

Abstract
Interleukin-10 (IL-10) is a potent deactivator of myeloid cells that limits the intensity and duration of immune and inflammatory responses. The activity of IL-10 can be suppressed during inflammation, infection, or after allogeneic tissue transplantation. We investigated whether inflammatory factors suppress IL-10 activity at the level of signal transduction. Out of many factors tested, only ligation of Fc receptors by immune complexes inhibited IL-10 activation of the Jak-Stat signaling pathway. IL-10 signaling was suppressed in rheumatoid arthritis joint macrophages that are exposed to immune complexes in vivo. Activation of macrophages with interferon-γ was required for Fc receptor–mediated suppression of IL-10 signaling, which resulted in diminished activation of IL-10–inducible genes and reversal of IL-10–dependent suppression of cytokine production. The mechanism of inhibition involved decreased cell surface IL-10 receptor expression and Jak1 activation and was dependent on protein kinase C delta. These results establish that IL-10 signaling is regulated during inflammation and identify Fc receptors and interferon-γ as important regulators of IL-10 activity. Generation of macrophages refractory to IL-10 can contribute to pathogenesis of inflammatory and infectious diseases characterized by production of interferon-γ and immune complexes.
Keywords: interleukin 10, Fc receptor, signal transduction, Jak-Stat, macrophage
Introduction
IL-10 is predominantly an immunosuppressive and antiinflammatory cytokine and is a potent deactivator of dendritic cells and macrophages (for a review, see reference 1). IL-10 plays a critical role in limiting tissue injury during infections by limiting the duration and intensity of immune and inflammatory reactions (1–5). In the absence of IL-10, mice spontaneously develop inflammatory bowel and skin disease (6). IL-10 has been shown to suppress inflammation in many experimental models of inflammatory disease, including collagen induced arthritis, pancreatitis, uveitis, keratitis, hepatitis, peritonitis, lung injury, and neural injury (1). IL-10 enhances heart allograft survival and suppresses graft versus host disease if given before bone marrow transplantation (7, 8).
It has been long appreciated that the amount and timing of IL-10 production is tightly regulated, consistent with its important role in determining the balance between effective clearance of infectious pathogens and collateral tissue damage (1, 9–11). More recently, evidence is emerging that the activity of IL-10 is also regulated during the course of an immune response. Examples include suppression of IL-10 activity during chronic infection with the LP-BM5 retrovirus (12) or during experimental endotoxemia (13). In contrast to treatment before transplantation, IL-10 treatment after transplantation is ineffective at suppressing heart allograft survival and graft versus host disease after bone marrow transplantation (14, 15). IL-10 activity also appears to be blunted during chronic inflammatory diseases, as IL-10 was not effective in suppressing cytokine production in monocytes and macrophages derived from patients with SLE or rheumatoid arthritis (RA)* (16, 17). Thus, modulation of IL-10 activity plays a role in regulation of the intensity and chronicity of immune and inflammatory reactions. The factors and mechanisms that regulate IL-10 activity are not known.
Fc receptors (FcRs) bind the Fc region of Igs and are activated by cross-linking that occurs after ligation by multimerized Igs, such as those present in immune complexes or on opsonized pathogens (for a review, see reference 18). Ligation of myeloid cell FcRs specific for IgG, termed FcγRs, activates cellular effector functions, including phagocytosis of opsonized pathogens, endocytic clearance of immune complexes, and production of cytokines, chemokines, and reactive oxygen intermediates. FcγRs are important effector molecules of humoral immunity and have been implicated in the pathogenesis of inflammatory disorders characterized by the presence of immune complexes (ICs), such as SLE (19). FcγRs contribute to SLE pathogenesis by mediating activation of cells by ICs, and secondary to defective clearance of ICs in patients with FcγR polymorphisms (19, 20). Lupus-prone mice deficient for functional activating FcγRs, FcγRI and FcγRIII, do not develop end organ damage or glomerulonephritis, despite high levels of IC deposition (21). ICs are present in joint fluid and cartilage in RA (22, 23), and recently there has been a revival of interest in the role of B cells, autoantibodies, and antibody-dependent effector mechanisms in the pathogenesis of RA (24). An important role for FcγRs in RA pathogenesis is supported by the linkage of FcγR polymorphisms with RA (25), and the striking effects of FcγR deficiency on disease incidence and severity in several animal models of inflammatory arthritis (18, 26–33).
At sites of inflammation, cells are exposed to multiple pro- and antiinflammatory factors. The current paradigm suggests that the balance between pro- and antiinflammatory factors regulates the severity of chronic inflammatory diseases such as RA. We investigated whether suppression of IL-10 signal transduction corresponded to a mechanism by which IL-10 activity, and thus cytokine balance, are regulated during inflammation. We found that IL-10 signal transduction is suppressed in RA and a block in IL-10 signaling is induced by the combination of IFN-γ and ICs, inflammatory factors that are expressed in several chronic inflammatory diseases and infections. These results suggest that dysregulation of IL-10 signaling contributes to pathogenesis of inflammatory/autoimmune diseases characterized by the production of IFN-γ and ICs, such as RA and SLE.
Materials and Methods
Cell Isolation and Tissue Culture.
Monocytes were obtained from peripheral blood or RA synovial fluid mononuclear cells or synovial tissue, using anti-CD14 magnetic beads, as recommended by the manufacturer (Miltenyi Biotec), and were >97% pure as verified using flow cytometry, as described previously (34). Monocytes were cultured for 2 d in RPMI 1640 medium supplemented with 10% fetal bovine serum (Hyclone) with either M-CSF (20 ng/ml) or IFN-γ (100 U/ml) (R&D Systems). RA synovial tissue macrophages were obtained by digesting tissues using collagenase and dispase, as described previously (35), and selection using anti-CD14 magnetic beads. All patients fulfilled revised ACR criteria for the diagnosis of RA, and the protocol was approved by the Hospital for Special Surgery institutional review board. Murine C57BL/6 thioglycollate-elicited macrophages were obtained by peritoneal lavage 5 d after injection of thioglycollate and purified by adherence. The protocol was approved by the Hospital for Special Surgery animal care and use committee.
Phagocytosis Assays.
E-IgG were prepared using oxen erythrocytes (Ox Es) that were opsonized with human IgG1, murine IgG, or rat mAb 2.4G2 against murine FcγRII and FcγRIII, using a streptavidin-biotin bridge system and labeled with the fluorescent dye PKH26, as described previously (36). Typically, each E-IgG showed a mean fluorescence of 100 channels over background. Es or E-IgGs were incubated with adherent macrophages for 1 h at a ratio of 20:1. Extracellular Es and E-IgGs were lysed using water and intracellular fluorescence of macrophages was measured using flow cytometry. Typically, >85% of macrophages were phagocytic. The phagocytosis index was calculated by multiplying the mean fluorescence intensity by the fraction of cells that were phagocytic, as described previously (36).
Electrophoretic Mobility Shift Assay and Immunoblotting.
