

Abstract
Lipopolysaccharide (LPS), a product of Gram-negative bacteria, is potent mediator of tumor necrosis factor (TNF)α production by myeloid/macrophage cells. Inhibitors capable of blocking the signaling events that result in TNFα production could provide useful therapeutics for treating septic shock and other inflammatory diseases. Broad spectrum tyrosine inhibitors are known to inhibit TNFα production, however, no particular family of tyrosine kinases has been shown to be essential for this process. Here we show that the Bruton's tyrosine kinase (Btk)-deficient mononuclear cells from X-linked agammaglobulinemia patients have impaired LPS-induced TNFα production and that LPS rapidly induces Btk kinase activity in normal monocytes. In addition, adenoviral overexpression of Btk in normal human monocytes enhanced TNFα production. We examined the role of Btk in TNFα production using luciferase reporter adenoviral constructs and have established that overexpression of Btk results in the stabilization of TNFα mRNA via the 3′ untranslated region. Stimulation with LPS also induced the activation of related tyrosine kinase, Tec, suggesting that the Tec family kinases are important components for LPS-induced TNFα production. This study provides the first clear evidence that tyrosine kinases of the Tec family, in particular Btk, are key elements of LPS-induced TNFα production and consequently may provide valuable therapeutic targets for intervention in inflammatory conditions.
Keywords: tyrosine kinase, adenovirus, TNFα, macrophage, X-linked agammaglobulinemia
Introduction
TNFα is a proinflammatory cytokine pivotal to the pathogenesis of chronic inflammatory diseases such as rheumatoid arthritis and Crohn's disease (1), and in inflammatory responses such as leukocyte migration, tissue resorption, the acute-phase response, and fever (2). The major producers of TNFα are cells of the mononuclear phagocyte lineage including macrophages, microglia, osteoclasts, and myeloid dendritic cells. Despite considerable efforts, relatively few signaling molecules, e.g., nuclear factor (NF)*-κB (3, 4) and p38 MAPK/MK2 (5, 6), have been shown to be essential for TNFα expression in macrophages. It is well documented that tyrosine kinase inhibitors are potent suppressors of LPS-induced TNFα production (7, 8) and that the activation of tyrosine kinase activity is one of the earliest detectable events after LPS stimulation of monocytic cells (9–11). However, defining a causal relationship between a particular tyrosine kinase and TNFα production has been difficult. LPS can activate the Src family kinases p58hck, p53lyn, and p59fgr, as well as p72syk (12–14), yet macrophages from mice deficient in these kinases do not show any impairment of LPS-induced TNFα production (15, 16). LPS has also been shown to activate the tyrosine kinase Pyk-2 (17), but its role, if any, in TNFα production has yet to be elucidated.
Another family of nonreceptor tyrosine kinases expressed by monocytic cells is the Tec family, and in particular, Bruton's tyrosine kinase (Btk). Recent studies have shown that the Tec family kinases are important components of both antigen receptor signaling and other cell surface receptors, resulting in the activation of numerous signal transduction pathways, regulation of the actin cytoskeleton, adhesion, migration, and transcriptional activation (18). Btk is found in all cells of the hematopoietic lineage except plasma and T cells (19), and is required for normal B cell development and signal transduction through cell surface molecules (20). Mutations in the btk gene result in aberrant B cell development, leading to the X-linked agammaglobulinemia (XLA) phenotype in humans and to the less severe X-linked immunodeficiency (xid) in mice. xid B cells show some hyporesponsiveness to LPS stimulation, although the precise cause has not been established (21, 22). Because the major phenotype of Btk deficiency is impaired B cell development and function, this cell type has been the major focus of interest to date. Earlier studies performed on xid mononuclear cells showed no obvious impairment of TNFα production in response to a series of inflammatory stimuli such as LPS and formalin-killed Staphylococcus aureus (23). However, bone marrow–derived cultured mast cells from xid mice have been shown to produce less TNFα in passive cutaneous anaphylactic reactions (24) and there is a recent report that xid peritoneal macrophages express reduced levels of TNFα and IL-1β in response to LPS (25). Additionally, xid mice are resistant to models of autoimmune diseases, e.g., collagen-induced arthritis (26, 27).
In humans, XLA is characterized by the absence of mature B cells in the periphery with a marked reduction in serum levels of all Ig isotypes resulting in a susceptibility to recurrent and severe bacterial infections (20, 28). Yet despite lacking Btk, XLA monocytes appear to develop normally and are present in expected numbers in the circulation. The importance of Btk in human monocyte/macrophages remains to be fully elucidated. As the XLA phenotype differs from that of the xid mice, we have examined the responses of XLA monocytes and macrophages to LPS stimulation. In addition, we have used an adenovirus expressing Btk in conjunction with TNFα luciferase reporter adenoviruses to analyze the contribution of Btk-dependent signaling events to the regulation of TNFα production.
Materials and Methods
Isolation and Culture of Cells
Isolation and Culture of PBMCs from XLA and Control Donors.
Human blood samples were collected into lithium heparin vacutainers. Each blood sample was mixed with an equal volume of HBSS. PBMCs were prepared by ficoll-hypaque centrifugation on a lymphoprep gradient. PBMCs were cultured in RPMI containing 100 units/ml penicillin/streptomycin and 10% heat-inactivated FCS at 37°C in a humidified atmosphere containing 5% CO2. Monocytes were isolated from the PBMCs by adherence to plastic for 1 h at 37°C in RPMI containing 100 units/ml penicillin/streptomycin and 10% FCS. Nonadherent cells were then washed off and the adherent monocytes were rested overnight and stimulated with LPS. B cells were depleted from PBMC using Dynabeads® coated with anti-CD19 antibodies. Cells were incubated with the beads for 1 h with constant agitation at 4°C. The beads were then magnetically removed and the remaining cells were subjected to FACS® analysis with FITC-conjugated anti-CD19 resulting in >90% B cell depletion.
Isolation of Monocytes by Elutriation.
