F-x(2)-G-[SA]-[LIVM]-C-x(4)-G-x-C-x-[LIVM]-S (14). Only one member of this family of proteases, PrtC of Porphyromonas gingivalis, has been characterized biochemically as a Ca2+-dependent metallo-protease with collagenolytic activity. No functional biochemical data were, however, present for HP0169, which reveals 29.4%/49.5% sequence identity/similarity with PrtC. Furthermore, the database search using the HP0169 sequence identified several other putative bacterial collagenases, such as PA5440 of Pseudomonas aeruginosa (41.0%/60.8%) and YdcP of E. coli O157:H7 (33.8%/55.8%) with significant sequence identity/similarity.
To obtain a first insight about a potential proteolytic activity of HP0169, total lysates of H. pylori P149 wild-type and the corresponding hp0169 mutant strain were compared using an Azocoll assay (15), which determines the release of an azo-dye due to proteolytic activity by a photometrical quantification. The wild-type strain revealed twice as much proteolytic activity as the mutant strain in this assay, but addition of EDTA reduced the proteolytic activity in the lysate to 25% of the wild type activity (Fig. 4 A), indicating that a proteolytic activity might be associated with HP0169.
Figure 4.

The essential gene hp0169, identified by the STM screen, encodes a secreted H. pylori collagenase. (A) Total protease activity of bacterial lysates of H. pylori P149 wt and P149::hp0169 mutant. Bacterial lysates (100 μg protein) of H. pylori wt and mutant strains were incubated with azocoll (14 h, 37°C) and the solubilization of the protein by proteolysis was determined by measuring the OD442 of the solution using a photometer. EDTA (10 mM) was used to block proteases dependent on divalent cations. The data are the results of three experiments. (B) Degradation of type 1 collagen by H. pylori P149 culture supernatant. Concentrated culture supernatants of H. pylori P149 wt and P149hp0169::TnHK9 (see Materials and Methods) were incubated with type I collagen and aliquots were analyzed at different time points by SDS-PAGE and immunoblotting using a collagen type 1-specific antiserum. (C) Absence of the cytoplasmic protein RecA in the supernatant fractions containing collagenase activity. Cleared lysates of H. pylori P149 (CL), collagenase-active supernatant of P149wt and of P149hp0169::TnHK9 (Sup) were analyzed by SDS-PAGE and immunoblotting using an H. pylori anti-RecA polyclonal antiserum (reference 37). (D) Degradation of type I collagen by the purified recombinant GST-HP0169 fusion protein. Purified GST-HP0169 fusion protein (2 μg) or purified GST alone was incubated with type I collagen (100 μg) for different length of time and analyzed by SDS-PAGE and immunoblotting using a type I collagen specific antiserum.
A putative collagenase activity of H. pylori would be expected to be on the surface of the bacteria, or secreted. We therefore isolated and concentrated the secreted proteins in the culture supernatant of a H. pylori P149 wt and the P149hp0169::TnHK9 mutant strain, as described in Material and Methods. The concentrated supernatant was incubated with type 1 collagen and analyzed by SDS-PAGE and immunoblotting using an anti-collagen type I antibody (Rockland). Interestingly, the collagen was completely degraded by the supernatant of the wt, but not the mutant strain after 15 h (Fig. 4 B). To verify that the activity was actually an actively secreted product, rather than the result of a general bacterial lysis during preparation of the supernatant, we analyzed the supernatant for the presence of RecA, a typical bacterial cytoplasmic protein and did not find any RecA protein in the supernatant by immunoblotting (Fig. 4 C). From these data we would conclude that the collagenase activity is actively secreted into the supernatant.
Finally, the hp0169 gene was fused to the glutathione-S-transferase gene (gst) and expressed in E. coli BL21 to produce the recombinant protein. The purified GST-HP0169 fusion protein, but not the purified GST protein alone, was active in degrading type 1 collagen (Fig. 4 D). Thus, we could show that HP0169, which was found to be essential for H. pylori to colonize in the gerbil stomach, is a secreted collagenase.
The In Vivo Essential Function of ComB4 Is Distinct from Its Role in Natural Transformation Competence.