Cells were stimulated with cytokines (IL-10, 20 ng/ml; IFN-γ, 8 ng/ml) for 10 min. Cell extracts were obtained, and protein levels quantitated using the Bradford assay (Bio-Rad Laboratories), as described previously (34). 5 μg of cell extracts were incubated for 15 min at room temperature with 0.5 ng of 32P-labeled double stranded high affinity sis-inducible element (hSIE) oligonucleotide 5′-GTCGACATTTCCCGTAAATC-3′ that binds Stat1 and Stat3, as confirmed by supershift experiments (37, 38), in a 15 μl binding reaction containing 40 mM NaCl and 2 μg of poly-dI-dC (Amersham Biosciences), and complexes were resolved on nondenaturing 4.5% polyacrylamide gels. For immunoblotting, 15 μg of cell extracts were fractionated on 7.5% polyacrylamide gels using SDS-PAGE, transferred to polyvinylidene fluoride membranes (Millipore), incubated with specific antibodies, and enhanced chemiluminescence was used for detection. mAbs against Stat1 (clone 1), Stat3 (clone 84), Jak1 (clone 73), and PKCδ (clone 14) were obtained from BD Transduction Laboratories, and anti–IL-10R1 antibodies were obtained from Santa Cruz Biotechnology, Inc. Phosphorylation-specific Stat1 (Tyr701), Stat3 (Tyr703), and PKCδ (Ser643) antibodies were purchased from Cell Signaling Technology. Phosphorylation specific anti-Jak1 (Tyr1022/1023) was from Biosource International.
ELISA.
Es or E-IgGs were added 1 h before addition of IL-10 (20 ng/ml) and LPS (100 ng/ml) and supernatants were harvested after an additional 16 h. ELISAs were performed using paired antibody sets, as recommended by the manufacturer (R&D Systems).
Analysis of mRNA.
For real time, quantitative PCR, DNA-free RNA was obtained using the RNeasy Mini Kit from QIAGEN with DNase treatment, and 1 μg of total RNA was reverse-transcribed using random hexamers and MMLV reverse transcriptase. Real time PCR was performed in triplicate using the iCycler iQ thermal cycler and detection system (Bio-Rad Laboratories), and the PCR Core Reagents kit (Applied Biosystems), with 500 nM primers; the final Mg2+ concentration was adjusted to 4 mM, as described previously (34). mRNA amounts were normalized relative to GAPDH mRNA. When reverse transcriptase was omitted, threshold cycle number increased by at least 10, signifying lack of genomic DNA contamination or nonspecific amplification, and the generation of only the correct size amplification products was confirmed using agarose gel electrophoresis. Oligonucleotide primers used are as following: SOCS3: 5′-CACTCTTCAGCATCTCTGTCGGAAG-3′ and 5′-CATAGGAGTCCAGGTGGCCGTTGAC-3′; TSG14: 5′-TACTAGACTTTATGCCATGGTGCTT-3′ and 5′-TTATCTGACAGAGACAGCATTAA-3′; KIAA0307: 5′-GACGGCTTGCTTCATCTAACAATCT-3′ and 5′-GTCCACTGACACCTGCTTATGAAAT-3′; GAPDH: 5′-GTGAA-GGTCGGAGTCAAC-3′ and 5′-TGGAATTTGCCATGG-GTG-3′.
Flow Cytometry.
Flow cytometry was performed as described previously (34). The binding of IL-10 to cell surface IL-10Rs was measured using a Fluorokine kit according to the instructions of the manufacturer (R&D Systems).
Results
IL-10 Signaling Is Inhibited by FcγR Ligation.
We reasoned that inflammatory factors may inhibit IL-10 activity by suppressing IL-10 signal transduction. Ligation of the heterodimeric IL-10 receptor (IL-10R), consisting of IL-10R1 and IL-10R2 subunits, results in the activation of receptor-associated Jak1 and Tyk2 protein tyrosine kinases, and subsequent tyrosine phosphorylation and activation of DNA binding of Stat3 and Stat1; in resting myeloid cells, IL-10 activates primarily Stat3 (1, 39, 40). The effects of various inflammatory factors, alone or in combination, on IL-10 activation of STATs (signal transducer and activator of transcription) were assessed using electrophoretic mobility shift assays (EMSAs) to measure STAT DNA binding and immunoblotting to measure STAT tyrosine phosphorylation in purified human monocytes that had been activated with IFN-γ. As expected, in control macrophages IL-10 activated STAT DNA binding to the hSIE oligonucleotide that binds Stat1 and Stat3, and activated tyrosine phosphorylation of Stat3 (Fig. 1 A, lane 2). IL-10 also consistently activated Stat1 tyrosine phosphorylation in macrophages that had been activated by IFN-γ, such as the cells used in these experiments (Fig. 1 A, third panel). The significance of Stat1 activation by IL-10 in macrophages is not clear but Stat1 activation provides a signaling event that can be monitored in addition to the activation of Stat3. Ligation of FcγRs during phagocytosis of IgG-opsonized erythrocytes (E-IgGs) nearly completely blocked IL-10 activation of STAT DNA binding and Stat3 and Stat1 tyrosine phosphorylation (Fig. 1 A, lane 3). When the ratio of E-IgGs to macrophages was decreased, inhibition of IL-10 signaling decreased in a dose dependent fashion that correlated with diminished phagocytosis (Fig. 1 A). Stat3 and Stat1 levels were comparable in all lanes (Fig. 1 A), indicating that E-IgGs induced a block in signal transduction, and not degradation of STATs. In contrast to FcγR ligation, TNF-α, IL-1β, LPS, GM-CSF, and TGF-β had minimal effect upon IL-10–activated STAT DNA binding or tyrosine phosphorylation (Fig. 1, B and C). Phagocytosis or endocytosis were not required for inhibition of IL-10 signaling, as inhibition occurred when FcγRs were cross-linked with plate-bound IgG (Fig. 1 D). GM-CSF activation of Stat5 was not affected by E-IgGs, indicating that FcγR ligation did not nonspecifically inhibit cytokine signaling via the Jak-STAT pathway (Fig. 1 E). Inhibition of IL-10 signaling by E-IgGs was consistently observed in over 50 independent experiments using monocytes derived from different donors; of the other inflammatory factors tested, none inhibited IL-10 signaling except for a weak and inconsistent effect of IL-1. Production of IL-10 after ligation of FcγRs was not detected in these experiments using an ELISA that could detect 2 pg/ml of IL-10 (unpublished data). The lack of IL-10 production does not contradict previous reports (41, 42) where FcγRs induced IL-10 production after LPS pretreatment, because we did not preactivate cells with LPS. These results demonstrate that FcγR ligation inhibits IL-10 signaling, and suggest that FcγRs trigger an inhibitory pathway that is not activated by TNF, IL-1, LPS, GM-CSF, or TGF-β.
Figure 1.

IL-10 signaling is inhibited after ligation of FcγRs. Macrophages were activated with IFN-γ (100 U/ml) for 2 d and then treated with various factors for 1 h before addition of IL-10 (20 ng/ml) for 10 min. (A) E-IgGs were added to macrophages in ratios that varied from 20:1 (1×) to 2.5:1, and STAT activation was measured using EMSA and immunoblotting. Phagocytosis index was measured using flow cytometry. E, erythrocytes; E-IgG, erythrocytes opsonized with human IgG1. (B) STAT activation was measured by EMSA using the hSIE oligonucleotide. (C) Cell extracts were analyzed for tyrosine-phosphorylated Stat3 (pY-Stat3), and total Stat3 protein using immunoblotting. (D) Cells were added to culture wells containing plate-bound IgG for 1 h before treatment with IL-10 for 10 min, and levels of pY-Stat3 and Stat3 were determined using immunoblotting. (E) Activation of Stat5 by GM-CSF was analyzed by immunoblotting.
Inhibition of IL-10 Signaling In Vivo.