PBMCs were prepared from buffy coat fractions of a unit of blood from a single donor using ficoll-hypaque. The monocytes were then isolated by centrifugal elutriation as previously described (4). Monocyte fractions of >85% purity were routinely collected in this manner. Monocytes were cultured in RPMI containing 100 units/ml penicillin/streptomycin and 10% heat-inactivated FCS at 37°C in a humidified atmosphere containing 5% CO2. For adenoviral infection, monocytes were treated with 100 ng/ml M-CSF (provided by G. Larsen, Genetics Institute, Boston, MA) for 72 h before viral infection.
Generation of Adenoviral Vectors and Cell Infection.
Recombinant, replication-deficient adenoviral constructs encoding wild-type human Btk (AdBtk; cDNA provided by C. Kinnon, Institute of Child Health, London, United Kingdom) were prepared using the AdEasy system as previously described (29). In short, recombinant viral DNA was transfected into 293 cells in 6-well tissue culture plates using lipofectamine (GIBCO BRL) according to the manufacturer's instructions. Cells were overlaid with 2% agarose/DMEM 24 h after transfection and viral plaques were picked after 9–10 d. Viral clones were propagated in 293 cells and screened for transgene expression by Western blotting. Clonal viruses were then prepared from 20 175 cm3 tissue culture flasks of 293 cells by ultracentrifugation through two caesium chloride gradients. Titres of viral stocks were determined by plaque assay and viral aliquots were stored at −70°C. The AdGFP adenovirus was prepared according to this protocol using AdTrackGFP with no insert. AdTrack0 (Ad0), pAdTrack-TNF 5′ promoter-Luc-3′ untranslated region (UTR; Ad5′3′luc), and pAdTrack-TNF 5′ promoter-Luc (Ad5′luc) were generated as previously described (30). The NF-κB luciferase adenovirus (AdNF-κB-luc) contains four tandem copies of the κ enhancer element located upstream of the firefly luciferase gene (31). This adenovirus was provided by P.B. McCray Jr. (University of Iowa, Iowa City, IA) and is a modification of the pNF-κB reporter vector (BD Clontech). Recombinant adenoviral vectors encoding Escherichia coli β-galactosidase (Adβgal) were provided by M. Woods and A. Bryne (Oxford University, Oxford, United Kingdom).
M-CSF–derived monocytes were plated in 96-well plates at 1.5 × 105 cells/well and allowed to settle for at least 4 h. The cells were washed in serum-free RPMI medium and then exposed to virus at different multiplicity of infection (m.o.i.) for 2 h in serum-free RPMI at 37°C, after which cells were washed in RPMI and cultured in complete medium for 24 h before stimulation with LPS as previously described (4).
Immunoprecipitation and In Vitro Kinase Assay.
Btk autokinase activity was measured in response to LPS in primary human monocytes. Cells were stimulated with LPS for various times as detailed. Cells were pelleted by centrifugation and lysed for 20 min on ice with 1% NP-40 lysis buffer containing 20 mM Tris, pH 8, 130 mM NaCl, 10 mM NaF, 1 mM DTT, 20 μM leupeptin, 100 μM sodium orthovanadate, 1 mM PMSF, 10 μM E64, and 2 mg/ml aprotinin. Lysates were microfuged for 10 min at 13,000 rpm at 4°C and supernatants were removed and precleared for 30 min at 4°C in 20 μl protein A–sepharose (previously washed in lysis buffer). After centrifuging the samples for 10 min at 1,300 rpm, the supernatants were removed and incubated with 2 μl rabbit polyclonal anti–Btk (provided C. Kinnon, Institute of Child Health, London, United Kingdom) for 1 h at 4°C. 30 μl protein A–sepharose was then added to each sample and incubated for an additional 1.5 h at 4°C. The beads were then washed three times in lysis buffer and once in kinase buffer (10 mM MgCl2; 10 mM MnCl2). Beads were mixed with kinase buffer containing γ32P[ATP] and incubated for 15 min at room temperature. The reaction was stopped by the addition of 4× Laemmli buffer and the samples were boiled for 10 min. Samples were electrophoresed on an 8% SDS polyacrylamide gel. The gel was stained, fixed, dried, and autoradiographed. Tec autokinase activity was measured as previously described (32). Tec protein was immunoprecipitated, either with rabbit anti–Tec serum (32) or goat polyclonal anti–Tec (Santa Cruz Biotechnology, Inc.).
Western Blot Analysis.
Cells were lysed in lysis buffer and debris pelleted as described above. Lysate protein concentration was assessed by Bradford assay and equivalent amounts of lysate protein were electrophoresed on 8% SDS polyacrylamide gels, followed by electrotransfer of proteins onto nitrocellulose membranes. Membranes were blocked in 10% Marvel/PBS/Tween (0.05%) and probed with either rabbit polyclonal anti–Btk (BD Biosciences) at 1 μg/ml in 5% Marvel/PBS/Tween (0.05%), anti–inhibitory protein κBα (IκBα; Santa Cruz Biotechnology, Inc.), anti-p54/JNK (Santa Cruz Biotechnology, Inc.), or rabbit anti–Tec for 2 h. After washing, membranes were incubated with anti–rabbit horseradish peroxidase at 1:5,000 in 5% Marvel/PBS/Tween (0.05%). After washing in PBS/Tween (0.05%), the membranes were developed using enhanced chemiluminescence according to the manufacturer's instructions (Amersham Biosciences).
ELISA.
Supernatants were harvested 18 h after stimulation of human macrophages or PBMCs. The concentration of TNFα (BD Biosciences) was determined by ELISA according to the manufacturer's instruction. Absorbance was read and analyzed at 450 nM on a spectrophotometric ELISA plate reader (Labsystems Multiskan Biochromic) using the Ascent 2.4.2 software program. Results are expressed as the mean concentration of triplicate cultures ±SD.
Luciferase Assays.
After LPS stimulation, cells were washed once in PBS and lysed with 100 μl CAT lysis buffer (0.65% [vol/vol] of NP-40, 10 mM Tris-HCl, pH 8, 0.1 mM EDTA, pH 8, 150 mM NaCl). 50 μl cell lysate were transferred into the well of a luminometer cuvette strip containing 120 μl luciferase assay buffer. Luciferase activity was measured with a Labsystem Luminometer by dispensing 30 μl luciferin (Bright-Glo luciferase assay system; Promega) per assay point. Cell lysates were assayed for protein concentration by Bradford assay and luciferase activity was adjusted accordingly.