The comB4 gene, which encodes a putative ATPase involved in natural transformation competence of H. pylori (16), was identified as essential for gastric colonization in the gerbil model (C.I. <0.0010, Table II). As the general screen resulted only in a single comB4 mutant, the mutation was reconstructed de novo in the wt strain background by transferring the mutated gene and was verified to be colonization defective. To prove experimentally whether or not natural transformation competence is the biological function determining the essential phenotype for in vivo colonization, we generated a deletion mutant in the comB8–10 genes of H. pylori P149 (see Materials and Methods). The ComB8–10 proteins are structural components of the type IV transport system involved in DNA transformation of H. pylori, probably by building a channel between inner and outer membrane (16, 17). Interestingly, the comB8–10 mutant strain, which was deficient in natural transformation competence, was still able to colonize the gerbil very efficiently in single infection experiments (Fig. 5) . From these data we would conclude that the ComB4 protein, besides its role in natural transformation competence, might have an additional essential function for H. pylori, which has not been identified yet.
Figure 5.
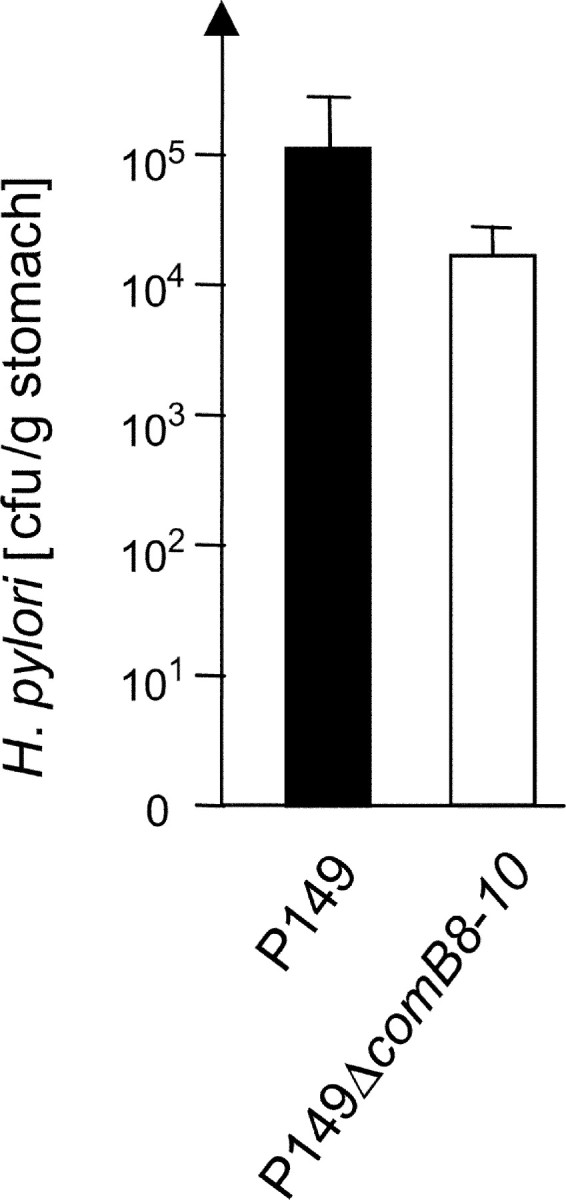
Determination of the reisolation rate of a ΔcomB8–10 mutant from the mongolian gerbil. Two gerbils were infected with H. pylori P149 wt strain and the P149ΔcomB8–10 mutant strain and a quantitative reisolation of H. pylori was performed 3 wk later.
Discussion
The present study reports for the first time on a more global approach on the identification of H. pylori genes essential for colonization of a model host, the mongolian gerbil. This model appears to be most suitable for screening of the STM mutants, as it has several advantages over the conventional mouse model, such as a lower gastric pH, the induction of typical gastritis, gastric ulceration, and carcinoma upon infection with H. pylori, similar to the situation in humans (18). The STM technique employed here is an extremely powerful tool, as shown by successful screens in a range of bacteria (11, 19). By adaptation and modification of the STM technique to the gerbil model we identified for the first time in a systematic way a larger number of genes essential for colonization of H. pylori. Although we cannot exclude the possibility of polar effects of TnHK9 on downstream genes within certain operons, the mutants still provide useful information, as they indicate a particular pathway important in colonization. Genetic in vivo complementation studies, which are not well established for H. pylori yet, should identify the responsible genes in such operons in future. We also cannot exclude a certain bias in the procedure to generate the H. pylori mutant collection. This is reflected by the fact that certain genes were found repeatedly mutagenized in different positions independently. This might be explained by the much better transformation efficiency of certain DNA fragments into H. pylori, as compared with others. Nevertheless, the independent reisolation of mutants in the same gene validates our model and finally proves that the screening is very efficient.