We investigated whether IL-10 signaling is inhibited after ligation of FcγRs in vivo in thioglycollate-elicited murine peritoneal macrophages. Elicited macrophages were activated in vivo by intraperitoneal injection of IFN-γ to achieve a fully activated state comparable to IFN-γ–activated human macrophages. When mice were injected intraperitoneally with E-IgGs, activation of STAT DNA binding and tyrosine phosphorylation by IL-10 in peritoneal macrophages was suppressed (Fig. 2 A). These results suggest that IL-10 signaling is modulated in vivo during the course of IC-mediated inflammatory processes. RA is a chronic inflammatory disease in which activated macrophages are exposed to immune complexes in joints. Therefore, we compared IL-10 signaling in freshly isolated RA synovial tissue and fluid macrophages with signaling in control blood-derived macrophages (Fig. 2 B, a representative experiment is shown). There were no significant differences in STAT activation by IL-10 between freshly isolated monocytes and blood-derived macrophages from normal donors that had been cultured in M-CSF for 2, 5, or 7 d, and thus blood-derived macrophages serve as a control for normal levels of IL-10 STAT activation in myeloid cells. IL-10 activated STAT DNA binding in control cells on all occasions tested (35/35). In contrast, IL-10 activated STAT DNA binding in RA synovial tissue-derived macrophages in only 1/11 samples and in synovial fluid macrophages in 3/12 samples. The differences in STAT activation were statistically significant (synovial tissue versus blood-derived macrophages, P < 0.001; synovial fluid versus blood-derived macrophages, P < 0.01). STAT activation by IL-6 was also consistently blocked in RA synovial macrophages (Fig. 2 B). IFN-γ signaling was preserved in RA synovial macrophages, demonstrating preferential suppression of IL-10 signaling (Fig. 2 B). Levels of Stat3 protein were comparable in RA and control cells (Fig. 2 C), and thus differences in DNA binding could not be explained on the basis of lower STAT protein levels. Diminished activation of STATs in RA synovial macrophages cannot be explained solely on the basis of desensitization of IL-10 pathways by IL-10 present in RA synovial fluid (<100 pg/ml; references 43 and 44) because: (a) IL-10 receptor occupancy by the levels of IL-10 present in RA synovial fluid would be very low, and ∼100–1,000-fold higher concentrations of IL-10 are required to saturate IL-10 receptors (43–45). (b) We did not detect desensitization of the IL-10 pathway when macrophages were cultured for 7 d in saturating concentrations of IL-10 (100 ng/ml, replenished every other day), isolated, and stimulated exactly as was done with RA macrophages. These results demonstrate that RA synovitis, which is characterized by activation of macrophages and high levels of synovial ICs, leads to suppression of IL-10 signaling.
Figure 2.
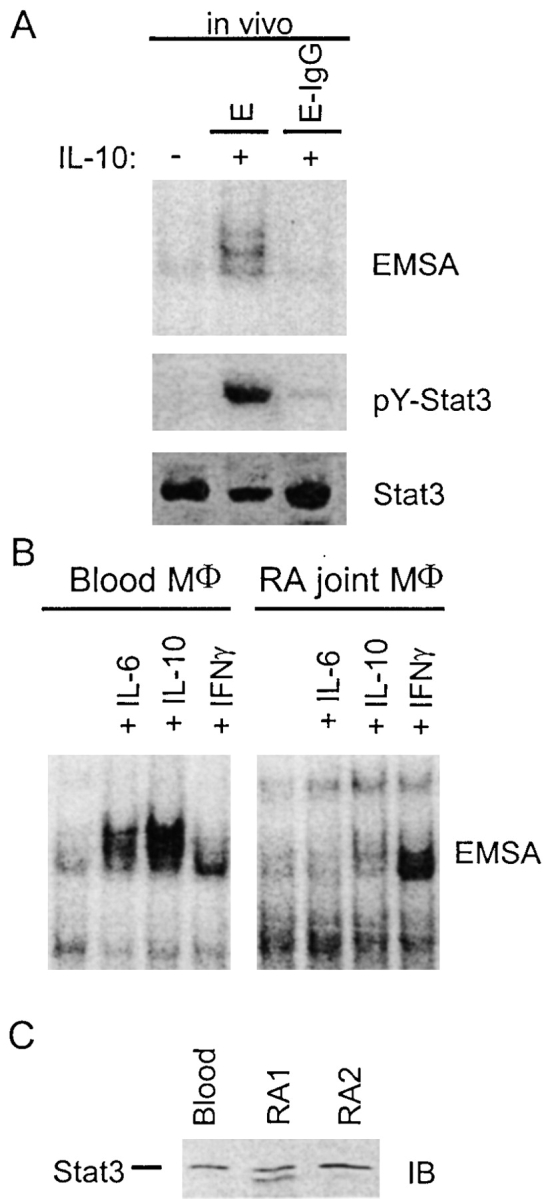
Inhibition of IL-10 signaling in vivo. (A) Murine thioglycollate-elicited peritoneal macrophages were activated by daily IP injections of 1,000 units of IFN-γ, and 2 d later 50 × 106 E-IgGs were injected IP and peritoneal cells were harvested after 4 h. IL-10 activation of STATs was assessed using EMSA and immunoblotting. (B) Activation of STAT DNA binding by IL-10 is suppressed in RA synovial macrophages. STAT activation in freshly isolated control and RA macrophages was measured by EMSA using the hSIE oligonucleotide. A representative experiment out of 23 RA samples analyzed is shown. (C) Stat3 protein levels were measured using immunoblotting.
FcγR Ligation Inhibits IL-10 Biological Activity.
An important antiinflammatory function of IL-10 is the suppression of cytokine production (1). We assessed the physiological significance of FcγR-dependent inhibition of IL-10 signaling by examining the effects of FcγR ligation on IL-10 inhibition of LPS-induced cytokine synthesis (Fig. 3 A). In control macrophages incubated with Es, IL-10 strongly inhibited TNF-α and IL-6 production. In contrast, when macrophages were incubated with E-IgGs, IL-10 had no detectable effect on TNF-α production. E-IgGs also suppressed the ability of IL-10 to inhibit IL-6 production (Fig. 3 A). We also assessed the effects of FcγR ligation on IL-10 induction of gene expression. The expression of the SOCS3, TSG14 (TNF-stimulated gene 14, also known as pentraxin-3 [46, 47]) and KIAA0307 genes that were consistently activated by IL-10 in control and IFN-γ–activated macrophages (unpublished data) was examined. Incubation of monocytes with E-IgGs completely blocked IL-10 induction of SOCS3, TSG14, and KIAA0307 mRNA (Fig. 3 B). Thus, inhibition of IL-10 signaling resulted in suppression of IL-10 biological activity and IL-10–dependent gene activation.
Figure 3.

Inhibition of IL-10 signaling results in suppression of IL-10 activity. (A) FcγR ligation suppresses IL-10-mediated inhibition of cytokine production. Es or E-IgGs were added to IFN-γ–activated macrophages 1 h before addition of IL-10 (20 ng/ml) and LPS (100 ng/ml). Culture supernatants were harvested 16 h later, and TNF-α and IL-6 were measured using ELISA. Three independent experiments were performed and the total LPS-induced TNFα and IL-6 production in the absence of IL-10 was 1.2 ng/ml and 3.4 ng/ml, respectively. (B) Inhibition of IL-10–induced gene activation. Es or E-IgGs were added to cells for 1 h followed by a 3 h stimulation with IL-10 (20 ng/ml). mRNA levels were measured using real time PCR and normalized relative to GAPDH mRNA.