Taqman RT-PCR.
M-CSF–treated monocytes were plated at 5 × 105 cells/well in 24-well plates and infected as described above. Total RNA was extracted using RNeasy Kit (QIAGEN) according to the manufacturer's instructions. All semiquantitative RT-PCR was performed using an ABI PRISM 7700 Sequence Detection System, Taqman One Step RT-PCR reagent, and TNFα, IL-6, and GAPDH predeveloped assay reagents (PerkinElmer). ABI PRISM 7700 Sequence detector was programmed for the RT step of 30 min at 48°C followed by a 5-min deactivation step at 95°C. Subsequent PCR amplification consisted of 40 cycles of denaturation at 94°C for 15 s and annealing/extension at 60°C for 60 s. The cycle number at which the amplification plot crosses a fixed threshold above baseline is defined as threshold cycle (Ct). To control variation in mRNA concentration, all results were normalized to the housekeeping gene, GAPDH. Relative quantitation was performed using the comparative ΔΔCt method according to the manufacturer's instructions.
Results
Btk Deficiency Results in Impaired TNFα Production.
PBMCs from XLA blood were used to see if Btk deficiency had any effect on LPS-induced TNFα production. When compared with PBMC from normal age and sex-matched controls, the LPS response of XLA cells was impaired. At the lowest dose of LPS (0.1 ng/ml), XLA and normal PBMCs produced similar levels of TNFα, but increasing concentrations of LPS failed to generate the increased response seen in the normal cells. At 10 ng/ml, LPS-induced TNFα production by XLA PBMCs was only 15% of the control cells, whereas at 100 ng/ml LPS the response was only 10% of control (Fig. 1 A). Although B cells are not known to produce TNFα in response to LPS, the involvement of B cells from the normal PBMCs in LPS-induced TNFα production was investigated to negate any secondary effects due to the absence of B cells in XLA PBMCs. Depleting B cells from normal PBMC showed no effect on LPS-induced TNFα production (Fig. 1 B). Furthermore, we found that the T cells, the major component of PBMCs, did not produce TNFα in response to LPS (unpublished data) and these cells do not express Btk (33). Although the amount of blood available made purifying the monocytes from XLA blood difficult, we managed to separate the monocytic cells using adherence. LPS-induced TNFα production was again significantly reduced, although at 50% of normal controls, the impairment was not as great as seen in PBMCs (Fig. 1 C).
Figure 1.
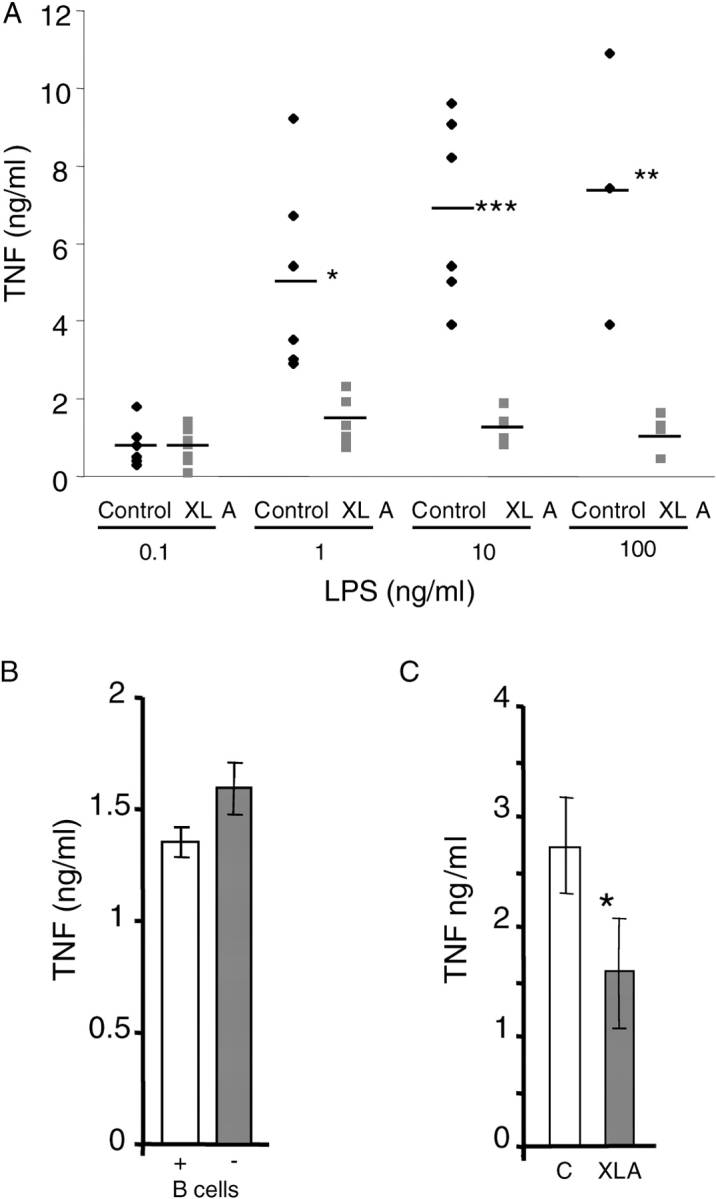
LPS-induced TNFα expression in PBMCs/monocytes from XLA and normal donors. (A) PBMCs from XLA and normal male donors (age range 17–38 yr). (B) Total PBMCs and PBMCs depleted of B cells from normal donors. (C) Monocytes prepared by adherence from normal and XLA PBMCs. Cells were stimulated with 10 ng/ml LPS (unless otherwise stated) for 18 h and supernatants were assayed for TNFα production by ELISA. In A, each point represents a donor, B is representative of three separate experiments, and C shows the combined results of five donors of each type (error bars ± SD). Results from Student's t test P values: *, P < 0.05; **, P < 0.01; ***, P < 0.001. TNFα production from unactivated cells was undetectable.
Stimulation with LPS Induces Btk Kinase Activity In Vitro.