Salama et al. defined recently a minimal functional core of 1281 H. pylori genes by comparing the gene content of 15 clinical isolates using a whole genome DNA microarray (20). Interestingly, 46/47 genes that we identified as essential for gastric colonization belong to this core group of H. pylori genes. The only exception was ureH, which was found in 93.3% of the strains (20). It is considered to be essential in vivo. This indicates that the genes we identified are in the pool of common H. pylori genes, which supports their essentiality.
A fairly large number of mutants in the essential group consists of genes affecting bacterial chemotaxis and motility. In addition to the major flagellin flaA, we identified mutants in genes, such as fliD, fliF, fliI, fliP, fliS, and flgE, which validated our method, as they have already been described to be involved in the assembly of a functional flagellar apparatus (21–25). In addition, we added several new genes, which are annotated as being involved in flagellar biosynthesis from their sequence homology, but which have not been verified experimentally.
The outer membrane proteins (OMP8, HP0788) might have a function as adhesins, as described for the AlpAB, the BabA, the HopZ (26), and other outer membrane proteins from in vitro studies. Alternatively, they might fulfill an essential role as porins, or in stabilizing the integrity of the outer membrane to resist the harsh conditions in the gastric mucus. The LPS of H. pylori is known to be essential for colonization in the mouse model (27). This explains the galE mutant to be colonization defective, supporting the essential role of the O-side chain and/or Lewisx/y mimicry of LPS as an important feature for survival of H. pylori within the host (28).
Several genes in the urease gene cluster were found to be essential for gastric colonization, thus validating again our screening model (29, 30). Only relatively few genes encoding enzymes of the central intermediary metabolism and for amino acid biosynthesis were disrupted by the Tn, indicating that many of these genes might be essential in vitro and therefore not present in our collection of mutants.
Another group of essential functions relates to the putative membrane transport systems. Surprisingly, two putative transport ATPases usually associated with type IV secretion systems, the VirB4 homologous ATPase ComB4 (hp0017; reference 16), and the product of hp1421, a VirB11-homologous ATPase, were essential for colonization. ComB4 was recently identified as an essential component of the natural transformation associated type IV transport system (16). The comB8–10 deletion mutant of H. pylori P149 was only weakly attenuated for gastric colonization (Fig. 5). In contrast, ComB4 seems to play a major role in the colonization process (see C.I., Table II) and might therefore have a further function in addition to mediating DNA uptake. This function has to be identified in future experiments.
One putative ATP-binding cassette transporter, as well as three permeases for glutamate, proline and α-ketoglutarate were found to be essential for H. pylori to colonize in vivo, indicating that H. pylori apparently relies on the exogenous uptake of these amino acids and precursors in the stomach mucosa, as shown previously for growth of H. pylori in vitro (31). Such an amino acid exchange between bacteria and host is frequently observed in primary and secondary symbionts of plants and animals. The squid Euprymna scolopes, for instance, provides at least 9 amino acids to support the growth of its auxotroph symbiont Vibrio fischeri, present in its light-emitting organ (32). The amino acid dependence demonstrated here might be in support of the view that H. pylori originated as a symbiont of humans (33).
The RpoN sigma factor and a stationary phase survival protein (SurE) are supposed to fulfill essential regulatory functions (Table II). Among others, RpoN is known to be involved in flagellin gene transcription, which might be one reason for its essential function. SurE is a member of a novel family of metal ion-dependent phosphatases responsible for survival of bacteria under certain stress conditions (34), as they may be present in the gastric mucosa. The VacA-paralogous protein HP0289 is an OMP expected to be exported by a putative autotransporter mechanism to the bacterial surface, analogous to the VacA cytotoxin, but its function or role in colonization is completely unknown. Another group of identified genes corresponds to those encoding hypothetical proteins. This group consists of 6 mutants (Table II; hp) encoding hypothetical genes, which are, according to their original annotation (3), only present in H. pylori. The corresponding proteins might be involved in specific functions to adapt H. pylori to its special niche, the gastric mucosa. Two genes encoded conserved hypothetical integral membrane proteins (chimp).