Inhibition of IL-10 Signaling Is Dependent upon Activation State of Macrophages and on Specific FcγRs.
In many physiological situations macrophages need to clear ICs or opsonized particles but remain responsive to IL-10, such that excessive inflammation is prevented (48). Therefore, we hypothesized that FcγR-mediated modulation of IL-10 signaling would be regulated, and would vary with the physiological state of the macrophage. This hypothesis was tested by comparing the effects of FcγR ligation in M-CSF–differentiated and IFN-γ–activated macrophages. In four independent experiments, E-IgGs were not effective in inhibiting IL-10 signaling in M-CSF–differentiated macrophages, relative to the near complete inhibition observed in IFN-γ–activated cells (Fig. 4 A; a representative experiment is shown). The M-CSF–cultured cells phagocytosed E-IgGs at a comparable, or slightly higher, rate than IFN-γ–activated cells (unpublished data), indicating that differences in rates of phagocytosis did not explain the differences in inhibition of IL-10 signaling. Comparable results were obtained when FcγRs were ligated by plate-bound IgG (Fig. 4 B). Thus, IFN-γ activation of macrophages was required for FcγR-mediated inhibition of IL-10 signaling in vitro. We have not determined if IFN-γ was required for inhibition of IL-10 signaling in vivo (Fig. 2 A), and it is possible that FcγR-mediated inhibition of IL-10 signaling can occur in different macrophage stages of differentiation or activation in the absence of IFN-γ.
Figure 4.

FcγR-induced inhibition of IL-10 signaling requires activation of macrophages with IFN-γ. (A) Macrophages were cultured with M-CSF or IFN-γ for 2 d, Es or E-IgGs were added for 1 h, and STAT activation was measured after a 10 min stimulation with IL-10 (20 ng/ml) using immunoblotting. C, no erythrocytes were added. (B) FcγRs were ligated for one hour using plate-bound IgG, and STAT activation was measured using immunoblotting. (C) Macrophages cultured with M-CSF or IFN-γ for 2 d were analyzed using flow cytometry. (D) Individual FcγRs were cross-linked on IFN-γ–activated macrophages using specific F(ab′)s followed by F(ab′)2 anti–mouse IgG, and STAT activation was measured using EMSA and immunoblotting.
As expected (18), flow cytometry showed that FcγRI expression was elevated in IFN-γ–activated macrophages, while expression of FcγRII and FcγRIII was comparable in IFN-γ–activated and M-CSF-cultured cells (Fig. 4 C). The roles of individual FcγRs in inhibition of IL-10 signaling were investigated by cross-linking FcγRs with isoform-specific F(ab′) fragments, followed by cross-linking with goat F(ab′)2 anti–mouse IgG. Cross-linking of FcγRI inhibited IL-10 induction of STAT DNA binding and Stat1 and Stat3 tyrosine phosphorylation (Fig. 4 D); the effects on STAT phosphorylation were modest, likely secondary to limited and transient ligation of FcγRI when F(ab′) fragments were used, but were consistently observed in five experiments. Inhibition of IL-10 signaling by FcγRIII was more limited, consistent with low level expression (Fig. 4 C), and cross-linking FcγRII had a minimal effect (Fig. 4 D, lane 4) that was not reproducibly detected in all experiments. Thus, it is likely that IFN-γ–induced increased expression of FcγRI contributed to inhibition of IL-10 signaling. These results indicate that crosstalk between FcγRs and IL-10Rs is regulated and that IFN-γ activation couples FcγRs to inhibition of IL-10 signaling.
Inhibition of IL-10 Signaling Occurs via a Rapid, Direct Pathway that Is Dependent upon Protein Kinase C.
The mechanism of inhibition of IL-10 signaling was investigated. We first determined whether FcγR ligation inhibited proximal events in the IL-10 signaling pathway. Cell surface expression of IL-10Rs was measured by flow cytometry using biotinylated IL-10. Cross-linking of FcγRs led to a rapid decrease in cell surface IL-10 binding sites on macrophages, whereas incubation with anti-FcγR mAbs in the absence of cross-linking had no effect (Fig. 5 A, and unpublished data). Similar results were obtained using a mAb specific for IL-10R1 (unpublished data), and surface expression of a control protein, HLA-DR, was not affected by FcγR cross-linking (Fig. 5 A). In contrast to cell surface IL-10R expression, total cellular expression of IL-10Rs did not change, as determined by immunoblotting of total cell lysates (Fig. 5 B). This result suggests that FcγR cross-linking induced internalization of IL-10Rs, and is consistent with previous work demonstrating rapid cycling of IL-10Rs between the cell surface and intracellular compartments (49). Down-regulation of cell surface IL-10R expression would be predicted to diminish the activation of IL-10R–associated Jak1. Indeed, activation of Jak1 was suppressed after FcγR ligation (Fig. 5 C). These results demonstrate that FcγR ligation inhibits IL-10 signaling at a proximal step, upstream of STATs.
Figure 5.

Inhibition occurs at a proximal step in IL-10 signal transduction. (A) FcγRs were cross-linked using specific mAbs and anti-mouse IgG for 1 h, and cell surface IL-10R and HLA DR expression was measured using biotinylated IL-10 or FITC-conjugated anti-HLA DR mAb and flow cytometry. (B) Total cell extracts from parallel wells were analyzed for IL-10R1 expression using immunoblotting. (C) Cell extracts were analyzed for pY-Jak1 and Jak1 by immunoblotting.
One major mechanism of inhibition of Jak-STAT signaling at the level of cytokine receptors or Jaks is de novo expression of suppressors of cytokine signaling (SOCS; reference 50). The requirement for de novo production of IL-10 signaling inhibitors was addressed by examining the kinetics of inhibition, and by using actinomycin D and cycloheximide to inhibit, respectively, de novo transcription and translation. E-IgGs induced inhibition of IL-10 signaling rapidly, within 20 min (Fig. 6 A), suggesting that inhibition was direct, and did not depend upon synthesis of inhibitory molecules. Inhibition of IL-10 signaling was preserved when de novo gene expression was blocked using actinomycin D (Fig. 6, B and C; actinomycin D essentially completely blocked transcription in these cells at the concentrations that were used [51]), and when de novo protein synthesis was blocked using cycloheximide (unpublished data). Additional experiments showed that culture supernatants from FcγR-stimulated macrophages did not inhibit IL-10 signaling, thus indicating that FcγR ligation did not result in the secretion of soluble inhibitors (unpublished data). Taken together, these results indicate that induction of inhibitory molecules, such as SOCS proteins, was not required for inhibition of IL-10 signaling. Instead, the results suggest that inhibition occurs by a direct mechanism.
Figure 6.

Fc receptor-mediated inhibition of IL-10 signaling occurs rapidly and is independent of de novo gene expression. (A) Time course of inhibition of IL-10 signaling. (B and C) Lack of reversal of inhibition of IL-10 signaling when de novo gene activation was blocked using actinomycin D (5 μg/ml). STAT activation was measured by EMSA (B) and immunoblotting (C).