To further establish a role for Btk in LPS-induced TNFα production, we attempted to determine whether LPS was able to regulate Btk activity. Autokinase assays of enzyme immunoprecipitated from human primary monocytes demonstrated that LPS could rapidly activate Btk (Fig. 2 A). Btk autokinase activity was also observed in the murine macrophage cell line RAW 264.7 after LPS stimulation (unpublished data). Additionally, the effect of overexpression of Btk was examined by infecting 48-h M-CSF–treated monocytes (4) with increasing amounts of adenovirus-encoding human Btk (AdBtk). Infection with AdBtk alone did not induce TNFα production (unpublished data). However, infection with increasing amounts of virus synergized with LPS to increase TNFα expression, resulting in a two- to threefold increase in TNFα at the highest m.o.i. of 100 (Fig. 2 B). This effect correlated with the increased amounts of Btk protein seen in the infected cells (Fig. 2 C). No effect on TNFα production was seen with control viruses encoding either β-galactosidase or Pyk2, another LPS-activated tyrosine kinase (17), at the highest m.o.i. of 100 (Fig. 2 B).
Figure 2.

Btk is activated by LPS stimulation and modulates TNFα production. (A) Primary monocytes were stimulated with LPS for the indicated intervals. Cell lysates were immunoprecipitated and immune complex kinase assays were performed. Autophosphorylated Btk proteins were detected by autoradiography. This result is representative of three separate experiments. (B) Monocytes were infected with adenoviruses encoding either wild-type Btk (Btk WT), β-galactosidase (β-Gal), or an unrelated tyrosine kinase, Pyk2. Cells were stimulated with LPS for 18 h and the supernatants were assayed for TNFα expression. Data is presented as fold activation compared with uninfected LPS stimulated control cells (±SD). *, P < 0.05; **, P < 0.01 compared with LPS-activated uninfected controls. (C) Cell lysates from the infected cells were examined for Btk expression by immuno-Western blotting. Data is representative of at least four different donors.
LPS-induced IκBα Degradation Is Intact in XLA PBMCs.
Overall, the data strongly suggested that there was a role for Btk in LPS-induced TNFα production. However, there was the possibility that this was the result of some development defect, rather than a direct effect of an absent kinase. As others have reported (34), FACS® analysis showed no impairment of CD14 expression, a component of the LPS binding on XLA monocytes (unpublished data). We also used the LPS-induced degradation of IκBα as a measure of signaling function, as this event does not appear to require tyrosine kinase activity (35, 36) and therefore should be intact. As shown in Fig. 3 A, there was a similar degradation of IκBα in XLA and normal monocytes in response to LPS, indicating no obvious impairment of the NF-κB activation pathway. We further investigated the involvement of the NF-κB signaling pathway by coinfecting M-CSF–treated monocytes with AdBtk and an NF-κB reporter virus that encodes luciferase under the control of a series of NF-κB binding sites (31). M-CSF monocytes were simultaneously infected with these viruses and controls, and the luciferase activity and TNFα production were measured after 18 h of LPS stimulation. Although we still observed an increase in TNFα production in these cultures (Fig. 3 C), there was no corresponding increase in luciferase activity (Fig. 3 B), indicating that Btk was able to promote TNFα production independently of the NF-κB activation.
Figure 3.


LPS-induced IκBα degradation in XLA and normal PBMCs. (A) PBMCs stimulated with 10 ng/ml LPS for 20 min were lysed and sequentially immunoblotted for the indicated proteins. This result is representative of three different experiments. (B and C) M-CSF monocytes were simultaneously infected with NF-κB luciferase reporter construct (m.o.i. of 100) alone, or coinfected with either AdBtk or Ad0 at various m.o.i. 24 h after adenoviral infection, cells were treated with LPS for 18 h before assay of (B) luciferase activity and (C) TNFα production. Data are means of triplicate cultures ± SD and are expressed as a percentage of the control level. This graph is representative of five experiments performed using different donors.
Overexpression of Btk Acts via the 3′ UTR of TNF mRNA.
To establish the mechanism of Btk action on TNFα production, the activity of the human TNF promoter in stimulated human macrophages was studied using 5′ promoter-luciferase and 5′ promoter-luciferase-3′ UTR adenoviral TNF gene reporter constructs (30). Primary human macrophages were simultaneously infected with reporter viruses at an m.o.i. of 40, whereas the Ad0 or AdBtk were used at an m.o.i. of 100. 24 h after adenoviral infection, macrophages were treated with LPS or left unstimulated for 18 h before assay of luciferase activity and ELISA for TNFα production. There was a threefold increase in luciferase activity in the cells containing the TNF5′ 3′ UTR reporter construct compared with the Ad0 control cells (Fig. 4 A). However, there was no enhancement of luciferase activity from the TNF 5′ promoter construct (Fig. 4 C). Overexpression of Btk enhanced LPS-stimulated TNFα production by threefold in the presence of either TNF reporter construct (Fig. 4, B and D). There was no significant difference in TNF production and luciferase activity when comparing Ad0-infected macrophages with controls infected with the reporter constructs alone. The lack of enhanced luciferase activity from the 5′ promoter construct after LPS stimulation indicated that the 3′ UTR of TNFα mRNA was required for the action of Btk.
Figure 4.

Btk overexpression enhances TNFα via the 3′ UTR. (A) Schematic representation of the human TNF 5′ promoter-luciferase-3′ UTR and TNF 5′ promoter-luciferase adenoviral constructs. Activity of the human TNF 5′ 3′ UTR (B and C) or TNF 5′ promoter (D and E) in stimulated human macrophages was tested by simultaneously infecting primary human macrophages with reporter virus at an m.o.i. of 40 and either Ad0 or AdBtk at various m.o.i. 24 h after adenoviral infection, macrophages were treated with LPS for 18 h before assay of luciferase activity (B and D) and TNFα production (C and E). Data are means of triplicate cultures ± SD and are expressed as a percentage of the control level. Each graph is representative of five experiments performed using different donors.
Overexpression of Btk Leads to Stabilization of TNFα mRNA.