Interestingly, a putative collagenase (HP0169) was identified by sequence homology to other bacterial collagenases, especially of the U32 protein family. Using a biochemical approach, we could verify that (a) the gene hp0169 actually encodes an active bacterial collagenase and that (b) HP0169 is actively transported to the cell surface or secreted. As HP0169 does not carry a typical hydrophobic signal sequence to enter the general secretory pathway, we assume that the protein is secreted via a specific secretion mechanism.
What might be the benefit for H. pylori to secrete a collagenase? Collagen type I and III are important components of the extracellular matrix of the stomach epithelium. The synthesis of type I collagen is induced in regions around gastric ulcers and collagens are of outmost importance for the process of ulcer healing (35). Thus, secretion of a collagen degrading enzyme by H. pylori could be responsible for the chronicity of gastric or duodenal ulcerogenesis and the delayed healing process. For H. pylori the degradation of collagen might be used as a source for the uptake of certain amino acids or short peptides. Alternatively, the enzyme might be important for the proteolytic degradation of components of the innate and acquired immune system, such as IgA antibodies or components of the complement system, to evade the immune response, as described for other mucosal pathogens (36). Both could explain the essential nature of the enzyme in vivo.
In conclusion, our approach allowed the identification of genes required for survival in the host, which has provided physiologically significant information about genes required for the adaptation of H. pylori to its specific niche, the gastric mucosa. Of particular significance, a more complete understanding of the role that the hypothetical proteins play in the infection process of gastric colonization of H. pylori may lead to a more rational understanding of the Helicobacter infection process and finally allow a specific intervention.
Acknowledgments
We thank H. Wirth and M.J. Blaser for providing the gerbil-adapted H. pylori strain G1.1.
This work was funded by grants from the Deutsche Forschungsgemeinschaft (HA 2697/1-4) and by Byk Gulden Konstanz, Germany. B.P. Burns was funded by a fellowship from the Alexander von Humboldt Foundation.
B.P. Burns' present address is School of Biotechnology and Biomolecular Sciences, The University of New South Wales, Sydney, 2052, NSW, Australia.
Footnotes
Abbreviations used in this paper: STM, signature tagged mutagenesis; Tn, transposon.
References
- 1.Blaser, M.J., and D.E. Berg. 2001. Helicobacter pylori genetic diversity and risk of human disease. J. Clin. Invest. 107:767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.ARC monographs on the evaluation of carcinogenic risks to humans. 1994. International Agency for Research on Cancer, World Health Organization, Lyon.
- 3.Tomb, J.-F., O. White, A.R. Kerlavage, R.A. Clayton, G.G. Sutton, R.D. Fleischmann, K.A. Ketchum, H.P. Klenk, S. Gill, B.A. Dougherty, et al. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 388:539–547. [DOI] [PubMed] [Google Scholar]
- 4.Alm, R.A., L.S. Ling, D.T. Moir, B.L. King, E.D. Brown, P.C. Doig, D.R. Smith, B. Noonan, B.C. Guild, B.L. deJonge, et al. 1999. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 397:176–180. [DOI] [PubMed] [Google Scholar]
- 5.Chalker, A.F., H.W. Minehart, N.J. Hughes, K.K. Koretke, M.A. Lonetto, K.K. Brinkman, P.V. Warren, A. Lupas, M.J. Stanhope, J.R. Brown, and P.S. Hoffman. 2001. Systematic identification of selective essential genes in Helicobacter pylori by genome prioritization and allelic replacement mutagenesis. J. Bacteriol. 183:1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jenks, P.J., C. Chevalier, C. Ecobichon, and A. Labigne. 2001. Identification of nonessential Helicobacter pylori genes using random mutagenesis and loop amplification. Res. Microbiol. 152:725–734. [DOI] [PubMed] [Google Scholar]
- 7.Hutchison, C.A., S.N. Peterson, S.R. Gill, R.T. Cline, O. White, C.M. Fraser, H.O. Smith, and J.C. Venter. 1999. Global transposon mutagenesis and a minimal Mycoplasma genome. Science. 286:2165–2169. [DOI] [PubMed] [Google Scholar]