Our lab and others have described rapid and direct inhibition of Jak-STAT signaling that is dependent upon mitogen-activated protein kinases (MAPKs), protein kinase C (PKC), or proteosomes (51–55). Inhibition has been proposed to occur by post-translational modification or degradation of Jak-STAT signaling components. The role of MAPKs, PKC, or proteosomes in inhibition of IL-10 signaling was investigated using inhibitors of these kinases or of proteosomes. Inhibitors of ERKs, p38, and proteosomes had no effect (unpublished data). In contrast, GF109203X, an inhibitor of PKC, reversed FcγR-mediated inhibition of STAT DNA binding (Fig. 7 A) and tyrosine phosphorylation (Fig. 7 B). Inhibition of PKC reversed inhibition of Jak1 tyrosine phosphorylation (Fig. 7 C), indicating that PKC inhibited a proximal step in the IL-10 signaling pathway. The ability of PKC to inhibit IL-10 signaling was further tested using PMA, which activates PKC directly by binding to the C1 zinc finger-containing domain (56). PMA strongly blocked IL-10 signaling, and this inhibition was reversed by GF109203X, which works at the PKC ATP binding site (Fig. 7 D). Similar to FcγR cross-linking, PMA suppressed cell surface IL-10R expression (Fig. 7 E). These results indicate that FcγR ligation inhibits IL-10 signaling by a direct pathway that utilizes PKC.
Figure 7.

Inhibition of IL-10 signaling is dependent upon PKC. (A–D) Reversal of inhibition of IL-10 signaling by the PKC inhibitor GF109203X. GF109203X (1 μM or as labeled) was added 20 min before adding Es or E-IgGs, and STAT activation was measured using EMSA with the hSIE oligonucleotide (A and D) and immunoblotting for pY-Stat3 (B). Activation of Jak1 was measured by immunoblotting for pY-Jak1 (C). In D, PKC was activated by adding PMA (50 ng/ml) 1 h before adding IL-10. (E) Cell surface IL-10R expression was measured using biotinylated IL-10 and flow cytometry 1 h after adding PMA (50 ng/ml). Positive control, control macrophages not treated with anti-FcγR antibodies or PMA.
There are at least 9 different isoforms of PKC that have been divided into conventional PKCs (α, β, γ; activated by diacyl glycerol [DAG] and calcium), novel PKCs (δ, ɛ, μ, θ; activated by DAG and not dependent on calcium), and atypical PKCs (ζ, λ; independent of DAG and calcium; reference 56). IFN-γ activation of macrophages alters FcγR signaling such that FcγRs activate PKCδ instead of PKCβ (57). Therefore, the role of PKCδ in FcγR regulation of IL-10 signaling was examined. PKCδ activity was low in M-CSF–cultured cells and was not increased by exposure to E-IgGs (Fig. 8 A). In contrast, baseline serine-phosphorylation of PKCδ was substantially higher in IFN-γ–cultured cells, and was further increased by exposure to E-IgGs (Fig. 8 A). E-IgGs also induced a striking activation of the 48 kD highly active catalytic fragment of PKCδ (reference 58; Fig. 8 A). IFN-γ activation of macrophage cell lines has been reported to couple FcγRI to activation of phosphatidylinositol 3-kinase (PI3K; reference 59), and PI3K has been linked to the activation of PKC, including by FcγRs (56, 59). Therefore, we investigated the role of PI3K in FcγR inhibition of IL-10 signaling using the PI3K inhibitors Wortmanin and LY294002. Inhibition of PI3K resulted in a reversal of FcγR-mediated inhibition of IL-10 signaling (Fig. 8 B). This result is consistent with IFN-γ–dependent coupling of FcγRs to the activation of PI3K and PKC, although PI3K may also act via PKC-independent pathways. The role of PKCδ was further examined using elicited peritoneal macrophages from PKCδ (60) deficient mice. Cross-linking FcγRs suppressed IL-10 signaling in wild-type and control PKCβ-deficient mice (61; Fig. 8 C), and inhibition was potentiated by the addition of PMA. In contrast, no inhibition of IL-10 signaling was detected in macrophages deficient in PKCδ. These results demonstrate that inhibition of IL-10 signaling by FcγRs is dependent upon PKCδ.
Figure 8.

Role of PKCδ in FcγR-induced inhibition of IL-10 signaling. (A) Macrophages were cultured with M-CSF or IFN-γ for 2 d, Es or E-IgGs were added for 5 min, and PKCδ activation was measured using immuno-blotting. (B) Wortmanin (30 nM) or LY294002 (10 μM) was added 20 min before adding E-IgGs, and STAT activation was measured using immunoblotting for pY-Stat3. (C) Elicited peritoneal macrophages from control wild-type mice and mice deficient in PKCβ or PKCδ were activated with IFN-γ for 2 d, incubated with Es opsonized with anti-FcγR mAb 2.4G2 (E-IgG) or with murine IgG (E-mIgG) and PMA (50 ng/ml) for 1 h, and stimulated with IL-10. STAT activation was measured using immunoblotting.
Discussion
IL-10 is a major deactivator of myeloid cells, and IL-10 production and activity need to be tightly regulated to ensure the appropriate balance between clearance of pathogens and avoidance of excessive tissue damage or autoimmunity (1). We report that IL-10 signal transduction was blocked in IFN-γ–activated macrophages when FcγRs were ligated, and in RA macrophages that exhibit an IFN-γ–activated phenotype and are exposed to ICs in synovial fluid and tissue (24, 62). These results establish that deactivation of macrophages is regulated at the level of IL-10 signal transduction and suggest that defects in IL-10 signal transduction may underlie previously reported diminished IL-10 activity in RA, SLE, and chronic infection (12, 16, 17). Inhibition of IL-10 signaling results in highly activated macrophages that are resistant to suppression and are able to carry out effector functions, even in the presence of high concentrations of IL-10. Excessive production of cytokines, chemokines and other inflammatory mediators by these macrophages would contribute to tissue damage and pathogenesis of disease. One example would be inappropriately high production of TNF-α in RA in the face of a homeostatic response that includes elevated IL-10 production.
The lack of inhibition of IL-10 signaling by IL-1, LPS, TNF-α, GM-CSF, and TGF-β suggested that inhibition was mediated by a pathway that is not activated by these factors, but is activated by ICs via FcγRs. Both pharmacologic and genetic data indicated that inhibition was mediated by PKC, specifically the δ isoform of PKC that is activated by FcγRs in IFN-γ–activated macrophages (56, 57), but not by the other inflammatory stimuli that were examined. PKCδ was modestly activated by IFN-γ alone, and it is possible that this pool of active PKCδ contributed to inhibition of IL-10 signaling by coactivating a FcγR-induced pathway. Inhibition mediated by PKCδ did not require de novo synthesis of inhibitory molecules, or secretion of preformed inhibitors. This suggests that PKC, or molecules activated downstream of PKC, directly modify and inactivate IL-10 Jak-STAT signaling components. Activation of PKC by either FcγRs or PMA resulted in decreased cell surface expression of IL-10R, but total cellular IL-10R levels did not change (Fig. 5, and unpublished data). The IL-10R1 subunit contains a consensus dileucine internalization motif preceded by a serine that corresponds to a consensus PKC phosphorylation site (1, 63), and the IL-10R has been shown to partition between the cell surface and an intracellular pool (49). Thus, the results suggest that PKC induces internalization of IL-10Rs by phosphorylation of the IL-10R1 internalization motif, similar to PKC-dependent phosphorylation and internalization of other cell surface receptors (64). Decreased Jak1 activation could be explained by decreased IL-10R cell surface expression. However, PKCδ has been shown to inactivate Jaks via phosphorylation (65), and it is also possible that direct phosphorylation of IL-10R–associated Jaks contributes to inhibition of IL-10 signaling. A role for PKC-dependent activation of ERKs or production of reactive oxygen intermediates was ruled out (unpublished data).