The importance of the 3′ UTR in the stability of TNFα mRNA has been studied extensively (37, 38). Because Btk appeared to be acting via the 3′ UTR, it seemed likely that it was also affecting the stability of TNFα mRNA. To further analyze the mechanism by which Btk determines the levels of TNFα, we blocked transcription in LPS-stimulated M-CSF monocytes by using actinomycin D and analyzed half-life of TNFα mRNA by Taqman RT-PCR analysis. As a control, GAPDH mRNA was detected and used to normalize the TNFα mRNA levels. For each of the controls, uninfected Ad0 and PykM, the half-life of TNFα mRNA was <45 min after actinomycin D treatment. However, in the presence of Btk, TNFα mRNA levels were maintained at 70–100% of the time 0 control for the duration of the 120-min time course (Fig. 5 A). These data demonstrate the ability of Btk to stabilize TNFα mRNA and consequently this may result in the elevated protein expression observed in these cultures.
Figure 5.


Btk overexpression stabilizes TNFα mRNA. (A) Cells were either uninfected or infected with Ad0, Ad PykM, or AdBtk (m.o.i. of 100) and activated with LPS for 4 h. 5 μg/ml actinomycin D was added to stop any further mRNA synthesis and the cells were incubated for an additional 0, 15, 30, 60, 90, or 120 min, after which time they were harvested in RNA lysis buffer and the supernatants were reserved for TNFα ELISA. Total mRNA was prepared and Taqman RT-PCR was used to access the quantity of TNFα. The results were normalized to 100% at the 0-min time point. The lines are representative of Control (♦), Ad0 (▪), Btk (▴), and PykM (x). Data are means of triplicate reactions ± SD expressed as a percentage of the control and are representative of three experiments performed using different donors. (B) PBMCs from XLA and normal male donors or (C) M-CSF monocytes infected with AdBtk or AdPyk2, were stimulated with LPS for 20 min, lysed, and sequentially immunoblotted for the indicated proteins. These results are representative of three different experiments.
As the stability of TNF mRNA is known to be associated with p38 mitogen-activated protein kinase (MAPK) activation, we examined the ability of PBMCs from XLA patients to phosphorylate p38 MAPK in response to LPS treatment. We were able to observe consistently low levels of p38 MAPK phosphorylation in XLA PBMCs (Fig. 5 B), however there was considerable variability in the degree of p38 MAPK phosphorylation from person to person in the normal controls. There was no difference in p38 MAPK phosphorylation after LPS treatment of normal PBMCs in either the presence or absence of B cells (unpublished data). Due to the limitation of patient samples we were unable to examine the kinetics of p38 MAPK activation in XLA patients compared with normal controls. To further establish the link between p38 MAPK and Btk, we used adenoviral overexpression of Btk to determine if there was any increase in p38 MAPK activity in normal M-CSF–treated monocytes. As shown in Fig. 5 C, we were able to observe a 1.5–2-fold increase in phosphorylated p38 MAPK.
Tec Is Phosphorylated in Response to LPS.
Although we have demonstrated that Btk is able to modulate TNFα expression, XLA patients do not show a serious impairment in their innate immune function. Treatment of XLA monocytes for 2 d with M-CSF greatly enhanced their production of TNFα in response to LPS to levels similar to those obtained with normal monocytes (Fig. 6 A). Consequently, we examined the mechanism of the increased responsiveness of monocytes after M-CSF treatment. Because members of the Tec family of kinases can functionally complement each other (39), we tested whether M-CSF can regulate the expression of an alternative Tec family kinase to Btk. An obvious candidate was Tec kinase, as this enzyme is expressed in myeloid cells and does, to some degree, complement Btk activity in xid B cells (40). Western immunoblot analysis showed that Tec kinase was expressed at low levels in untreated monocytes from both normal donors and XLA patients (Fig. 6 B), however, after treatment with M-CSF for 48 h the expression of Tec protein was increased. Counter blots confirmed that the expression of Btk was restricted to the normal cells (Fig. 6 B). Next, the response of Tec kinase to LPS in M-CSF–treated monocytes was investigated using autokinase assays of immunoprecipitated enzyme. Like Btk, Tec kinase activity was increased in response to LPS treatment of human M-CSF–treated monocytes (Fig. 6 C).
Figure 6.


The effect of M-CSF treatment on LPS-stimulated TNFα production and Tec expression. (A) Adherent monocytes from XLA and normal donors were either stimulated with LPS for 2 h, or incubated with M-CSF for 48 h before LPS stimulation for 2 h. Supernatants were assayed for TNFα production. (B) Adherent monocytes from normal (lanes 1 and 2) and XLA donors (lanes 3 and 4) were either lysed immediately, or after M-CSF treatment for 48 h. Western blot analysis was performed using either α-TecSH3, α-Btk, or α-actin antibody. (C) Elutriated monocytes from normal donors were stimulated with LPS for the indicated time periods. Tec was immunoprecipitated and in vitro autokinase assay was performed. Each study is representative of at least three separate experiments.
Discussion
Our data has shown that stimulation of human monocytes with LPS triggers the activation of the nonreceptor tyrosine kinase, Btk. Moreover, in XLA monocytes that lack functional Btk, there is a failure to respond to LPS treatment as determined by their reduced TNFα production after stimulation. Our initial findings led us to further examine the contribution of Btk in controlling TNFα production. Adenoviral gene transfer has been used extensively to investigate the role of different transgenes in primary cells due to the ability of adenoviruses to infect both quiescent and dividing cells at high efficiency (4). Overexpression of Btk resulted in a consistent two- to threefold increase in TNFα production from M-CSF monocytes. In B cells, activation of the NF-κB transcription complex by signals derived from the B cell antigen receptor act via Btk to control B cell development, survival, and antigenic responses. However, the actions of Btk in macrophages appear to be independent of NF-κB activation and there is no defect in IκBα degradation in response to LPS in XLA monocytes.