- 8.Akerley, B.J., E.J. Rubin, V.L. Novick, K. Amaya, N. Judson, and J.J. Mekalanos. 2002. A genome-scale analysis for identification of genes required for growth or survival of Haemophilus influenzae. Proc. Natl. Acad. Sci. USA. 99:966–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eaton, K.A. 1999. Animal models of Helicobacter gastritis. Curr. Top. Microbiol. Immunol. 241:123–154. [DOI] [PubMed] [Google Scholar]
- 10.Guo, B.P., and J.J. Mekalanos. 2002. Rapid genetic analysis of Helicobacter pylori gastric mucosal colonization in suckling mice. Proc. Natl. Acad. Sci. USA. 99:8354–8359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hensel, M., J.E. Shea, C. Gleeson, M.D. Jones, E. Dalton, and D.W. Holden. 1995. Simultaneous identification of bacterial virulence genes by negative selection. Science. 269:400–403. [DOI] [PubMed] [Google Scholar]
- 12.Fischer, W., D. Schwan, E. Gerland, G.E. Erlenfeld, S. Odenbreit, and R. Haas. 1999. A plasmid-based vector system for the cloning and expression of Helicobacter pylori genes encoding outer membrane proteins. Mol. Gen. Genet. 262:501–507. [DOI] [PubMed] [Google Scholar]
- 13.Kahrs, A.F., S. Odenbreit, W. Schmitt, D. Heuermann, T.F. Meyer, and R. Haas. 1995. An improved TnMax mini-transposon system suitable for sequencing, shuttle mutagenesis and gene fusions. Gene. 167:53–57. [DOI] [PubMed] [Google Scholar]
- 14.Rawlings, N.D., and A.J. Barrett. 1993. Evolutionary families of peptidases. Biochem. J. 290:205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chavira, R., Jr., T.J. Burnett, and J.H. Hageman. 1984. Assaying proteinases with azocoll. Anal. Biochem. 136:446–450. [DOI] [PubMed] [Google Scholar]
- 16.Hofreuter, D., S. Odenbreit, and R. Haas. 2001. Natural transformation competence in Helicobacter pylori is mediated by the basic components of a type IV secretion system. Mol. Microbiol. 41:379–391. [DOI] [PubMed] [Google Scholar]
- 17.Fischer, W., R. Haas, and S. Odenbreit. 2002. Type IV secretion systems in pathogenic bacteria. Int. J. Med. Microbiol. 292:159–168. [DOI] [PubMed] [Google Scholar]
- 18.Ikeno, T., H. Ota, A. Sugiyama, K. Ishida, T. Katsuyama, R.M. Genta, and S. Kawasaki. 1999. Helicobacter pylori-induced chronic active gastritis, intestinal metaplasia, and gastric ulcer in Mongolian gerbils. Am. J. Pathol. 154:951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mecsas, J. 2002. Use of signature-tagged mutagenesis in pathogenesis studies. Curr. Opin. Microbiol. 5:33–37. [DOI] [PubMed] [Google Scholar]
- 20.Salama, N., K. Guillemin, T.K. McDaniel, G. Sherlock, L. Tompkins, and S. Falkow. 2000. A whole-genome microarray reveals genetic diversity among Helicobacter pylori strains. Proc. Natl. Acad. Sci. USA. 97:14668–14673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim, J.S., J.H. Chang, S.I. Chung, and J.S. Yum. 1999. Molecular cloning and characterization of the Helicobacter pylori fliD gene, an essential factor in flagellar structure and motility. J. Bacteriol. 181:6969–6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allan, E., N. Dorrell, S. Foynes, M. Anyim, and B.W. Wren. 2000. Mutational analysis of genes encoding the early flagellar components of Helicobacter pylori: evidence for transcriptional regulation of flagellin A biosynthesis. J. Bacteriol. 182:5274–5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jenks, P.J., S. Foynes, S.J. Ward, C. Constantinidou, C.W. Penn, and B.W. Wren. 1997. A flagellar-specific ATPase (FliI) is necessary for flagellar export in Helicobacter pylori. FEMS Microbiol. Lett. 152:205–211. [DOI] [PubMed] [Google Scholar]
- 24.Josenhans, C., K.A. Eaton, T. Thevenot, and S. Suerbaum. 2000. Switching of flagellar motility in Helicobacter pylori by reversible length variation of a short homopolymeric sequence repeat in fliP, a gene encoding a basal body protein. Infect. Immun. 68:4598–4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Toole, P.W., M. Kostrzynska, and T.J. Trust. 1994. Non-motile mutants of Helicobacter pylori and Helicobacter mustelae defective in flagellar hook production. Mol. Microbiol. 14:691–703. [DOI] [PubMed] [Google Scholar]
- 26.Gerhard, M., S. Hirno, T. Wadström, H. Miller-Podraza, S. Teneberg, K.-A. Karlsson, B. Appelmelk, S. Odenbreit, R. Haas, A. Arnquvist, and T. Borén. 2001. Helicobacter pylori, an adherent pain in the stomach. Helicobacter pylori: Molecular and Cellular Biology. M. Achtman, editors. Horizon Scientific Press, Wymondham, UK. 185–206.