An interesting question is how activation of macrophages with IFN-γ couples FcγRs to inhibition of IL-10 signaling. Ligation with FcγR isoform-specific reagents showed that FcγRI and, to a lesser extent, FcγRIII, inhibited IL-10 signaling. Because FcγRI and FcγRIII signal via the receptor-associated ITAM-containing FcRγ common signaling subunit (18), and thus activate similar signal transduction pathways, it is possible that the weak inhibitory effect mediated by FcγRIII was secondary to low cell surface expression that was not increased by IFN-γ (Fig. 4 C). IFN-γ treatment resulted in increased expression of FcγRI and thus quantitatively increased signaling via FcγRI after IFN-γ activation likely contributed to inhibition of IL-10 signaling. In addition to modulating FcγRI expression, IFN-γ has been reported to increase coupling of FcγRI with ITAM-containing FcRγ (18, 66), with concomitant activation PI3K and PKCδ (57, 59). Thus, a qualitative change in FcγR signaling that induced activation of PKCδ was also important for inhibition of IL-10 signaling.
Inhibition of IL-10 signaling required exposure of macrophages to products of both cellular and humoral immunity, IFN-γ and antibodies complexed with antigens. Thus, during the course of a normal immune response, generation of macrophages refractory to inhibition by IL-10 will be transient and such cells will only be generated when the immune system is fully active and maximal effector function is required. In contrast, several autoimmune and inflammatory diseases, such as SLE and RA (19, 24), are characterized by chronic production of IFN-γ and ICs. Our results suggest that chronic production of IFN-γ and ICs will lead to the ongoing generation of macrophages with defects in IL-10 signal transduction. These defects in IL-10 signaling and activity will result in diminished ability of IL-10 to suppress TNF-α production, thus shifting cytokine balance toward increased TNF-α and inflammation. FcγRs have been strongly implicated in the pathogenesis of arthritis in animal models of RA (24, 26–33), and RA joint macrophages show evidence of activation by IFN-γ and are exposed to high levels of immune complexes (62). While it is possible that several mechanisms contribute to suppressed IL-10 signaling in RA, our data suggest that inhibition mediated by FcγRs plays a role, especially as IL-10R expression is low in RA macrophages (unpublished data), similar to low IL-10R expression after FcγR ligation (Fig. 5). Diminished responsiveness to IL-10, and thus inadequate suppression of macrophage function, may contribute to chronicity and severity of inflammation in RA. In addition, diminished ability of joint macrophages to respond to IL-10 helps explain the lack of efficacy of exogenous IL-10 in the treatment of RA (67).
Acknowledgments
We thank Peggy Crow and Jillian Zhang for critically reviewing the manuscript and Jacqueline Bromberg and Raphael Clynes for helpful discussions.
Supported by National Institutes of Health grants AI44938 and AR46713 (to L.B. Ivashkiv), AR47106 (to L. Pricop), and the Kirkland Center for Lupus Research.
Footnotes
Abbreviations used in this paper: DAG, diacyl glycerol; EMSA, electrophoretic mobility shift assay; IC, immune complex; PKC, protein kinase C; RA, rheumatoid arthritis; SOCS, suppressors of cytokine signaling; STAT, signal transducer and activator of transcription.
References
- 1.Moore, K.W., R. de Waal Malefyt, R.L. Coffman, and A. O'Garra. 2001. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 19:683–765. [DOI] [PubMed] [Google Scholar]
- 2.Berg, D.J., R. Kuhn, K. Rajewsky, W. Muller, S. Menon, N. Davidson, G. Grunig, and D. Rennick. 1995. Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. J. Clin. Invest. 96:2339–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown, J.P., J.F. Zachary, C. Teuscher, J.J. Weis, and R.M. Wooten. 1999. Dual role of interleukin-10 in murine Lyme disease: regulation of arthritis severity and host defense. Infect. Immun. 67:5142–5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howard, M., T. Muchamuel, S. Andrade, and S. Menon. 1993. Interleukin 10 protects mice from lethal endotoxemia. J. Exp. Med. 177:1205–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin, M.T., D.R. Hinton, B. Parra, S.A. Stohlman, and R.C. van der Veen. 1998. The role of IL-10 in mouse hepatitis virus-induced demyelinating encephalomyelitis. Virology. 245:270–280. [DOI] [PubMed] [Google Scholar]
- 6.Kuhn, R., J. Lohler, D. Rennick, K. Rajewsky, and W. Muller. 1993. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 75:263–274. [DOI] [PubMed] [Google Scholar]
- 7.Bacchetta, R., M. Bigler, J.L. Touraine, R. Parkman, P.A. Tovo, J. Abrams, R. de Waal Malefyt, J.E. de Vries, and M.G. Roncarolo. 1994. High levels of interleukin 10 production in vivo are associated with tolerance in SCID patients transplanted with HLA mismatched hematopoietic stem cells. J. Exp. Med. 179:493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li, W., F. Fu, L. Lu, S.K. Narula, J.J. Fung, A.W. Thomson, and S. Qian. 1999. Recipient pretreatment with mammalian IL-10 prolongs mouse cardiac allograft survival by inhibition of anti-donor T cell responses. Transplant. Proc. 31:115–121. [DOI] [PubMed] [Google Scholar]
- 9.Fiorentino, D.F., M.W. Bond, and T.R. Mosmann. 1989. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J. Exp. Med. 170:2081–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Powell, M.J., S.A. Thompson, Y. Tone, H. Waldmann, and M. Tone. 2000. Posttranscriptional regulation of IL-10 gene expression through sequences in the 3′-untranslated region. J. Immunol. 165:292–296. [DOI] [PubMed] [Google Scholar]
- 11.Brightbill, H.D., S.E. Plevy, R.L. Modlin, and S.T. Smale. 2000. A prominent role for Sp1 during lipopolysaccharide-mediated induction of the IL-10 promoter in macrophages. J. Immunol. 164:1940–1951. [DOI] [PubMed] [Google Scholar]
- 12.Avdiushko, R., D. Hongo, H. Lake-Bullock, A. Kaplan, and D. Cohen. 2001. IL-10 receptor dysfunction in macrophages during chronic inflammation. J. Leukoc. Biol. 70:624–632. [PubMed] [Google Scholar]
- 13.Pajkrt, D., L. Camoglio, M.C. Tiel-van Buul, K. de Bruin, D.L. Cutler, M.B. Affrime, G. Rikken, T. van der Poll, J.W. ten Cate, and S.J. van Deventer. 1997. Attenuation of proinflammatory response by recombinant human IL-10 in human endotoxemia: effect of timing of recombinant human IL-10 administration. J. Immunol. 158:3971–3977. [PubMed] [Google Scholar]