TNF biosynthesis is under the control of multiple and complex regulatory mechanisms. Because the Tec family is known to interact with a wide range of signal transduction molecules (18), we took the alternate approach of examining the effect of Btk on the UTRs of TNFα. Using the 5′ promoter-luciferase and 5′ promoter-luciferase-3′ UTR adenoviral reporter constructs, we were able to show that the ability of Btk to enhance TNFα production required the 3′ UTR. There is considerable evidence indicating that some of these may occur at the translational level and be mediated by AU-rich elements (AREs) in the 3′ UTR of the mRNA. Certain inflammatory gene mRNAs, including cyclooxygenase-2, IL-6, IL-8, and TNFα mRNAs, are stabilized by activation of the p38 MAPK pathway by stimuli such as IL-1 and LPS (37, 41–43). Studies with mRNA reporter constructs have shown that the p38 MAPK-mediated stabilization directly involves AREs (38). Thus, AREs confer instability on mRNAs. However, after activation of the p38 MAPK pathway, they allow mRNA stabilization and hence increased protein expression. There is also a strong association between Btk and p38 MAPK in B cells showing that the activation of p38 MAPK was completely inhibited in cells deficient in Lyn and Btk, and the introduction of wild-type Btk, but not kinase-inactive Btk, restored the p38 MAPK activation in response to 280 nm UV irradiation in chicken DT40 B cells (44). The precise regions of the TNF 3′ UTR involved in this interaction with Btk and the potential involvement of p38 MAPK and other intermediary molecules is a field of ongoing research in our laboratory.
The proposition that Btk is essential for LPS-induced TNFα production is not easily reconciled with the XLA phenotype that shows no obvious gross defect of the innate immune system. However, there have been limited reports that XLA patients have reduced inflammatory responses (45). This is in contrast to patients with common variable immunodeficiency who are prone to multiorgan granulomatous disease, although they too have a type of antibody deficiency similar to the XLA patients (28, 46). In humans, Btk is absolutely required for the progression of developing B cells through the pro-B to pre-B stage. However, in xid mice the absence of Btk alone does not result in the same phenotype and it is only when Tec is also ablated that the same B cell deficiency is observed (40). It would be of interest to investigate LPS responsiveness in monocyte/macrophages from these mice. This suggests that Tec might be able to substitute for the absence of Btk in mouse pro-B cells, but at this stage of development in man, Tec is either unavailable or is incapable of substituting for Btk (47). It is of interest to note that monocyte differentiation toward macrophages ablates the differences observed between the XLA patients and normal donors (Fig. 6 A) and that this is accompanied by an increase in Tec levels (Fig. 6 B). The observation that Tec kinase is expressed in resting monocytes from XLA and normal individuals, although at a lower level, may explain why there is some response to LPS in the Btk-deficient cells. The data obtained here would suggest that without an increase in Tec kinase expression induced by M-CSF, there is insufficient kinase activity in the resting XLA monocytes to support a full LPS response. This hypothesis is supported by the observation that at the lowest LPS concentration used in this study (0.1 ng/ml), TNFα production by XLA and normal cells was similar (Fig. 1 A). The majority of the studies in mice (23, 24), with the exception of the study by Mukhopadhyay et al. (25), have been unable to find any differences in LPS responsiveness between xid and normal macrophages. We also examined the responsiveness of peritoneal macrophages from xid mice versus normal mice and were unable to observe any differences in TNFα production and NF-κB DNA binding activity in response to LPS (unpublished data). Taken together, these data suggest that rather than a difference between murine and human cells, that it is in fact a difference between monocytes and macrophages. It is therefore possible that the up-regulation of Tec, or other Tec kinase family members, in human macrophages may explain why there is no major impairment of innate immunity in XLA patients although further work is required to confirm this hypothesis.
In summary, this study has demonstrated that Btk is not only another tyrosine kinase activated by LPS, but also that deficiency in its expression is associated with impairment of TNFα expression. Furthermore, overexpression of Btk in macrophages synergizes with LPS to induce TNFα production by stabilizing TNFα mRNA. Although these data imply that Btk is the first described tyrosine kinase having a key role in LPS-induced production of TNFα by monocytes, this role is probably not restricted to Btk alone. The intriguing question remains as to what role Tec kinases may have in mediating TNFα production by myeloid cells to other stimuli, particularly those involved in inflammatory conditions like rheumatoid arthritis and Crohn's disease, where TNFα is a validated therapeutic target.
Acknowledgments
The authors would like to thank Dr. V. Tybulewicz and Ms. L. Vanes (National Institute for Medical Research, Millhill, United Kingdom) for providing access to the xid mice. Many thanks to Prof. C. Kinnon and Dr. M. Tomlinson for providing access to the Btk and Tec family antibodies used in this study. The authors would also like to thank Dr. A. Clark for constructive criticism of the manuscript.
N.J. Horwood is a Howard Florey Research Fellow. T. Mahon is a Training and Mobility of Researchers Marie Curie Fellow. These studies were supported by a block grant from the Arthritis Research Campaign, UK.
N.J. Horwood, T. Mahon, and J.P. McDaid contributed equally to this work.
Footnotes
Abbreviations used in this paper: AdBtk, adenovirus-encoding human Bruton's tyrosine kinase; ARE, AU-rich element; Btk, Bruton's tyrosine kinase; IκBα, inhibitory protein κBα; MAPK, mitogen-activated protein kinase; m.o.i., multiplicity of infection; NF, nuclear factor; UTR, untranslated region; xid, X-linked immunodeficiency; XLA, X-linked agammaglobulinemia.