- 27.Moran, A.P., E. Sturegard, H. Sjunnesson, T. Wadström, and S.O. Hynes. 2000. The relationship between O-chain expression and colonisation ability of Helicobacter pylori in a mouse model. FEMS Immunol. Med. Microbiol. 29:263–270. [DOI] [PubMed] [Google Scholar]
- 28.Appelmelk, B.J., M.A. Monteiro, S.L. Martin, A.P. Moran, and C.M. Vandenbroucke-Grauls. 2000. Why Helicobacter pylori has Lewis antigens. Trends Microbiol. 8:565–570. [DOI] [PubMed] [Google Scholar]
- 29.Weeks, D.L., S. Eskandari, D.R. Scott, and G. Sachs. 2000. A H+-gated urea channel: the link between Helicobacter pylori urease and gastric colonization. Science. 287:482–485. [DOI] [PubMed] [Google Scholar]
- 30.Bury-Mone, S., S. Skouloubris, A. Labigne, and H. De Reuse. 2001. The Helicobacter pylori UreI protein: role in adaptation to acidity and identification of residues essential for its activity and for acid activation. Mol. Microbiol. 42:1021–1034. [DOI] [PubMed] [Google Scholar]
- 31.Stark, R.M., M.S. Suleiman, I.J. Hassan, J. Greenman, and M.R. Millar. 1997. Amino acid utilisation and deamination of glutamine and asparagine by Helicobacter pylori. J. Med. Microbiol. 46:793–800. [DOI] [PubMed] [Google Scholar]
- 32.Graf, J., and E.G. Ruby. 1998. Host-derived amino acids support the proliferation of symbiotic bacteria. Proc. Natl. Acad. Sci. USA. 95:1818–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blaser, M.J. 1997. The versatility of Helicobacter pylori in the adaptation to the human stomach. J. Physiol. Pharmacol. 48:307–314. [PubMed] [Google Scholar]
- 34.Lee, J.Y., J.E. Kwak, J. Moon, S.H. Eom, E.C. Liong, J.D. Pedelacq, J. Berendzen, and S.W. Suh. 2001. Crystal structure and functional analysis of the SurE protein identify a novel phosphatase family. Nat. Struct. Biol. 8:789–794. [DOI] [PubMed] [Google Scholar]
- 35.Gillessen, A., M. Shahin, T. Pohle, E. Foerster, T.H. Krieg, and W. Domschke. 1995. Evidence of de novo collagen synthesis in healing human gastric ulcers. Scand. J. Gastroenterol. 30:515–518. [DOI] [PubMed] [Google Scholar]
- 36.Pohlner, J., R. Halter, K. Beyreuther, and T.F. Meyer. 1987. Gene structure and extracellular secretion of Neisseria gonorrhoeae IgA protease. Nature. 325:458–462. [DOI] [PubMed] [Google Scholar]
- 37.Schmitt, W., S. Odenbreit, D. Heuermann, and R. Haas. 1995. Cloning of the Helicobacter pylori recA gene and functional characterization of its product. Mol. Gen. Genet. 248:563–572. [DOI] [PubMed] [Google Scholar]