- 14.Li, W., L. Lu, Y. Li, F. Fu, J.J. Fung, A.W. Thomson, and S. Qian. 1997. High-dose cellular IL-10 exacerbates rejection and reverses effects of cyclosporine and tacrolimus in mouse cardiac transplantation. Transplant. Proc. 29:1081–1082. [DOI] [PubMed] [Google Scholar]
- 15.Blazar, B.R., P.A. Taylor, S. Smith, and D.A. Vallera. 1995. Interleukin-10 administration decreases survival in murine recipients of major histocompatibility complex disparate donor bone marrow grafts. Blood. 85:842–851. [PubMed] [Google Scholar]
- 16.Mongan, A.E., S. Ramdahin, and R.J. Warrington. 1997. Interleukin-10 response abnormalities in systemic lupus erythematosus. Scand. J. Immunol. 46:406–412. [DOI] [PubMed] [Google Scholar]
- 17.Hart, P.H., M.J. Ahern, M.D. Smith, and J.J. Finlay-Jones. 1995. Comparison of the suppressive effects of interleukin-10 and interleukin-4 on synovial fluid macrophages and blood monocytes from patients with inflammatory arthritis. Immunology. 84:536–542. [PMC free article] [PubMed] [Google Scholar]
- 18.Ravetch, J.V., and S. Bolland. 2001. IgG Fc receptors. Annu. Rev. Immunol. 19:275–290. [DOI] [PubMed] [Google Scholar]
- 19.Salmon, J.E., and L. Pricop. 2001. Human receptors for immunoglobulin G: key elements in the pathogenesis of rheumatic disease. Arthritis Rheum. 44:739–750. [DOI] [PubMed] [Google Scholar]
- 20.Salmon, J.E., S. Millard, L.A. Schachter, F.C. Arnett, E.M. Ginzler, M.F. Gourley, R. Ramsey-Goldman, M.G. Peterson, and R.P. Kimberly. 1996. Fc gamma RIIA alleles are heritable risk factors for lupus nephritis in African Americans. J. Clin. Invest. 97:1348–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clynes, R., C. Dumitru, and J.V. Ravetch. 1998. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science. 279:1052–1054. [DOI] [PubMed] [Google Scholar]
- 22.Zvaifler, N.J. 1973. The immunopathology of joint inflammation in rheumatoid arthritis. Adv. Immunol. 16:265–336. [DOI] [PubMed] [Google Scholar]
- 23.Pope, R.M., D.C. Teller, and M. Mannik. 1975. Intermediate complexes formed by self-association of IgG-rheumatoid factors. Ann. NY Acad. Sci. 256:82–87. [DOI] [PubMed] [Google Scholar]
- 24.Hirano, T. 2002. Revival of the autoantibody model in rheumatoid arthritis. Nat. Immunol. 3:342–344. [DOI] [PubMed] [Google Scholar]
- 25.Morgan, A.W., B. Griffiths, F. Ponchel, B.M. Montague, M. Ali, P.P. Gardner, H.C. Gooi, R.D. Situnayake, A.F. Markham, P. Emery, and J.D. Isaacs. 2000. Fcgamma receptor type IIIA is associated with rheumatoid arthritis in two distinct ethnic groups. Arthritis Rheum. 43:2328–2334. [DOI] [PubMed] [Google Scholar]
- 26.Barnes, N., A.L. Gavin, P.S. Tan, P. Mottram, F. Koentgen, and P.M. Hogarth. 2002. FcgammaRI-deficient mice show multiple alterations to inflammatory and immune responses. Immunity. 16:379–389. [DOI] [PubMed] [Google Scholar]
- 27.Corr, M., and B. Crain. 2002. The role of FcgammaR signaling in the K/B x N serum transfer model of arthritis. J. Immunol. 169:6604–6609. [DOI] [PubMed] [Google Scholar]
- 28.Diaz de Stahl, T., M. Andren, P. Martinsson, J.S. Verbeek, and S. Kleinau. 2002. Expression of FcgammaRIII is required for development of collagen-induced arthritis. Eur. J. Immunol. 32:2915–2922. [DOI] [PubMed] [Google Scholar]
- 29.Ioan-Facsinay, A., S.J. de Kimpe, S.M. Hellwig, P.L. van Lent, F.M. Hofhuis, H.H. van Ojik, C. Sedlik, S.A. da Silveira, J. Gerber, Y.F. de Jong, et al. 2002. FcgammaRI (CD64) contributes substantially to severity of arthritis, hypersensitivity responses, and protection from bacterial infection. Immunity. 16:391–402. [DOI] [PubMed] [Google Scholar]
- 30.Ji, H., K. Ohmura, U. Mahmood, D.M. Lee, F.M. Hofhuis, S.A. Boackle, K. Takahashi, V.M. Holers, M. Walport, C. Gerard, et al. 2002. Arthritis critically dependent on innate immune system players. Immunity. 16:157–168. [DOI] [PubMed] [Google Scholar]
- 31.Kaplan, C.D., S.K. O'Neill, T. Koreny, M. Czipri, and A. Finnegan. 2002. Development of inflammation in proteoglycan-induced arthritis is dependent on Fc gamma R regulation of the cytokine/chemokine environment. J. Immunol. 169:5851–5859. [DOI] [PubMed] [Google Scholar]
- 32.van Lent, P.L., K. Nabbe, A.B. Blom, A.E. Holthuysen, A. Sloetjes, L.B. van de Putte, S. Verbeek, and W.B. van den Berg. 2001. Role of activatory Fc gamma RI and Fc gamma RIII and inhibitory Fc gamma RII in inflammation and cartilage destruction during experimental antigen-induced arthritis. Am. J. Pathol. 159:2309–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuasa, T., S. Kubo, T. Yoshino, A. Ujike, K. Matsumura, M. Ono, J.V. Ravetch, and T. Takai. 1999. Deletion of fcgamma receptor IIB renders H-2(b) mice susceptible to collagen-induced arthritis. J. Exp. Med. 189:187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu, X., C. Herrero, W.-P. Li, E. Falck-Pedersen, A.E. Koch, J.M. Woods, G.K. Haines, III, and L.B. Ivashkiv. 2002. Sensitization of IFN-γ Jak-STAT signaling during macrophage activation. Nat. Immunol. 3:859–866. [DOI] [PubMed] [Google Scholar]
- 35.Deon, D., S. Ahmed, K. Tai, N. Scaletta, C. Herrero, I.H. Lee, A. Krause, and L.B. Ivashkiv. 2001. Cross-talk between IL-1 and IL-6 signaling pathways in rheumatoid arthritis synovial fibroblasts. J. Immunol. 167:5395–5403. [DOI] [PubMed] [Google Scholar]
- 36.Pricop, L., J.E. Salmon, J.C. Edberg, and A.J. Beavis. 1997. Flow cytometric quantitation of attachment and phagocytosis in phenotypically-defined subpopulations of cells using PKH26-labeled Fc gamma R-specific probes. J. Immunol. Methods. 205:55–65. [DOI] [PubMed] [Google Scholar]
- 37.Zhong, Z., Z. Wen, and J.E. Darnell, Jr. 1994. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 264:95–98. [DOI] [PubMed] [Google Scholar]
- 38.Sengupta, T.K., A. Chen, Z. Zhong, J.E. Darnell, Jr., and L.B. Ivashkiv. 1995. Activation of monocyte effector genes and STAT family transcription factors by inflammatory synovial fluid is independent of interferon gamma. J. Exp. Med. 181:1015–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Finbloom, D.S., and K.D. Winestock. 1995. IL-10 induces the tyrosine phosphorylation of tyk2 and Jak1 and the differential assembly of STAT1 alpha and STAT3 complexes in human T cells and monocytes. J. Immunol. 155:1079–1090. [PubMed] [Google Scholar]