References
- 1.Feldmann, M., F.M. Brennan, and R.N. Maini. 1996. Rheumatoid arthritis. Cell. 85:307–310. [DOI] [PubMed] [Google Scholar]
- 2.Beutler, B., and A. Cerami. 1988. Tumor necrosis, cachexia, shock, and inflammation: a common mediator. Annu. Rev. Biochem. 57:505–518. [DOI] [PubMed] [Google Scholar]
- 3.Bondeson, J., B. Foxwell, F. Brennan, and M. Feldmann. 1999. Defining therapeutic targets by using adenovirus: blocking NF-kappaB inhibits both inflammatory and destructive mechanisms in rheumatoid synovium but spares anti-inflammatory mediators. Proc. Natl. Acad. Sci. USA. 96:5668–5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foxwell, B., K. Browne, J. Bondeson, C. Clarke, R. de Martin, F. Brennan, and M. Feldmann. 1998. Efficient adenoviral infection with IkappaB alpha reveals that macrophage tumor necrosis factor alpha production in rheumatoid arthritis is NF-kappaB dependent. Proc. Natl. Acad. Sci. USA. 95:8211–8215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee, J.C., J.T. Laydon, P.C. McDonnell, T.F. Gallagher, S. Kumar, D. Green, D. McNulty, M.J. Blumenthal, J.R. Heys, S.W. Landvatter, et al. 1994. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 372:739–746. [DOI] [PubMed] [Google Scholar]
- 6.Kotlyarov, A., A. Neininger, C. Schubert, R. Eckert, C. Birchmeier, H.D. Volk, and M. Gaestel. 1999. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat. Cell Biol. 1:94–97. [DOI] [PubMed] [Google Scholar]
- 7.Novogrodsky, A., A. Vanichkin, M. Patya, A. Gazit, N. Osherov, and A. Levitzki. 1994. Prevention of lipopolysaccharide-induced lethal toxicity by tyrosine kinase inhibitors. Science. 264:1319–1322. [DOI] [PubMed] [Google Scholar]
- 8.Geng, Y., B. Zhang, and M. Lotz. 1993. Protein tyrosine kinase activation is required for lipopolysaccharide induction of cytokines in human blood monocytes. J. Immunol. 151:6692–6700. [PubMed] [Google Scholar]
- 9.Han, J., J.D. Lee, P.S. Tobias, and R.J. Ulevitch. 1993. Endotoxin induces rapid protein tyrosine phosphorylation in 70Z/3 cells expressing CD14. J. Biol. Chem. 268:25009–25014. [PubMed] [Google Scholar]
- 10.Weinstein, S.L., J.S. Sanghera, K. Lemke, A.L. DeFranco, and S.L. Pelech. 1992. Bacterial lipopolysaccharide induces tyrosine phosphorylation and activation of mitogen-activated protein kinases in macrophages. J. Biol. Chem. 267:14955–14962. [PubMed] [Google Scholar]
- 11.Knapp, K.M., and B.K. English. 2000. Ceramide-mediated stimulation of inducible nitric oxide synthase (iNOS) and tumor necrosis factor (TNF) accumulation in murine macrophages requires tyrosine kinase activity. J. Leukoc. Biol. 67:735–741. [DOI] [PubMed] [Google Scholar]
- 12.Stefanova, I., M.L. Corcoran, E.M. Horak, L.M. Wahl, J.B. Bolen, and I.D. Horak. 1993. Lipopolysaccharide induces activation of CD14-associated protein tyrosine kinase p53/56lyn. J. Biol. Chem. 268:20725–20728. [PubMed] [Google Scholar]
- 13.Beaty, C.D., T.L. Franklin, Y. Uehara, and C.B. Wilson. 1994. Lipopolysaccharide-induced cytokine production in human monocytes: role of tyrosine phosphorylation in transmembrane signal transduction. Eur. J. Immunol. 24:1278–1284. [DOI] [PubMed] [Google Scholar]
- 14.Crowley, M.T., S.L. Harmer, and A.L. DeFranco. 1996. Activation-induced association of a 145-kDa tyrosine-phosphorylated protein with Shc and Syk in B lymphocytes and macrophages. J. Biol. Chem. 271:1145–1152. [DOI] [PubMed] [Google Scholar]
- 15.Crowley, M.T., P.S. Costello, C.J. Fitzer-Attas, M. Turner, F. Meng, C. Lowell, V.L. Tybulewicz, and A.L. DeFranco. 1997. A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J. Exp. Med. 186:1027–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meng, F., and C.A. Lowell. 1997. Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. J. Exp. Med. 185:1661–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams, L.M., and A.J. Ridley. 2000. Lipopolysaccharide induces actin reorganization and tyrosine phosphorylation of Pyk2 and paxillin in monocytes and macrophages. J. Immunol. 164:2028–2036. [DOI] [PubMed] [Google Scholar]
- 18.Takesono, A., L.D. Finkelstein, and P.L. Schwartzberg. 2002. Beyond calcium: new signaling pathways for Tec family kinases. J. Cell Sci. 115:3039–3048. [DOI] [PubMed] [Google Scholar]
- 19.Smith, C.I., B. Baskin, P. Humire-Greiff, J.N. Zhou, P.G. Olsson, H.S. Maniar, P. Kjellen, J.D. Lambris, B. Christensson, L. Hammarstrom, et al. 1994. Expression of Bruton's agammaglobulinemia tyrosine kinase gene, BTK, is selectively down-regulated in T lymphocytes and plasma cells. J. Immunol. 152:557–565. [PubMed] [Google Scholar]
- 20.Desiderio, S. 1997. Role of Btk in B cell development and signaling. Curr. Opin. Immunol. 9:534–540. [DOI] [PubMed] [Google Scholar]
- 21.Huber, B., and F. Melchers. 1979. Frequencies of mitogen-reactive B cells in the mouse. Lipopolysaccharide-, lipoprotein- and Nocardia mitogen-reactive B cells in CBA/N mice. Eur. J. Immunol. 9:827–829. [DOI] [PubMed] [Google Scholar]
- 22.Scher, I. 1982. The CBA/N mouse strain: an experimental model illustrating the influence of the X-chromosome on immunity. Adv. Immunol. 33:1–71. [DOI] [PubMed] [Google Scholar]
- 23.Zhao, Y.X., A. Abdelnour, R. Holmdahl, and A. Tarkowski. 1995. Mice with the xid B cell defect are less susceptible to developing Staphylococcus aureus-induced arthritis. J. Immunol. 155:2067–2076. [PubMed] [Google Scholar]
- 24.Hata, D., Y. Kawakami, N. Inagaki, C.S. Lantz, T. Kitamura, W.N. Khan, M. Maeda-Yamamoto, T. Miura, W. Han, S.E. Hartman, et al. 1998. Involvement of Bruton's tyrosine kinase in FcepsilonRI-dependent mast cell degranulation and cytokine production. J. Exp. Med. 187:1235–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukhopadhyay, S., M. Mohanty, A. Mangla, A. George, V. Bal, S. Rath, and B. Ravindran. 2002. Macrophage effector functions controlled by Bruton's tyrosine kinase are more crucial than the cytokine balance of T cell responses for microfilarial clearance. J. Immunol. 168:2914–2921. [DOI] [PubMed] [Google Scholar]
- 26.Golding, B., H. Golding, P.G. Foiles, and J.I. Morton. 1983. CBA/N X-linked defect delays expression of the Y-linked accelerated autoimmune disease in BXSB mice. J. Immunol. 130:1043–1046. [PubMed] [Google Scholar]
- 27.Jansson, L., and R. Holmdahl. 1993. Genes on the X chromosome affect development of collagen-induced arthritis in mice. Clin. Exp. Immunol. 94:459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Webster, A.D.B. 2001. Humoral immunodeficiencies. Immunology and Allergy Clinics of North America. C.M. Roifman, editor. MD Consult (Elsevier), St. Louis, MO. 1–34.