- 40.Weber-Nordt, R.M., J.K. Riley, A.C. Greenlund, K.W. Moore, J.E. Darnell, and R.D. Schreiber. 1996. Stat3 recruitment by two distinct ligand-induced, tyrosine-phosphorylated docking sites in the interleukin-10 receptor intracellular domain. J. Biol. Chem. 271:27954–27961. [DOI] [PubMed] [Google Scholar]
- 41.Sutterwala, F.S., G.J. Noel, P. Salgame, and D.M. Mosser. 1998. Reversal of proinflammatory responses by ligating the macrophage Fcgamma receptor type I. J. Exp. Med. 188:217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grazia Cappiello, M., F.S. Sutterwala, G. Trinchieri, D.M. Mosser, and X. Ma. 2001. Suppression of Il-12 transcription in macrophages following Fc gamma receptor ligation. J. Immunol. 166:4498–4506. [DOI] [PubMed] [Google Scholar]
- 43.Katsikis, P.D., C.Q. Chu, F.M. Brennan, R.N. Maini, and M. Feldmann. 1994. Immunoregulatory role of interleukin 10 in rheumatoid arthritis. J. Exp. Med. 179:1517–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feldmann, M., F.M. Brennan, and R.N. Maini. 1996. Role of cytokines in rheumatoid arthritis. Annu. Rev. Immunol. 14:397–440. [DOI] [PubMed] [Google Scholar]
- 45.Ding, Y., L. Qin, D. Zamarin, S.V. Kotenko, S. Pestka, K.W. Moore, and J.S. Bromberg. 2001. Differential IL-10R1 expression plays a critical role in IL-10-mediated immune regulation. J. Immunol. 167:6884–6892. [DOI] [PubMed] [Google Scholar]
- 46.Garlanda, C., E. Hirsch, S. Bozza, A. Salustri, M. De Acetis, R. Nota, A. Maccagno, F. Riva, B. Bottazzi, G. Peri, et al. 2002. Non-redundant role of the long pentraxin PTX3 in anti-fungal innate immune response. Nature. 420:182–186. [DOI] [PubMed] [Google Scholar]
- 47.Dias, A.A., A.R. Goodman, J.L. Dos Santos, R.N. Gomes, A. Altmeyer, P.T. Bozza, M.F. Horta, J. Vilcek, and L.F. Reis. 2001. TSG-14 transgenic mice have improved survival to endotoxemia and to CLP-induced sepsis. J. Leukoc. Biol. 69:928–936. [PubMed] [Google Scholar]
- 48.Ezekowitz, R.A. 2002. Local opsonization for apoptosis? Nat. Immunol. 3:510–512. [DOI] [PubMed] [Google Scholar]
- 49.Elbim, C., H. Reglier, M. Fay, C. Delarche, V. Andrieu, J. El Benna, and M.A. Gougerot-Pocidalo. 2001. Intracellular pool of IL-10 receptors in specific granules of human neutrophils: differential mobilization by proinflammatory mediators. J. Immunol. 166:5201–5207. [DOI] [PubMed] [Google Scholar]
- 50.Yasukawa, H., A. Sasaki, and A. Yoshimura. 2000. Negative regulation of cytokine signaling pathways. Annu. Rev. Immunol. 18:143–164. [DOI] [PubMed] [Google Scholar]
- 51.Ahmed, S.T., and L.B. Ivashkiv. 2000. Inhibition of IL-6 and IL-10 signaling and Stat activation by inflammatory and stress pathways. J. Immunol. 165:5227–5237. [DOI] [PubMed] [Google Scholar]
- 52.Sengupta, T.K., E.S. Talbot, P.A. Scherle, and L.B. Ivashkiv. 1998. Rapid inhibition of interleukin-6 signaling and Stat3 activation mediated by mitogen-activated protein kinases. Proc. Natl. Acad. Sci. USA. 95:11107–11112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee, I.H., W.P. Li, K.B. Hisert, and L.B. Ivashkiv. 1999. Inhibition of interleukin 2 signaling and signal transducer and activator of transcription (STAT)5 activation during T cell receptor-mediated feedback inhibition of T cell expansion. J. Exp. Med. 190:1263–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu, J., H. Huang, L. Guo, T. Stonehouse, C.J. Watson, J. Hu-Li, and W.E. Paul. 2000. Transient inhibition of interleukin 4 signaling by T cell receptor ligation. J. Exp. Med. 192:1125–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tian, Z., X. Shen, H. Feng, and B. Gao. 2000. IL-1beta attenuates IFN-alphabeta-induced antiviral activity and STAT1 activation in the liver: involvement of proteasome-dependent pathway. J. Immunol. 165:3959–3965. [DOI] [PubMed] [Google Scholar]
- 56.Mellor, H., and P.J. Parker. 1998. The extended protein kinase C superfamily. Biochem. J. 332:281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Melendez, A.J., M.M. Harnett, and J.M. Allen. 1999. Differentiation-dependent switch in protein kinase C isoenzyme activation by FcgammaRI, the human high-affinity receptor for immunoglobulin G. Immunology. 96:457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emoto, Y., Y. Manome, G. Meinhardt, H. Kisaki, S. Kharbanda, M. Robertson, T. Ghayur, W.W. Wong, R. Kamen, R. Weichselbaum, et al. 1995. Proteolytic activation of protein kinase C delta by an ICE-like protease in apoptotic cells. EMBO J. 14:6148–6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Melendez, A.J., D.J. Gillooly, M.M. Harnett, and J.M. Allen. 1998. Aggregation of the human high affinity immunoglobulin G receptor (FcgammaRI) activates both tyrosine kinase and G protein-coupled phosphoinositide 3-kinase isoforms. Proc. Natl. Acad. Sci. USA. 95:2169–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mecklenbrauker, I., K. Saijo, N.Y. Zheng, M. Leitges, and A. Tarakhovsky. 2002. Protein kinase Cdelta controls self-antigen-induced B-cell tolerance. Nature. 416:860–865. [DOI] [PubMed] [Google Scholar]
- 61.Leitges, M., C. Schmedt, R. Guinamard, J. Davoust, S. Schaal, S. Stabel, and A. Tarakhovsky. 1996. Immunodeficiency in protein kinase cbeta-deficient mice. Science. 273:788–791. [DOI] [PubMed] [Google Scholar]
- 62.Ivashkiv, L.B. 1996. Cytokine expression and cell activation in inflammatory arthritis. Adv. Immunol. 63:337–376. [DOI] [PubMed] [Google Scholar]
- 63.Marsh, M., and H.T. McMahon. 1999. The structural era of endocytosis. Science. 285:215–220. [DOI] [PubMed] [Google Scholar]
- 64.Guinamard, R., N. Signoret, I. Masamichi, M. Marsh, T. Kurosaki, and J.V. Ravetch. 1999. B cell antigen receptor engagement inhibits stromal cell-derived factor (SDF)-1alpha chemotaxis and promotes protein kinase C (PKC)-induced internalization of CXCR4. J. Exp. Med. 189:1461–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kovanen, P.E., I. Junttila, K. Takaluoma, P. Saharinen, L. Valmu, W. Li, and O. Silvennoinen. 2000. Regulation of Jak2 tyrosine kinase by protein kinase C during macrophage differentiation of IL-3-dependent myeloid progenitor cells. Blood. 95:1626–1632. [PubMed] [Google Scholar]
- 66.Cameron, A.J., M.M. Harnett, and J.M. Allen. 2001. Differential recruitment of accessory molecules by FcgammaRI during monocyte differentiation. Eur. J. Immunol. 31:2718–2725. [DOI] [PubMed] [Google Scholar]
- 67.St Clair, E.W. 2000. Interleukin-10: therapeutic prospects in rheumatoid arthritis. Curr. Dir. Autoimmun. 2:126–149. [DOI] [PubMed] [Google Scholar]