- 29.He, T.C., S. Zhou, L.T. da Costa, J. Yu, K.W. Kinzler, and B. Vogelstein. 1998. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA. 95:2509–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Denys, A., I.A. Udalova, C. Smith, L.M. Williams, C.J. Ciesielski, J. Campbell, C. Andrews, D. Kwaitkowski, and B.M. Foxwell. 2002. Evidence for a dual mechanism for IL-10 suppression of TNF-alpha production that does not involve inhibition of p38 mitogen-activated protein kinase or NF-kappa B in primary human macrophages. J. Immunol. 168:4837–4845. [DOI] [PubMed] [Google Scholar]
- 31.Sanlioglu, S., C.M. Williams, L. Samavati, N.S. Butler, G. Wang, P.B. McCray, Jr., T.C. Ritchie, G.W. Hunninghake, E. Zandi, and J.F. Engelhardt. 2001. Lipopolysaccharide induces Rac1-dependent reactive oxygen species formation and coordinates tumor necrosis factor-alpha secretion through IKK regulation of NF-kappa B. J. Biol. Chem. 276:30188–30198. [DOI] [PubMed] [Google Scholar]
- 32.Mano, H., Y. Yamashita, K. Sato, Y. Yazaki, and H. Hirai. 1995. Tec protein-tyrosine kinase is involved in interleukin-3 signaling pathway. Blood. 85:343–350. [PubMed] [Google Scholar]
- 33.Vihinen, M., P.T. Mattsson, and C.I.E. Smith. 1997. BTK, the tyrosine kinase affected in X-linked agammaglobulinemia. Front. Biosci. 2:d27–d42. [DOI] [PubMed] [Google Scholar]
- 34.Cambronero, R., W.A. Sewell, M.E. North, A.D. Webster, and J. Farrant. 2000. Up-regulation of IL-12 in monocytes: a fundamental defect in common variable immunodeficiency. J. Immunol. 164:488–494. [DOI] [PubMed] [Google Scholar]
- 35.Yoza, B.K., J.Y. Hu, and C.E. McCall. 1996. Protein-tyrosine kinase activation is required for lipopolysaccharide induction of interleukin 1beta and NFkappaB activation, but not NFkappaB nuclear translocation. J. Biol. Chem. 271:18306–18309. [DOI] [PubMed] [Google Scholar]
- 36.Delude, R.L., M.J. Fenton, R. Savedra, Jr., P.Y. Perera, S.N. Vogel, R. Thieringer, and D.T. Golenbock. 1994. CD14-mediated translocation of nuclear factor-kappa B induced by lipopolysaccharide does not require tyrosine kinase activity. J. Biol. Chem. 269:22253–22260. [PubMed] [Google Scholar]
- 37.Rutault, K., C.A. Hazzalin, and L.C. Mahadevan. 2001. Combinations of ERK and p38 MAPK inhibitors ablate tumor necrosis factor-alpha (TNF-alpha) mRNA induction. Evidence for selective destabilization of TNF-alpha transcripts. J. Biol. Chem. 276:6666–6674. [DOI] [PubMed] [Google Scholar]
- 38.Brook, M., G. Sully, A.R. Clark, and J. Saklatvala. 2000. Regulation of tumour necrosis factor alpha mRNA stability by the mitogen-activated protein kinase p38 signalling cascade. FEBS Lett. 483:57–61. [DOI] [PubMed] [Google Scholar]
- 39.Tomlinson, M.G., T. Kurosaki, A.E. Berson, G.H. Fujii, J.A. Johnston, and J.B. Bolen. 1999. Reconstitution of Btk signaling by the atypical tec family tyrosine kinases Bmx and Txk. J. Biol. Chem. 274:13577–13585. [DOI] [PubMed] [Google Scholar]
- 40.Ellmeier, W., S. Jung, M.J. Sunshine, F. Hatam, Y. Xu, D. Baltimore, H. Mano, and D.R. Littman. 2000. Severe B cell deficiency in mice lacking the tec kinase family members Tec and Btk. J. Exp. Med. 192:1611–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dean, J.L., M. Brook, A.R. Clark, and J. Saklatvala. 1999. p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J. Biol. Chem. 274:264–269. [DOI] [PubMed] [Google Scholar]
- 42.Winzen, R., M. Kracht, B. Ritter, A. Wilhelm, C.Y. Chen, A.B. Shyu, M. Muller, M. Gaestel, K. Resch, and H. Holtmann. 1999. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 18:4969–4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang, S.W., J. Pawlowski, S.T. Wathen, S.D. Kinney, H.S. Lichenstein, and C.L. Manthey. 1999. Cytokine mRNA decay is accelerated by an inhibitor of p38-mitogen-activated protein kinase. Inflamm. Res. 48:533–538. [DOI] [PubMed] [Google Scholar]
- 44.Kabuyama, Y., M.K. Homma, T. Kurosaki, and Y. Homma. 2002. Early signaling events induced by 280-nm UV irradiation. Eur. J. Biochem. 269:664–670. [DOI] [PubMed] [Google Scholar]
- 45.Webster, A.D.B. 1978. Mycoplasma osteomyelitis and pneumonia associated with primary hypogammaglobulinaemia. Skeletal Radiolog. 3:131–132. [Google Scholar]
- 46.Smith, C.I.E., and O.N. Witte. 1998. Primary Immunodeficiency Diseases: A Molecular and Genetic Approach. H. Ochs, C.D.E. Smith, and J. Puck, editors. Oxford University Press, Oxford. 263–284.
- 47.Pillai, S., and S.T. Moran. 2002. Tec kinase pathways in lymphocyte development and transformation. Biochim. Biophys. Acta. 1602:162–167. [DOI] [PubMed] [Google Scholar]